Enzyme Immobilization for Solid-Phase Catalysis


New England Biolabs, Inc., Ipswich, MA 01938, USA
*
Author to whom correspondence should be addressed.
Catalysts 2019, 9(9), 732; https://doi.org/10.3390/catal9090732
Submission received: 29 July 2019
/
Revised: 22 August 2019
/
Accepted: 24 August 2019
/
Published: 29 August 2019
(This article belongs to the Special Issue Immobilization of Enzymes)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The covalent immobilization of an enzyme to a solid support can broaden its applicability in various workflows. Immobilized enzymes facilitate catalyst re-use, adaptability to automation or high-throughput applications and removal of the enzyme without heat inactivation or reaction purification. In this report, we demonstrate a step-by-step procedure to carry out the bio-orthogonal immobilization of DNA modifying enzymes employing the self-labelling activity of the SNAP-tag to covalently conjugate the enzyme of interest to the solid support. We also demonstrate how modifying the surface functionality of the support can improve the activity of the immobilized enzyme. Finally, the utility of immobilized DNA-modifying enzymes is depicted through sequential processing of genomic DNA libraries for Illumina next-generation sequencing (NGS), resulting in improved read coverage across AT-rich sequences.
1. Introduction
Enzymes are macromolecular biocatalysts, displaying high levels of stereo-, chemo- and regioselectivity in a variety of chemical or biochemical reactions under mild conditions. Enzymes are routinely leveraged to enable unique applications throughout research, industrial production and molecular diagnostics as they are known to accelerate reactions by >1017 fold [1]. Enzyme immobilization has been an area of great interest since it was first reported in 1916 [2], with many reviews available [3,4,5,6,7,8,9,10,11]. Immobilization is achieved by anchoring enzymes to or within solid supports in order to facilitate efficient separation from the reaction media in heterogeneous systems and increase the reusability of enzymes. In addition, immobilization of enzymes on the support materials usually leads to better enzymatic stability under both storage and operational conditions [7]. Generation of an immobilized enzyme requires a thorough understanding of the biochemical/physical properties of the enzyme (i.e., homogeneity, availability, stability, conformational flexibility, molecular weight, size, surface charge, pH and temperature profile, and specific activity) and the support materials (available functional groups, chemical and physical properties) so that an appropriate immobilization process can be tailored to maximize the specific enzyme activity and stability throughout the intended application.
Generating a standard methodology that is universally amenable to the immobilization of any enzyme is not possible; and finding the optimal strategy often requires iterative improvements through trial and error. The most frequently employed immobilization techniques can be described by one of the following strategies: non-covalent adsorption and deposition, immobilization via ionic interactions, covalent attachment, cross-linking of an enzyme, or entrapment within a solid support via a polymer gel lattice [3,7,12,13,14,15]. For simplicity, this article will broadly categorize methodologies as either covalent or non-covalent immobilization; the focus herein is on covalent immobilization. Covalently immobilized enzymes are typically considered to be superior to non-covalently immobilized enzymes as the potential for enzyme leaching from the surface is minimized, thereby avoiding protein contamination of the product.
The reactions employed to effectively immobilize the enzyme depend on the chemical and physical properties of the support, as well as the environment in which it is applied [16]. In this regard, we believe it is advantageous to utilize stable, pre-existing solids as core support materials, such as agarose, chitin, silicates and synthetic (co-)polymer encapsulated magnetic beads because of their known physical properties, chemical functionality and compatibility within molecular biology workflows (i.e., immunoprecipitation, affinity purification, or pull-down reactions).
Advancements in chemical/molecular biology and material science have provided the means to easily produce large amounts of recombinant protein and enable unique strategies for enzyme immobilization. Researchers can easily manipulate the gene of interest, clone it in optimized expression vectors, transform them into the host of choice, induce protein expression and then, the protein is ready for purification, characterization and immobilization.
Traditionally, covalent immobilization was performed through amino acids containing an ionizable side chain (aspartic acid, glutamic acid, lysine, cysteine, histidine, tyrosine and arginine) or the N-terminal α-amine. Such covalent immobilization strategies require tedious experimentation and analysis for selection of the appropriate reaction conditions, as loss of enzymatic activity can result from sub-optimal orientation of the enzyme active site (steric hindrance/blocking of substrate accessibility/product release), reduced conformational flexibility of the enzyme, or the resulting chemical modifications imparted on the amino acid constituents, prosthetic groups/cofactors of the enzyme [17,18]. Recently, advancements in site-specific protein for characterization and immobilization of enzymes have been driven by the development of bio-orthogonal reactions that proceed under physiological conditions between chemical groups that are absent in, and do not cross-react with, endogenous functionalities in proteins [19,20].
These new site-specific protein immobilization approaches by taking advantages of bio-orthogonal reactions, enable better control of both the site and level of enzyme modification (i.e., one can append bio-orthogonal reactive handles to the enzyme as opposed to leveraging the native amino acid sequence) as well as direct the enzyme immobilization to preserve the optimal orientation of the active site, although some disadvantages such as alteration of enzyme 3D structure and complicated preparation process were also reported [21]. Introducing these bio-orthogonal groups into proteins typically relies on the cellular incorporation of unnatural amino acids for which the site-specific modification can be performed [22]. An alternative approach for site specific enzyme immobilization relies on the expression of recombinant enzyme as a fusion protein with the SNAP-tag. The SNAP-tag is a small protein (20 kDa) based on human O6-alkylguanine-DNA-alkyltransferase (hAGT). During the self-labelling reaction of the SNAP-tag with its substrate, O6-benzylguanine (BG), a stable thioether bond is formed between the reactive cysteine of the tag and the label/support with high specificity and defined stoichiometry [23,24]. Therefore, use of a fusion partner immobilization strategy results in each enzyme molecule carrying only a single conjugation site. In this regard, we have succeeded in immobilizing several DNA modifying enzymes on BG-functionalized magnetic beads by taking advantage of self-labeling SNAP-tag as a fusion partner of the relevant enzymes [25]. These immobilized enzymes have been employed in construction of human genomic libraries by performing sequential manipulation of DNA fragments, resulting in improved sequence coverage and reduced GC-bias in Illumina next-generation sequencing [26]. Employing immobilized enzymes throughout library construction removes the requirement for high temperature treatment and permits easy separation of the end-polished library DNA from the enzymes linked to magnetic beads at ambient temperature.
This article describes a methodology to express, purify and site-specifically immobilize (through bio-orthogonal reactivity) a fusion protein of interest onto various microbead supports. Attempts to optimize the support surfaces, to enable the application of the immobilized enzymes for next-generation sequencing are also reported. We dedicate the first part of this article to outlining the basic steps to generate and validate site-specific immobilized SNAP-tagged fusion proteins, using recombinant SNAP-tagged T4 DNA ligase as a model. Then, we show that the support material (i.e., agarose, chitin, magnetic and Si-magnetic chitin beads) is of tremendous influence on the performance of an immobilized enzyme by performing gel electrophoresis for its DNA ligation evaluation. Different strategies to modify the bead surface such as using different concentrations of PEG spacers and coatings are shown to impact the performance of the immobilized enzyme. This is followed by highlighting the utility of immobilized enzymes for construction of a genomic DNA library of AT-rich Clostridium acetobutylicum for the Illumina sequencing platform. The goal of this research article is to provide the researcher with a broad picture of the merits and challenges of covalent enzyme immobilization and its potential applications in molecular workflows.
2. Results and Discussion
2.1. Production of SNAP-Tagged Enzyme
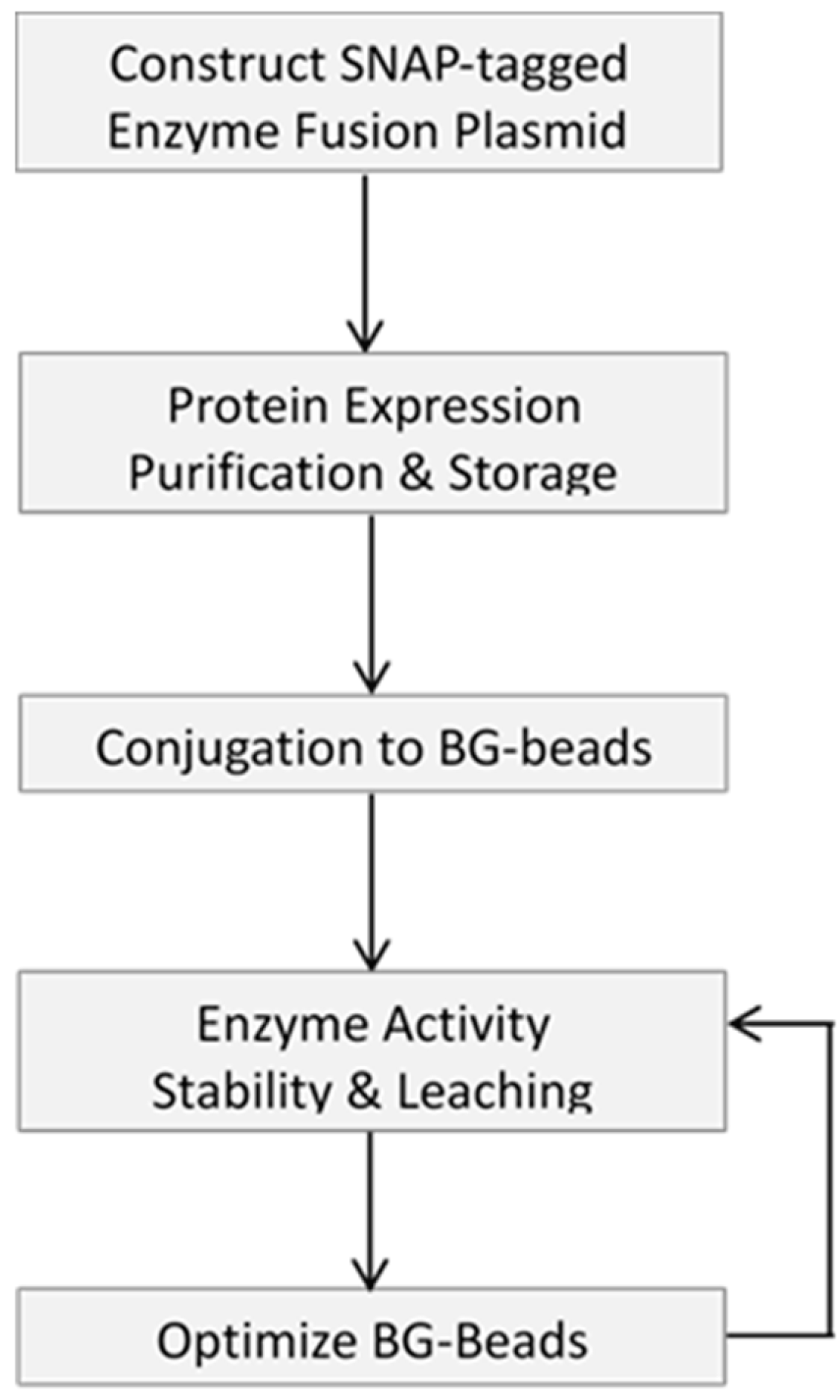
The initial step to generate an immobilized enzyme of interest is to prepare an enzyme of appropriate purity and specific activity, and the schematic flowchart of the work is shown in Figure 1.
