Proteases Immobilization for In Situ Time-Limited Proteolysis on MALDI Chips



1
Department of Biochemistry, Faculty of Science, Charles University, 128 43 Prague, Czech Republic
2
Institute of Microbiology, v.v.i., Czech Academy of Sciences, 142 20 Prague, Czech Republic
3
Institute of Biotechnology, v.v.i., Czech Academy of Sciences, 252 50 Vestec, Czech Republic
*
Author to whom correspondence should be addressed.
Catalysts 2019, 9(10), 833; https://doi.org/10.3390/catal9100833
Submission received: 9 September 2019
/
Revised: 23 September 2019
/
Accepted: 30 September 2019
/
Published: 3 October 2019
(This article belongs to the Special Issue Immobilization of Enzymes)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:A large number of different enzyme immobilization techniques are used in the field of life sciences, clinical diagnostics, or biotechnology. Most of them are based on a chemically mediated formation of covalent bond between an enzyme and support material. The covalent bond formation is usually associated with changes of the enzymes’ three-dimensional structure that can lead to reduction of enzyme activity. The present work demonstrates a potential of an ambient ion-landing technique to effectively immobilize enzymes on conductive supports for direct matrix-assisted laser desorption/ionization (MALDI) mass spectrometry analyses of reaction products. Ambient ion landing is an electrospray-based technique allowing strong and stable noncovalent and nondestructive enzyme deposition onto conductive supports. Three serine proteolytic enzymes including trypsin, α-chymotrypsin, and subtilisin A were immobilized onto conductive indium tin oxide glass slides compatible with MALDI mass spectrometry. The functionalized MALDI chips were used for in situ time-limited proteolysis of proteins and protein–ligand complexes to monitor their structural changes under different conditions. The data from limited proteolysis using MALDI chips fits to known or predicted protein structures. The results show that functionalized MALDI chips are sensitive, robust, and fast and might be automated for general use in the field of structural biology.
1. Introduction
Enzymes immobilized on various support materials are frequently used for their improved enzyme stability, activity, and possibility to effectively control an enzymatic reaction. Most of the immobilization protocols are based on the formation of a covalent bond, affinity interaction, and electrostatic or physical adsorption [1,2].
Immobilization of enzymes by ion soft landing was reported to be an appropriate technique for in situ digestion of proteins [3]. Ion soft landing refers to the process where the hyperthermal molecular ions are nondestructively captured on conductive target supports [4]. This approach was first described by Cooks in 1977. Originally, the ion soft-landing process was demonstrated by deposition of carbon disulfide ion beam onto lead target support using an evacuated ion-generating and transiting apparatus with mass selection [5].
Since the 1990s, several ion soft-landing techniques were developed such as polyatomic ion soft-landing deposition on FSAM [6], nucleic acid landing on a nitrocellulose membrane [7], and organic clusters deposition using modified mass spectrometer [8]. Ouyang et al. reported retained activity of enzymes separated by different mass to charge (m/z) ratio and soft-landed on a gold support covered with a glycerol/fructose layer [9]. A direct protein soft landing on a metal support surface was demonstrated in 2005 by Turecek’s group [10,11], where a plasma-treated stainless steel support was used for protease immobilization. The catalytic activities of soft-landed enzymes were monitored by a solution assay that was based on cleavage of VLK-p-nitroaniline. [10].
The surfaces functionalized by ambient soft-landed molecules represent novel possibilities of their use in life sciences and diagnostics [12]. The standard soft landing is performed under the vacuum and requires a sophisticated and expensive instrumentation, whereas an ambient soft landing is operated at atmospheric pressure conditions. Different variants of ambient soft landing were recently applied for deposition of titanium propoxide for phosphopeptide enrichment [13], nanomaterials preparation [14], deposition of antibodies for immunoaffinity assays [15], deposition of lectins [16], or deposition of proteolytic enzymes for in situ protein digestion [3]. The native conditions during ion beam deposition were proved by a scanning tunneling microscopy [17].
Limited proteolysis reactions naturally initiate many biochemical regulation processes such as blood coagulation or enzyme maturation [18]. In structural biology, limited proteolysis has been used for low-resolution three-dimensional protein structure characterization [19,20]. The proteolytic fragments of studied proteins were originally analyzed by gel electrophoresis [21]. The invention of soft ionization techniques for mass spectrometric analysis of biomolecules allowed detection of proteolysis products using both the electrospray ionization (ESI) [22] and matrix-assisted laser desorption/ionization (MALDI) [23] ion sources. Later, Schopper et al. presented an analysis of a highly complex sample resulting from protein-limited digestion utilizing the combination of HPLC separation and ESI MS [24]. Subsequently, applications of limited proteolysis and mass spectrometry were reported for antibody epitope mapping [25,26], mapping of protein–DNA interaction [27], or characterization of protein structural changes in a whole yeast cell proteome [28].
This study represents a novel approach for monitoring alterations in protein tertiary structure, which combines in situ limited proteolysis on functionalized chips with MALDI mass spectrometry.
