
Structural Properties and Dielectric Hysteresis of Molecular Organic Ferroelectric Grown from Different Solvents

 , ,
, ,
Abstract
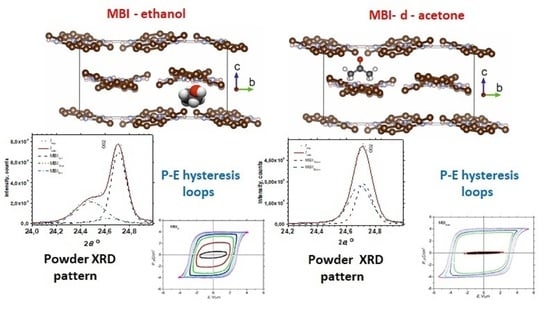
:
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Raman Spectroscopy
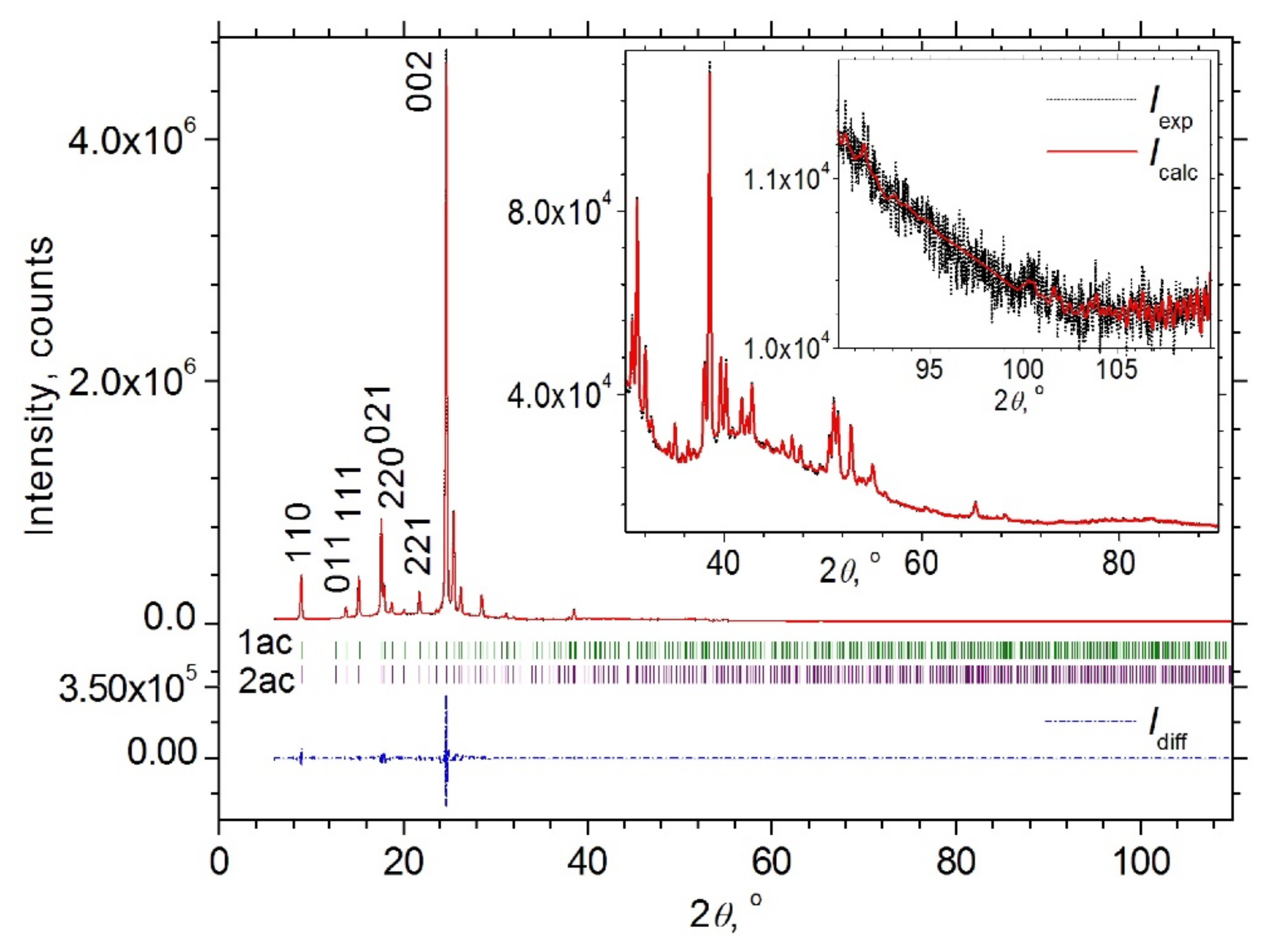
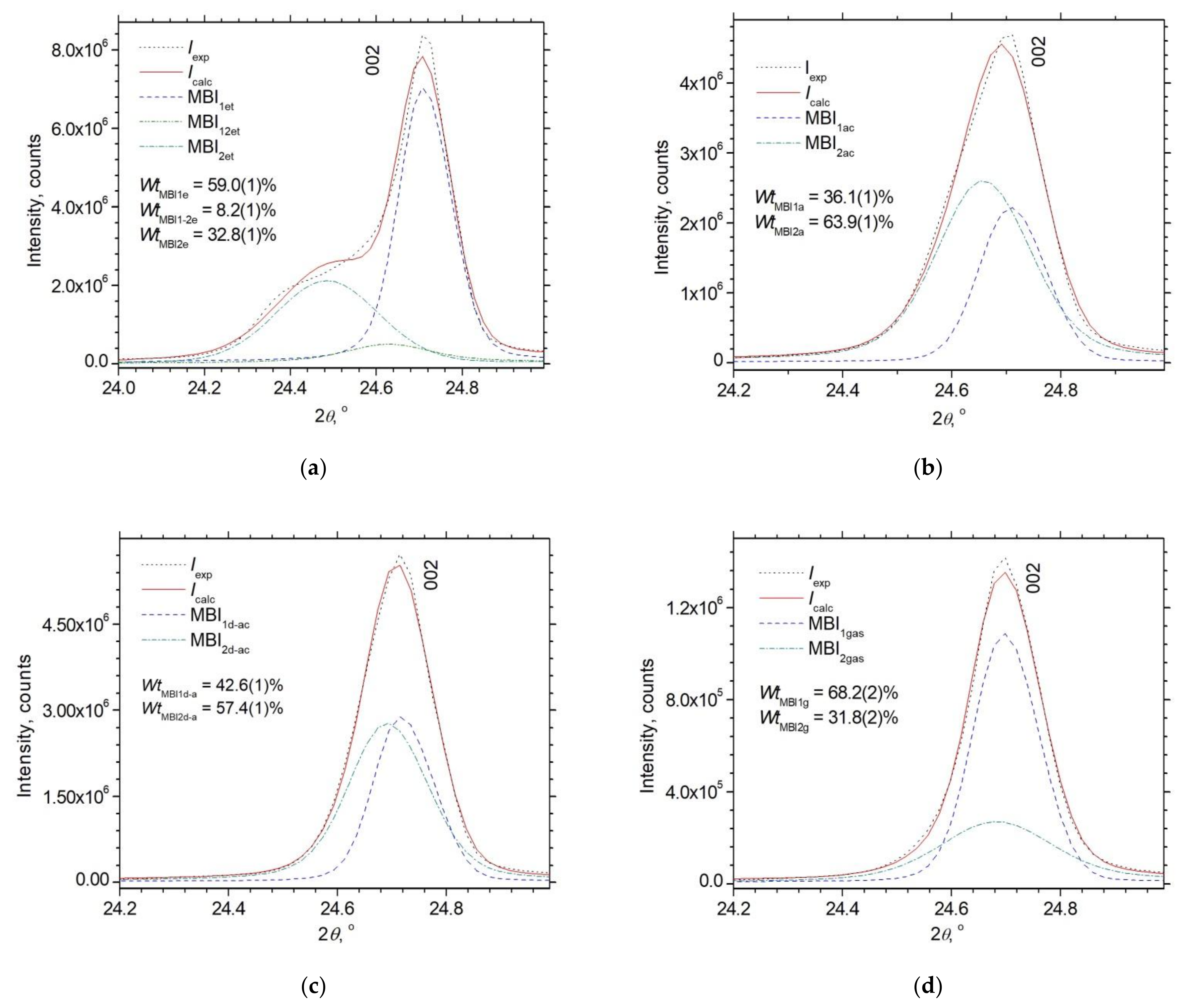
3.2. Powder XRD
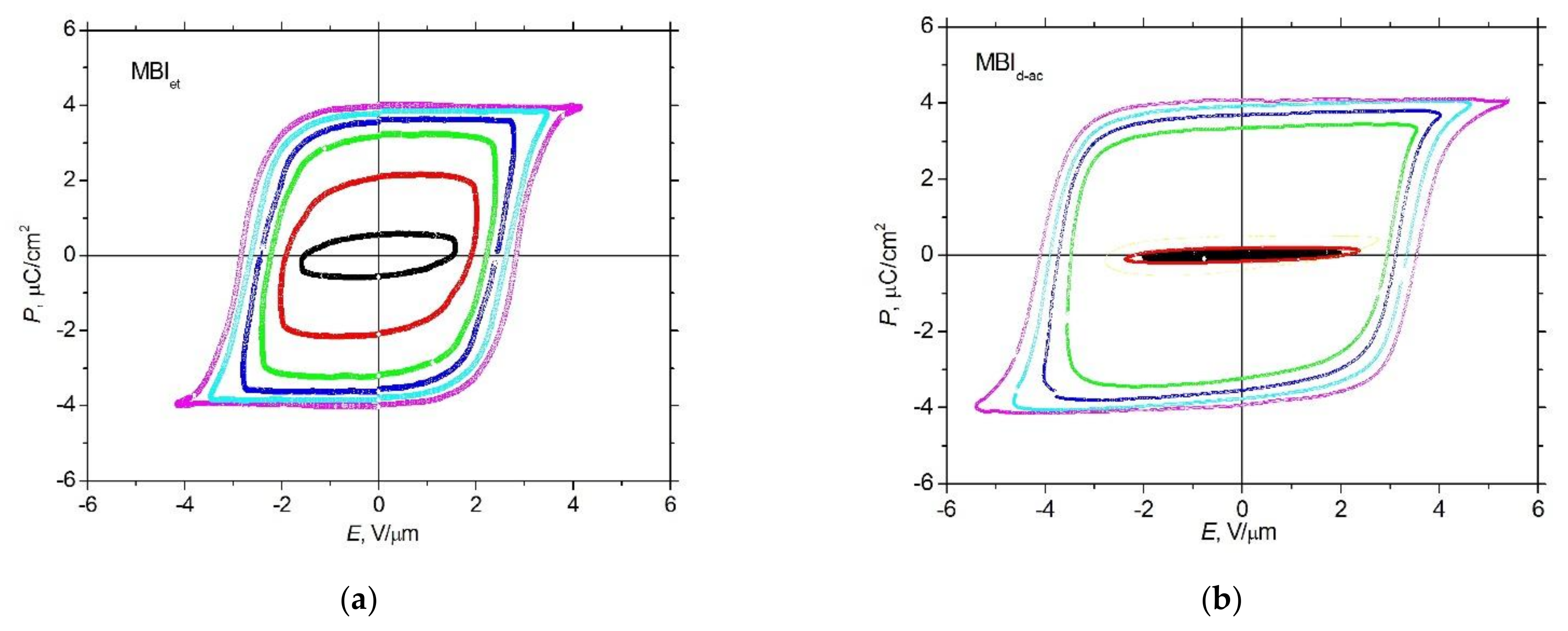
3.3. P-E Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Park, C.; Lee, K.; Koo, M.; Park, C. Soft Ferroelectrics Enabling High Performance Intelligent Photo Electronics. Adv. Mater. 2020, 2004999. [Google Scholar] [CrossRef]
- Horiuchi, S.; Ishibashi, S. Hydrogen-Bonded Small-Molecular Crystals Yielding Strong Ferroelectric and Antiferroelectric Polarizations. J. Phys. Soc. Jpn. 2020, 89, 051009. [Google Scholar] [CrossRef]
- Kawai, H. The Piezoelectricity of Poly (vinylidene Fluoride). Jpn. J. Appl. Phys. 1969, 8, 975–976. [Google Scholar] [CrossRef]