Our approach was to prepare a SNAP-tagged enzyme of high purity via Ni-NTA resin, which was used for His-tagged protein purification by immobilized metal affinity chromatography (IMAC). We took advantage of the NEBuilder technique to seamlessly join DNA fragments, independent of the sequence content and cleavage sites of restriction enzymes [27]. In this study, we constructed a recombinant plasmid capable of producing a chimeric protein consisting of T4 DNA ligase, SNAP-tag and a C-terminal His-tag (six histidine residues). The expression of this chimeric fusion protein was conducted overnight at relatively low temperature to ensure production of soluble protein, followed by purification on Ni-NTA resin. The purified protein was dialyzed against a buffer possessing 50% glycerol, assayed for activity and subjected to long-term storage at −20 °C. SNAP-tag mediated immobilization is a simple, straightforward protocol. The enzyme of interest, after dilution to a desired concentration, was incubated with an aliquot of BG-functionalized support material, i.e., agarose, chitin, magnetic and Si-magnetic chitin (SiM) beads in the presence of buffered saline and 1 mM DTT at 4 °C for 12–24 h with the reaction gently mixed on a rotator. For initial screening, four types of support materials were included to compare their effect on conjugation efficiency, enzyme activity and leaching level as described below. After immobilization, the support requires extensive washing, and the enzyme-conjugated beads were dialyzed into a storage buffer containing 50% glycerol and stored at −20 °C.
2.2. Characterization of Immobilized Enzyme
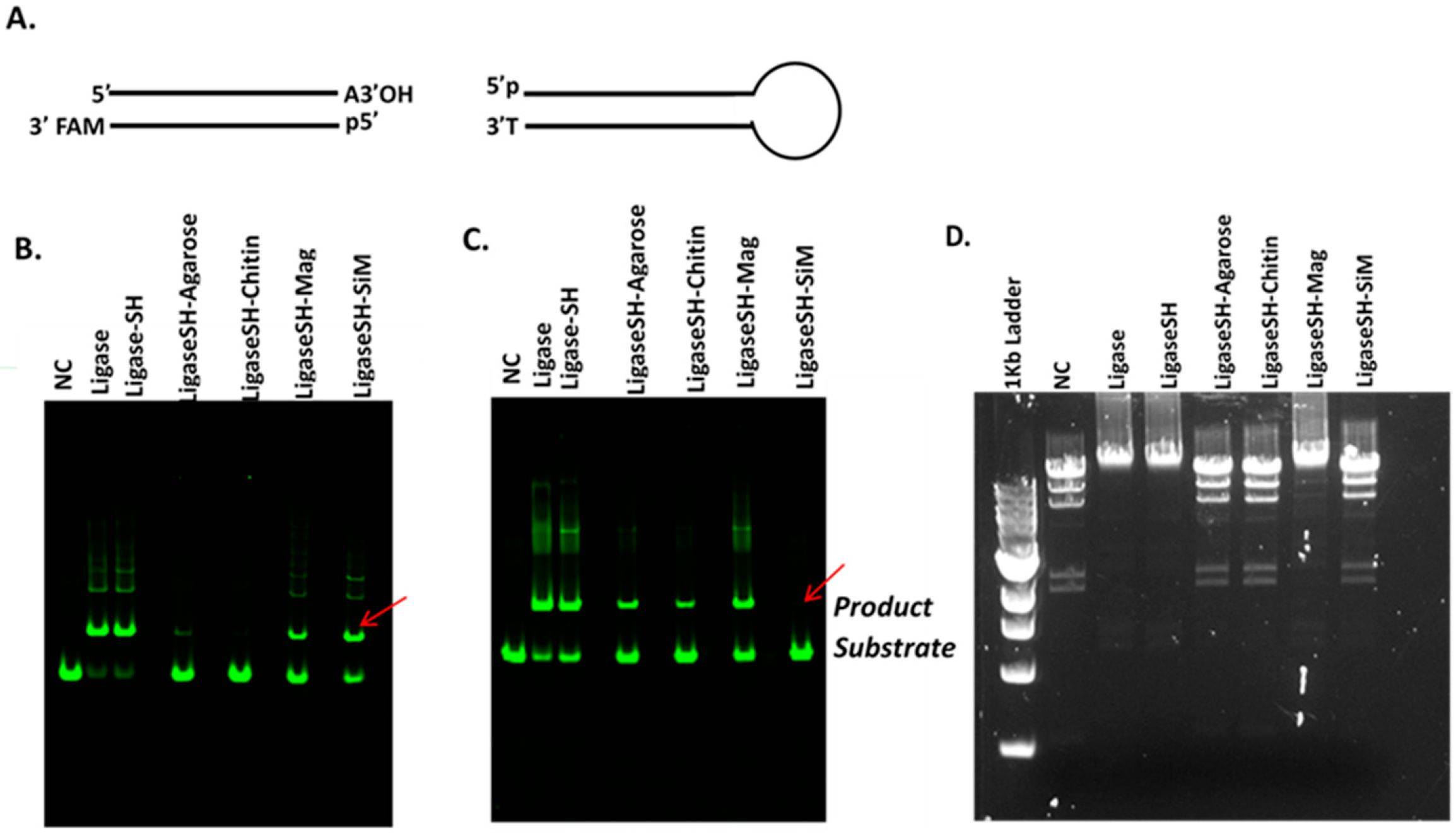
In this study DNA ligase from bacteriophage T4 was selected for immobilization owing to its wide applications since the early years of recombinant DNA technology [28,29]. One of the important tasks in the initial phase of this study was to ensure that the presence of SNAP-tag (or His-tag) in the chimeric fusion protein does not interfere with enzyme activity. The placement of each tag can be varied between the N- or C-terminus of the fusion protein. Various fusion protein constructs can be screened for activity, then thoroughly characterized. The SNAP-tag self-labelling is bio-orthogonal and stoichiometric; therefore, does not impart any off-target modifications to the enzyme itself. This allows one to simply reduce the impacts that the solid supports have on enzyme activity, as opposed to traditional conjugation techniques where off-target reactivity during immobilization can confound such analysis. Four types of pre-existing microbeads, i.e., agarose, chitin, magnetic and Si-magnetic chitin (SiM) microbeads were functionalized with BG ligand and used for the immobilization of the SNAP-T4 DNA ligase fusion protein. Ligase activity was initially assayed by joining two synthetic DNA duplexes with a single A/T base pair overhang (Figure 2A). Gel electrophoretic analysis was performed to compare ligation efficiencies of the native soluble T4 DNA ligase, the soluble SNAP-T4 DNA ligase fusion protein, and various immobilized versions (Figure 2B–D).
The SNAP-tagged fusion protein displayed ligase activity in both Blunt/TA ligation buffer and T4 DNA ligase reaction buffer; and regarding to ligation of HindIII-digested Lambda DNA. Of the four immobilized samples tested, the ligaseSH-Mag version retained ligase activity across the tested substrates/buffer conditions, with a smaller loss of activity compared to the soluble enzyme counterparts (the positive controls) (Figure 2B–D). The presence of polyethylene glycol (PEG6500) in Blunt/TA ligation buffer appeared to decrease the product yield for the immobilized forms of T4 DNA ligase except for the LigaseSH-SiM sample. LigaseSH-SiM displayed little activity in the standard T4 DNA ligase reaction buffer (Figure 2C) but a noticeably higher ligase activity in Blunt/TA ligation buffer (Figure 2B). This shows that buffer composition can influence the activity of the immobilized enzyme, and that buffer optimization may be able to modulate the enzyme activity of various immobilized enzymes. The immobilized enzyme samples were further tested to determine if they are capable of ligating large DNA fragments, such as lambda DNA pre-digested with restriction enzyme HindIII. Agarose gel electrophoresis in Figure 2D indicates that the LigaseSH-Mag microbead sample performed well in ligation of the large HindIII restriction fragments of more than 10 Kb, whereas the other three immobilized versions displayed little, if any, ligase activity. The data show that the substrate specificity and the activity of T4 DNA ligase can be significantly influenced by the support material and buffer composition. The loss of enzymatic activity may be caused by the intrinsic hydrophobic property of the support surface resulting in conformational change, or even denaturation of immobilized protein [30]. The large bead size (100–200 µm) in agarose and chitin beads may adversely affect the enzyme activity due to poor colloidal stability during the reaction. In this case, the LigaseSH-Mag version, which is based on a 1 µm non-porous polymeric magnetic bead displayed optimal activity, probably owing to its good colloidal stability.