2. Results and Discussion
2.1. Protease Immobilization
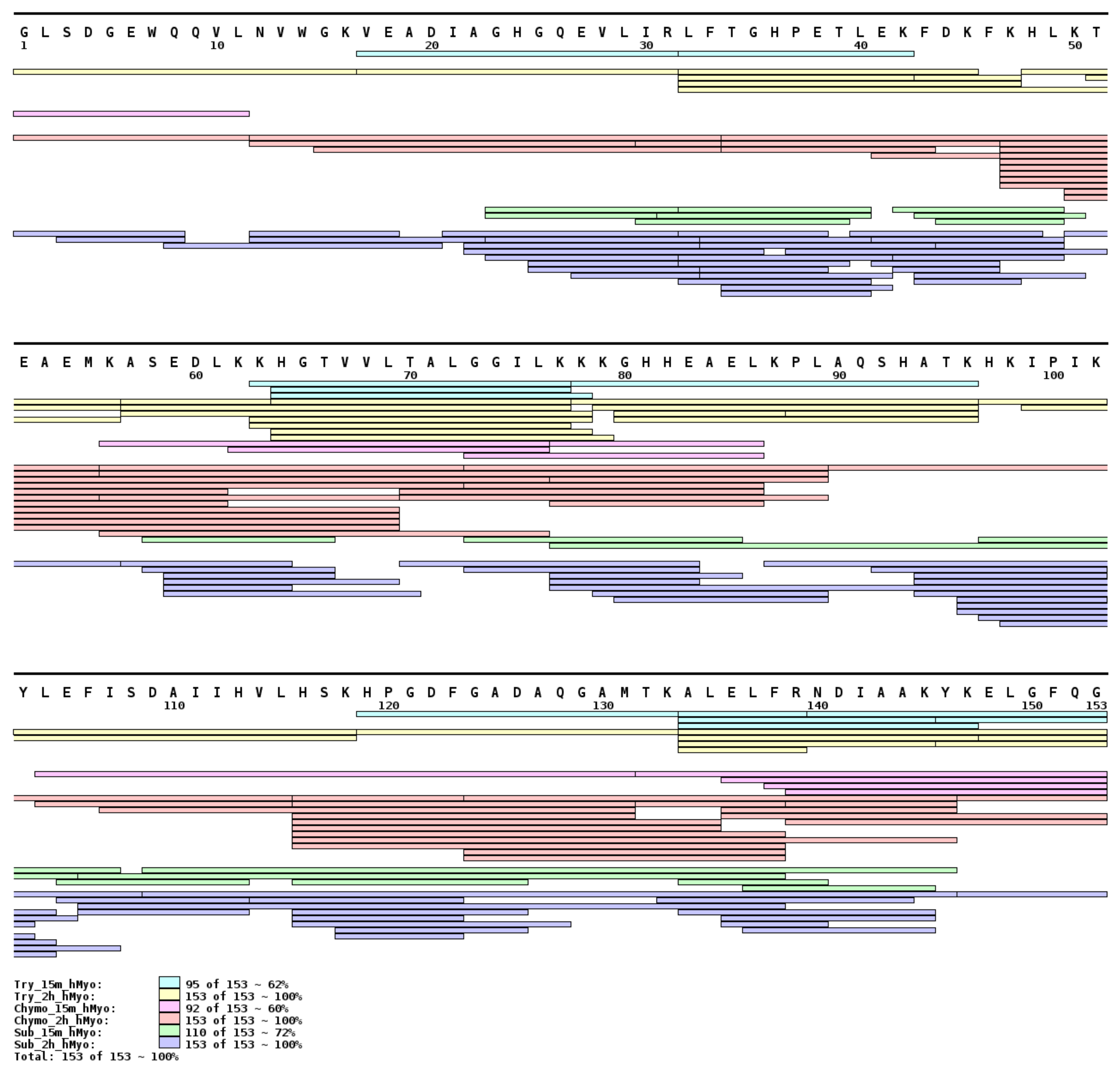
The proteolytic activity of enzymes trypsin, α-chymotrypsin, and subtilisin A immobilized on indium tin oxide (ITO) glass by ambient ion soft landing was tested by in situ holomyoglobin (hMyo, heme-bound myoglobin) digestion performed at two different reaction times. The products of the reaction, peptides, were identified by MALDI mass spectrometry. The complete protein sequence coverage was achieved for all three proteases after two hours of reaction on the functionalized surfaces. This was in contrast with 15 min reaction time, where different peptide distributions and sequence coverages were observed (Figure 1). Immobilized trypsin and α-chymotrypsin digested the hMyo to peptides with optimal length for limited proteolysis experiment after 15 min, whereas subtilisin A reached the optimal peptide length in shorter time (5 min, Figure S1). Selection of time points for in situ limited proteolysis is a critical parameter, which depends on the type of enzyme and requires a careful optimization.
2.2. Data Interpretation Procedure
An absence of processing tools for time-limited proteolysis in combination with MALDI detection and missing tools for visualization of the results led to designing novel computational operations for their interpretation and evaluation. A procedure leading to butterfly plot-based presentation (Figure 2a) was chosen for results visualization. A new value called Relative Amino Acid Cleavage Incidence (RCI), which expresses the frequency of protease activity at specific cleavage sites, was plotted on the Y axis. The Python script (see RCI calculation script source code in Supplementary Materials) was developed to calculate each cleavage site’s RCI value using all assigned peptides (see Section 3.7).
The RCI value computing script combines two simple equations (1) and (2) below:
where the amino acidn intensity index is the sum of intensities of all peptides, whose C-terminal or N-terminal foregoing amino acid (P1 cleavage site nomenclature [29]) is the amino acidn. The resulting amino acidn RCI value was calculated as a simple ratio of amino acidn intensity index over the sum of intensities of all assigned peptides in the spectrum. Exemplar RCI value calculation is described in Supplementary Materials.
For easier graphical visualization of the results, RCI values of one protein form got a negative operator during the script processing. Such a graphical outcome enabled simple evaluation of protein conformational rearrangement induced by the local environment. Moreover, this simple averaging eliminated the potential false positive differences between two MALDI spectra.