- Horiuchi, S.; Kobayashi, K.; Kumai, R.; Ishibashi, S. Proton tautomerism for strong polarization switching. Nat. Commun. 2017, 8, 14426. [Google Scholar] [CrossRef]
- Horiuchi, S.; Kagawa, F.; Hatahara, K.; Kobayashi, K.; Kumai, R.; Murakami, Y.; Tokura, Y. Above-room-temperature ferroelectricity and antiferroelectricity in benzimidazoles. Nat. Commun. 2012, 3, 1308. [Google Scholar] [CrossRef] [Green Version]
- Noda, Y.; Yamada, T.; Kobayashi, K.; Kumai, R.; Horiuchi, S.; Kagawa, F.; Hasegawa, T. Few-Volt Operation of Printed Organic Ferroelectric Capacitor. Adv. Mater. 2015, 27, 6475. [Google Scholar] [CrossRef]
- Balashova, E.V.; Krichevtsov, B.B.; Svinarev, F.B.; Zaitseva, N.V. Thin films of a new organic single-component ferroelectric 2-methylbenzimidazole. Sci. Tech. J. Inf. Technol. 2016, 16, 801–808. [Google Scholar] [CrossRef]
- Balashova, E.V.; Krichevtsov, B.B.; Svinarev, F.B.; Zaitseva, N.V.; Pankova, G.A. Structural and Dielectric Properties of Organic Ferroelectric 2-Methylbenzimidazole. J. Surf. Invest. X-ray Synchrotron Neutron Tech. 2018, 12, 233–239. [Google Scholar] [CrossRef]
- Balashova, E.V.; Svinarev, F.B.; Ankudinov, A.V.; Pankova, G.A.; Lityagin, G.A.; Kunkel, T.S.; Krichevtsov, B.B. Polarization switching, dielectric, structural and elastic properties of 2-Methylbenzimidazole crystals and films. Ferroelectrics 2019, 538, 74–82. [Google Scholar] [CrossRef]
- Svinarev, F.B.; Balashova, E.V.; Krichevtsov, B.B. Dielectric properties of self-assembled spherulite films of organic ferroelectric 2-methylbenzimidazole. Ferroelectrics 2019, 543, 167–174. [Google Scholar] [CrossRef]
- Yuan, Y. Growth and Characterization of Molecular Ferroelectric Thin Films. A Dissertation for the Degree of Dr. Ph., Lincoln, Nebraska. 2021. Available online: https://digitalcommons.unl.edu/mechengdiss/165 (accessed on 18 April 2021).
