Crystal Structure of 2-Ethylimidazole-1-sulfonyl Azide: A New Azidation Reagent


Abstract
:1. Introduction
2. Results and Discussion
2.1. Design and Synthesis
- a) the solid hydrochloride is clearly not suitable for use with lithiated species, a free base is desired
- b) imidazole-1-sulfonyl azide is liquid, but a solid is preferred for improved handling
- c) the acidic 2-position of the imidazole should be blocked to facilitate its use with strong bases
- d) solubility of the reagent in aprotic solvents is requested
2.2. Crystal Structures of 1 and 2
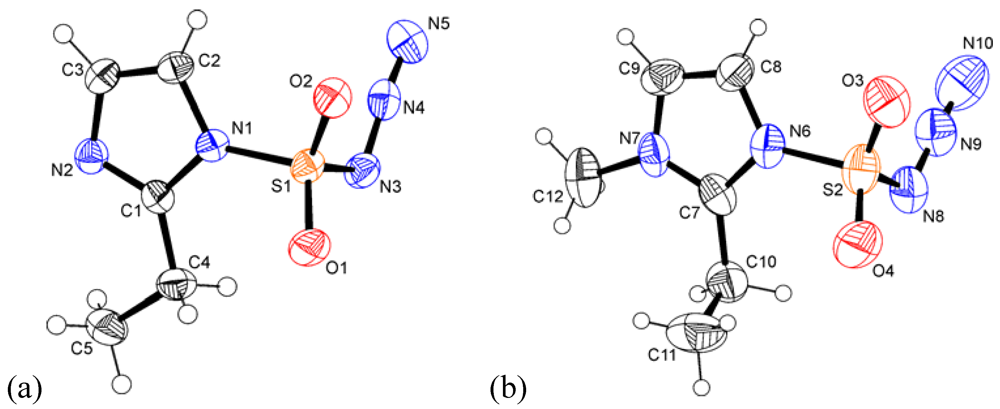



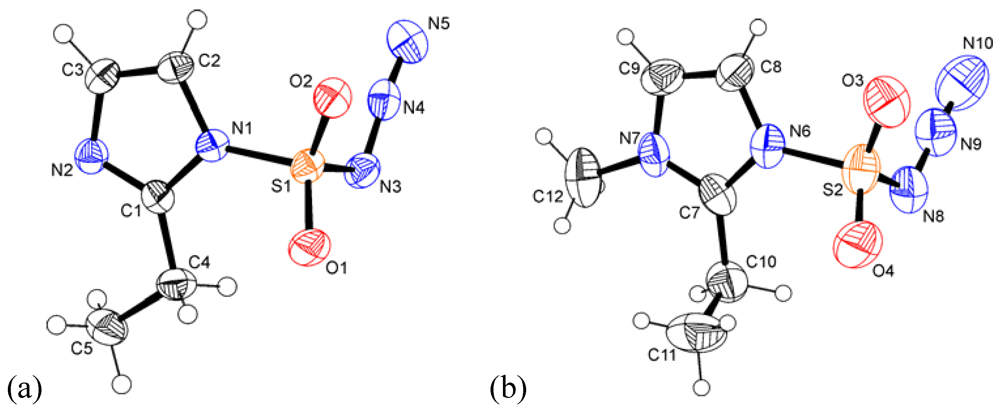{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction | H...F | C...F | C–H...F | Symmetry code (F) |
| C3–H...F | 2.399(8) | 3.14(1) | 135.3(6) | x, –1/2 + y, –1/2 + z |
| C4–H...F | 2.56(2) | 3.44(2) | 148.9(6) | –1/4 + x,–1/4 – y,–3/4 + z |
| C5–H...F | 2.57(2) | 3.26(2) | 128.3(8) | –1/4 + x,–1/4 – y,–3/4 + z |
| C6–H...F | 2.29(1) | 3.19(1) | 155.3(7) | 1/4 – x,–1/4 + y,–1/4 + z |
| C9–H...F | 2.47(2) | 3.29(2) | 146.0(7) | – |
| C10–H...F | 2.404(8) | 3.14(1) | 131.3(6) | 1/4 – x,–1/4 + y,3/4 + z |
| C11–H...F | 2.57(1) | 3.22(2) | 124.3(7) | 1/4 – x,–1/4 + y,–1/4 + z |
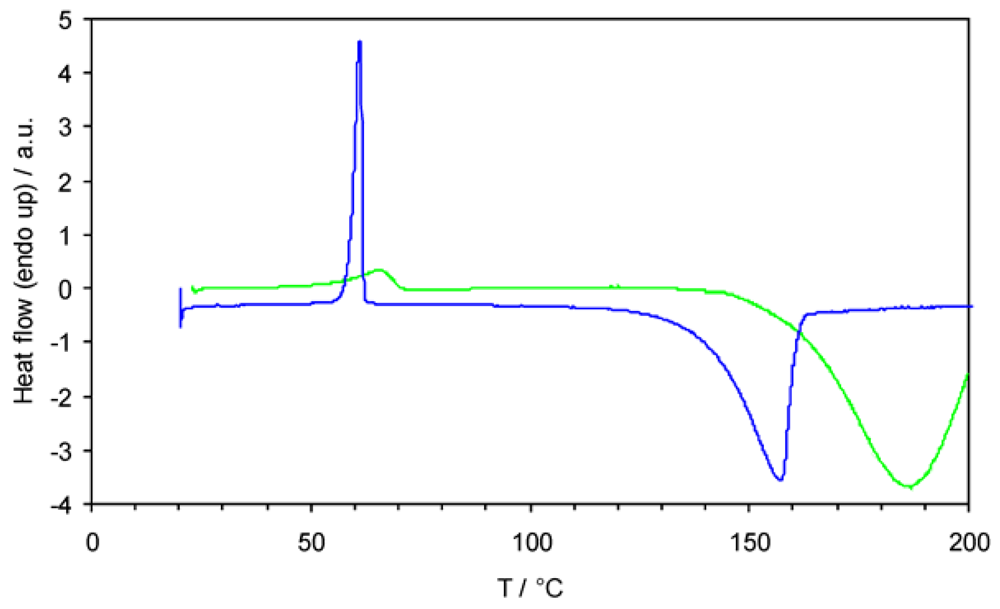
2.3. Reactivity and Stability

3. Experimental Section
3.1. Synthesis of 2-Ethylimidazole-1-sulfonyl Azide (1)
3.2. Synthesis of 1-Azidosulfonyl-3-alkyl-2-ethylimidazolium Tetrafluoroborates (2 and 3)
3.3. Synthesis of 1-Azidosulfonyl-3-alkyl-2-ethylimidazolium Triflimides (2a and 3a)
3.4. Azidation of 4-Methoxybiphenyl
3.5. X-ray Data Collection and Structure Refinement
4. Conclusions
Acknowledgments
References and Notes
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic Azides: An Exploding Diversity of a Unique Class of Compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar]