2.3. Method Feasibility
Method feasibility was verified by in situ limited proteolysis of two myoglobin conformers—hMyo and aMyo (apomyoglobin, heme-free myoglobin). Limited proteolysis was performed directly on MALDI-compatible supports functionalized by trypsin and subtilisin A. hMyo and aMyo were digested in situ for 5, 15, and 30 min and for 2 h (see Figure S1 of Supplementary Materials).
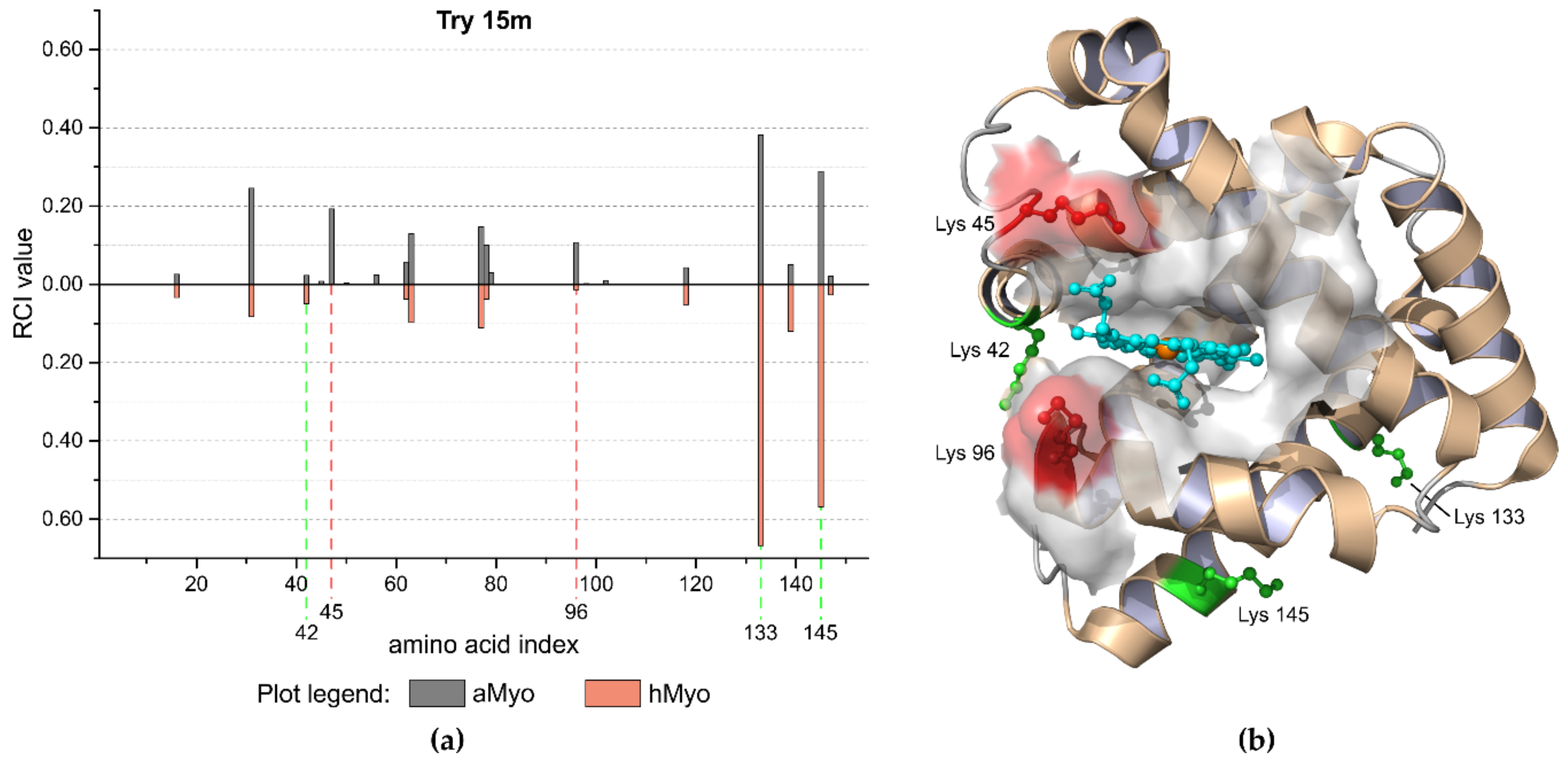
aMyo tertiary structure was described as molten globule [30] with retained secondary structure motifs, whose relative positions are less rigid compared to hMyo. Observed faster proteolysis of aMyo correlated with its lower rigidity. Different RCI values distinguishing aMyo and hMyo forms are shown in the butterfly plot in Figure 2a. In situ limited proteolysis results reflected a three-dimensional structure of hMyo (PDB ID: 1WLA) and are in good correlation with known structural data for aMyo [31] (Figure 2b).
2.4. Limited Proteolysis of FOXO4 DNA Complex
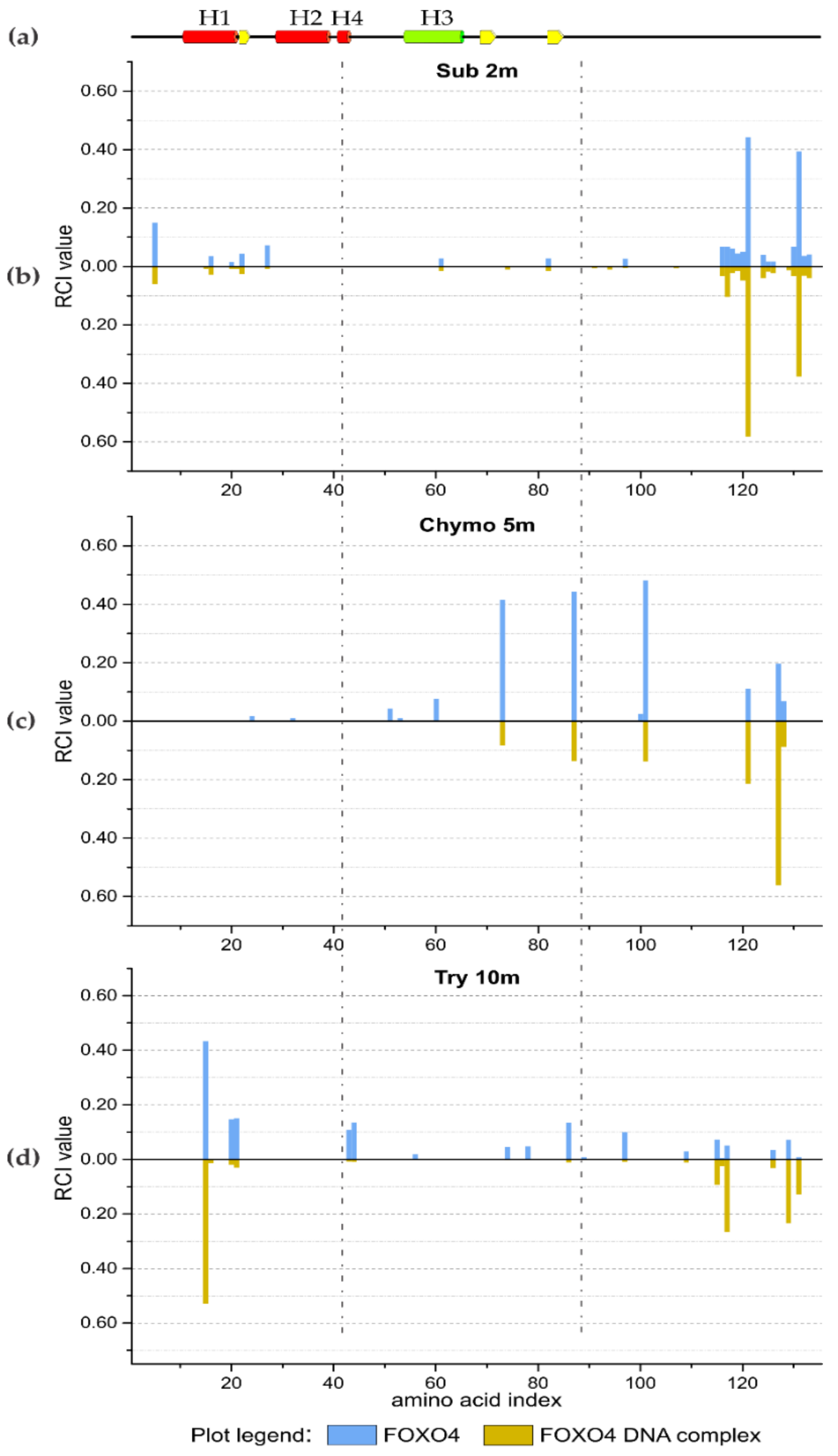
The initial experiments with hMyo and aMyo forms showed the feasibility of the method to monitor protein structural changes. In structural proteomics, the study of interaction interface between protein and DNA usually requires advanced instrumentation and carefully optimized workflows. The strong negative charge of DNA influences the performance of analytical methods such as chromatographic separation prior to mass spectrometry measurement. In this study, the transcription factor Forkhead box protein O4 (FOXO4) DNA binding domain and its interacting partner DAF16 DNA were analyzed by in situ limited proteolysis and MALDI mass spectrometry. The in situ limited proteolysis experiments were performed with three different proteases (subtilisin A, α-chymotrypsin, and trypsin) at six time points. Three selected butterfly plots for each protease at different time points, by which the most differences in RCI values were observed, are shown in Figure 3. All butterfly plots are available in Supplementary Materials (Figures S2, S3, and S4). The results of limited proteolysis indicated that the region between amino acids Lys 43 and Phe 87 is the most resistant to proteolysis in the presence of DNA molecules. It is in accordance with the 3L2C crystal structural model for the FOXO4 bound to the DNA duplex [32]. The H3 helix and its neighboring parts are responsible for the protein–DNA interaction. The high RCI values for peptides from both N- and C-termini of the FOXO4 protein for short reaction times indicated that these parts of the protein are not in contact with the DNA molecule (Figure 3 and Figures S2, S3, and S4).