- Balashova, E.V.; Krichevtsov, B.B.; Kunkel, T.S.; Ankudinov, A.V. AFM Visualization of Ferroelastic and Feroelectric Domains in 2-Methylbenzimidazole C8H8N2 Crystals. J. Surf. Invest. X-ray Synchrotron Neutron Tech. 2021, 11, 58–61. (In Russian) [Google Scholar] [CrossRef]
- Kinoshita, Y.; Sotome, M.; Miyamoto, T.; Uemura, Y.; Arai, S.; Horiuchi, S.; Hasegawa, T.; Okamoto, H.; Kida, N. Observation of the Three-Dimensional Polarization Vector in Films of Organic Molecular Ferroelectrics Using Terahertz Radiation Emission. Phys. Rev. Appl. 2020, 14, 054002. [Google Scholar] [CrossRef]
- Uemura, Y.; Matsuoka, S.; Tsutsumi, J.; Horiuchi, S.; Arai, S.; Hasegawa, T. Birefrigent field-modulation imaging of transparent ferroelectrics. Phys. Rev. Appl. 2020, 14, 024060. [Google Scholar] [CrossRef]
- Griesser, U.J. The Importance of Solvates. In Polymorphism: In the Pharmaceutical Industry; Hilfiker, R., Ed.; Wiley-VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2006; Chapter 8; pp. 211–233. [Google Scholar] [CrossRef]
- Losev, E.A.; Mikhailenko, M.A.; Achkasov, A.F.; Boldyreva, E.V. The effect of carboxylic acids on glycine polymorphism, salt and co-crystal formation. A comparison of different crystallisation techniques. New J. Chem. 2013, 37, 1973–1981. [Google Scholar] [CrossRef]
- Lemanov, V.V.; Yarmarkin, V.K.; Egorov, V.M.; Pankova, G.A.; Zaitseva, N.V.; Markova, L.A. Phase transitions in crystals of protein amino acids with trapped drops of water solutions. Phys. Solid State 2012, 54, 346–349. [Google Scholar] [CrossRef]
- Klitou, P.; Pask, C.M.; Onoufriadi, L.; Rosbottom, I.; Simone, E. Solid-State Characterization and Role of Solvent Molecules on the Crystal Structure, Packing, and Physiochemical Properties of Different Quercetin Solvates. Cryst. Growth Des. 2020, 20, 6573–6584. [Google Scholar] [CrossRef]
- Balashova, E.V.; Svinarev, F.B.; Zolotarev, A.A.; Levin, A.A.; Brunkov, P.N.; Davydov, V.Y.; Smirnov, A.N.; Redkov, A.V.; Pankova, G.A.; Krichevtsov, B.B. Crystal structure, Raman spectroscopy and dielectric properties of new semi-organic crystals based on 2-methylbenzimidazole. Crystals 2019, 9, 573. [Google Scholar] [CrossRef] [Green Version]
- Rosbottom, I.; Ma, C.Y.; Turner, T.D.; O’Connell, R.A.; Loughrey, J.; Sadiq, G.; Davey, R.J.; Roberts, K.J. Influence of Solvent Composition on the Crystal Morphology and Structure of p-Aminobenzoic Acid Crystallized from Mixed Ethanol and Nitromethane Solutions. Cryst. Growth Des. 2017, 17, 4151–4161. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.R.; Oswald, I.D.H. Hidden Solvates and Transient Forms of Trimesic Acid. Crystals 2020, 10, 1098. [Google Scholar] [CrossRef]
- Jiang, M.; Ni, X.-W. Effects of solvents and impurity on crystallization kinetics and crystal properties in a reactive crystallization of paracetamol. J. Cryst. Growth 2019, 523, 125150. [Google Scholar] [CrossRef]
- Davey, R.J.; Mullin, J.W.; Whitting, M.J.L. Habit Modification of Succinic Acid Crystals Grown from Different Solvents. J. Cryst. Growth 1982, 58, 304–312. [Google Scholar] [CrossRef]
- Hod, I.; Mastai, Y.; Medina, D.D. Effect of solvents on the growth morphology of DL-alanine crystals. Cryst. Eng. Comm. 2011, 13, 502–509. [Google Scholar] [CrossRef]
- Tanaka, Y.; Matsuoka, M. Selection of Solvents for Organic Crystal Growth from Solution. J. Cryst. Growth 1990, 99, 1130–1133. [Google Scholar] [CrossRef]
- Chewle, S.; Emmerling, F.; Weber, M. Effect of Choice of Solvent on Crystallization Pathway of Paracetamol: An Experimental and Theoretical Case Study. Crystals 2020, 10, 1107. [Google Scholar] [CrossRef]
- Zieliński, W.; Katrusiak, A. Colossal Monotonic Response to Hydrostatic Pressure in Molecular Crystal Induced by a Chemical Modification. Cryst. Growth Des. 2014, 14, 4247–4253. [Google Scholar] [CrossRef]
- Levin, A.A.; Levichkova, M.; Hildebrandt, D.; Klisch, M.; Weiss, A.; Wynands, D.; Elschner, C.; Pfeiffer, M.; Leo, K.; Riede, M. Effect of film thickness, type of buffer layer, and substrate temperature on the morphology of dicyanovinyl-substituted sexithiophene films by X-ray methods. Thin Solid Films 2012, 520, 2479–2487. [Google Scholar] [CrossRef]
- Maunders, C.; Etheridge, J.; Wright, N.; Whitfield, H.J. Structure and microstructure of hexagonal Ba3Ti2RuO9 by electron diffraction and microscopy. Acta. Cryst. B 2005, 61, 154–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diffrac Suite Eva Version 5.1.0.5; Brucker AXS: Karlsruhe, Germany, 2019.
