Direct Catalytic Conversion of CO2 to Cyclic Organic Carbonates under Mild Reaction Conditions by Metal—Organic Frameworks


Department of Chemistry and Protein Research Center for Bio-Industry, Hankuk University of Foreign Studies, Yongin 17035, Korea
Catalysts 2019, 9(1), 34; https://doi.org/10.3390/catal9010034
Submission received: 20 November 2018
/
Revised: 16 December 2018
/
Accepted: 21 December 2018
/
Published: 2 January 2019
(This article belongs to the Special Issue Catalysis by Metal-Organic Frameworks)
Abstract
:The reduction of the representative greenhouse gas, carbon dioxide (CO2), is significantly an important theme for the current research in the modern chemical world. For the last two decades, the development of new metal-organic framework (MOF) systems with highly selective capture of CO2, in the presence of other competing gaseous molecules, has flourished to capture or separate CO2 for environmental protection. Nonetheless, the ultimate resolution to lessen the atmospheric CO2 concentration may be in the chemical or electrochemical conversion of CO2 to other compounds. In this context, the catalytic cycloaddition reaction of CO2 into organic epoxides to produce cyclic carbonates is a more attractive method. MOFs are being proven as efficient heterogeneous catalytic systems for this important reaction. In this review, we collected very recent progress in MOF-based catalytic systems, fully operable under very mild reaction conditions (room temperature and 1 atm CO2).
1. Introduction
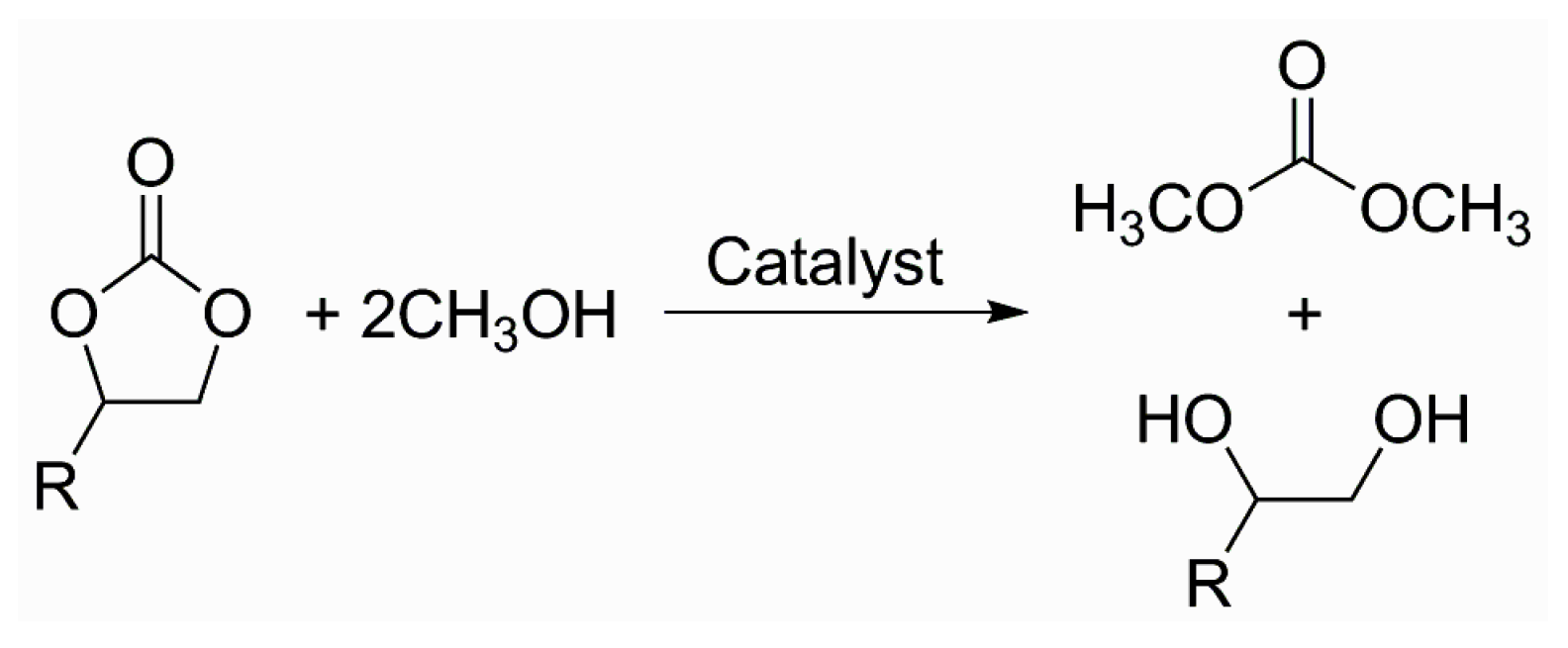
The selective uptake of CO2 by a range of porous materials is continuously attracting many researchers to seek for better sorbent materials for CO2 at high sorption temperatures and under humid conditions [1,2,3,4,5,6,7,8,9]. The abrupt climate change mainly caused by the increase of CO2 concentration and subsequent atmospheric temperature is a crucial issue for the protection of the global environment [10]. In this regard, there are numerous review articles used to collect the MOFs exhibiting selective sorption ability for CO2 over other competing gases with high efficacy [1,2,3,4,5,6,7,8,9]. The MOFs with large available void spaces sometimes outperform other porous materials in CO2 sorption applications. MOFs can also be shaped as membranes or thin films for effective separation of the CO2 from other gases [11,12,13]. Nonetheless, the direct conversion of CO2 into more valuable organic compounds may be an alternative option for the reduction and utilization of CO2. CO2 is considered as a more useful and environmentally benign C1 source than other C1 feedstocks in the industry. Cyclic organic carbonates can be directly synthesized from various epoxides and gas phase CO2 in the presence of the Lewis acid catalyst and tetra-n-butylammonium bromide (nBu4NBr, TBABr) as a cocatalyst, as shown in Scheme 1 [14,15,16]. The reaction is a direct incorporation of CO2 into epoxides to lead to cyclic organic carbonates. Cyclic carbonates are value-added compounds because they are good and greener precursors for the preparation of dimethyl carbonate (DMC), a very useful compound in the oil industry and battery technology, as shown in Scheme 2 [17]. Furthermore, no byproducts were usually observed for the cycloaddition reaction.
Although there are many heterogeneous catalytic systems for the cycloaddition reaction, the reaction conditions usually require high temperature or pressure. Therefore, better catalytic systems operable under mild conditions are sought for recently. Interestingly, it has been demonstrated that MOF-based catalytic systems are an ideal catalytic system for the cycloaddition of CO2 with epoxides, under various reaction conditions. There are several excellent review articles in this field [18,19,20,21]. Therefore, in this contribution we mainly focus on very recently reported MOF-based catalytic systems that are fully operable at room temperature and 1 atm CO2. The sophisticated design of the MOF-based catalytic systems can lead to very competing, economic, and recyclable CO2 conversion processes.
2. Mechanism of the Cycloaddition of CO2 with Epoxides
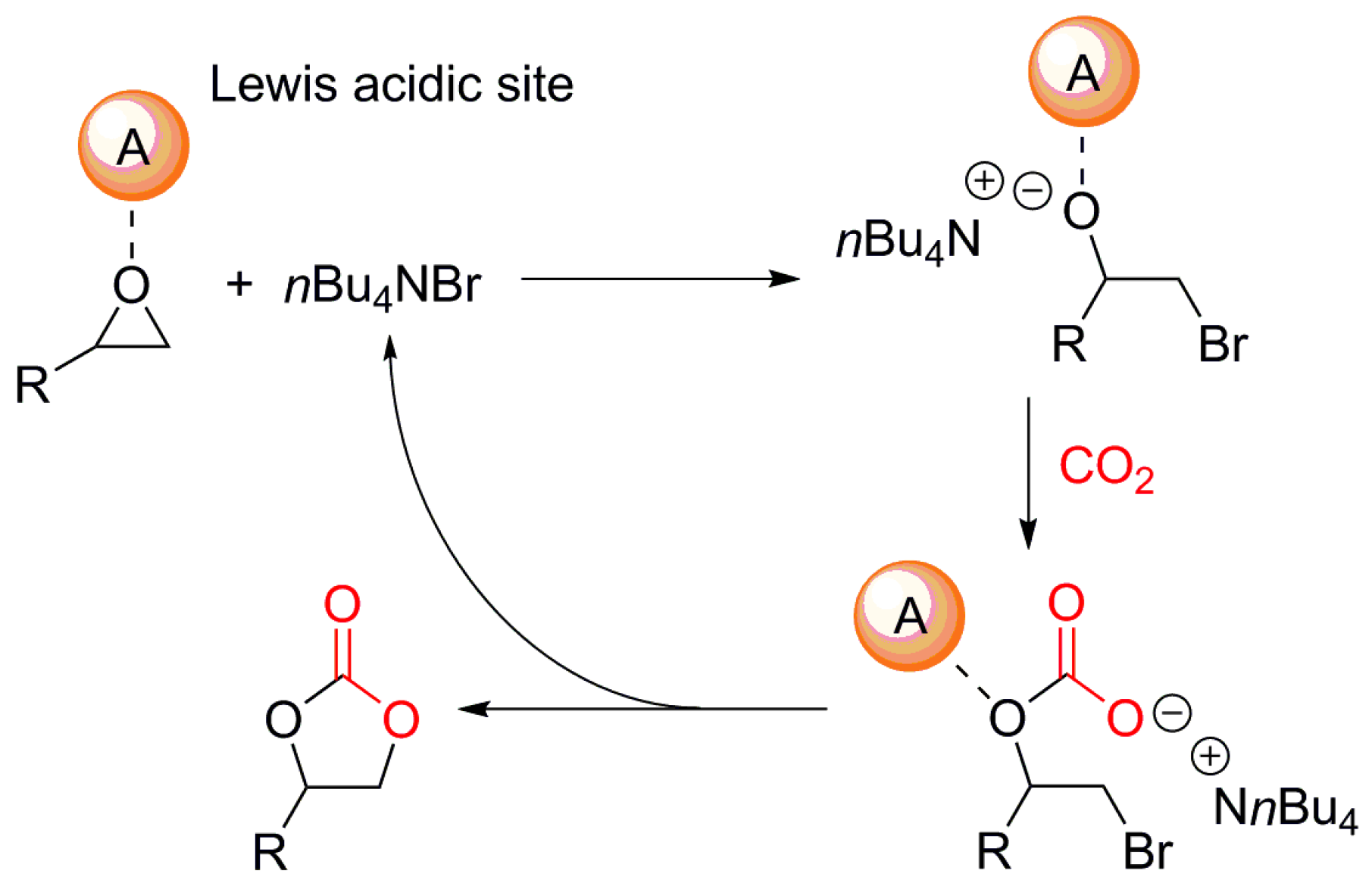
In order to develop efficient MOF-based catalytic systems, we needed to get familiar with the mechanistic information of the cycloaddition reaction between the epoxides and CO2. The catalytic incorporation of CO2 into epoxides in the presence of both Lewis acid catalyst and TBABr cocatalyst tends to proceed through the following three key reaction steps [22]:
- (i)
- Lewis acid catalyst activates the epoxide.