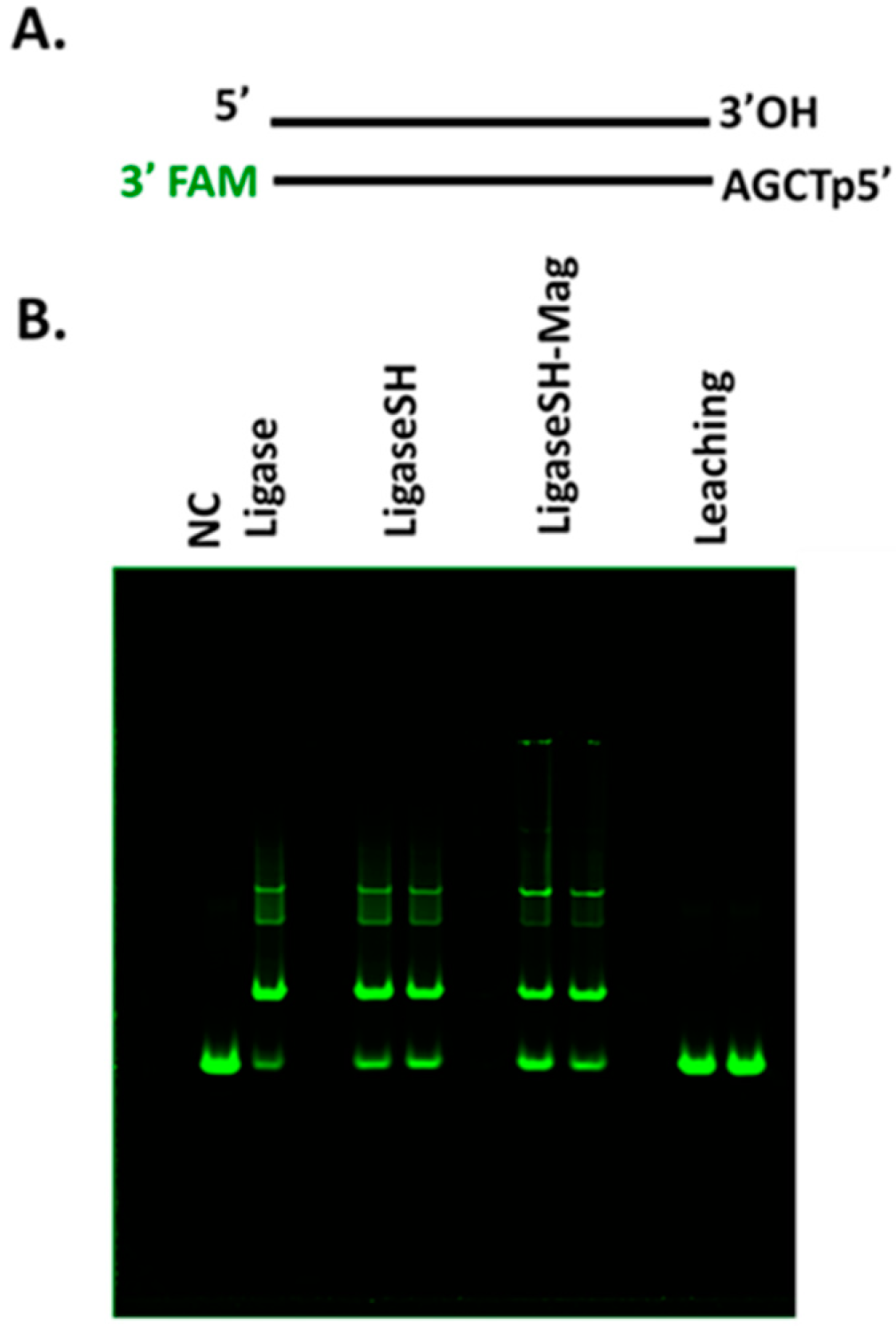
Next, we focused our efforts on analysis of immobilization and characterization of SNAP-tagged T4 DNA ligase on 1 µm magnetic bead. The enzymatic activity was analyzed by employing an assay where a fluorescent probe-labeled synthetic DNA substrate containing a protruding four-nucleotide (AGCT) overhang was used (Figure 3A). Ligation of the DNA substrate by LigaseSH-Mag and soluble ligases proceeded similarly, as can be seen in the distribution of concatemers present upon polyacrylamide gel electrophoresis of the crude reactions (Figure 3B). The absence of ligation products in the leaching lanes of Figure 3B demonstrates that the immobilization of the ligase to the BG-Mag beads is stable, with no indication of significant enzyme leaching over time; if leaching were an issue, T4 DNA ligase activity would be expected to be present in the supernatant. Therefore, of the supports tested, the LigaseSH-Mag version was determined to perform most similarly to the soluble version; and was the version we decided to progress from screening (enzyme activity, stability and leaching, Figure 1) to optimization.
2.3. Optimization of Enzyme Performance
Once the activity of immobilized enzymes has been confirmed to be adequate in traditional/intended workflows, the focus can shift toward modifying the selected solid supports to maximize the protein load, test the impact of spacers between the protein and bead surface, or to alter the chemical properties of the beads—in order to further increase activity, stability and limit protein aggregation/non-specific binding.
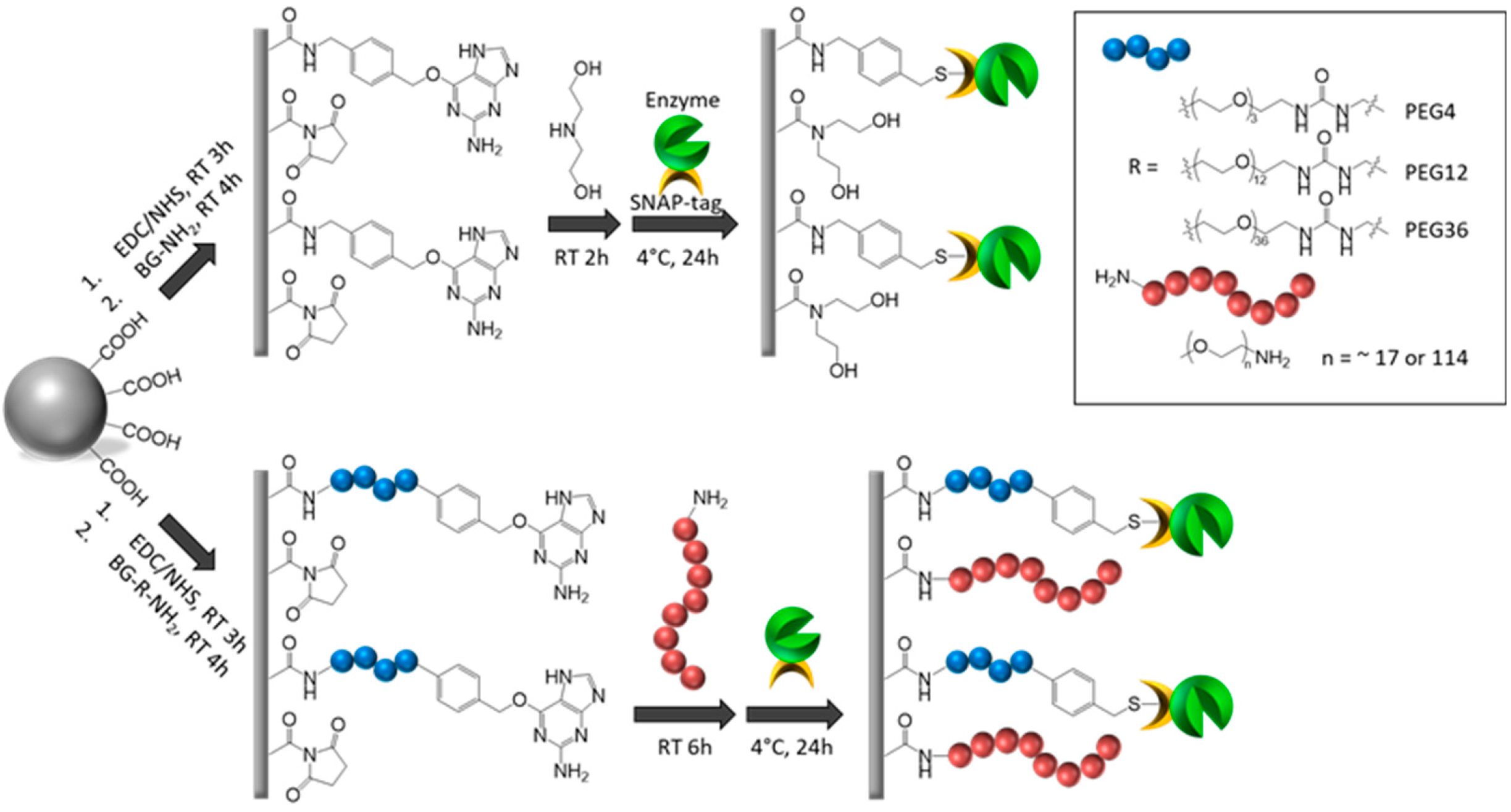
As shown in Figure 2B,C, the LigaseSH-Mag version exhibits slightly less activity compared to its soluble counterpart (LigaseSH). Initially, we varied the amount of BG moieties presents on the bead surface, to increase the ligase load, this was accomplished by altering the concentration of BG used during bead synthesis from 1 mM to 3.7 mM (experimental procedure section for details). Next, the BG moiety was modified to incorporate a short PEG spacer (PEG4) between the beads and the enzyme, this modification was tested with both concentrations (1 mM and 3.7 mM) in an attempt to determine if reduced flexibility or steric hindrance, due to modification of PEG4 spacer, was impacting enzyme performance. We also altered the surface coating of the magnetic microbeads with a hydrophilic PEG750 in conjunction with the different concentrations of BG (1 mM and 3.7 mM) moiety with and without possessing the short PEG4 spacer as shown in Figure 4 to change the hydrophobic property of the BG beads [31].