- NSRDS. Tables of molecular vibrational frequencies consolidated Volume 1. Nat. Stand. Ref. Data Ser. Nat. Bur. Stand. 1972, 39, 164. [Google Scholar]
- Güllüoglu, M.T.; Özduran, M.; Kurt, M.; Kalaichelvan, S.; Sundaraganesan, N. Molecular structure and vibrational spectra of 2- and 5-methylbenzimidazole molecules by density functional theory. Spectrochim. Acta A 2010, 76, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Obodovskaya, A.E.; Starikova, Z.A.; Belous, S.N.; Pokrovskaya, I.E. Crystal and molecular structure of 2-methylbenzimidazole. J. Struct. Chem. 1991, 32, 421–422. [Google Scholar] [CrossRef]
- Horiuchi, S.; Tsutsumi, J.; Kobayashi, K.; Kumai, R.; Ishibashi, S. Piezoelectricity of Strongly Polarized Ferroelectrics in Prototropic Organic Crystals. J. Mater. Chem. C 2018, 6, 4714–4719. [Google Scholar] [CrossRef]
- Kraus, W.; Nolze, G. Powder Cell—A program for the representation and manipulation of crystal structures and calculation of the resulting X-ray powder patterns. J. Appl. Cryst. 1996, 29, 301–303. [Google Scholar] [CrossRef]
- Langford, J.I.; Cernik, R.J.; Louer, D. The Breadth and Shape of Instrumental Line Profiles in High-Resolution Powder Diffraction. J. Appl. Phys. 1991, 24, 913–919. [Google Scholar] [CrossRef]
- Terlan, B.; Levin, A.A.; Börrnert, F.; Simon, F.; Oschatz, M.; Schmidt, M.; Cardoso-Gil, R.; Lorenz, T.; Baburin, I.A.; Joswig, J.-O.; et al. Effect of Surface Properties on the Microstructure, Thermal, and Colloidal Stability of VB2 Nanoparticles. Chem. Mater. 2015, 27, 5106–5115. [Google Scholar] [CrossRef]
- Terlan, B.; Levin, A.A.; Börrnert, F.; Zeisner, J.; Kataev, V.; Schmidt, M.; Eychmüller, A. A Size-Dependent Analysis of the Structural, Surface, Colloidal, and Thermal Properties of Ti1−xB2 (x = 0.03–0.08) Nanoparticles. Eur. J. Inorg. Chem. 2016, 6, 3460–3468. [Google Scholar] [CrossRef]
- Rehani, B.R.; Joshi, P.B.; Lad, K.N.; Pratap, A. Crystallite size estimation of elemental and composite silver nano-powders using XRD principles. Indian J. Pure Appl. Phys. 2006, 44, 157–161. [Google Scholar]
- Scherrer, P. Bestimmung der Grösse und der inneren Struktur von Kolloidteilchen mittels Röntgenstrahlen. Nachr. Königl. Ges. Wiss. Göttingen. 1918, 26, 98–100. [Google Scholar]
- Stokes, A.R.; Wilson, A.J.C. The diffraction of X-rays by distorted crystal aggregates. Proc. Phys. Soc. Lond. 1944, 56, 174–181. [Google Scholar] [CrossRef]
- Le Bail, A.; Duroy, H.; Fourquet, J.L. Ab-initio structure determination of LiSbWO6 by X-ray powder diffraction. Mat. Res. Bull. 1988, 23, 447–452. [Google Scholar] [CrossRef]
- TOPAS Version 5; Technical reference; Brucker AXS: Karlsruhe, Germany, 2014.