- (ii)
- Nucleophilic attack by a cocatalyst (usually Br− ion from TBABr) to lead to an alkoxide.
- (iii)
- The intermediate alkoxide reacts with CO2 to provide the required cyclic carbonate.
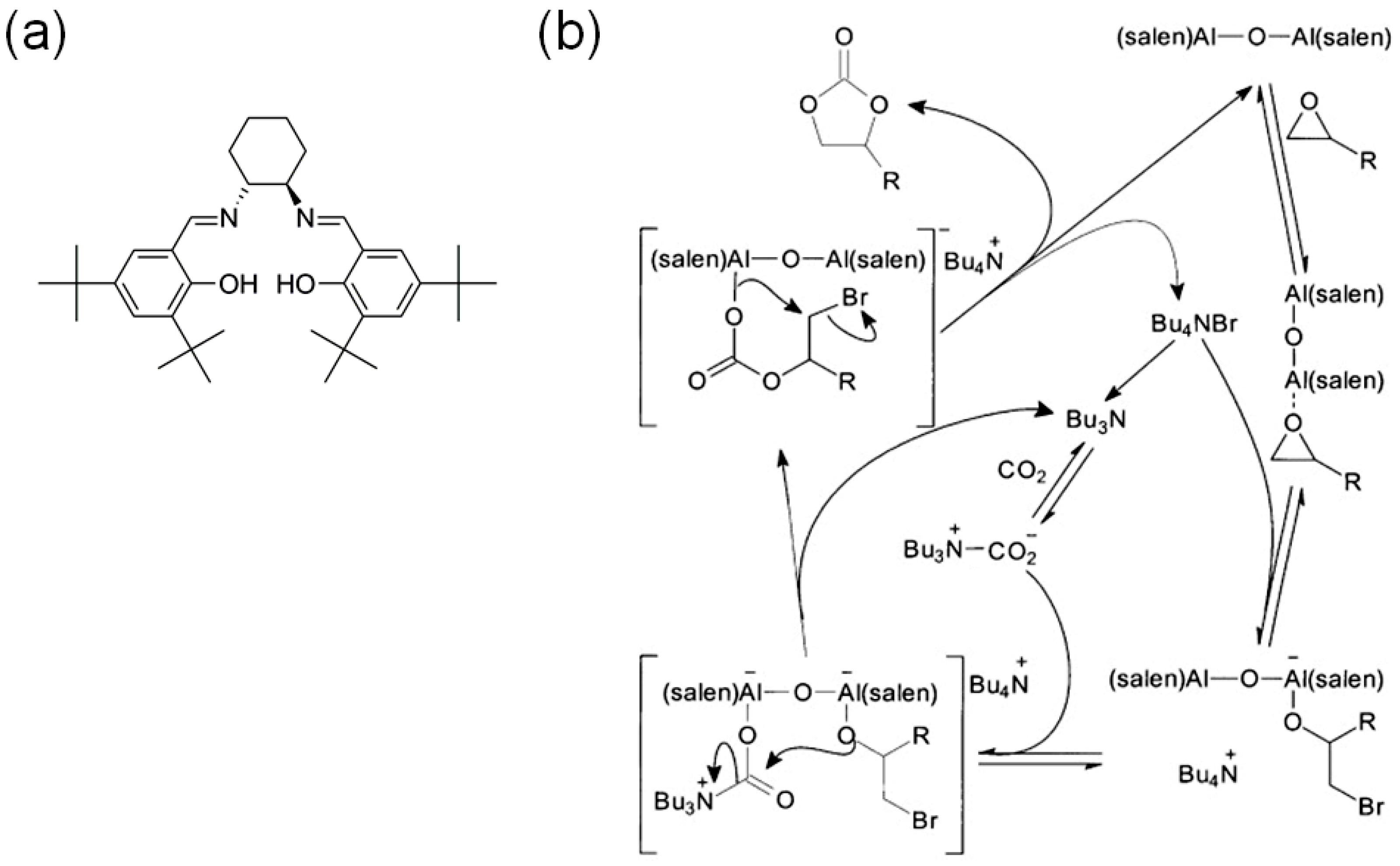
In step (i), the Lewis acid catalyst can accelerate the reaction through coordination of epoxide. Then, Br− nucleophile from TBABr cocatalyst can activate epoxides to give a ring-opened bromoalkoxide in step (ii). The ring-opened bromoalkoxide can be further reacted with CO2 in the final step (iii). The subsequent cyclization step provides cyclic carbonates and TBABr cocatalyst as shown in Scheme 1. However, the additional role of TBABr has been revealed by North and Pasquale through a series of controlled kinetic experiments through the use of an oxo-bridged dinuclear Al-salen catalyst, (salen)Al-O-Al(salen) [22]. Notably, the bimolecular rate dependence on the concentration of TBABr clearly suggested that the Lewis base nBu3N possibly derived from TBABr can play a key role in the activation of CO2 to form carbamate salt, which can often be considered as an activated form of CO2:
rate = k[epoxide][CO2][Catalyst][TBABr]2 (where Catalyst is a dinuclear Al-salen complex)
As a result, the proposed catalytic cycle in this case is rather complex than the previous one as shown in Scheme 3. Although this example is a homogeneous catalytic system, it can be inferred that the cooperative activation of both the epoxide and CO2 by bifunctional Lewis acid-base catalysts may be responsible for the good catalytic performances. The solvent-free cycloaddition reaction of propylene oxide with CO2 was catalyzed by (salen)Al-O-Al(salen) at room temperature and 1 atm CO2 [23]. The conversion was 77% for 3 h (TON = 30.8, TOF = 10.3 h−1). The conversion of phenylethylene oxide under the same conditions for 24 h reached up to 98% (TON = 39.2, TOF = 1.63 h−1).
3. MOF-Based Catalytic Systems
3.1. Acid Site-Rich MOF Catalytic Systems
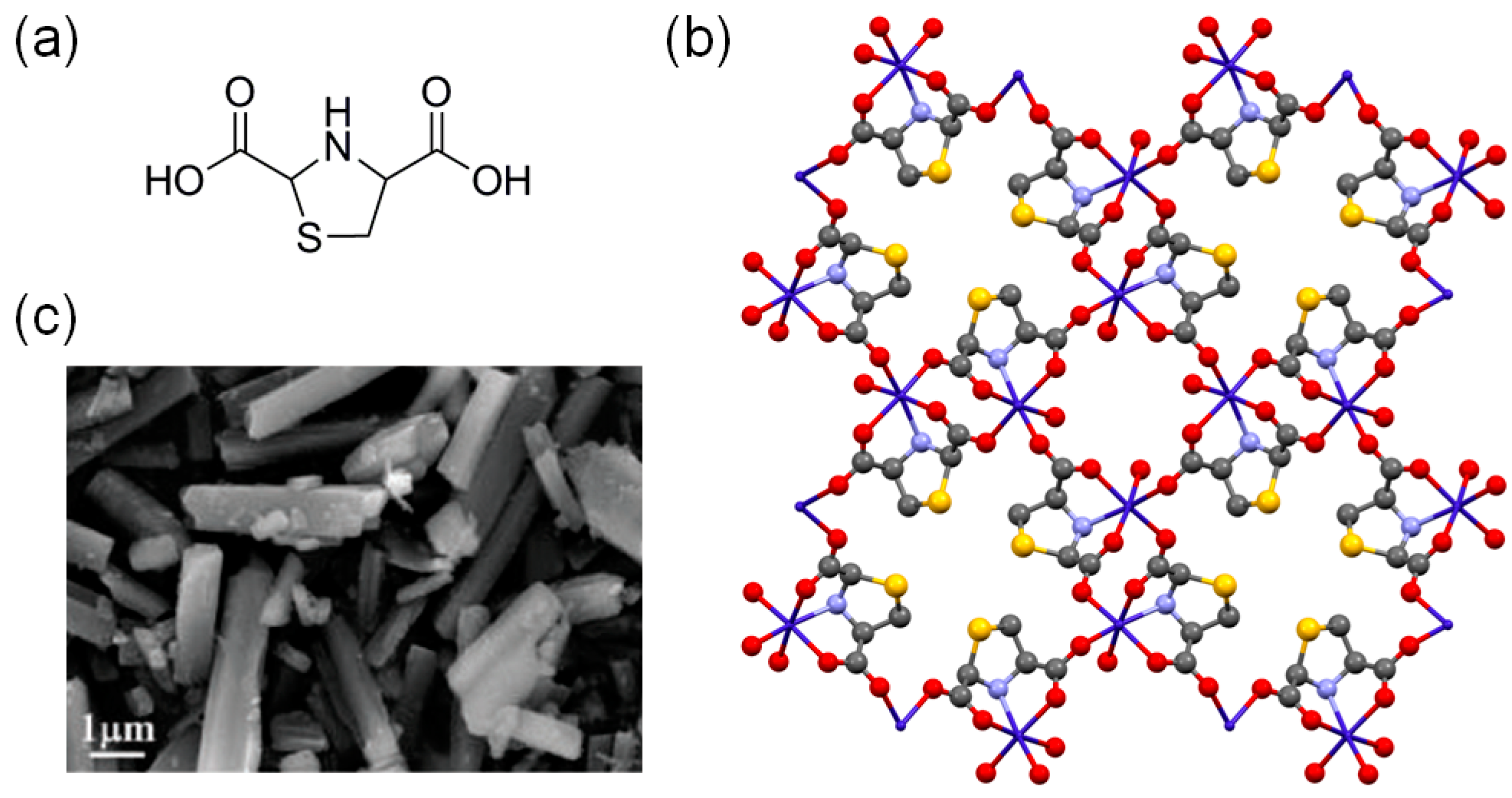
As it can be seen in the earlier section, the Lewis acid sites are active centers for the cycloaddition of CO2 into various epoxide substrates. The acid reaction centers can be systematically generated in the MOFs by a simple dehydration of coordinated aqua ligands under suitable conditions. For instance, the dehydration of HKUST-1, [Cu3(BTC)2(H2O)3]n, where BTC is 1,3,5-benzenetricarboxylate, has been successfully demonstrated to lead to well-defined openly accessible Lewis acid sites [24]. Similarly, the dehydrated form of the Lewis acid site-rich Co-MOF, {Co(μ3-L)(H2O)]·0.5H2O}n, where L is thiazolidine 2,4-dicarboxylate (Figure 1), showed 98.2% of conversion efficiency for converting propylene oxide into propylene carbonate at 50 °C and 1 atm CO2 [25]. The Co-MOF contains well-defined one-dimensional (1D) channels with pore dimensions of ~8.32 × 8.32 Å2. The Co-MOF could be recycled at least five times without a significant loss of activity. Although the reaction conversion at 25 °C was relatively low (19.3% for 48 h), its nano-sized analogs showed enhanced catalytic activities compared to the bulk Co-MOF. The nano-sized Co-MOFs were easily prepared by employing two conventional surface capping ligands: polyvinylpyrrolidone (PVP) or n-hexadecyltrimethylammonium bromide (CTABr).