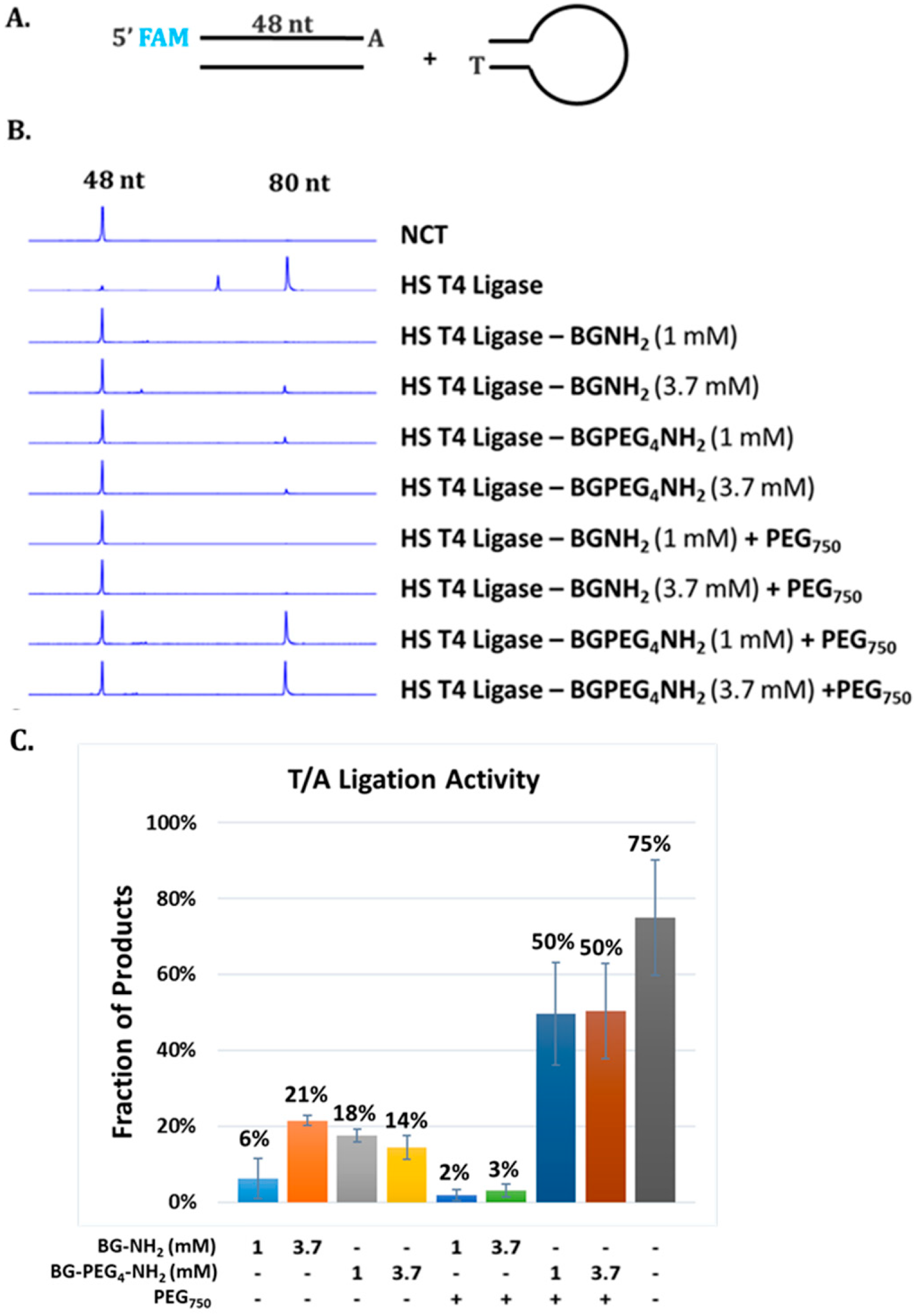
In order to quantitatively analyze how these factors impact the activity of the immobilized enzyme, we adopted a relatively high throughput approach employing capillary gel electrophoresis (CE) [32]. Ligase activity was evaluated by ligation of FAM-labeled double-stranded DNA (dsDNA) and DNA adaptor for Illumina library construction as shown in Figure 5A. Coating with PEG polymers presumably crafts a hydrophilic microenvironment surrounding the conjugated enzyme molecules whereas a spacer between the bead surface and the BG group introduces a greater distance between the enzyme and surface [25,33]. These modifications appeared to have dramatic effect on the performance of the immobilized T4 DNA ligase as shown in Figure 5B,C.
As displayed in Figure 5B, a modestly higher ligase activity was detected corresponding to the use of a higher concentration of BG moiety (3.7 mM) compared to a lower concentration of BG moiety (1 mM). The similar trend was also observed when PEG4 spacer was used to increase the distance between the beads surface and the enzyme molecules. The largest increase in enzymatic activity resulted from the application of both PEG750 coating and PEG4 spacer (both 1 mM and 3.7 mM) as shown in Figure 5B. Thus, we conclude that this combination provides adequate flexibility and reduced steric hindrance through the PEG4 spacer, and as well as a more hydrophilic surface environment due to the PEG750 coating which may stabilize the immobilized T4 DNA ligase and provide a better environment for enzymatic catalysis. The immobilized enzyme with the combination PEG coating and spacer still displayed approximately 67% activity of the free form under the conditions tested. Further investigation is required to optimize performance of the immobilized enzyme. In our previous work, the effects of PEG coating and spacer on enzymatic activity of immobilized DNA modifying enzymes are enzyme-specific [25]. T4 DNA polymerase immobilized on BG beads modified with PEG coating showed a significant increase in enzyme activity compared to BG beads coated with diethanolamine (control) whereas use of PEG spacers of various length or combinations of both PEG coating and spacers exhibited a lower activity. We observed longer PEG spacers between the BG moiety and the surface reactive amine group correlated with increased loss of enzyme activity. Thus, we argue that the use of PEG spacer may allow enzyme molecules to fold back towards the bead surface resulting in non-productive interactions with the surface or even the deactivation of enzymes [25]. Therefore, optimization with PEG coating and spacers, as well as other polymers, must be validated for each enzyme of interest.
2.4. Illumina Library Construction Using Immobilized Enzymes
In the following two sections, we will describe how this enzyme immobilization strategy can be leveraged to improve important molecular workflows, such as library construction for next-generation sequencing. Previously we reported successful immobilization of T4 DNA polymerase, T4 polynucleotide kinase (PNK) and Taq DNA polymerase to BG-functionalized magnetic microbeads and use of these immobilized enzymes in library construction for Illumina next-generation sequencing [26]. A typical protocol of library preparation for the Illumina platform consists of several enzymatic steps, end repair (blunting and 5′ phosphorylation), 3′A-tailing and adaptor ligation [34]. We found that this GC-bias, i.e., under-representation of AT-rich regions, is likely caused by processing DNA ends by DNA modifying enzymes, such as T4 DNA polymerase and Taq DNA polymerase, resulting in preferential depletion of AT-rich DNA in the DNA pool [26]. We demonstrated that sequence coverage GC-bias in sequencing human DNA library on Illumina platform can be significantly improved by substituting the relevant soluble enzymes with their immobilized counterparts at the end repair and A-tailing steps, by avoiding heat treatment of library DNA with the enzymes involved. This was the first time that immobilized enzymes were applied to a widely used workflow for library construction.
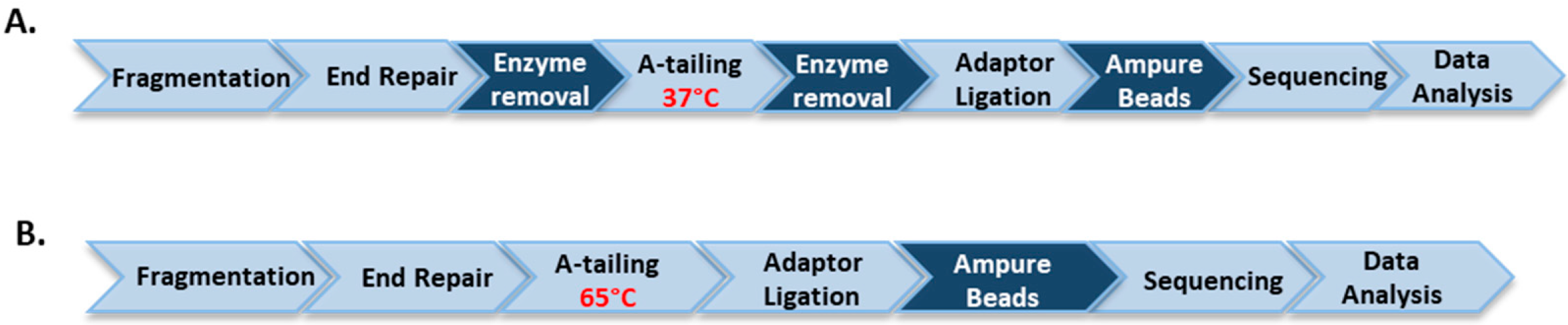
In this report we sought to further validate if this strategy can be applied to other genomes, in particular, AT-rich genomes. We compared GC-Bias in Illumina sequence coverage from the Clostridium acetobutylicum libraries prepared using the soluble and immobilized enzyme methods. We used T4 DNA polymerase, T4 PNK and Taq DNA polymerase immobilized to BG-functionalized magnetic microbeads to replace their soluble counterparts. The immobilized versions of these enzymes were generated through the SNAP-tag as previously described [26], in a manner similar to that outlined for T4 DNA ligase. For library preparation, randomly sheared genomic DNA was first end-repaired at 20 °C (or 37 °C) with immobilized T4 DNA polymerase and immobilized T4 PNK; the enzyme-coated beads were then readily separated and removed from the reaction on a magnet before A-tailing at 37 °C using immobilized Taq DNA polymerase as shown in Figure 6A. For the soluble enzyme protocol fragmented DNA was end-repaired at 20 °C followed by heat treatment at 65 °C as shown in Figure 6B.