It is also important to perform control experiments. The proteolytic activities of immobilized proteases on the MALDI chips were verified using both forms of protein. The in situ enzymatic reaction was also performed for a significantly longer period (two hours) than the last expected time point of the limited proteolysis experiment. The resulting RCI values for FOXO4 and FOXO4/DNA complex were almost identical (see last plots in Figures S2, S3, and S4 of Supplementary Materials). Thus, on the chips, proteolytic activity was not affected by the presence of DNA. Spots with immobilized proteases only were used to identify the product of protease auto proteolysis. Only low intense signals were observed in the spectra which did not influence the quality of peptide signals in real samples and had no impact on the experiment (see Figures S2, S3, and S4 of Supplementary Materials).
Another important step prior to the limited proteolysis experiment was the verification of protein integrity, which was performed by spotting both protein forms on spots without immobilized proteases (Figure S5b). The degradation products were identified by the LinX software [33]. Several peptides originating from the C-terminal region were observed in the sample (see Figure S5b of Supplementary Materials) prior to the experiment. The low intense signals of spontaneous FOXO4 degradation had no effect on limited proteolysis results (see Figures S2, S3, and S4 of Supplementary Materials). The protein integrity should be considered during data interpretation, mainly when studied proteins are fragile or sensitive to experimental conditions.
3. Materials and Methods
3.1. Proteolytic Enzymes
Lyophilized proteolytic enzymes trypsin (treated by tosylphenylalanylchloromethyl ketone, TCPK), α-chymotrypsin, and subtilisin A were purchased from Sigma-Aldrich (St. Louis, MO, USA). All proteases were reconstituted in LCMS-grade water (Merck, Darmstadt, Germany) to the 200 µM concentration. Reconstituted proteases were 20x diluted with 20 mM ammonium bicarbonate buffer (AMBIC, Sigma-Aldrich, MO, USA), pH 7.8, and immediately used.
3.2. Protein Systems
3.2.1. Apomyoglobin and Holomyoglobin Model System
Equine heart myoglobin with bound heme group (hMyo) was purchased from Sigma-Aldrich (St. Louis, MO, USA). The heme extraction was carried out according to the previously published procedure [34] to obtain myoglobin apo-form. Apomyoglobin (aMyo) was dialyzed to the 20 mM AMBIC, pH 7.8 buffer and lyophilized from this solution on the SpeedVac vacuum concentrator (Thermo Fisher Scientific, Waltham, MA, USA). Both forms were resuspended in 50 mM AMBIC, pH 7.8, to final 10 µM concentration.
3.2.2. FOXO4 Transcription Factor and Oligonucleotide Duplex
A DNA binding domain of FOXO4 was recombinantly produced as an N-terminal His-tag fusion protein in BL21 competent Escherichia coli (DE3). The pET-15b plasmid carrying His-tag, thrombin cleavage site, and FOXO4 DNA binding domain sequence was used as a transformation vector. After initial growth at 37 °C, protein expression was induced by ITPG addition (1 mM final concentration) to the cell culture cooled at 30 °C. Affinity purification was performed on a TALON Superflow Resin column (Clontech Laboratories, Mountain View, CA, USA) according to the manufacturer’s protocol. The His-tag was cleaved out overnight at 4 °C by human thrombin (2 units per 1 mg of recombinant protein). Buffer exchange to the 50 mM AMBIC buffer, pH 7.8, as well as the thrombin and cleaved His-tag removal steps, were carried out by a gel permeation chromatography using ENrich SEC 70, 10 × 300 column (Bio-Rad Laboratories, Hercules, CA, USA).
Both forward and reverse complementary oligonucleotide strands were purchased from Integrated DNA Technologies (Coralville, IA, USA). Both strands were dissolved in LCMS-grade water, mixed in an equimolar ratio, and heated up to 95 °C for 1 min. The mixture was left to cool down at room temperature to form a13 bp DNA duplex.
FOXO4 was diluted to the final concentration of 10 µM in 50 mM AMBIC buffer, pH 7.8, and an equimolar 10 µM mixture of FOXO 4 and oligonucleotide duplex was prepared in the same buffer as well.
3.3. Ambient Ion-Landing Immobilization
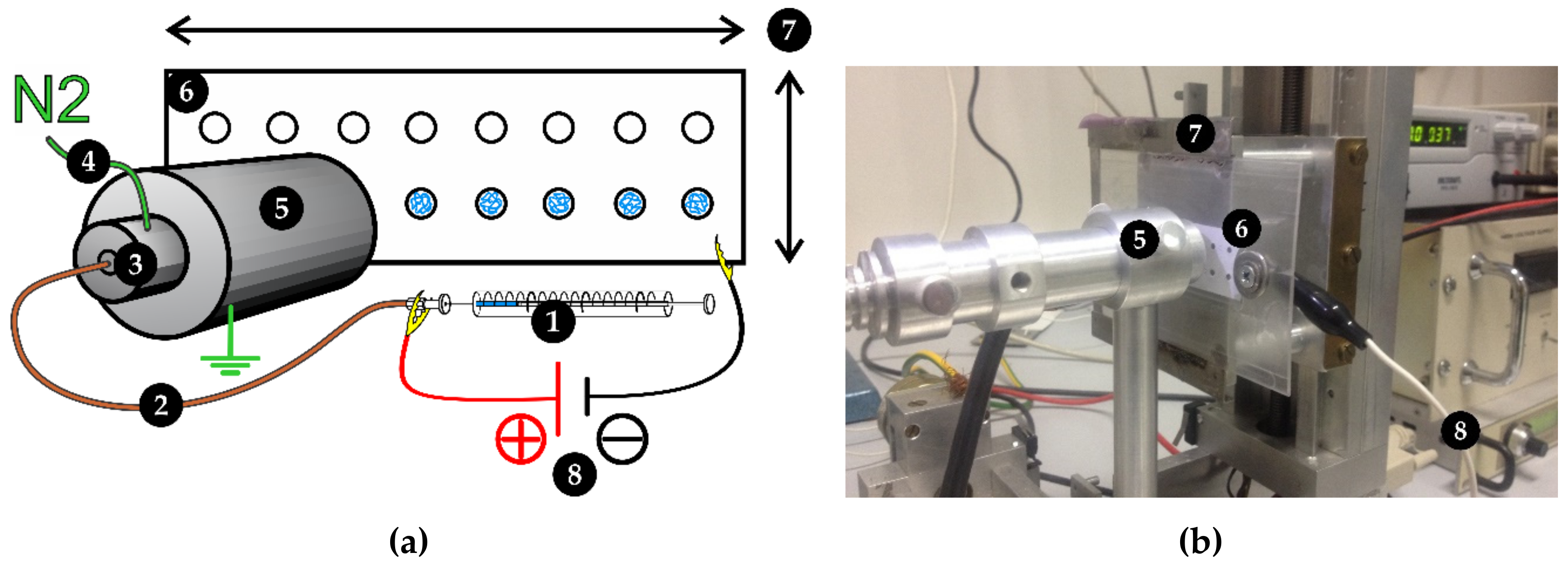
All three serine proteases were immobilized by the ambient ion-landing technique using an in-house built electrospray device [13], as shown in Figure 4. The protease solution was delivered to the ion emitter through a 40 μm inner diameter fused silica capillary (Polymicro Technologies, Phoenix, AZ, USA) by the syringe linear pump (flow rate 1 µL·min−1). A positive +1.5 kV voltage was applied to the metal syringe port. Preheated (45 °C) nitrogen gas was used to further improve nebulization and desolvation of ions. The positively charged aerosol was dried in a grounded 10 cm long desolvation tube, which was externally heated up to 60 °C. Dried ions were landed to the negatively charged (−1.5 kV) ITO (indium tin oxide) glass slide support (Bruker Corporation, Billerica, MA, USA). The uniform spot size and geometry were achieved by application of sticky mask with engraved 2 mm circular spots. All proteases were landed for 3 min per spot.