- Coelho, A.A. TOPAS and TOPAS-Academic: An optimization program integrating computer algebra and crystallographic objects written in C++. J. Appl. Cryst. 2018, 51, 210–218. [Google Scholar] [CrossRef] [Green Version]
- Cheary, R.W.; Coelho, A.A. A fundamental parameters approach to X-ray line-profile fitting. J. Appl. Cryst. 1992, 25, 109–121. [Google Scholar] [CrossRef]
- Berger, H. Study of the K alpha emission spectrum of copper. X-ray Spectrom. 1986, 15, 241–243. [Google Scholar] [CrossRef]
- Hill, R.J.; Fischer, R.X. Profile Agreement Indices in Rietveld and Pattern-Fitting Analysis. J. Appl. Cryst. 1990, 23, 462–468. [Google Scholar] [CrossRef]
- Levin, A.A.; Filatov, S.K.; Paufler, P.; Bubnova, R.S.; Krzhizhanovskaya, M.; Meyer, D.C. Temperature-dependent evolution of RbBSi2O6 glass into crystalline Rb-boroleucite according to X-ray diffraction data. Z. Kristallogr. Cryst. Mater. 2013, 228, 259–270. [Google Scholar] [CrossRef]
- Bérar, J.-F.; Lelann, P.J. ESD’s and estimated probable error obtained in Rietveld refinements with local correlations. J. Appl. Cryst. 1991, 2, 1–5. [Google Scholar] [CrossRef]
- Langford, J.I.; Delhez, A.R.; de Keijser, T.H.; Mittemeijer, E.J. Profile Analysis for Microcrystalline Properties by the Fourier and Other Method. Aust. J. Phys. 1988, 41, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Balzar, D. Voigt-function model in diffraction line-broadening analysis. In Defect and Microstructure Analysis by Diffraction; Snyder, R.L., Fiala, J., Bunge, H.J., Eds.; IUCr, Oxford Uni, Press: Oxford, UK, 1999; pp. 94–126. [Google Scholar]
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Cryst. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Van der Bruggen, B.; Schaep, J.; Wilms, D.; Vandecasteele, C. Influence of molecular size, polarity and charge on the retention of organic molecules by nanofiltration. J. Membr. Sci. 1999, 156, 29–41. [Google Scholar] [CrossRef]
- Nadykto, A.B.; Yu, F. Uptake of neutral polar vapor molecules by charged clusters/particles: Enhancement due to dipole-charge interaction. J. Geophys. Res. 2003, 108, 4717. [Google Scholar] [CrossRef] [Green Version]
- Table of experimental nuclear ground state charge radii: An update. At. Data Nucl. Data Tables 2013, 99, 69–95. [CrossRef]
- Xiangli, F.; Chen, Y.; Jin, W.; Xu, N. Polydimethylsiloxane (PDMS)/Ceramic Composite Membrane with High Flux for Pervaporation of Ethanol-Water Mixtures. Ind. Eng. Chem. Res. 2007, 46, 2224–2230. [Google Scholar] [CrossRef]
- Kolmogorov, A.N. K statisticheskoi teorii kristallizacii metallov [To the statistical theory of the crystallization of metals]. Izv. Akad. Nauk SSSR Ser. Mat. 1937, 1, 355–359. (In Russian) [Google Scholar]
- Avrami, M. Kinetics of Phase Change. J. Chem. Phys. 1939, 7, 1103. [Google Scholar] [CrossRef]
- Ishibashi, Y.; Takagi, Y. Note on ferroelectric domain switching. J. Phys. Soc. Jpn. 1971, 31, 506–510. [Google Scholar] [CrossRef]
- Merz, W.J. Domain formation and domain wall motions in ferroelectric BaTiO3 single crystals. Phys. Rev. 1954, 95, 690–698. [Google Scholar] [CrossRef]
- Svinarev, F.B.; Balashova, E.V.; Pankova, G.A.; Krichevtsov, B.B. Polarization switching in single crystals and films of 2-methylbenzimidazole. J. Phys. Conf. Ser. 2018, 1038, 012117. [Google Scholar] [CrossRef] [Green Version]
- Janta, J. The influence of the shape of domains on the ferroelectric hysteresis loop. Ferroelectrics 1971, 2, 299–302. [Google Scholar] [CrossRef]