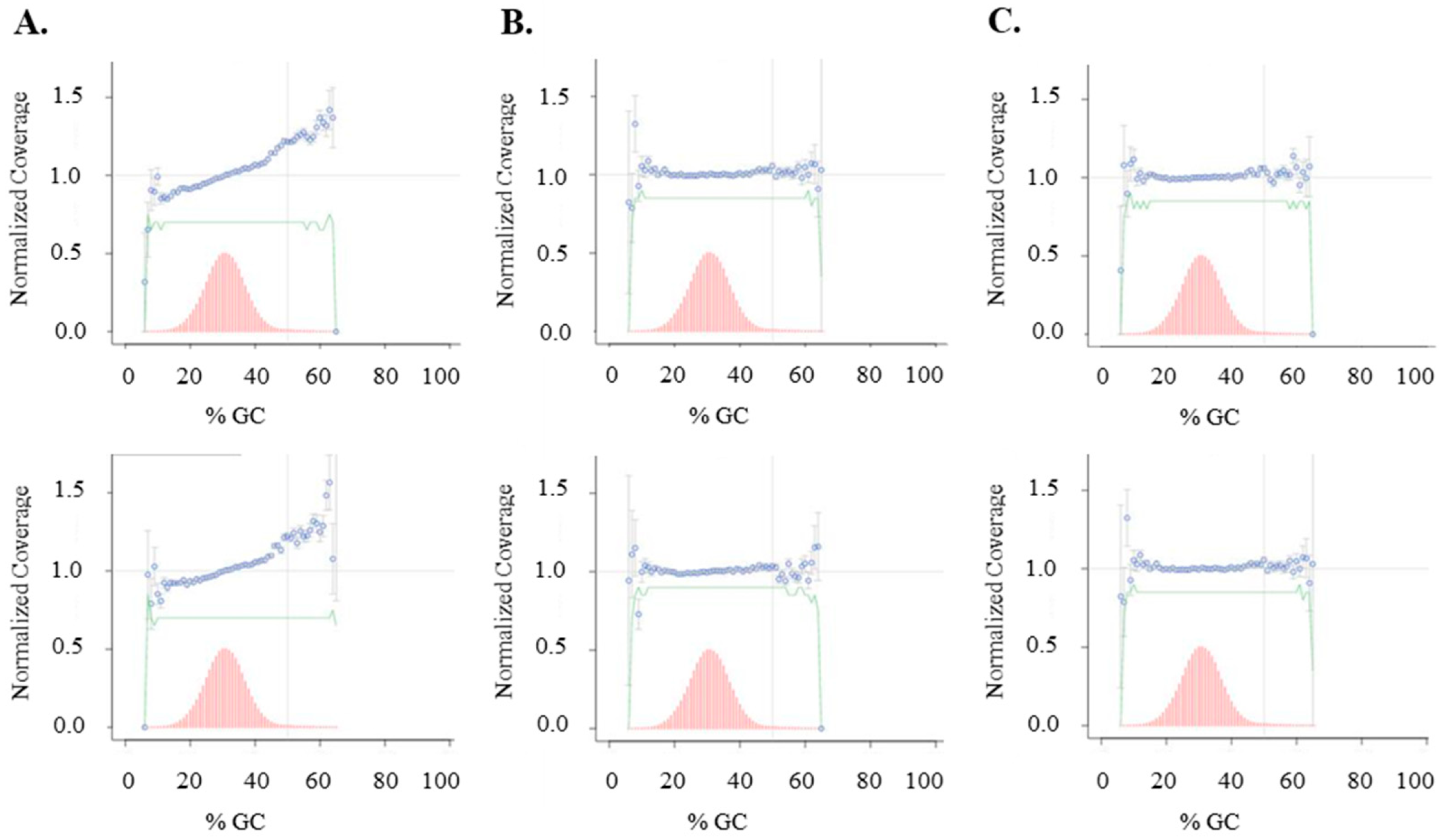
When the soluble enzyme workflow was employed, Illumina MiSeq sequencing data shows GC-bias in the AT-rich Clostridium acetobutylicum libraries, consistent with our observations from sequencing human genomic DNA libraries as shown in Figure 7. In contrast, the use of the immobilized enzyme workflow resulted in more even sequence coverage across various GC-content in the genome. The data are consistent with our earlier observation with human DNA library preparation that removal of the immobilized end-repair enzymes without heat treatment of the library DNA at 65 °C can reduce sequence coverage GC-bias.
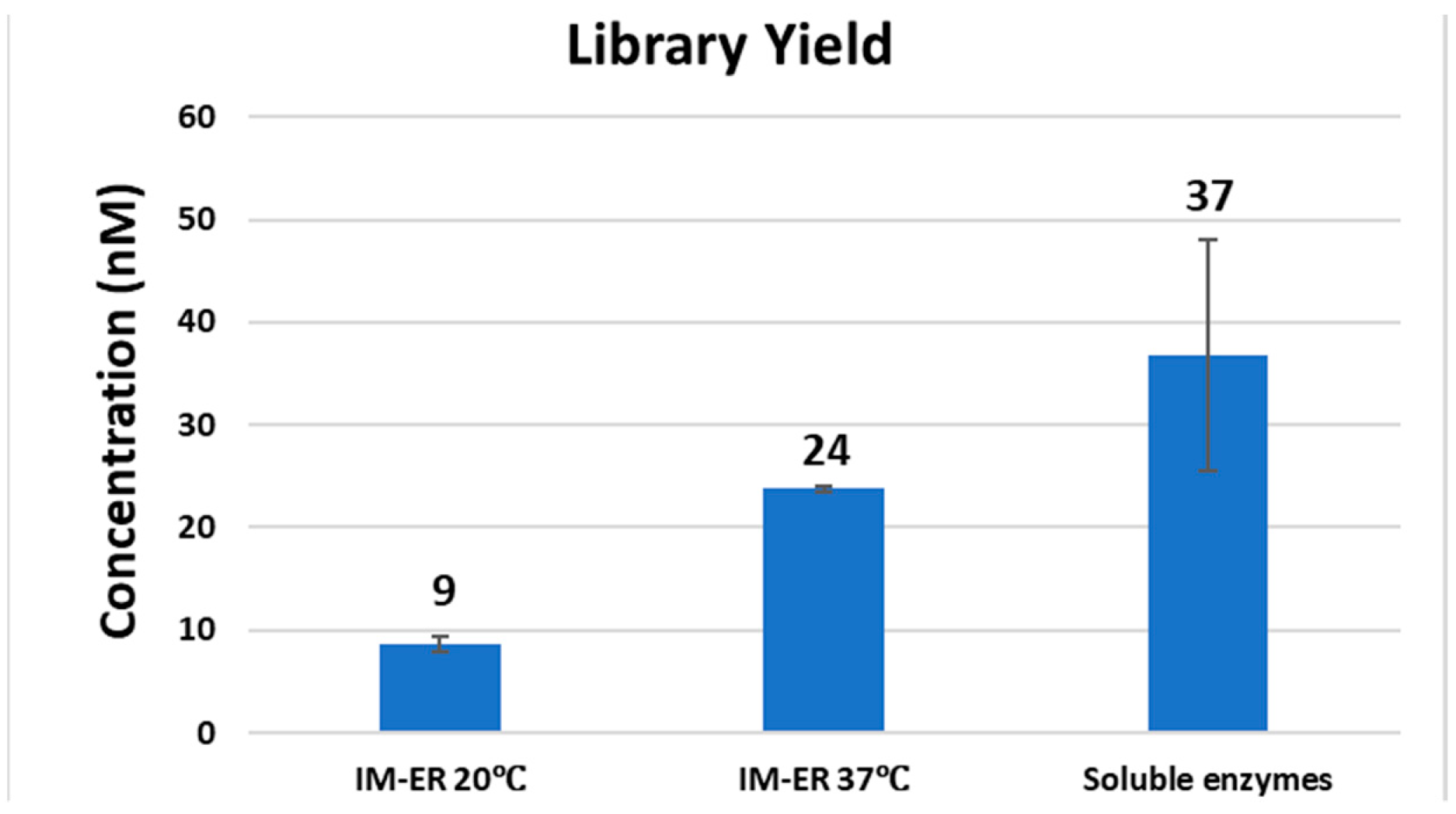
We noticed that the library yield is lower than the method using soluble enzymes when the end-repair step was performed with immobilized enzymes at 20 °C (Figure 8). We performed end-repair at 37 °C instead of 20 °C with the immobilized enzyme method and found that the yield was substantially improved (Figure 8). These immobilized enzymes displayed lower specific activity than their soluble form thereby demanding higher temperature to improve efficiency [25]. We conclude that immobilized enzymes can be applied to preparation of genomic DNA libraries of various GC-content with improved sequence coverage on the Illumina NGS platform.
2.5. Enzyme Immobilization in Perspective
In this report we describe the basic procedure to effectively produce, immobilize and characterize an enzyme of interest by employing the SNAP-tag self-labelling methodology, detailed for the immobilization of T4 DNA ligase to various solid supports. This methodology ensures that the immobilization proceeds in a directed fashion, leading to a homogeneous presentation of the enzyme through a stable covalent linkage. We compare the performance of the immobilized T4 DNA ligase to its soluble counterpart, through T/A and sticky end ligation efficiency, to screen which solid supports impact activity the least. We then employ immobilized DNA modifying enzymes (PNK, T4 DNA Polymerase, and Taq DNA polymerase) in a library preparation workflow for the Illumina NGS platform to validate that this enzyme immobilization strategy can be utilized to improve the traditional/current workflows.