3.4. In Situ Limited Proteolysis Workflow
All functionalized surfaces were immersed in deionized water and washed for 5 min on an oscillating stirrer. The surfaces were dried in a vacuum desiccator under weak (–0.08 MPa) vacuum. One microliter of 10 µM protein solution was directly applied on the proteolytic spot. An incubation was carried out in a 15 cm3 humidity chamber to minimize evaporation; the incubation was carried out at room temperature with endpoints at time range 2 min to 2 h. The enzymatic reaction was quenched by addition of 1 µL of 2% TFA (trifluoroacetic acid, Sigma-Aldrich, St. Louis, MO, USA). The whole ITO glass slide was placed in the vacuum desiccator after the quench. One microliter of HCCA matrix solution was deposited onto each dried sample spot and allowed to crystalize spontaneously on the bench. The matrix solution was prepared by mixing a saturated HCCA (α-Cyano-4-hydroxycinnamicacid, Sigma-Aldrich, St. Louis, MO, USA) solution in methanol, acetonitrile, and 0.1% TFA water solution in the ratio 1:1:1. The scheme of the workflow is summarized in Figure 5.
3.5. Control Spots
Two spots of the total 16 were used as controls of digestion performance. The positive control spots were allowed to digest substrate for 2 h. Another two spots, each in a separate row, were dedicated for protease auto proteolysis control and directly overlaid by HCCA matrix solution. Protein homogeneity was tested by applying a protein sample on the chips not carrying immobilized protease and overlaid with HCCA matrix solution. The quenching efficacy was also tested on the myoglobin model system. A 1:1 ratio mixture of quench solution and protein sample was prepared on the spots and allowed to dry after 30 min of incubation at room temperature.
3.6. Mass Spectrometry
High-resolution mass spectra were acquired using Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometer 15TsolariX XR FTICR equipped with dual ESI/MALDI ion source (Bruker Corporation, Billerica, MA, USA). Mass spectra were acquired in a positive ion mode with 2M data points at m/z 250 using a SmartBeam II laser MALDI source (100 laser shots accumulation, 12 averaged scans per acquired spectrum using random walk feature during acquisition). The data were acquired in the 250 to 5000 m/z range. Spectra were externally calibrated using a Peptide Calibration Standard II mixture (Bruker Corporation, Billerica, MA, USA) and subsequently, single peak correction was applied during an acquisition (m/z 922 Da). The ESI tuning mix (Sigma-Aldrich, St. Louis, MO, USA) was continuously evaporating from the ESI ion source orifice during the MALDI acquisition.
3.7. Data Interpretation
Processing of raw data was performed using Data Analysis 4.4 (Bruker Corporation, Billerica, MA, USA). The Mascot Generic Format files (MGF), containing peak m/z, intensity, and charge information, were exported from all spectra using the build-in SNAP 2.0 algorithm. The MGF files serve as an input for protein sequence-based peak assignment performed by the LinX software (Version, Company, City, State Abbr., Country, Year) [33] developed in-house for fast high-resolution MS data processing. LinX software assignment criteria were set as follows: Protease cleavage rules were set according to the used immobilized protease. C-terminal side of Lys, Arg, and Tyr if Pro was not a subsequent amino acid was considered for the trypsin, C-terminal side of Phe, Leu, Met, Tyr, and Trp if Pro was not a following amino acid was considered for α-chymotrypsin, and nonspecific cleavage was set for subtilisin A. No potential post-translational modifications were added to the assignment calculation. The maximal acceptable error between theoretical and experimental mass was set to 2 ppm. Unassigned masses were automatically omitted from the list using the filtering module and the list was saved as an output file for the Python script processing. This process was applied for all acquired spectra including control spots. The Python script itself, as well as example analysis files, are available in Supplementary Materials.
3.8. Data Visualization
Data from all LinX output files was processed by the Python script (see Supplementary Materials) which recognized all identified cleavage positions in the studied protein sequence. Based on peptide intensities, the script calculated the relative amino acid cleavage incidence (RCI) value. The resulting butterfly plots were created using OriginPro 2015 software (OriginLab Corporation, Northampton, MA, USA, 2015). The summarizing peptide map (Figure 1) was created using MS Tools web application [35]. The three-dimensional structure of hMyo 1WLA [36] was visualized and processed in PyMol [37] (version 1.9.0, Schrödinger, LLC, New York, NY, USA).
4. Conclusions
Lab-on-chip technology represents the future direction of analytical chemistry. Current techniques for protein coupling to solid substrates are based on formation of a covalent bond between enzyme and stationary phase. This often led to reduction or loss of enzyme activity. Ion soft landing offers an alternative approach for immobilization of enzymes to conductive supports and enables direct detection of reaction products by MALDI mass spectrometry. Surfaces prepared by ion soft landing can serve as a chip array for various kinds of biochemical assays. The immobilization of serine proteases (trypsin, chymotrypsin, and subtilisin A) on a conductive support enables manufacturing of a chip array, which was successfully used for monitoring of protein structural changes by in situ limited proteolysis and MALDI mass spectrometry. Three different serine proteases and several time points were used for monitoring of protein structural changes. While subtilisin A revealed structural information in minutes, chymotrypsin and trypsin required medium and longer incubation times, respectively. Thus, selection of time points for in situ limited proteolysis is a key parameter for each protease. Time-controlled hydrolysis of standard protein and protein/DNA complex reflects the structural rearrangement induced by cofactor removal or ligand binding.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/9/10/833/s1, RCI value calculation, Figure S1: Characterization of proteolytically active chips compatible with MALDI MS, Figure S2: FOXO4 and FOXO4-DNA complex trypsin (Try) limited proteolysis results, Figure S3: FOXO4 and FOXO4-DNA complex α-chymotrypsin (Chymo) limited proteolysis results, Figure S4: FOXO4 and FOXO4-DNA complex subtilisin A (Sub) limited proteolysis results, Figure S5: Control spots results illustration, RCI calculation script source code (Python 2.7).