- Scriven, E.F.V.; Turnbull, K. Azides: Their Preparation and Synthetic Uses. Chem. Rev. 1988, 88, 297–368. [Google Scholar]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise HuisgenCycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Bock, V.D.; Hiemstra, H.; van Maarseveen, J.H. CuI-Catalyzed Alkyne-Azide“Click”Cycloadditions from a Mechanistic and Synthetic Perspective. Eur. J. Org. Chem. 2006, 51–68. [Google Scholar]
- Amblard, F.; Cho, J.H.; Schinazi, R.F. Cu(I)-Catalyzed HuisgenAzide-Alkyne 1,3-Dipolar Cycloaddition Reaction in Nucleoside, Nucleotide, and Oligonucleotide Chemistry. Chem. Rev. 2009, 109, 4207–4220. [Google Scholar] [CrossRef]
- Nyffeler, P.T.; Liang, C.-H.; Koeller, K.M.; Wong, C.-H. The Chemistry of Amine-AzideInterconversion: Catalytic Diazotransfer and RegioselectiveAzide Reduction. J. Am. Chem. Soc. 2002, 124, 10773–10778. [Google Scholar]
- Regitz, M. New Methods of Preparative Organic Chemistry. Transfer of Diazo Groups. Angew. Chem. Int. Ed. Engl. 1967, 6, 733–749. [Google Scholar] [CrossRef]
- Curphey, T.J. Preparation of p-toluenesulfonylazide. A cautionary note. Org. Prep. Proc. Int. 1981, 13, 112–115. [Google Scholar] [CrossRef]
- Cavender, C.J.; Shiner, V.J., Jr. Trifluoromethanesulfonyl Azide. Its Reaction with Alkyl Amines to Form Alkyl Azides. J. Org. Chem. 1972, 37, 3567–3569. [Google Scholar] [CrossRef]
- Liu, Q.; Tor, Y. Simple Conversion of Aromatic Amines into Azides. Org. Lett. 2003, 14, 2571–2572. [Google Scholar]
- Zaloom, J.; Roberts, D.C. Preparation of Azido Derivatives from Amino Acids and Peptides by Diazo Transfer. J. Org. Chem. 1981, 46, 5173–5176. [Google Scholar] [CrossRef]
- Taber, D.F.; Ruckle, R.E.; Hennessy, M.J. MesylAzide: A Superior Reagent for Diazo Transfer. J. Org. Chem. 1986, 51, 4077–4078. [Google Scholar]