- Nakatani, N. The dependence of the coercive field of tri-glycine sulfate on frequency, amplitude and temperature. J. Phys. Soc. Jpn. 1972, 32, 1556–1559. [Google Scholar] [CrossRef]
- Dontsova, L.I.; Popov, E.S.; Shil’nikov, A.V.; Bulatova, L.G.; Tikhomirova, N.A.; Shuvalov, L.A. Influence of temperature changes on the domain structure and surface energy density of domain walls in triglycine sulfate crystals. Kristallografiya 1981, 26, 758. (In Russian) [Google Scholar]
- Tikhomirova, N.A.; Pikin, S.A.; Shuvalov, L.A.; Dontsova, L.I.; Popov, E.S.; Shilnikov, A.V.; Balatova, L.G. Visualization of static and dynamics of domain structure in triglycine sulfate by liquid crystals. Ferroelectrics 1980, 29, 145. [Google Scholar] [CrossRef]
- Savage, A.; Miller, R.C. Temperature Dependence of the Velocity of Sidewise 180° Domain-Wall Motion in BaTiO3. J. Appl. Phys. 1960, 31, 1546–1549. [Google Scholar] [CrossRef]
- Hatano, J.; Suda, F.; Futama, H. Domain-Wall Orientations and Domain Shapes of Ferroelectric TGS and TGSe Crystals. Ferroelectrics 1978, 20, 265–266. [Google Scholar] [CrossRef]
- Tybell, T.; Paruch, P.; Giamarchi, T.; Triscone, J.-M. Domain Wall Creep in Epitaxial Ferroelectric PbZr0.2Ti0.8O3 Thin Films. Phys. Rev. Lett. 2002, 89, 097601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.J.; Juo, D.-M.; You, L.; Wang, J.; Chen, Y.C.; Chu, Y.H.; Wu, T. Universal Ferroelectric Switching Dynamics of Vinylidene Fluoride-trifluoroethylene Copolymer Films. Sci. Rep. 2013, 4, 4772. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.C.; Weinreich, G. Mechanism for the sidewise motion of 180° domain walls in barium titanate. Phys Rev. 1960, 117, 1460–1466. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | MBIet | MBIac | MBId-ac | Assignment [19,32] |
|---|---|---|---|---|
| 1 | 3065m, 3075m | 3065m, 3077m | 3066s, 3077s | νCH |
| 2 | 2968m | 2970m | 2970m | MνCH |
| 3 | 2919m, 2929m | 2919m, 2929m | 2921m | MνCH |
| 4 | 1589m | 1591m | 1591s | νCC |
| 5 | 1545s | 1546s | 1547s | νCN (double bond) + νCC |
| 6 | 1472w, 1488m | 1483m, 1493m | 1483w, 1491w | MδCH2 + ΓCCCN + δCCH |
| 7 | 1438m, 1452s | 1440w, 1453s | 1440m, 1454m | δCCH + νCC |
| 8 | 1418m, 1425w | 1420m, 1425m | 1420m, 1424m | δCNH + νCN + νCC + δCCH |
| 10 | 1388m | 1389w | 1390w | MδCH2 + δCCH |
| 11 | 1363m | 1365m | 1364m | νCC + νCN + MδCH2 |
| 12 | 1322m | 1322m | 1323m | δCCH + νCN |
| 13 | 1272s | 1274s, 1279sh | 1274s, 1279sh | νCN + νCC + δCCH |
| 14 | 1222m | 1223m | 1223m | δCCH + νCN + νCC |
| 15 | 1155m | 1155m | 1155m | δCCH |
| 16 | 1113m, 1120m | 1113m | 1113m | δCCH + νCC |
| 17 | 1044m | 1045m | 1046s | δCCH + ΓNCCH |
| 18 | 1030s | 1031s | 1031s | νCC + νCN |
| 19 | 1004s | 1006s | 1005s | νCC + δCCH |
| 20 | 960w | 963w | 962w | ΓHCCH + ΓCCCH |
| 21 | 921w | 921w | ΓHCCH + ΓCCCH | |
| 22 | 893m | 894w | 894w | δCCC + δCCH |
| 23 | 868m | 872w | 871w | ΓCCCH +ΓHCCN + ΓHCCH+ ΓCCCH |
| 24 | 839s | 840w | 841s | νCC |
| 25 | 764w | 765w | ΓCCCH + ΓCCCC | |
| 26 | 741m | 742m | 742m | ΓCCCH + ΓHCCN |
| 27 | 677s | 678s | 677s | νCC + δCCC+ δCCN |
| 28 | 657m | 660m | 659s | ΓCNCN + ΓHCCN + ΓCCCN + ΓHNCN |
| 29 | 623w, 628m | 623w, 628w | 623m, 628m | δCCN + δCCC + νCC |
| 30 | 582s | 584m | 584m | ΓCCCC + ΓCCCN + ΓCCCH |
| 31 | 499s | 501s | 501s | δCCC + νCC + νCN |
| 32 | 483m | 485m | 485m | δCCN + δCCC + νCC |
| 33 | 435m | 436m | 436m | ΓCCCC + ΓCCCH + ΓCCNH + ΓCCNC |
| 34 | 329w, 334m | 330m, 334m | 330w, 335w | ΓCCCN + ΓCCNH |
| 35 | 274m | 283w | 277w,281w | δCCN |
| 36 | 123vs | 123vs | 123vs | ΓCCCN + ΓHCCN, lattice modes? |
| Program | Celsiz | SizeCr | ||