Author Contributions
Methodology, M.R., P.P., and P.N.; software, M.R.; validation, M.R.; formal analysis, M.R.; investigation, M.R.; resources, P.D. and L.S.; data curation, M.R.; writing—original draft preparation, M.R.; writing—review and editing, P.P. and P.N.; visualization, M.R.; supervision, P.P.; project administration, P.N.; funding acquisition, M.R., P.D., P.P., and P.N.
Funding
This work was mainly supported by the Czech Science Foundation (grant number 16-24309S) and Grant Agency of Charles University (grant numbers 932316 and 1042217), the Ministry of Education of the Czech Republic (project LH15010, program “NPU II” project LQ1604), European Commission H2020 (EPIC-XS—grant agreement ID: 823839), and, in part, by the Czech Academy of Sciences (RVO61388971).
Acknowledgments
We acknowledge the Centre of molecular structure Core Facility at BIOCEV, a facility funded by European Regional Development Funds (CZ.1.05/1.1.00/02.0109 BIOCEV) and supported by the Czech Infrastructure for Integrative Structural Biology (LM2015043 CIISB for CMS BIOCEV funded by MEYS CR).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rusmini, F.; Zhong, Z.; Feijen, J. Protein Immobilization Strategies for Protein Biochips. Biomacromolecules 2007, 8, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.; Christena, L.R.; Rajaram, Y.R. Enzyme immobilization: An overview on techniques and support materials. 3 Biotech 2013, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pompach, P.; Benada, O.; Rosulek, M.; Darebna, P.; Hausner, J.; Ruzicka, V.; Volny, M.; Novak, P. Protein Chips Compatible with MALDI Mass Spectrometry Prepared by Ambient Ion Landing. Anal. Chem. 2016, 88, 8526–8534. [Google Scholar] [CrossRef] [PubMed]
- Grill, V.; Shen, J.; Evans, C.; Cooks, R.G. Collisions of ions with surfaces at chemically relevant energies: Instrumentation and phenomena. Rev. Sci. Instrum. 2001, 72, 3149. [Google Scholar] [CrossRef]
- Franchetti, V.; Solka, B.H.; Baitinger, W.E.; Amy, J.W.; Cooks, R.G. Soft landing of ions as a means of surface modification. Int. J. Mass Spectrom. Ion Phys. 1977, 23, 29–35. [Google Scholar] [CrossRef]
- Miller, S.A.; Luo, H.; Pachuta, S.J.; Cooks, R.G. Soft-Landing of Polyatomic Ions at Fluorinated Self-Assembled Monolayer Surfaces. Science 1997, 275, 1447–1450. [Google Scholar] [CrossRef]
- Feng, B.; Wunschel, D.S.; Masselon, C.D.; Pasa-Tolic, L.; Smith, R.D. Retrieval of DNA Using Soft-Landing after Mass Analysis by ESI-FTICR for Enzymatic Manipulation. J. Am. Chem. Soc. 1999, 121, 8961–8962. [Google Scholar] [CrossRef]
- Geiger, R.; Melnyk, M.; Busch, K.; Bartlett, M. Modifications to an analytical mass spectrometer for the soft-landing experiment. Int. J. Mass Spectrom. 1999, 182–183, 415–422. [Google Scholar] [CrossRef]
- Ouyang, Z.; Takats, Z.; Blake, T.A.; Gologan, B.; Guymon, A.J.; Wiseman, J.M.; Oliver, J.C.; Davisson, V.J.; Cooks, R.G. Preparing protein microarrays by soft-landing of mass-selected ions. Science 2003, 301, 1351–1354. [Google Scholar] [CrossRef]
- Volny, M.; Elam, W.T.; Branca, A.; Ratner, B.D.; Turecek, F. Preparative soft and reactive landing of multiply charged protein ions on a plasma-treated metal surface. Anal. Chem. 2005, 77, 4890–4896. [Google Scholar] [CrossRef]
- Volny, M.; Elam, W.T.; Ratner, B.D.; Turecek, F. Preparative soft and reactive landing of gas-phase ions on plasma-treated metal surfaces. Anal. Chem. 2005, 77, 4846–4853. [Google Scholar] [CrossRef] [PubMed]
- Badu-Tawiah, A.K.; Wu, C.; Cooks, R.G. Ambient ion soft landing. Anal. Chem. 2011, 83, 2648–2654. [Google Scholar] [CrossRef] [PubMed]
- Krasny, L.; Pompach, P.; Strohalm, M.; Obsilova, V.; Strnadova, M.; Novak, P.; Volny, M. In-situ enrichment of phosphopeptides on MALDI plates modified by ambient ion landing. J. Mass Spectrom. 2012, 47, 1294–1302. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zheng, Q.; Badu-Tawiah, A.K.; Xiong, C.; Guan, C.; Chen, S.; Nie, Z.; Wang, D.; Wan, L. Electrospray soft-landing for the construction of non-covalent molecular nanostructures using charged droplets under ambient conditions. Chem. Commun. (Camb) 2016, 52, 13660–13663. [Google Scholar] [CrossRef] [PubMed]
- Pompach, P.; Novakova, J.; Kavan, D.; Benada, O.; Ruzicka, V.; Volny, M.; Novak, P. Planar Functionalized Surfaces for Direct Immunoaffinity Desorption/Ionization Mass Spectrometry. Clin. Chem. 2015, 68, 270–278. [Google Scholar] [CrossRef]
- Darebna, P.; Spicka, J.; Kucera, R.; Topolcan, O.; Navratilova, E.; Ruzicka, V.; Volny, M.; Novak, P.; Pompach, P. Detection and Quantification of Carbohydrate-Deficient Transferrin by MALDI-Compatible Protein Chips Prepared by Ambient Ion Soft Landing. Clin. Chem. 2018, 64, 1319–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Z.; Thontasen, N.; Malinowski, N.; Rinke, G.; Harnau, L.; Rauschenbach, S.; Kern, K. A close look at proteins: Submolecular resolution of two- and three-dimensionally folded cytochrome c at surfaces. Nano Lett. 2012, 12, 2452–2458. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, M. Induction of biological activity by limited proteolysis. Annu. Rev. Biochem. 1967, 36, 55–76. [Google Scholar] [CrossRef]
- Schejter, A.; Goldkorn, T.; Sokolovsky, M. Limited Proteolysis of Horse Heart Cytochrome c. Eur. J. Biochem. 1971, 20, 414–419. [Google Scholar] [CrossRef]
- Foster, J.F.; Wilson, W.D. Conformation-dependent limited proteolysis of bovine plasma albumin by an enzyme present in commercial albumin preparations. Biochemistry 1971, 10, 1772–1780. [Google Scholar] [CrossRef]
- Cleveland, D.W.; Fischer, S.G.; Kirschner, M.W.; Laemmli, U.K. Peptide mapping by limited proteolysis in sodium dodecyl sulfate and analysis by gel electrophoresis. J. Biol. Chem. 1977, 252, 1102–1106. [Google Scholar] [PubMed]