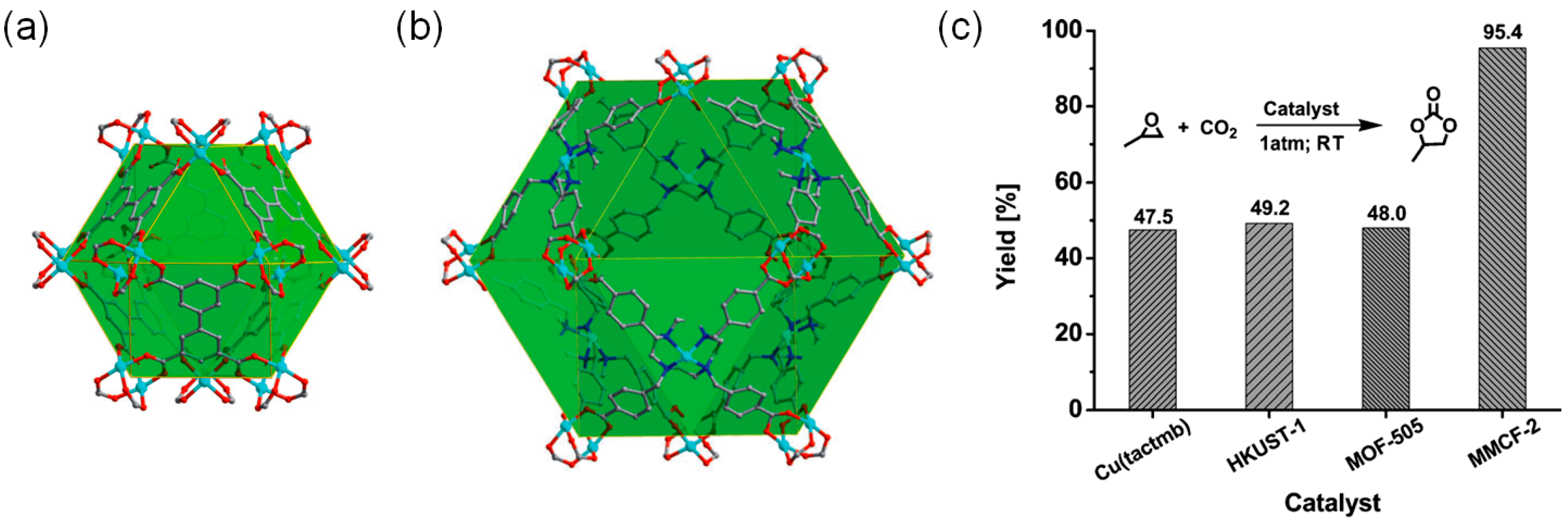
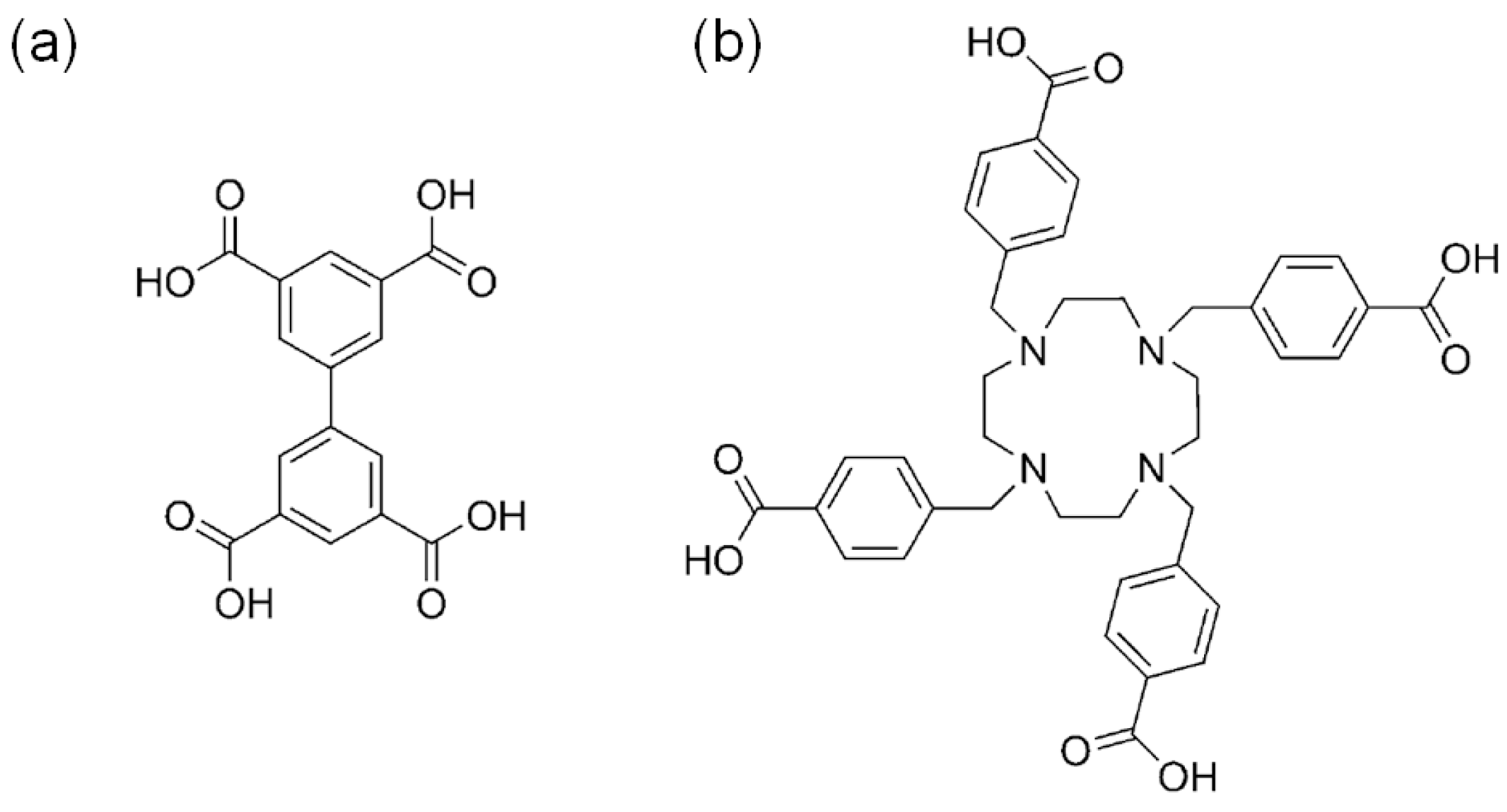
Ma and coworkers also developed a Cu-MOF catalytic system, [Cu2(Cu-tactmb)(H2O)3(NO3)2] (MMCF-2), with well-defined Cu(II) acidic sites by using a tetratopic tactmb (1,4,7,10-tetrazacyclododecane-N,N′,N″,N‴-tetra-p-methylbenzoate) bridging linker as given in Scheme 4 [26]. The free aza macrocycles in the initially prepared Cu(tactmb)-MOF were further coordinated to the Cu(II) ions to form Cu-MOF, having rich Lewis acidic sites fully open toward cage-like pores. Interestingly, the nbo topology previously shown by MOF-505, in which the bridging linker was tetratopic 3,3′,5,5′-biphenyltetracarboxylate (bptc), was maintained in the MMCF-2. Both frameworks contained cuboctahedral cage-like pore structures as depicted in Figure 2. The pore dimension of the MMCF-2 was larger than that of the MOF-505. Interestingly, the cycloaddition reaction between the propylene oxide and CO2 was possible under mild conditions (room temperature and 1 atm CO2) in the presence of TBABr cocatalyst with a conversion of 95.4% (48 h). Especially, MMCF-2 is a superior catalytic system than the Cu(tactmb)-MOF, HKUST-1 and MOF-505 under the same conditions as depicted in Figure 2c. Although the catalytic activity of Cu(tactmb)-MOF may come from the cooperation of acid sites from defects and free aza macrocycles, the conversion of Cu(tactmb)-MOF (47.5%) is still much lower than that of the MMCF-2. These results clearly exemplified the merit of well-defined rich acidic sites in MMCF-2.