|---|---|---|---|---|
| WHP | SSP | |||
| Phase | a, Å c, Å | Vcell, Å3 | D, nm εs, % | D, nm εs, % |
| MBI from [33] (CCDC code 1199885, Tmeas = 295 K) | ||||
| MBI | 13.950(9) 7.192(3) | 1399.6(1.4) | - | - |
| MBI grown in ethanol (MBIet), 3 phases | ||||
| MBI1et | 13.958(3) 7.202(2) | 1403.1(6) | 86(23) 0 | 96(10) 0.10(3) |
| MBI2et | 14.056(10) 7.252(5) | 1432.8(1.7) | 44(12) 0 | 44(12) 0 |
| MBI3et | 14.002(6) 7.223(3) | 1416.1(1.0) | 53(11) 0 | 68(7) 0.17(4) |
| MBI grown in acetone (MBIac), 2 phases | ||||
| MBI1ac | 13.939(4) 7.198(3) | 1398.5(8) | 73(13) 0 | 84(2) 0.08(1) |
| MBI2ac | 13.961(5) 7.209(4) | 1405.1(1.1) | 57(5) 0 | 57(5) 0 |
| MBI grown in d-acetone (MBId-ac), 2 phases | ||||
| MBI1d-ac | 13.963(2) 7.201(1) | 1403.9(3) | 103(31) 0 | 107(8) 0.06(3) |
| MBI2d-ac | 13.975(2) 7.208(1) | 1407.7(5) | 79(22) 0 | 79(22) 0 |
| MBI grown from gas (MBIgas), 2 phases | ||||
| MBI1gas | 13.950(4) 7.199(3) | 1401.1(8) | 61(16) 0 | 79(5) 0.12(2) |
| MBI2gas | 13.973(5) 7.211(3) | 1408.1(9) | 56(12) 0 | 62(2) 0.09(1) |
| TOPAS, LB Method | ||||||
|---|---|---|---|---|---|---|
| Phase | Wt, wt.% Vcell, Å3 | a, Å c, Å | Lvol−FWHM, nm e, % a | Rwp, % b Rp, % | cRwp, % b cRp, % | RB, % b |
| MBI from Ref. [33] (CCDC code 1199885, Tmeas = 295 K) | ||||||
| MBI | 100 1399.6(1.4) | 13.950(9) 7.192(3) | - - | 5.2 c | - - | 5.9 c |
| MBI grown in ethanol (MBIet), 3 phases, me.s.d.= 2.964 d | ||||||
| MBI1et | 59.0(1) 1399.8(1) | 13.949(1) 7.1944(2) | 112.3(9) 0.055(1) | 6.36 4.49 | 7.52 5.63 | 0.25 |
| MBI2et | 32.8(1) 1437.2(3) | 14.070(2) 7.2601(4) | 33.0(2) 0.003 (2) | 0.25 | ||
| MBI3et | 8.2(1) 1415.1(3) | 14.001(2) 7.2187(9) | 68(2) 0.111(4) | 0.27 | ||
| MBI grown in acetone (MBIac), 2 phases, me.s.d.= 3.637 d | ||||||
| MBI1ac | 36.1(1) 1399.3(2) | 13.943(1) 7.1980(3) | 90(2) 0.035(5) | 4.29 2.93 | 7.20 6.53 | 0.26 |
| MBI2ac | 63.9(1) 1407.9(1) | 13.971(1) 7.2132(2) | 50.2(4) 0.000(3) | 0.23 | ||
| MBI grown in d-acetone (MBIdac), 2 phases, me.s.d.= 2.911 d | ||||||
| MBI1d-ac | 42.6(1) 1403.4(1) | 13.9600(5) 7.2013(2) | 113.1(7) 0.027(5) | 5.81 3.36 | 9.69 7.61 | 0.48 |
| MBI2d-ac | 57.4(1) 1409.3(3) | 13.981(2) 7.2096(2) | 63.7(4) 0.000(9) | 0.16 | ||
| MBI grown from gas, 2 phases (MBIgas), me.s.d.= 4.000 d | ||||||
| MBI1gas | 68.2(2) 1401.4(3) | 13.950(2) 7.2017(2) | 86(2) 0.0(3) | 6.36 3.95 | 8.43 5.85 | 0.34 |
| MBI2gas | 31.8(2) 1405.7(2) | 13.966(1) 7.2068(4) | 51.9(7) 0.09(1) | 0.38 | ||
| Sample | Phase1 | Phase2 | ΔVcell/Vcell_Phase1 (%) |
|---|---|---|---|
| MBIgas | MBI1gas | MBI2gas | 0.31(3) |
| MBIdac | MBI1d-ac | MBI2d-ac | 0.42(2) |
| MBIac | MBI1ac | MBI2ac | 0.61(1) |
| MBIet | MBI1et | MBI2et | 2.67(2) |
| MBIet | MBI1et | MBI3et | 1.09(2) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balashova, E.; Levin, A.A.; Fokin, A.; Redkov, A.; Krichevtsov, B. Structural Properties and Dielectric Hysteresis of Molecular Organic Ferroelectric Grown from Different Solvents. Crystals 2021, 11, 1278. https://doi.org/10.3390/cryst11111278
Balashova E, Levin AA, Fokin A, Redkov A, Krichevtsov B. Structural Properties and Dielectric Hysteresis of Molecular Organic Ferroelectric Grown from Different Solvents. Crystals. 2021; 11(11):1278. https://doi.org/10.3390/cryst11111278
Chicago/Turabian StyleBalashova, Elena, Aleksandr A. Levin, Alexander Fokin, Alexey Redkov, and Boris Krichevtsov. 2021. "Structural Properties and Dielectric Hysteresis of Molecular Organic Ferroelectric Grown from Different Solvents" Crystals 11, no. 11: 1278. https://doi.org/10.3390/cryst11111278