- Brockerhoff, S.E.; Edmonds, C.G.; Davis, T.N. Structural analysis of wild-type and mutant yeast calmodulins by limited proteolysis and electrospray ionization mass spectrometry. Protein Sci. Publ. Protein Soc. 1992, 1, 504–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karas, M.; Hillenkamp, F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal. Chem. 1988, 60, 2299–2301. [Google Scholar] [CrossRef] [PubMed]
- Schopper, S.; Kahraman, A.; Leuenberger, P.; Feng, Y.; Piazza, I.; Muller, O.; Boersema, P.J.; Picotti, P. Measuring protein structural changes on a proteome-wide scale using limited proteolysis-coupled mass spectrometry. Nat. Protoc. 2017, 12, 2391–2410. [Google Scholar] [CrossRef]
- Suckau, D.; Kohl, J.; Karwath, G.; Schneider, K.; Casaretto, M.; Bitter-Suermann, D.; Przybylski, M. Molecular epitope identification by limited proteolysis of an immobilized antigen-antibody complex and mass spectrometric peptide mapping. Proc. Natl. Acad. Sci. USA 1990, 87, 9848–9852. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chait, B.T. Protein Epitope Mapping By Mass Spectrometry. Anal. Chem. 1994, 66, 3723–3726. [Google Scholar] [CrossRef]
- Cohen, S.L.; Ferre-D’Amare, A.R.; Burley, S.K.; Chait, B.T. Probing the solution structure of the DNA-binding protein Max by a combination of proteolysis and mass spectrometry. Protein Sci. Publi. Protein Soc. 1995, 4, 1088–1099. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; De Franceschi, G.; Kahraman, A.; Soste, M.; Melnik, A.; Boersema, P.J.; de Laureto, P.P.; Nikolaev, Y.; Oliveira, A.P.; Picotti, P. Global analysis of protein structural changes in complex proteomes. Nat. Biotechnol. 2014, 32, 1036–1044. [Google Scholar] [CrossRef]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Lin, L.; Pinker, R.J.; Forde, K.; Rose, G.D.; Kallenbach, N.R. Molten globular characteristics of the native state of apomyoglobin. Nat. Struct. Mol. Biol. 1994, 1, 447–452. [Google Scholar] [CrossRef]
- Kim, Y.J.; Kim, Y.A.; Park, N.; Son, H.S.; Kim, K.S.; Hahn, J.H. Structural characterization of the molten globule state of apomyoglobin by limited proteolysis and HPLC-mass spectrometry. Biochemistry 2005, 44, 7490–7496. [Google Scholar] [CrossRef] [PubMed]
- Boura, E.; Rezabkova, L.; Brynda, J.; Obsilova, V.; Obsil, T. Structure of the human FOXO4-DBD-DNA complex at 1.9 A resolution reveals new details of FOXO binding to the DNA. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 1351–1357. [Google Scholar] [CrossRef] [PubMed]
- Laboratory of Structural Biology and Cell Signaling, Institute of Microbiology of the CAS. Linx: Software for Interpretation of High-Resolution MS Data Obtained after Protein Chemical Cross-Linking. 2019. Available online: http://peterslab.org/downloads.php (accessed on 9 August 2019).
- Mrazova, B.; Martinkova, M.; Martinek, V.; Frei, E.; Stiborova, M. Optimalization of preparation of apo-cytochrome b(5) utilizing apo-myoglobin. Interdiscip. Toxicol. 2008, 1, 190–192. [Google Scholar] [CrossRef] [PubMed]
- Kavan, D.; Man, P. MSTools—Web based application for visualization and presentation of HXMS data. Int. J. Mass Spectrom. 2011, 302, 53–58. [Google Scholar] [CrossRef]
- Maurus, R.; Overall, C.M.; Bogumil, R.; Luo, Y.; Mauk, A.G.; Smith, M.; Brayer, G.D. A myoglobin variant with a polar substitution in a conserved hydrophobic cluster in the heme binding pocket. Biochim. Biophys. Acta 1997, 1341, 1–13. [Google Scholar] [CrossRef]
- Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 1.8; Schrodinger, LLC: New York, NY, USA, 2015. [Google Scholar]
Figure 1.
hMyo sequence coverage map. A full sequence coverage was obtained after two hours of digestion for all proteases (trypsin: yellow, α-chymotrypsin: red, subtilisin A: dark blue). Sequence coverage of hMyo after 15 min of digestion by trypsin (light blue) and α-chymotrypsin (pink). Subtilisin A (green) digestion of the hMyo after 15 min provided higher number of overlapping peptides compared to trypsin and chymotrypsin.
Figure 1.
hMyo sequence coverage map. A full sequence coverage was obtained after two hours of digestion for all proteases (trypsin: yellow, α-chymotrypsin: red, subtilisin A: dark blue). Sequence coverage of hMyo after 15 min of digestion by trypsin (light blue) and α-chymotrypsin (pink). Subtilisin A (green) digestion of the hMyo after 15 min provided higher number of overlapping peptides compared to trypsin and chymotrypsin.

Figure 2.
Limited tryptic 15-min proteolysis of aMyo and hMyo forms. (a) Butterfly plot visualization of the limited proteolysis experiment for aMyo and hMyo forms. The cleavage sites 42, 133, and 145 were preferentially cleaved in hMyo, whereas sites 45 and 96 were compromised. (b) The structure of hMyo with preferred cleavage sites is highlighted in green. The highlighted regions in red color, which include residues 45 and 96, are located close to the heme binding region and are less accessible for protease when heme is present. Heme molecule is highlighted in blue.
Figure 2.
Limited tryptic 15-min proteolysis of aMyo and hMyo forms. (a) Butterfly plot visualization of the limited proteolysis experiment for aMyo and hMyo forms. The cleavage sites 42, 133, and 145 were preferentially cleaved in hMyo, whereas sites 45 and 96 were compromised. (b) The structure of hMyo with preferred cleavage sites is highlighted in green. The highlighted regions in red color, which include residues 45 and 96, are located close to the heme binding region and are less accessible for protease when heme is present. Heme molecule is highlighted in blue.