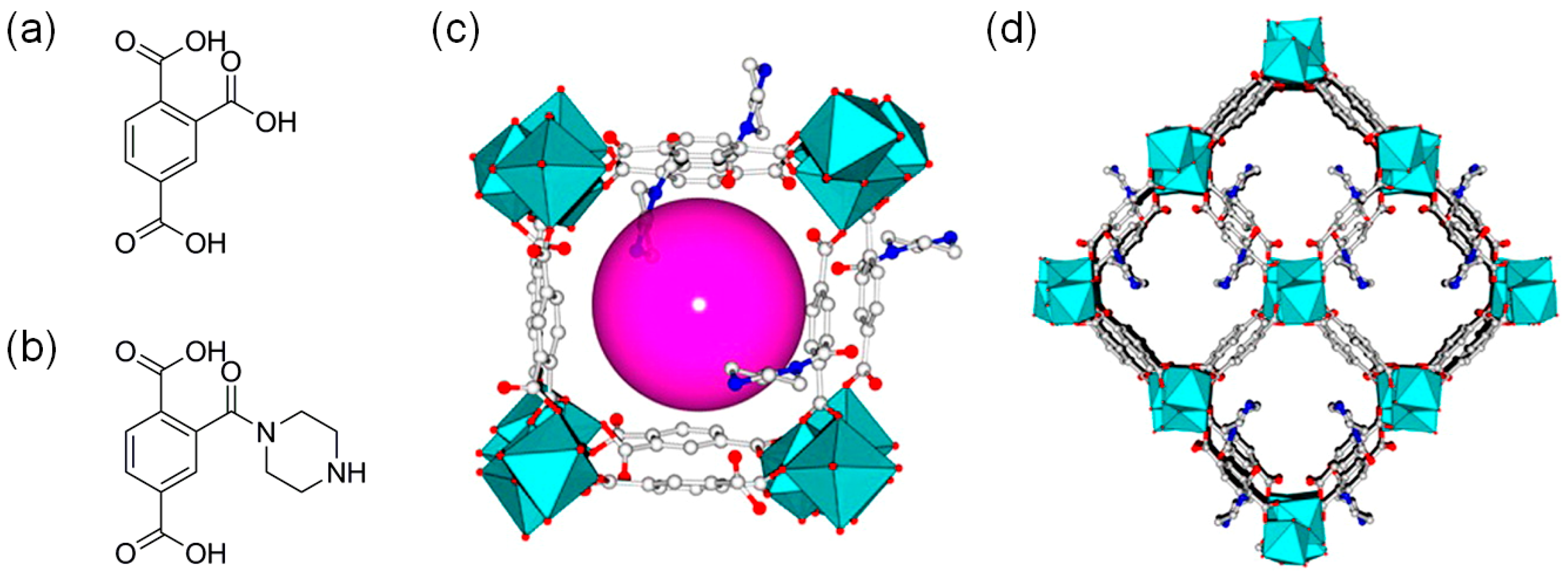
Both the Brønsted and Lewis acid sites can activate epoxides. In this context, Jiang and coworkers recently reported a robust In-MOF, In2(OH)(btc)(Hbtc)0.4(L)0.6·3H2O where btc is 1,2,4-benzenetricarboxylate and the ligand L is an in-situ generated derivative of btc with piperazine as shown in Figure 3 [27]. The 3D In-MOF contained binuclear In-OH-In subunits with 1D tubular channels. The In-MOF exhibited high CO2 uptake capacity together with the rich Lewis and Brønsted acidic sites ideal for cycloaddition reaction. The presence of both acidic sites was confirmed by the diffuse reflectance infrared Fourier transform (DRIFT) spectra using CO adsorption at 173 K. The calculated void volume of the In-MOF was 34.6%. Propylene carbonate was formed by In-MOF at room temperature and 1 atm CO2 with a conversion of 77.9% (48 h) as shown in Table 1. Other large epoxides showed decreased conversions due to the substrate diffusion issue. Five times of recycling experiments did not show a loss of activity. The synergistic effect of Lewis and Brønsted acid sites has been proposed for the confined space of In-MOF.
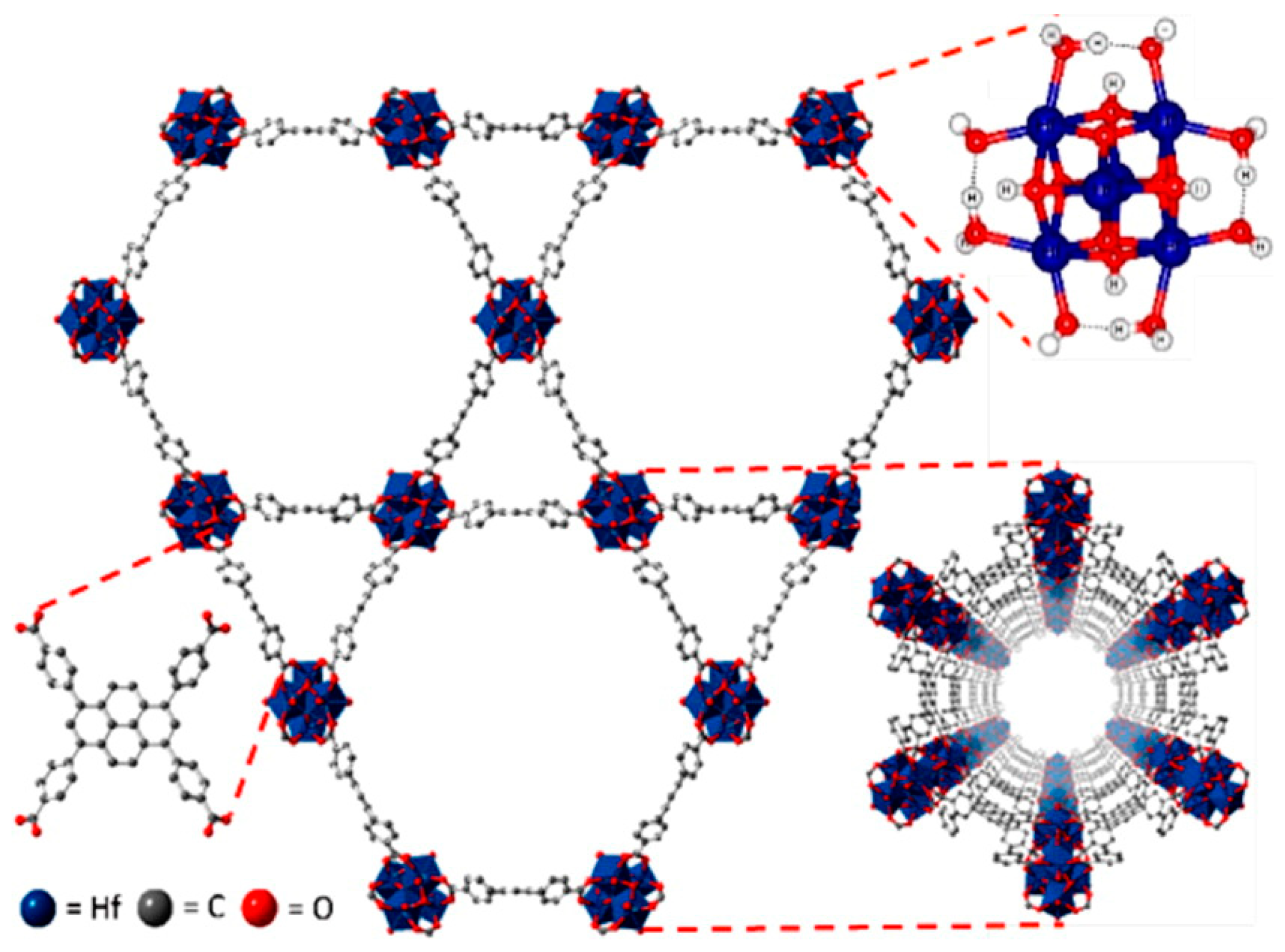
The Hf6-cluster-based MOF (Hf-NU-1000), with 1,3,6,8-tetrakis(p-benzoic acid)pyrene bridging linker having a Zr-based NU-1000 structure, was found to be a very active catalyst for the cycloaddition reaction of a range of epoxides under mild conditions (room temperature and 1 atm CO2) as shown in Table 2 [28]. Interestingly, the Hf-NU-1000 displayed bimodal porosity (13 and 29 Å) as shown in Figure 4. The large pore belongs to the mesoporous range of pore size, which is very useful for the easy diffusion of the substrates, CO2, and product. As a result, the large pore dimensions of the Hf-NU-1000 with good Brønsted acidic centers, such as -OH and aqua ligands, led to a good catalytic system. Both -OH group and aqua ligand made Hf6-cluster node to be a good proton donor for the ring opening of an epoxide. The Hf-NU-1000 was also very active in the cycloaddition of bifunctional epoxide with CO2 at a slightly elevated temperature (55 °C) as summarized in Table 2. Additionally, the Hf-NU-1000 showed good activities in the regioselective synthesis of chiral β-azidohydrins and β-alkoxy alcohols.
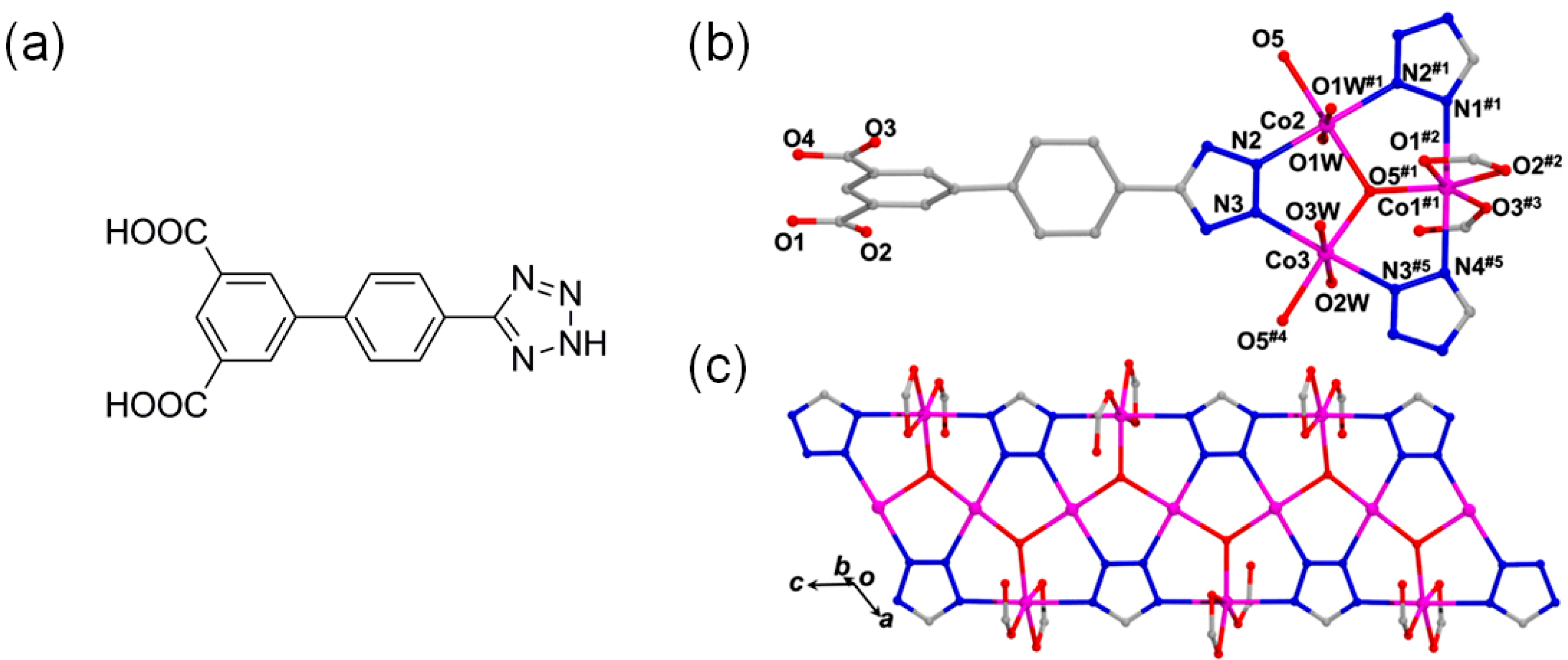
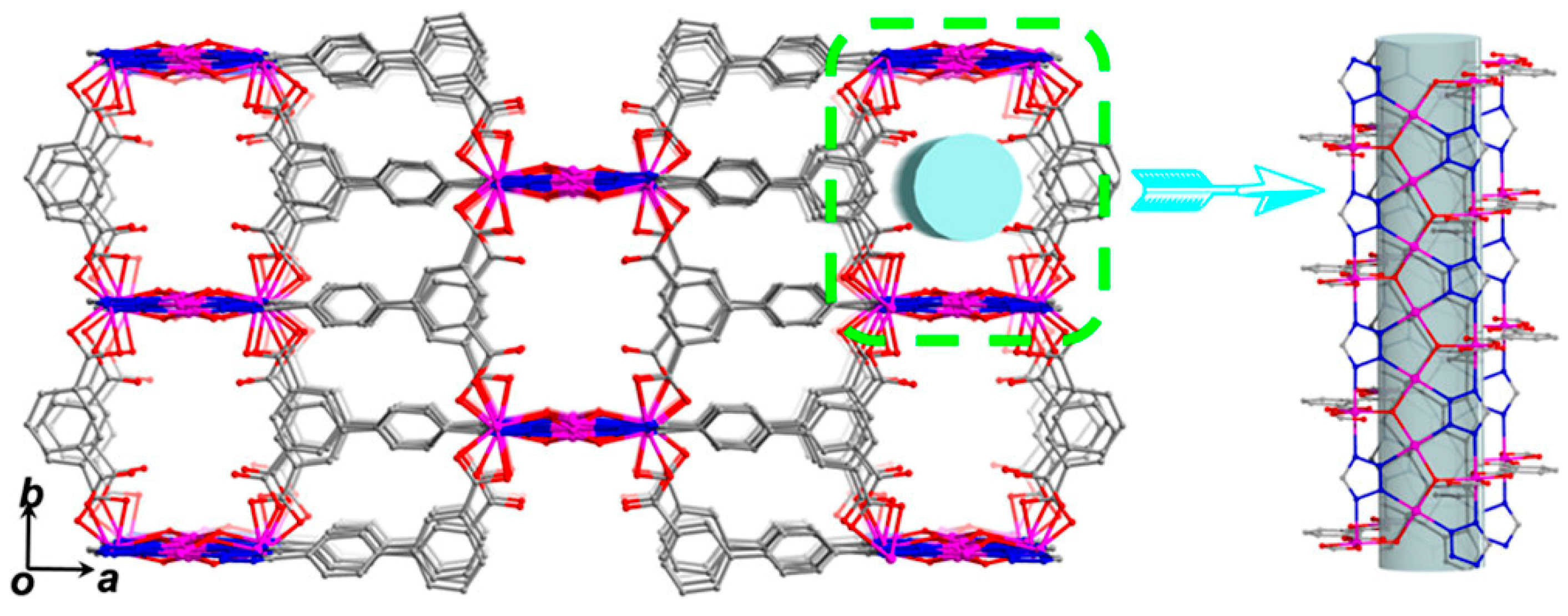