In our hands SNAP-tag based enzyme immobilization results in a solid support exhibiting acceptable levels of activity and low levels of leaching from all enzymes used for library preparation; employing these immobilized enzymes in a slightly modified library preparation workflow did not adversely affect the library yield or other measurements in Illumina NGS and resulted in more even sequence coverage across various GC-content in the genome. The reduction in GC bias observed for the immobilized enzyme workflow can be attributed to the rapid removal of enzymes under mild conditions, which allowed us to omit the conventional heat treatment of enzymes. Incubation of enzymes and substrate/product at high temperature may result in side-products thereby affecting the quality of enzyme-catalyzed process. This feature may be advantageous in designing streamlined workflows based on multi-enzyme cascade reactions, without the need to perform product purification following each reaction. The higher cost in production of enzyme may be more than offset by potential savings to handling and further miniature of reactions. Particularly, in conjunction with microfluidic techniques, immobilized enzymes may play important roles in transformation and automation of workflows in laboratories and industry.
3. Materials and Methods
3.1. Materials
All DNA modifying enzymes, including T4 DNA ligase, T4 DNA Polymerase, T4 Polynucleotide Kinase, and Taq DNA Polymerase were from New England Biolabs (NEB, Ipswich, MA, USA). Carboxylate-modified magnetic beads (average diameter 1 µm) were from GE Healthcare Bio-Sciences (Pittsburgh, PA, USA). HPLC-purified synthetic single stranded oligonucleotides were obtained from Integrated DNA Technologies (IDT, Coralville, IA, USA) as lyophilized solids. Oligonucleotides were dissolved in nuclease-free water prior to use. T4 DNA Ligase Reaction Buffer, Quick Ligation Buffer, NEBNext® End Repair Reaction Buffer and NEB Buffer 2 were from NEB. pSNAPf-tag(T7) vector was used for construction of recombinant plasmids expressing SNAP-tagged enzymes and is commercially available (NEB). In this report four microbeads were functionalized with BG ligand and used for immobilization of T4 DNA ligase. The sources and average bead size are described below. Carboxylate-modified magnetic beads (average diameter 1 µm), obtained from GE Healthcare Bio-Sciences (Pittsburgh, PA, USA), were used to generate BG-Mag beads. BG-SiMag (1 µm, SiM in Figure 2) was derived from SiMag-Chitosan (Product no. 14095, Chemicell, Berlin, Germany). BG-Agarose (100–200 µm) was commercially available (SNAP-capture, NEB); BG-Chitin (100–200 µm) was made by BG-modification of Chitin Beads (NEB). 1 µm beads can typically stay in suspension during reaction without agitation. BG-NH2 and BG-PEG4-NH2 (sold under the name BG-PEG-NH2) were from NEB. Methoxypolyethylene glycol amines with average molecular weight 750 Da, referred to as PEG750, was purchased from Sigma-Aldrich (St Louis, MO, USA) and used for coating BG-Mag beads.
3.2. Beads Surface Modifications
Surface modifications of beads were previously reported [25]. 2 mL of a 50 mg/mL suspension of carboxylate magnetic beads (average diameter 1 µm) in water were separated with aid of an external magnet and were washed three times with 10 mL of water followed with 3 washes with 10 mL of a 50 mM 2-(N-morpholino) ethanesulfonic acid (MES) buffer pH 6.0. The supernatant was removed and a mixture containing 50 mM N-(3-Dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) and 50 mM NHS in 10 mL of 50 mM MES buffer pH 6.0 was added to the magnetic beads. The suspension mixture was gently shaken at room temperature for 3 h. The supernatant was removed, and the beads were washed three times with ice-cold 50 mM MES. After bead washing, a solution of 3.7 mM BG-NH2 or BG-PEGn-NH2 (wherein n = 4) in 10 mL of isopropanol/water 1:1 was added. The reaction mixture was shaken for 4 h. The supernatant was removed and a solution of 0.2 M ethanolamine in isopropanol/water 1:1 was added. The reaction mixture was shaken at room temperature for 2 h. For the PEG-coated beads, instead of diethanolamine, a solution of 20 mM methoxypolyethylene glycol amines PEG750 in isopropanol/water 1:1 solution was added to the beads and shaken at room temperature for 24 h. The supernatant was then removed, and the beads were washed with water (3 × 10 mL) and isopropanol/water 1:1 (3 × 10 mL). The beads were stored in isopropanol/water 1:1 at 10 mg/mL concentration.
3.3. Protein Immobilization
SNAP-tagged enzymes were conjugated to benzylguanine-functionalized magnetic beads by mixing 18.8 nmol of protein with 100 mg beads resuspended in 500 µL of a 10 mM phosphate-buffered saline (PBS) pH 7.4 (10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, and 137 mM NaCl) with 1 mM dithiothreitol (DTT). The mixture was shaken at 4 °C for 24 h. Beads were washed 8 times with 1 mL of 10 mM PBS buffer pH 7.4. The immobilized enzymes were stored at −20 °C in a storage buffer containing 1 mM DTT, 0.1% Triton® X-100, 10 mM Tris-HCl, 0.1 mM EDTA, 100 mM KCl, 50% glycerol, pH 7.4.
3.4. Plasmids and Expression of SNAP-Tagged Enzymes
Construction of SNAP-tagged T4 DNA polymerase, T4 polynucleotide kinase and Taq DNA polymerase were previously described using pSNAPf-tag (T7) [25]. pSNAPf-tag(T7) vector was used to subclone the genes encoding T4 DNA ligase, resulting in a construct termed T4 DNA ligase-SNAP-6× His expresses a fusion protein consisting of an N-terminal T4 DNA ligase, SNAP-tag and C-terminal six histidine residues. E. coli NEB T7 Express strain was used for inducible expression of SNAP-tagged enzymes, typically at 16 °C for 16 h. Soluble fusion proteins were purified from clarified cell lysate by affinity chromatography using Ni-NTA agarose (Qiagen, Hilden, Germany), after extensive wash with 800 mL of buffer containing 10 mM Tris-HCl, pH 8.0, 20 mM Imidazole, 1 mM DTT and 0.3 M NaCl, per 10 mL of Ni-NTA agarose resin. Purified protein was dialyzed into a storage buffer (1 mM DTT, 0.1% Triton® X-100, 10 mM Tris-HCl, 0.1 mM EDTA, 100 mM KCl, pH 7.4) containing 50% glycerol for long-term storage at −80 °C.
3.5. Enzyme Immobilization
SNAP-tagged enzymes were conjugated to BG-functionalized magnetic beads by mixing approximately 18.8 nmol of protein with 10 mg of beads resuspended in 500 µL of a 10 mM phosphate-buffered saline (PBS) pH 7.4 (10 mM Na2HPO4, 1.8 mM KH2PO4, 2.7 mM KCl, and 135 mM NaCl) with 1 mM dithiothreitol (DTT). The mixture was shaken at 4 °C for 24 h. Beads were washed 8 times with 1 mL of 10 mM PBS buffer (pH 7.4). The immobilized enzymes were stored at −20°C in a storage buffer containing 1 mM DTT, 0.1% Triton® X-100, 10 mM Tris-HCl, 0.1 mM EDTA, 100 mM KCl, 50% glycerol, pH 7.4.
3.6. Ligase Activity Assays
Synthetic DNA duplexes possessing a fluorescent 5′-FAM probe were used to monitor ligation efficiency via capillary gel electrophoresis (CE) analysis. DNA duplex was prepared by mixing each pair of complementary oligos at 10 µM final concentration in 1× NEB Buffer 2. The mixture was heated to 80 °C and cooled to room temperature slowly. 5′ FAM-54mer/50mer and 5′ FAM-66mer/70mer substrates, with complementary four nucleotide overhangs, were used for standard activity assays using 1× T4 DNA Ligase Reaction Buffer (NEB) to determine the unit in comparison to that of soluble T4 DNA ligase. A 5′ FAM-labeled 48-bp synthetic DNA containing a single 3′A and an Illumina adaptor (NEB) containing a 3′T were ligated using Blunt/TA Ligation Buffer provided by NEB. Lambda DNA digested with restriction enzyme HindIII (NEB) was used to assess ligation of large DNA fragments via agarose gel electrophoresis. All DNA fragments are 5′ phosphorylated. Ligation reactions were typically performed at 25 °C for 2 h in 1× T4 DNA Ligation Buffer or at 25 °C for 15 min in T/A Ligation Buffer or Quick Ligation Buffer prior to CE analysis as described below.
3.7. Capillary Gel Electrophoresis Assays
T4 DNA ligase activity was determined using DNA duplex end-labeled with a fluorescent FAM probe. Briefly, 100 nM DNA substrate and 1.5 nM soluble or immobilized T4 DNA ligase were incubated in 10 µL of 1× T4 DNA ligation Buffer, T/A Ligation Buffer or Quick Ligation Buffet at 20 °C for 30 min. The reaction was terminated by addition of 40 µL of 12.5 mM EDTA solution (10 mM final concentration). The resulting mixture was diluted to a 5 nM final concentration of FAM-labeled substrate. Capillary gel electrophoresis (CE) analysis was performed on an Applied Biosystems 3730 xl Genetic Analyzer to measure substrate depletion and product formation. DNA fragment analysis and quantification were performed using Peak Scanner software v1.0 (Applied Biosystems, Foster City, CA, USA).