Figure 3.
The secondary structure motifs of FOXO4 (a) are displayed at the top; dash-dotted lines cover the region Lys 45–Phe 87 which remains resistant to proteolysis in the presence of DNA. Relative Amino Acid Cleavage Incidence (RCI) value butterfly plots of FOXO4 and FOXO4/DAF16 DNA complex at different time points: 2 min for subtilisin A (b), 5 min for α-chymotrypsin (c), and 10 min for trypsin (d) where major differences were observed.
Figure 3.
The secondary structure motifs of FOXO4 (a) are displayed at the top; dash-dotted lines cover the region Lys 45–Phe 87 which remains resistant to proteolysis in the presence of DNA. Relative Amino Acid Cleavage Incidence (RCI) value butterfly plots of FOXO4 and FOXO4/DAF16 DNA complex at different time points: 2 min for subtilisin A (b), 5 min for α-chymotrypsin (c), and 10 min for trypsin (d) where major differences were observed.

Figure 4.
Schematic representation (a) and real-life picture (b) of the ambient ion-landing device. (1) linear Hamilton syringe sample loading; (2) fused silica capillary; (3) nebulizer; (4) heated nebulizing and drying gas source; (5) heated desolvation tube; (6) ITO glass MALDI-compatible support; (7) movable support holder controlled by stepper motors; (8) high-voltage power supply.
Figure 4.
Schematic representation (a) and real-life picture (b) of the ambient ion-landing device. (1) linear Hamilton syringe sample loading; (2) fused silica capillary; (3) nebulizer; (4) heated nebulizing and drying gas source; (5) heated desolvation tube; (6) ITO glass MALDI-compatible support; (7) movable support holder controlled by stepper motors; (8) high-voltage power supply.

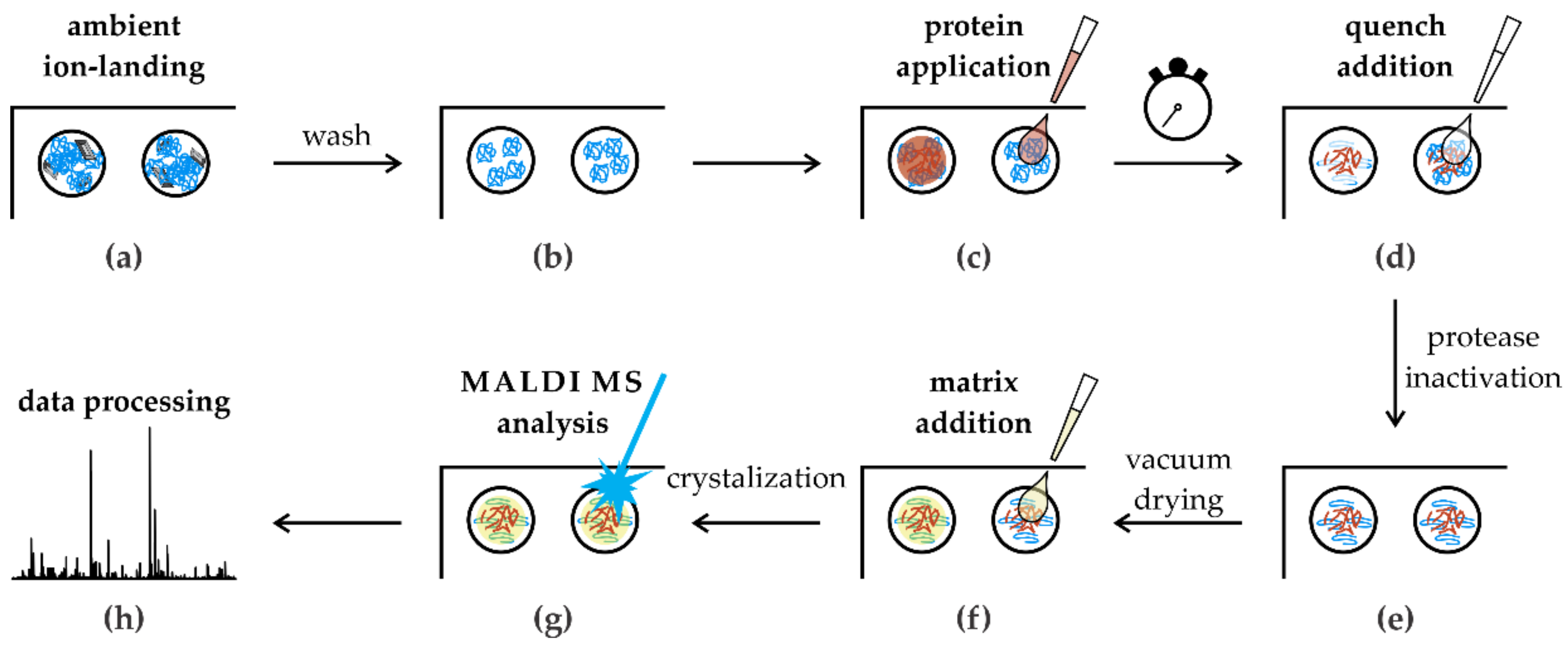
Figure 5.
Scheme of the in situ limited proteolysis workflow. (a) Protease spots containing solvent impurities; (b) only ion-landed molecules remained on spots after the wash; (c) protein sample deposition on the dried spots; (d) proteolytic reaction was stopped after the incubation time by acidification; (e) quenched reaction mixture was dried in vacuum; (f) all spots were overlaid by the MALDI matrix solution; (g) MALDI MS analysis; (h) semiautomatic data processing.
Figure 5.
Scheme of the in situ limited proteolysis workflow. (a) Protease spots containing solvent impurities; (b) only ion-landed molecules remained on spots after the wash; (c) protein sample deposition on the dried spots; (d) proteolytic reaction was stopped after the incubation time by acidification; (e) quenched reaction mixture was dried in vacuum; (f) all spots were overlaid by the MALDI matrix solution; (g) MALDI MS analysis; (h) semiautomatic data processing.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Rosulek, M.; Darebna, P.; Pompach, P.; Slavata, L.; Novak, P. Proteases Immobilization for In Situ Time-Limited Proteolysis on MALDI Chips. Catalysts 2019, 9, 833. https://doi.org/10.3390/catal9100833
AMA Style
Rosulek M, Darebna P, Pompach P, Slavata L, Novak P. Proteases Immobilization for In Situ Time-Limited Proteolysis on MALDI Chips. Catalysts. 2019; 9(10):833. https://doi.org/10.3390/catal9100833
Chicago/Turabian StyleRosulek, Michal, Petra Darebna, Petr Pompach, Lukas Slavata, and Petr Novak. 2019. "Proteases Immobilization for In Situ Time-Limited Proteolysis on MALDI Chips" Catalysts 9, no. 10: 833. https://doi.org/10.3390/catal9100833
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.