Another interesting bifunctional Co-MOF catalytic system, {[Co2(tzpa)(OH)(H2O)2]·DMF}n (H2tzpa = 5-(4-(tetrazol-5-yl)phenyl)isophthalic acid), with both Lewis and Brønsted acid sites was reported by Wang and coworkers [29]. The Co-MOF assembled from Co(II) ions and tzpa bridging ligands formed a 3D framework in which trinuclear Co cluster bridged by a μ3-OH group was further interconnected each other by three tetrazolate groups from tzpa linkers to form a ribbon-like SBU as depicted in Figure 5. The 3D framework contains well-defined 1D tubular channels with a dimension of 10.4 × 7.5 Å2 (Figure 6). The activated Co-MOF after dehydration and desolvation at 220 °C showed Brunauer-Emmett-Teller (BET) surface area of 455 m2 g−1 with a pore dimension of 4.1–7.1 Å. The removal of aqua ligands generated active Lewis acid sites on Co2 and Co3 atoms. The activated Co-MOF displayed a good cycloaddition activity for propylene oxide at room temperature and 1 atm CO2 for 48 h (yield ~93.8%). It was proposed that propylene oxide was additionally activated by Brønsted acidic μ3-OH groups.
3.2. Cooperative Bifunctional Acid-Base Catalytic Systems
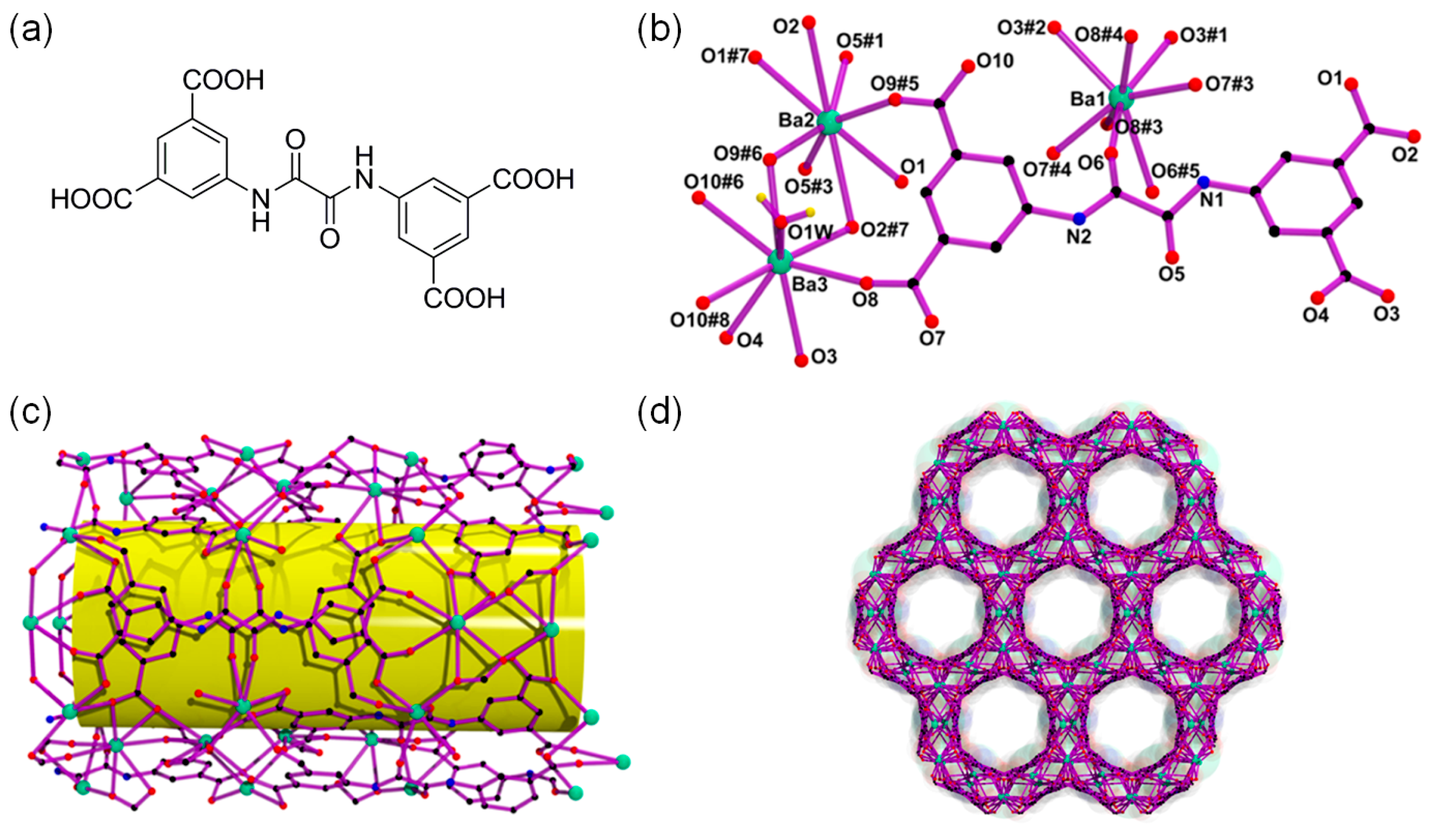
Even though the cycloaddition of CO2 into epoxides is primarily catalyzed by the Lewis acid sites, a growing number of reports revealed that the cooperative activation of both epoxide substrate and CO2 by the bifunctional Lewis acid-base catalytic system is a very favorable case for mild reactions [30,31,32,33,34]. For example, Hou and coworkers described the 3D Ba-MOF formulated as [Ba2(BDPO)(H2O)] consisting of Lewis basic oxalamide-containing bridging linkers using N,N′-bis(isophthalic acid)-oxalamide (H4BDPO) as depicted in Figure 7 [30]. The Ba-MOF contains hexagonally oriented 1D honeycomb channels with a window dimension of 8.2 Å. The Ba-MOF exhibited excellent catalytic CO2 cycloaddition activities under mild reaction conditions due to the presence of both Lewis acidic metal sites and basic oxalamide groups as given in Table 3. The high catalytic activities under the mild conditions were attributed to the rodlike metal-organic chains, which can be ideal for the activation of epoxides. Meanwhile, the CO2-philic oxalamide groups tended to interact with CO2 molecules to effectively assist the catalytic reaction. However, the conversion became lower for the larger epoxides. This means the reaction mainly occurred inside the MOF pores. The recyclability of the Ba-MOF was tested using the cycloaddition of CO2 into epichlorohydrin. Five consecutive runs did not show loss of activity.