3.8. DNA Library Preparation and Sequencing
Clostridium acetobutylicum genomic DNA, purchased from ATCC (Manassas, VA, USA), was diluted to a final concentration of 100 µg/mL in 10 mM Tris-HCl, 1 mM EDTA, pH 7.5 and processed for ultrasonication shearing using Covaris AFA S2 system (Covaris, Woburn, MA, USA) with the settings of 5% duty cycle, intensity 10 and 200 cycles per burst for 6 min. For library construction, 1 µg of genomic DNA fragments was treated with either soluble untagged T4 DNA pol, T4 PNK and Taq DNA pol or the corresponding immobilized enzymes according to the workflow of the NEBNext Ultra DNA Library Prep Kit. DNA was first treated at 20 °C for 30 min with soluble end-repair enzymes or at 20 °C or 37 °C for 30 min with immobilized enzymes. For the workflow of soluble enzymes, end-repaired DNA was treated for A-tailing at 65 °C for 30 min. For the workflow of immobilized enzymes, after the removal of immobilized T4 DNA pol and T4 PNK, an aliquot of Taq DNA polymerase conjugated to magnetic beads was added to the end-repaired library and A-tailing was performed at 37 °C for 30 min. Each library was ligated to pre-annealed full-length paired-end Illumina adaptors according to the protocol of TruSeq DNA PCR-free LT Library Preparation Kit (Illumina, San Diego, CA, USA). DNA libraries were size-selected and analyzed to determine the size distribution using an Agilent High Sensitivity DNA Kit on a Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA) and by qPCR using the NEBNext Library Quant Kit for Illumina.
The libraries were sequenced on an Illumina MiSeq in paired-end mode (2 × 75 bp). Reads were adapter trimmed (SeqPrep, v1.1 https://github.com/jstjohn/SeqPrep, GitHub, San Francisco, CA, USA) before alignment to the GRCh37 reference genome (bowtie 2.3.2, end-to-end, -X 1000, GitHub, San Francisco, CA, USA) [35]. GC Bias was assessed using Picard’s Collect GC Bias Metrics (Picard 2.7.1, GitHub, San Francisco, CA, USA). Relevant low-GC regions were identified by intersecting 100 bp windows (Bedtools v2.25.0, Quinlan Lab, University of Utah, Salt Lake City, UT, USA) having GC fractions <0.2 with 80% overlap and with features in the Gencode v26 basic genes. Coverage of low-GC regions was assessed using Bedtools genomecov, Quinlan Lab, University of Utah, Salt Lake City, UT, USA.
4. Conclusions
In this research, we successfully expressed and purified the SNAP-tagged T4 DNA ligase as the model enzyme to study the effect of immobilization strategies with various microbead supports, the optimization of the microbead surface using PEG4 as spacer and PEG750 as coating, and the application of the immobilized T4 DNA ligase for Illumina next-generation sequencing. The purified SNAP-tagged T4 DNA ligase was used for immobilization with agarose, chitin, magnetic and SiM microbeads as the support material, demonstrating the BG-modified magnetic beads can be used as the basic immobilization model with little enzyme leaching in gel electrophoresis. Further experiments also demonstrate the application of PEG4 as spacer between the BG-microbeads and the enzyme, and PEG750 as bead coating for higher hydrophilic environment, significantly improve the behavior of immobilized enzyme. The optimized immobilized T4 DNA ligase successfully demonstrates its potential application in DNA library preparation for Illumina next-generation sequencing by minimizing GC-bias compared to its soluble enzyme counterpart.
Author Contributions
Writing—original draft preparation, review and editing, Y.F., A.Z., M.S., M.-Q.X.; Conceptualization and methodology, S.L., M.S., M.-Q.X.; data curation, formal analysis and investigation, A.Z., S.L.; Supervision, M.-Q.X.
Funding
This research is supported by New England Biolabs, Inc.
Acknowledgments
We thank J. McFarland, J. Buswell and M. Sproviero for bead preparation; L. Mazzola, J. Bybee, and D. Rivizzigno for CE analysis. We also thank R. J. Roberts, W. Jack, T. Evans and A. Gardner, E. Dimalanta, T. Davis, J. Ellard, D. Comb and New England Biolabs for research support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Richard, J.P. Enzymatic rate enhancements: A review and perspective. Biochemistry 2013, 52, 2009–2011. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.; Griffin, E.G. ADSORPTION OF INVERTASE. J. Am. Chem. Soc. 1916, 38, 1109–1115. [Google Scholar] [CrossRef]
- Hanefeld, U.; Gardossi, L.; Magner, E. Understanding enzyme immobilisation. Chem. Soc. Rev. 2009, 38, 453–468. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Kostrov, X. Immobilization of enzymes on porous silicas–benefits and challenges. Chem. Soc. Rev. 2013, 42, 6277–6289. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, N.R.; Marzuki, N.H.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Zucca, P.; Fernandez-Lafuente, R.; Sanjust, E. Agarose and its derivatives as supports for enzyme immobilization. Molecules 2016, 21, 1577. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Enzyme immobilization: The quest for optimum performance. Adv. Synth. Catal. 2007, 349, 1289–1307. [Google Scholar] [CrossRef]
- Zdarta, J.; Meyer, A.; Jesionowski, T.; Pinelo, M. A general overview of support materials for enzyme immobilization: Characteristics, properties, practical utility. Catalysts 2018, 8, 92. [Google Scholar] [CrossRef]
- Zdarta, J.; Meyer, A.S.; Jesionowski, T.; Pinelo, M. Multi-faceted strategy based on enzyme immobilization with reactant adsorption and membrane technology for biocatalytic removal of pollutants: A critical review. Biotechnol. Adv. 2019. [Google Scholar] [CrossRef]
- Guzik, U.; Hupert-Kocurek, K.; Wojcieszyńska, D. Immobilization as a strategy for improving enzyme properties-application to oxidoreductases. Molecules 2014, 19, 8995–9018. [Google Scholar] [CrossRef]
- Hitaishi, V.; Clement, R.; Bourassin, N.; Baaden, M.; De Poulpiquet, A.; Sacquin-Mora, S.; Ciaccafava, A.; Lojou, E. Controlling redox enzyme orientation at planar electrodes. Catalysts 2018, 8, 192. [Google Scholar] [CrossRef]
- Fang, Y.; Bullock, H.; Lee, S.A.; Sekar, N.; Eiteman, M.A.; Whitman, W.B.; Ramasamy, R.P. Detection of methyl salicylate using bi-enzyme electrochemical sensor consisting salicylate hydroxylase and tyrosinase. Biosens. Bioelectron. 2016, 85, 603–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Fang, Y.; Ramasamy, R.P. Non-covalent functionalization of carbon nanotubes for electrochemical biosensor development. Sensors 2019, 19, 392. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Umasankar, Y.; Ramasamy, R.P. A novel bi-enzyme electrochemical biosensor for selective and sensitive determination of methyl salicylate. Biosens. Bioelectron. 2016, 81, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, Y.; Ramasamy, R.P. Detection of p-ethylphenol, a major plant volatile organic compound, by tyrosinase-based electrochemical biosensor. ECS J. Solid State Sci. 2016, 5, M3054–M3059. [Google Scholar] [CrossRef]
- Guisan, J.M. Immobilization of Enzymes and Cells; Springer: Totowa, NJ, USA, 2006; Volume 22. [Google Scholar]
- Hermanson, G.T. Bioconjugate Techniques; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Xu, M.-Q.; Evans, T.C., Jr. Recent advances in protein splicing: Manipulating proteins in vitro and in vivo. Curr. Opin. Biotechnol. 2005, 16, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Meldal, M.; Schoffelen, S. Recent advances in covalent, site-specific protein immobilization. F1000Research 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [Google Scholar] [CrossRef]
- Liu, W.; Wang, L.; Jiang, R. Specific enzyme immobilization approaches and their application with nanomaterials. Top. Catal. 2012, 55, 1146–1156. [Google Scholar] [CrossRef]
- Lang, K.; Chin, J.W. Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem. Rev. 2014, 114, 4764–4806. [Google Scholar] [CrossRef]
- Kindermann, M.; George, N.; Johnsson, N.; Johnsson, K. Covalent and selective immobilization of fusion proteins. J. Am. Chem. Soc. 2003, 125, 7810–7811. [Google Scholar] [CrossRef] [PubMed]
- Recker, T.; Haamann, D.; Schmitt, A.; Küster, A.; Klee, D.; Barth, S.; Müller-Newen, G. Directed covalent immobilization of fluorescently labeled cytokines. Bioconj. Chem. 2011, 22, 1210–1220. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, A.; Zatopek, K.; Parvez, S.; Gardner, A.F.; Correa, I.R., Jr.; Noren, C.J.; Xu, M.Q. Enhancing Multistep DNA Processing by Solid-Phase Enzyme Catalysis on Polyethylene Glycol Coated Beads. Bioconj. Chem. 2018, 29, 2316–2324. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Li, S.; Apone, L.; Sun, X.; Chen, L.; Ettwiller, L.M.; Langhorst, B.W.; Noren, C.J.; Xu, M.Q. Solid-phase enzyme catalysis of DNA end repair and 3′ A-tailing reduces GC-bias in next-generation sequencing of human genomic DNA. Sci. Rep. 2018, 8, 15887. [Google Scholar] [CrossRef] [PubMed]
- Birla, B.S.; Chou, H.-H. Rational design of high-number dsDNA fragments based on thermodynamics for the construction of full-length genes in a single reaction. PLoS ONE 2015, 10, e0145682. [Google Scholar] [CrossRef] [PubMed]
- Engler, M.; Richardson, C. The Enzymes; Boyer, P.D., Ed.; Academic Press: San Diego, CA, USA, 1982; Volume 5, p. 3. [Google Scholar]
- Remaut, E.; Tsao, H.; Fiers, W. Improved plasmid vectors with a thermoinducible expression and temperature-regulated runaway replication. Gene 1983, 22, 103–113. [Google Scholar] [CrossRef]
- Talbert, J.N.; Goddard, J.M. Enzymes on material surfaces. Colloids Surf. B Biointerfaces 2012, 93, 8–19. [Google Scholar] [CrossRef]
- Pegg, A.E.; Kanugula, S.; Edara, S.; Pauly, G.T.; Moschel, R.C.; Goodtzova, K. Reaction ofO 6-Benzylguanine-resistant Mutants of Human O 6-Alkylguanine-DNA Alkyltransferase with O 6-Benzylguanine in Oligodeoxyribonucleotides. J. Biol. Chem. 1998, 273, 10863–10867. [Google Scholar] [CrossRef] [Green Version]
- Greenough, L.; Schermerhorn, K.M.; Mazzola, L.; Bybee, J.; Rivizzigno, D.; Cantin, E.; Slatko, B.E.; Gardner, A.F. Adapting capillary gel electrophoresis as a sensitive, high-throughput method to accelerate characterization of nucleic acid metabolic enzymes. Nucleic Acids Res. 2015, 44, e15. [Google Scholar] [CrossRef]
- Kohler, N.; Fryxell, G.E.; Zhang, M. A bifunctional poly (ethylene glycol) silane immobilized on metallic oxide-based nanoparticles for conjugation with cell targeting agents. J. Am. Chem. Soc. 2004, 126, 7206–7211. [Google Scholar] [CrossRef]
- Head, S.R.; Komori, H.K.; LaMere, S.A.; Whisenant, T.; Van Nieuwerburgh, F.; Salomon, D.R.; Ordoukhanian, P. Library construction for next-generation sequencing: Overviews and challenges. Biotechniques 2014, 56, 61–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Framework for study of enzyme immobilization using SNAP-tag technology.