- Besenyei, G.; Parkanyi, L.; Foch, I.; Simandi, L.I. Sterically controlled pathways in the reaction of 2,4,6-tris(isopropyl)benzenesulfonylazide and [Pd2Cl2(dppm)2]. Angew. Chem. Int. Ed. 2000, 39, 956–958. [Google Scholar] [CrossRef]
- Tuma, L.D. Identification of a safe diazotransfer reagent. Thermochim. Acta 1994, 243, 161–167. [Google Scholar] [CrossRef]
- Goddard-Borger, E.D.; Stick, R.V. An Efficient, Inexpensive, and Shelf-Stable Diazotransfer Reagent: Imidazole-1-sulfonyl Azide Hydrochloride. Org. Lett. 2007, 9, 3797–3800, Supporting Information.. [Google Scholar] [CrossRef]
- Benati, L.; Nanni, D.; Spagnolo, P. Reactions of Benzocyclic β-Keto Esters with SulfonylAzides. Further Insight into the Influence of Azide Structure and Solvent on the Reaction Course. J. Org. Chem. 1999, 64, 5132–5138. [Google Scholar] [CrossRef]
- Kumar, J.S.D.; Dupradeau, F.-Y.; Strouse, M.J.; Phelps, M.E.; Toyokuni, T. ElectrophilicAzidation for Stereoselective Synthesis of 2-Azido-2-deoxyaldono-1,5-lactones. J. Org. Chem. 2001, 66, 3220–3223. [Google Scholar]
- Batch, A.; Dodd, R.H. Ortho-Directed Metalation of 3-Carboxy-β-carbolines: Use of the SmI2-Cleavable 9-N-(N',N'-Dimethylsulfamoyl) Blocking Group for the Preparation of 9-N-Deprotected 4-Amino Derivatives via Azide Introduction or a Palladium-Catalyzed Cross-Coupling Reaction. J. Org. Chem. 1998, 63, 872–877. [Google Scholar] [CrossRef]
- Hanessian, S.; Bennani, Y.L. ElectrophilicAmination and Azidation of Chiralα-Alkyl Phosphonamides: Asymmetric Syntheses of α-Amino α-Alkyl Phosphonic Acids. Synthesis 1994, 1272–1274. [Google Scholar] [CrossRef]
- Denmark, S.E.; Chatani, N.; Pansare, S.V. Asymmetric electrophilicamination of chiral phosphorus-stabilized anions. Tetrahedron 1992, 48, 2191–2208. [Google Scholar]
- Ionic Liquids in Synthesis, 2nd; Wasserscheid, P.; Welton, T. (Eds.) Wiley-VCH: Weinheim, Germany, 2007.