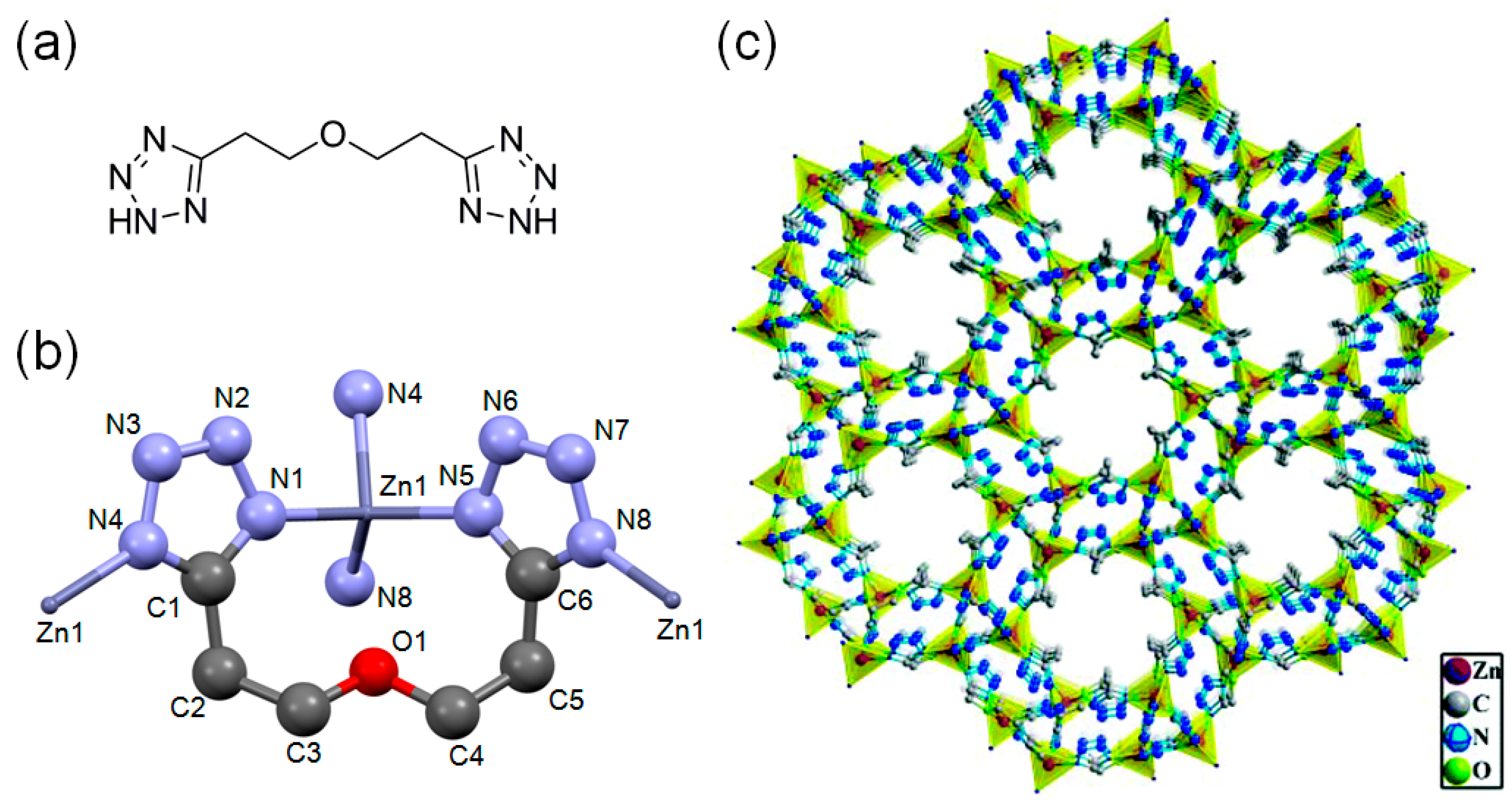
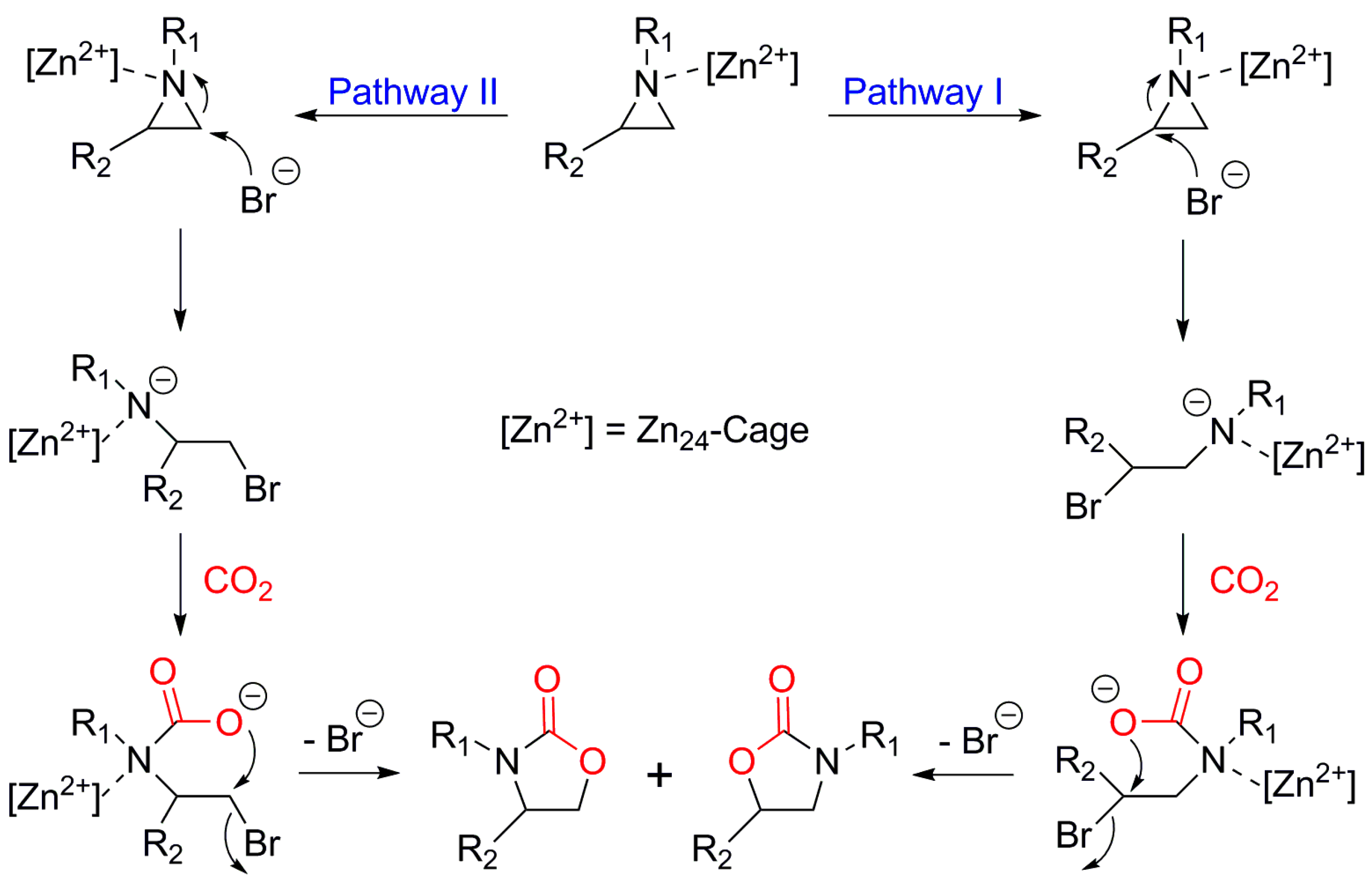
Zeolite-like microporous Zn-btz-MOF, {[Zn(btz)]·DMF·0.5H2O}n (H2btz = 1,5-bis(5-tetrazole)-3-oxapentane), showed very excellent stability in several common organic solvents and solutions of the pH range of 1–14 [31]. The Zn-btz-MOF also showed very high CO2 uptake capacity at 273 K and 1 atm of CO2 (8.09 mmol g−1) with a large BET surface area of 1151 m2 g−1. Figure 8 shows the chemical structure of H2btz ligand and crystal structure of the Zn-btz-MOF, which is indicative of plenty of openly accessible Lewis basic sites in the tetrazole rings of btz linkers. In the reaction of phenylethylene oxide with CO2 at 30 °C under 1 atm CO2, the conversion was relatively low (39%). If we consider the bulkiness of the phenylethylene oxide, the conversion of the smaller propylene oxide would be much higher under the same conditions. Upon the increase of the reaction temperature to 70 °C, the reaction was almost completed (> 99%) within 12 h for phenylethylene oxide. This excellent reactivity of the Zn-btz-MOF was attributed to; (i) nanoscale [Zn24] cages for efficient enrichment of epoxides and CO2, and (ii) prevention of direct coordination of the Br− nucleophile to Zn(II) ion due to the sterically demanding Zn(II) ion sites. Additionally, the rich Lewis basic sites also contributed in the acceleration of the reactions through effective acid-base interactions with quadrupolar CO2. The authors successfully demonstrated 10 times of recycling of the Zn-btz-MOF. Interestingly, the Zn-btz-MOF could also catalyze the cycloaddition of a series of aziridines with CO2 at elevated temperatures and pressure. The proposed reaction mechanism for the reaction of aziridines with CO2 is depicted in Figure 9. In this case, there were two proposed reaction pathways, I and II, that led to two regioisomers. Based on the distribution of the two regioisomers, it was concluded that pathway I was a main reaction pathway.
The microporous anionic 3D Zn-MOF containing 4,4′,4″-(pyridine-2,4,6-triyl)tribenzoate (PTB) linker contained well-defined square channels as depicted in Figure 10 [32]. The Zn-MOF exhibited excellent catalytic activities. For instance, in the case of the reaction between the propylene oxide and CO2, the conversion was as high as 92.1% at room temperature under 1 atm CO2 for 48 h. This high activity of Zn-MOF was attributed to the cooperative activation process of the Lewis acidic site for epoxide and anionic framework for nBu4N+ ion. The Lewis basic pyridyl sites were also thought to be involved in the favorable interactions with CO2 to enrich the CO2 molecules near the acid reaction centers. Therefore, the Zn-MOF catalytic system featured a very effective cooperativity in the cycloaddition reaction of the epoxides and CO2. The substrate size-dependent activities were also observed as summarized in Table 4. The larger the dimensions of the epoxide substrates, the lower the conversions. These results clearly confirm that the catalytic sites were mainly located inside the Zn-MOF channels.