Figure 2.
Comparison of enzymatic activity of soluble T4 DNA ligase versus the soluble SNAP-tagged T4 DNA ligase (Ligase SH) and various beads immobilized versions. Ligation activity was assayed by joining two synthetic DNA duplexes with a single A/T base pair overlap (A) in the presence of 1× Blunt/TA ligation buffer at 23 °C for 15 min (B) or 1× T4 DNA ligase reaction buffer at 23 °C for 2 h (C) and visualized by fluorescent scanning following polyacrylamide gel electrophoresis. The arrows indicate the presence or absence of the ligation product in the Ligase-SiM reactions. Ligation of HindIII-digested Lambda DNA fragments was carried out in 1× T4 DNA ligase reaction buffer at 23 °C for 2 h and examined by agarose gel electrophoretic separation (D).
Figure 2.
Comparison of enzymatic activity of soluble T4 DNA ligase versus the soluble SNAP-tagged T4 DNA ligase (Ligase SH) and various beads immobilized versions. Ligation activity was assayed by joining two synthetic DNA duplexes with a single A/T base pair overlap (A) in the presence of 1× Blunt/TA ligation buffer at 23 °C for 15 min (B) or 1× T4 DNA ligase reaction buffer at 23 °C for 2 h (C) and visualized by fluorescent scanning following polyacrylamide gel electrophoresis. The arrows indicate the presence or absence of the ligation product in the Ligase-SiM reactions. Ligation of HindIII-digested Lambda DNA fragments was carried out in 1× T4 DNA ligase reaction buffer at 23 °C for 2 h and examined by agarose gel electrophoretic separation (D).

Figure 3.
Comparison of sticky end ligation efficiency between soluble and immobilized T4 DNA ligases. Soluble Ligases (Ligase and LigaseSH) are compared to the immobilized (LigaseSH-Mag) and bead leachate for sticky end ligation. The supernatant of the LigaseSH-Mag bead slurry (leachate) was collected and assayed for ligase activity. (A) A fluorophore-labeled synthetic DNA duplex with a four-nucleotide overhang (AGCT) was used for ligation reactions. (B) Ligation reactions were performed in 1× T4 DNA Ligase Reaction Buffer at 23 °C for 2 h and analyzed by polyacrylamide gel electrophoresis of the samples followed by fluorescent scanning. The assays of the SNAP-tagged T4 DNA ligase were performed in duplicate.
Figure 3.
Comparison of sticky end ligation efficiency between soluble and immobilized T4 DNA ligases. Soluble Ligases (Ligase and LigaseSH) are compared to the immobilized (LigaseSH-Mag) and bead leachate for sticky end ligation. The supernatant of the LigaseSH-Mag bead slurry (leachate) was collected and assayed for ligase activity. (A) A fluorophore-labeled synthetic DNA duplex with a four-nucleotide overhang (AGCT) was used for ligation reactions. (B) Ligation reactions were performed in 1× T4 DNA Ligase Reaction Buffer at 23 °C for 2 h and analyzed by polyacrylamide gel electrophoresis of the samples followed by fluorescent scanning. The assays of the SNAP-tagged T4 DNA ligase were performed in duplicate.

Figure 4.
Schematic illustration of enzyme immobilization via SNAP-tag covalent conjugation onto bead surface functionalized with BG moiety and PEG coating.
Figure 4.
Schematic illustration of enzyme immobilization via SNAP-tag covalent conjugation onto bead surface functionalized with BG moiety and PEG coating.

Figure 5.
Optimization of activity of immobilized T4 DNA ligase by PEG spacer and coating. SNAP-tagged T4 DNA ligase was immobilized to magnetic beads loaded with 1 mM or 3.7 mM BG ligand with or without PEG4 spacer and PEG750 coating. (A) Ligase activity was evaluated by ligation of two synthetic duplex DNA substrates possessing a single 3′A and 3′T overhang, respectively. The reactions were carried out by incubation of 100 nM of synthetic substrates and 200 nM enzyme in 1× Blunt/TA buffer (NEB) at 20 °C for 15 min. A 49-nt oligomer with a 3′A overhang and a 5′ FAM probe was used to monitor ligation by CE analysis. (B) Representative CE data showing an increase of 80-nt product catalyzed by T4 DNA ligase immobilized to PEG750 coated and PEG4 spaced beads. (C) Quantitative analysis of CE data showing efficiency of product formation in each combination of chemical modifications.
Figure 5.
Optimization of activity of immobilized T4 DNA ligase by PEG spacer and coating. SNAP-tagged T4 DNA ligase was immobilized to magnetic beads loaded with 1 mM or 3.7 mM BG ligand with or without PEG4 spacer and PEG750 coating. (A) Ligase activity was evaluated by ligation of two synthetic duplex DNA substrates possessing a single 3′A and 3′T overhang, respectively. The reactions were carried out by incubation of 100 nM of synthetic substrates and 200 nM enzyme in 1× Blunt/TA buffer (NEB) at 20 °C for 15 min. A 49-nt oligomer with a 3′A overhang and a 5′ FAM probe was used to monitor ligation by CE analysis. (B) Representative CE data showing an increase of 80-nt product catalyzed by T4 DNA ligase immobilized to PEG750 coated and PEG4 spaced beads. (C) Quantitative analysis of CE data showing efficiency of product formation in each combination of chemical modifications.

Figure 6.
Workflows for Illumina library construction using immobilized or soluble DNA modifying enzymes. (A) End repair reactions including DNA blunting and phosphorylation are carried out by immobilized T4 DNA polymerase and T4 polynucleotide kinase, respectively, which are covalently conjugated to magnetic beads and are removed prior to 3′A-tailing using immobilized Taq DNA polymerase at 37 °C. (B) DNA end repair and 3′A-tailing are catalyzed by a mixture of the soluble enzymes described above. Following the end repair step 3′A-tailing is performed at 65 °C. The use of immobilized enzymes circumvents the requirement of high temperature treatment of DNA library.
Figure 6.
Workflows for Illumina library construction using immobilized or soluble DNA modifying enzymes. (A) End repair reactions including DNA blunting and phosphorylation are carried out by immobilized T4 DNA polymerase and T4 polynucleotide kinase, respectively, which are covalently conjugated to magnetic beads and are removed prior to 3′A-tailing using immobilized Taq DNA polymerase at 37 °C. (B) DNA end repair and 3′A-tailing are catalyzed by a mixture of the soluble enzymes described above. Following the end repair step 3′A-tailing is performed at 65 °C. The use of immobilized enzymes circumvents the requirement of high temperature treatment of DNA library.

Figure 7.
Comparison of Illumina library construction of AT-rich Clostridium acetobutylicum genome. Sequence coverage GC-bias curves obtained from three sets of PCR-free DNA libraries generated with untagged soluble enzymes (A) or enzymes immobilized on BG-beads with end repair performed at 20 °C (B) or 37 °C (C). Each set of libraries was prepared in duplicate (top and bottom). Sequencing reactions were carried out on an Illumina MiSeq.
Figure 7.
Comparison of Illumina library construction of AT-rich Clostridium acetobutylicum genome. Sequence coverage GC-bias curves obtained from three sets of PCR-free DNA libraries generated with untagged soluble enzymes (A) or enzymes immobilized on BG-beads with end repair performed at 20 °C (B) or 37 °C (C). Each set of libraries was prepared in duplicate (top and bottom). Sequencing reactions were carried out on an Illumina MiSeq.

Figure 8.
Comparing library yield. The preparation conditions of Clostridium acetobutylicum DNA libraries with immobilized enzymes (IM) or soluble enzymes were described in Figure 7 legend.
Figure 8.
Comparing library yield. The preparation conditions of Clostridium acetobutylicum DNA libraries with immobilized enzymes (IM) or soluble enzymes were described in Figure 7 legend.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fang, Y.; Zhang, A.; Li, S.; Sproviero, M.; Xu, M.-Q. Enzyme Immobilization for Solid-Phase Catalysis. Catalysts 2019, 9, 732. https://doi.org/10.3390/catal9090732
AMA Style
Fang Y, Zhang A, Li S, Sproviero M, Xu M-Q. Enzyme Immobilization for Solid-Phase Catalysis. Catalysts. 2019; 9(9):732. https://doi.org/10.3390/catal9090732
Chicago/Turabian StyleFang, Yi, Aihua Zhang, Shaohua Li, Michael Sproviero, and Ming-Qun Xu. 2019. "Enzyme Immobilization for Solid-Phase Catalysis" Catalysts 9, no. 9: 732. https://doi.org/10.3390/catal9090732
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.