- Laus, G.; Hummel, M.; Adamer, V.; Bentivoglio, G.; Wurst, K.; Griesser, U.; Schottenberger, H. Approaches to Azidometallocenes and Related Azidoarenes. In Proceedings of 7th Ferrocene Colloquium, Düsseldorf, Germany, 2009.
- Katritzky, A.R.; El Khatib, M.; Bol’shakov, O.; Khelashvili, L.; Steel, P.J. Benzotriazol-1-yl-sulfonyl Azide for Diazotransfer and Preparation of Azidoacylbenzotriazoles. J. Org. Chem. 2010, 75, 6532–6539. [Google Scholar]
- Besenyei, G.; Parkanyi, L.; Foch, I.; Simandi, L.I.; Kalman, A. Crystallographic characterisation of arenesulfonyl azides. Structural and kinetic effects induced by ortho- and para-substituents. J. Chem. Soc. Perkin Trans. 2000, 2, 1798–1802. [Google Scholar]
- Vicarel, M.L.; Norris, P.; Zeller, M. 4-Acetamidobenzenesulfonyl azide. Acta Crystallogr. 2006, E62, o1751–o1753. [Google Scholar]
- Basanagouda, M.; Nayak, S.K.; Row, T.N.G.; Kulkarni, M.V. 4-Azidomethyl-7-methyl-2-oxo-2H-chromene-6-sulfonyl azide. Acta Crystallogr. 2010, E66, o2780. [Google Scholar]
- Parkanyi, L.; Besenyei, G. Crystallographic characterisation of benzoylazides. A comparative study. J. Mol. Struct. 2004, 691, 97–106. [Google Scholar] [CrossRef]
- Herberhold, M.; Ayazi, A.; Milius, W.; Wrackmeyer, B. Silyl derivatives of ferrocene with pending indenyl or fluorenylsubstituents at silicon. J. Organomet. Chem. 2002, 656, 71–80. [Google Scholar]
- Trost, B.M.; Pearson, W.H. Azidomethyl Phenyl Sulfide. A Synthon for NH2+. J. Am. Chem. Soc. 1981, 103, 2483–2485. [Google Scholar]
- Walla, P.; Arion, V.B.; Brinker, U.H. Solvent- and Temperature-Tuned Orientation of FerrocenylAzide Inside β-Cyclodextrin. J. Org. Chem. 2006, 71, 3274–3277. [Google Scholar] [CrossRef]
- Burla, M.C.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Polidori, G. More power for direct methods: SIR2002. Z. Kristallogr. 2002, 217, 629–635. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Laus, G.; Adamer, V.; Hummel, M.; Kahlenberg, V.; Wurst, K.; Nerdinger, S.; Schottenberger, H. Crystal Structure of 2-Ethylimidazole-1-sulfonyl Azide: A New Azidation Reagent. Crystals 2012, 2, 118-126. https://doi.org/10.3390/cryst2010118
Laus G, Adamer V, Hummel M, Kahlenberg V, Wurst K, Nerdinger S, Schottenberger H. Crystal Structure of 2-Ethylimidazole-1-sulfonyl Azide: A New Azidation Reagent. Crystals. 2012; 2(1):118-126. https://doi.org/10.3390/cryst2010118
Chicago/Turabian StyleLaus, Gerhard, Verena Adamer, Michael Hummel, Volker Kahlenberg, Klaus Wurst, Sven Nerdinger, and Herwig Schottenberger. 2012. "Crystal Structure of 2-Ethylimidazole-1-sulfonyl Azide: A New Azidation Reagent" Crystals 2, no. 1: 118-126. https://doi.org/10.3390/cryst2010118
APA StyleLaus, G., Adamer, V., Hummel, M., Kahlenberg, V., Wurst, K., Nerdinger, S., & Schottenberger, H. (2012). Crystal Structure of 2-Ethylimidazole-1-sulfonyl Azide: A New Azidation Reagent. Crystals, 2(1), 118-126. https://doi.org/10.3390/cryst2010118