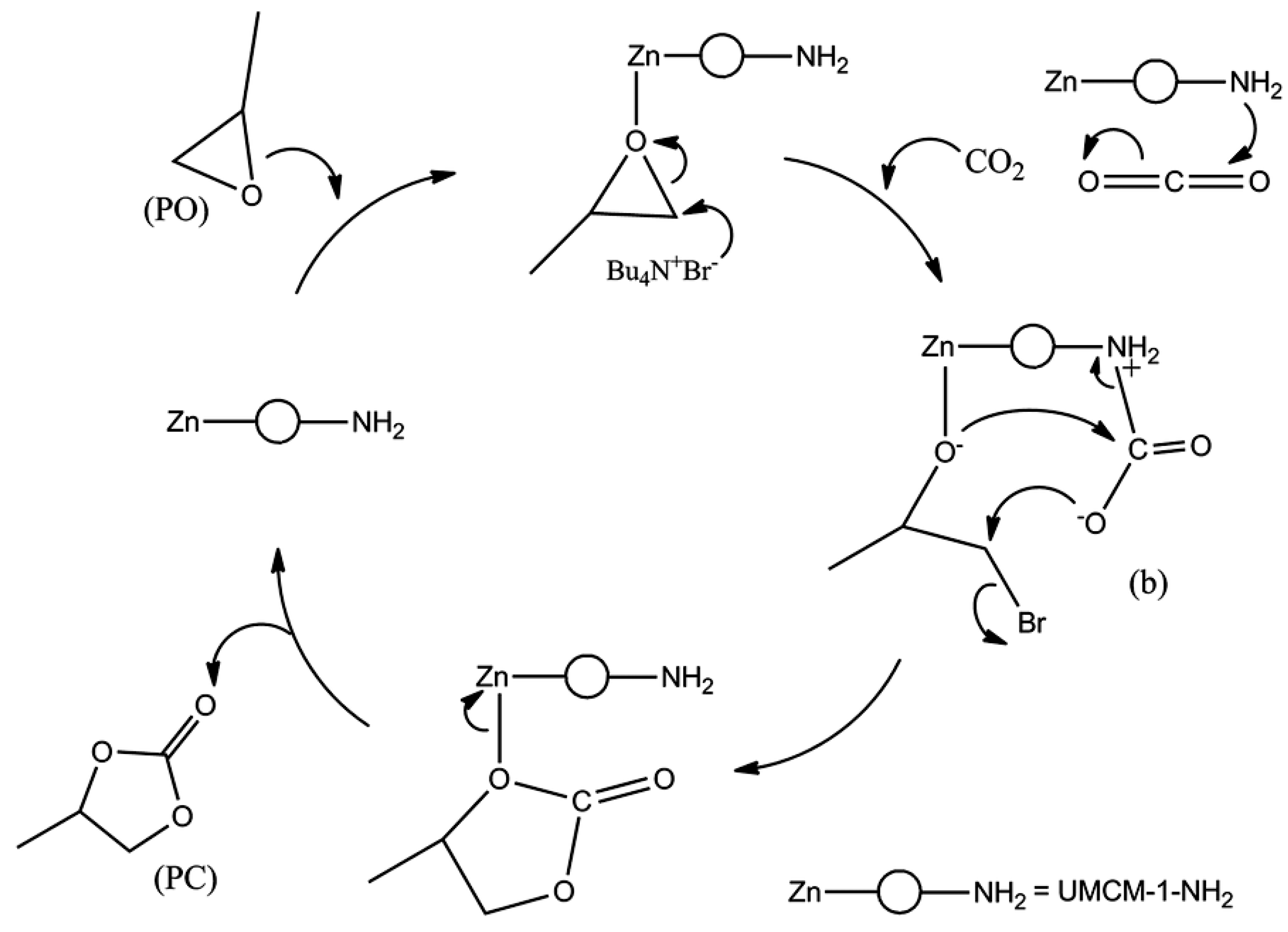
Park and coworkers reported an amine-functionalized micro-/mesoporous UMCM-1-NH2 system, and their catalytic activities for the cycloaddition reaction of epoxides with CO2 in the presence or absence of TBABr cocatalyst [33]. The Lewis basic primary amine site as well as large bimodal porosity was beneficial for the high-performance catalysis. The reaction catalyzed by the UMCM-1-NH2/TBABr system at room temperature under increased CO2 pressure (3.95 atm) for 4 h, showed the conversion of 78% with high selectivity for propylene oxide. It was noticed that the Lewis basic sites cooperatively captured and activated CO2 very effectively as illustrated in Scheme 5. The proposed synergism of the Lewis acid-base sites are well illustrated in Figure 11. Nonetheless, the amine-free control UMCM-1/TBABr system also showed a marginally smaller conversion (85%) than the UMCM-1-NH2/TBABr system (90%) at room temperature under 11.84 atm CO2. This result was mainly attributed to the larger pore size of the UMCM-1 compared to UMCM-1-NH2. Therefore, MOFs with a delicate balance between pore size and accessible Lewis basic sites can be very efficient catalytic systems operable under milder conditions.
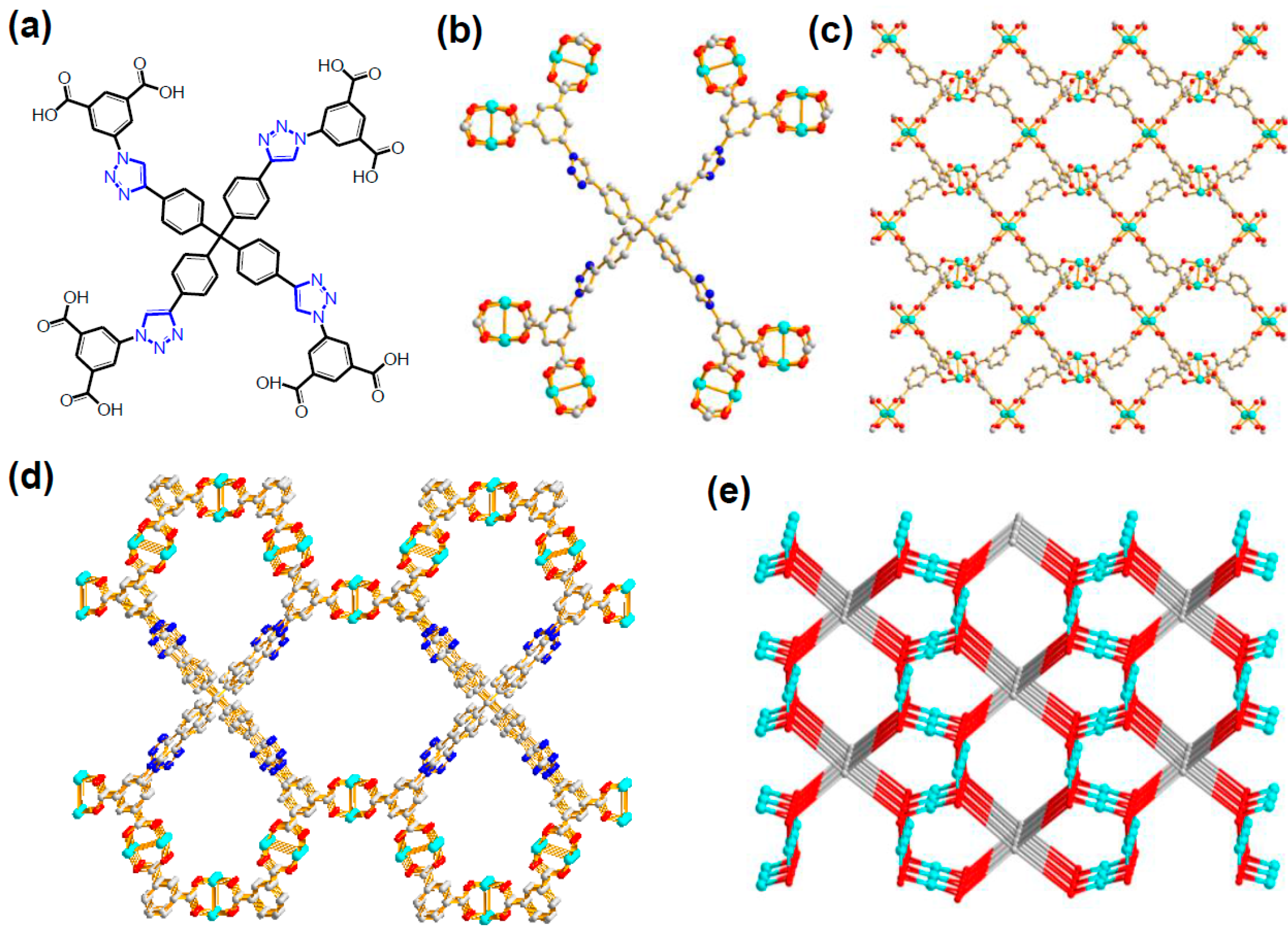
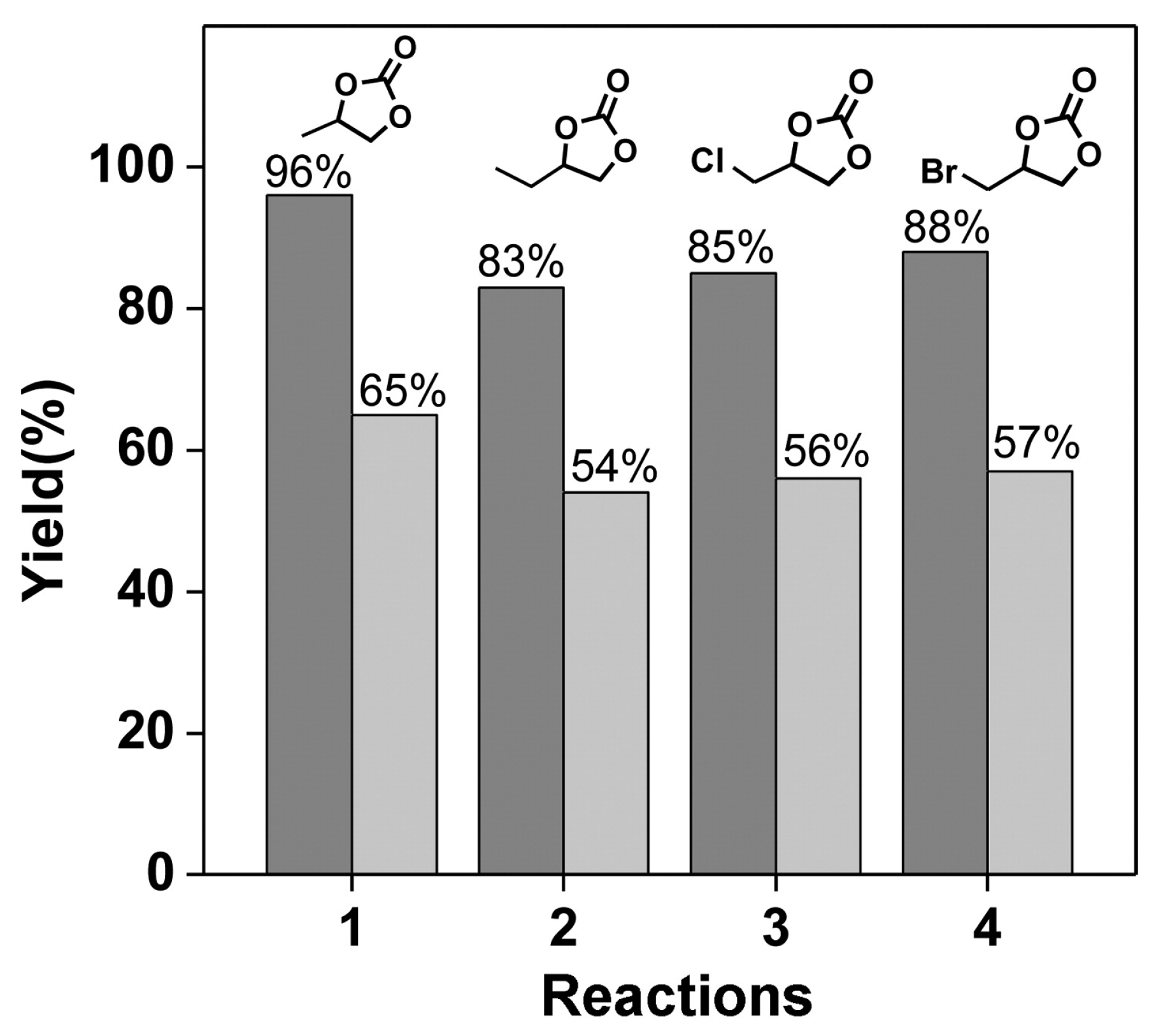
Zhou and coworkers prepared a new Cu-MOF with a paddlewheel SBU that had a large octatopic carboxylate bridging ligand (L1) obtained from the “click” reaction as given in Figure 12 [34]. As a result, the open channels of the Cu-MOF contained the Lewis basic 1,2,3-triazole rings, as well as very well-defined openly accessible Lewis acidic Cu(II) sites. The presence of open metal sites was unequivocally confirmed from the Raman spectroscopy. The desolvated Cu-MOF indicated that the symmetric C=O stretching mode at 1377 cm−1 for the coordinated CO2 molecule in Raman spectrum under CO2 atmosphere. Contrarily, the free gas phase CO2 displays the stretching mode at 1388 cm−1. The Cu-MOF showed bimodal porosity, 7.9 and 12.6 Å. These features led to a very active catalytic system for the cycloaddition of CO2 into epoxides at room temperature and 1 atm CO2 in the presence of TBABr. It was also noted that the Cu-MOF was a more active catalyst than the control HKUST-1, containing open metal sites without additional Lewis basic sites, as shown in Figure 13. In the case of the smallest propylene oxide, the conversion by Cu-MOF was as high as 96% for 48 h.
3.3. Direct Incorporation of Br- Nucleophile into MOF Channels
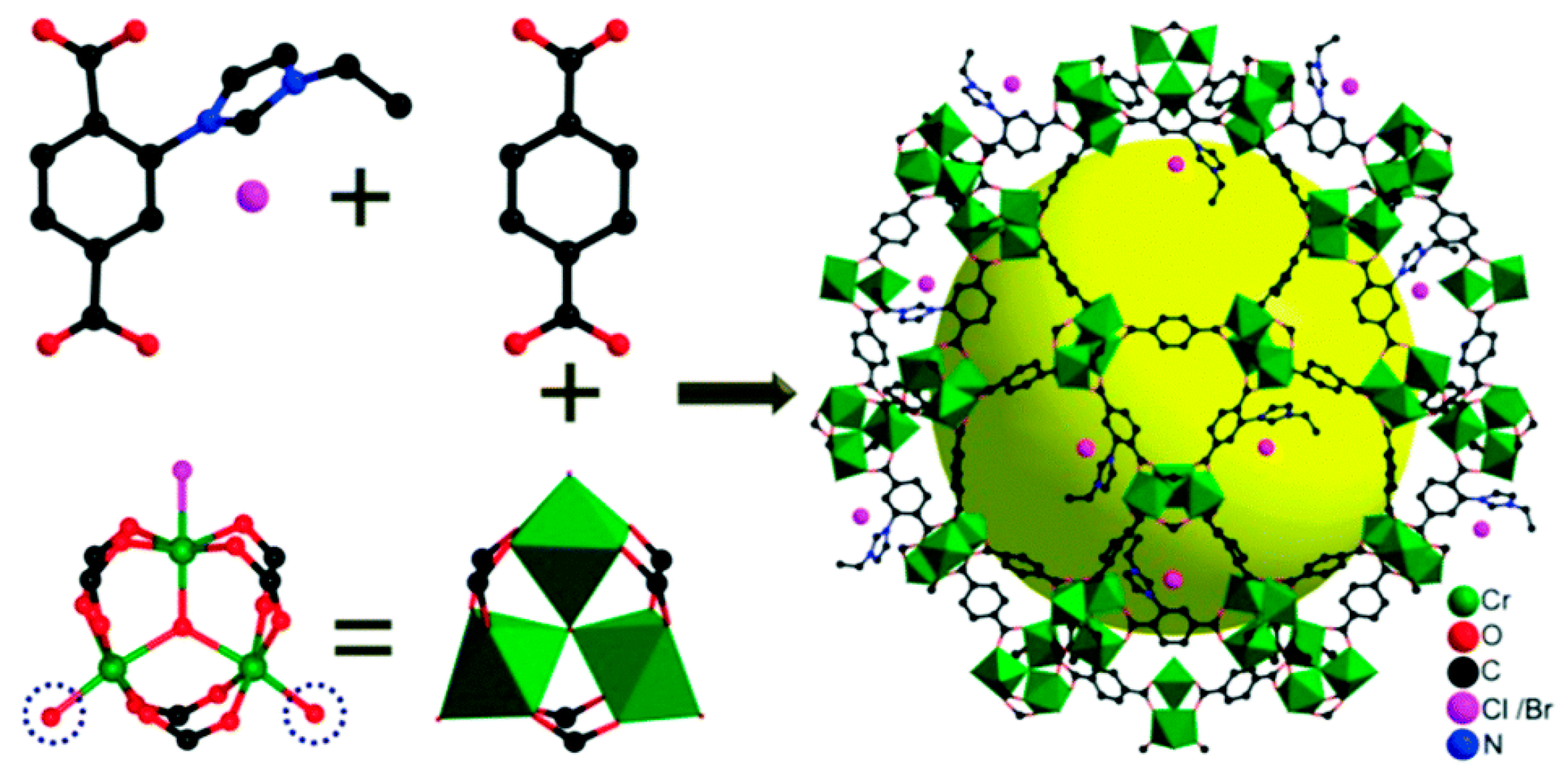
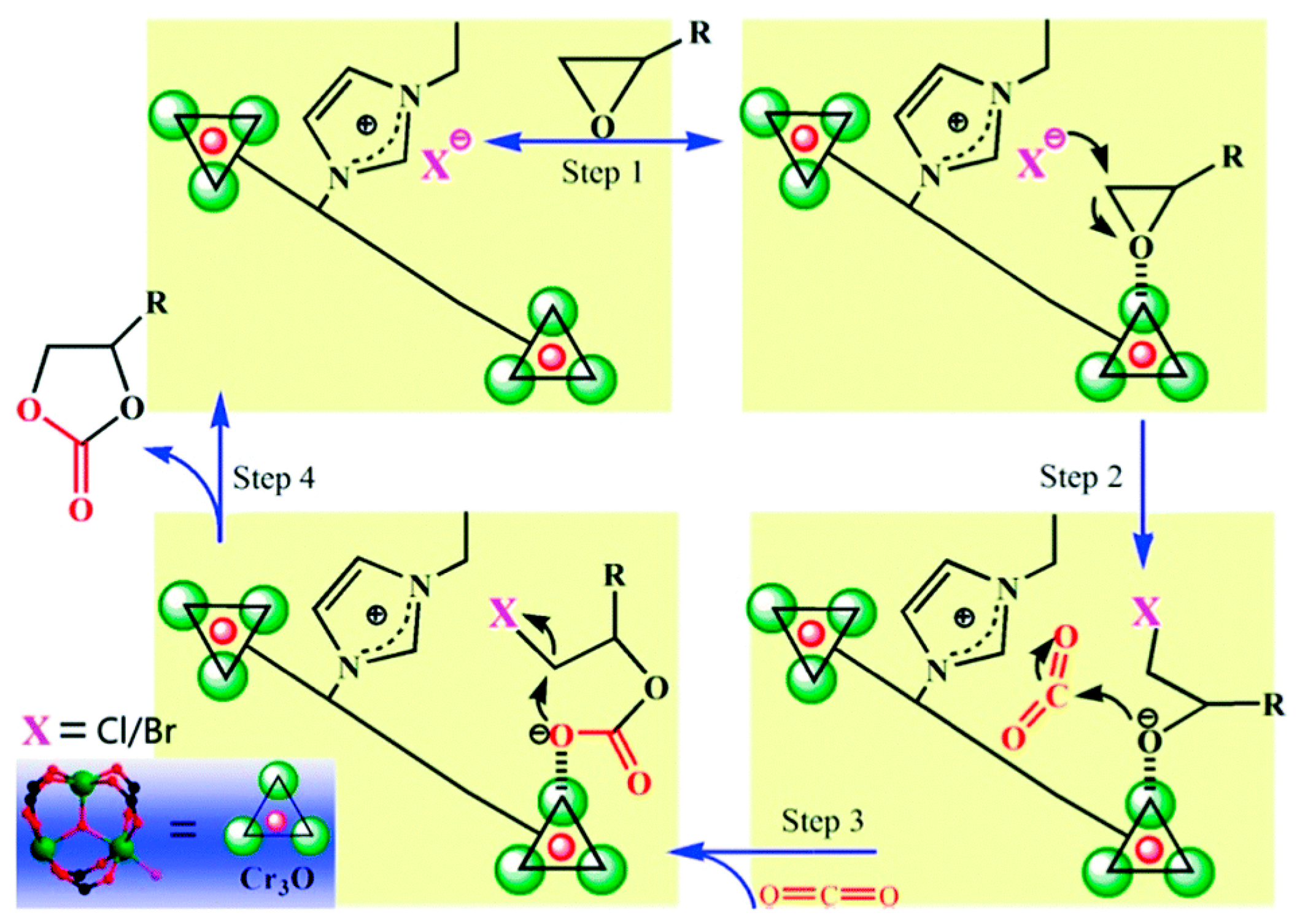
Since the early stage of the cycloaddition of CO2 into the epoxide includes the nucleophilic attack of Br− ion to epoxide to lead to ring-opening of epoxide as shown in Scheme 1, it may be a good strategy to permanently locate Br− ions near the acidic reaction centers. Thus, it is reasonable to imagine that once the Br− nucleophiles are permanently located near the Lewis acid reaction centers, it may not be necessary to add the TBABr cocatalyst into reaction mixture. The process can thus be simpler and more cost-effective. Furthermore, the close proximity of the Br− nucleophiles to the reaction centers in the confined spaces may significantly accelerate the cycloaddition reaction to proceed under mild conditions. Recently, Huang and coworkers prepared a similar system using a mesoporous Cr-based MOF (FJI-C10) based on the MIL-101 structure completely operable in the absence of TBABr cocatalyst by incorporating an ionic ligand, 2-(3-ethylimidazol-1-yl)terephthalic acid [(Etim-H2BDC)Br] linker (Figure 14) [35]. The ionic surface of the framework was beneficial for efficient interactions with CO2 due to the dipole-quadrupole interaction. Furthermore, additional TBABr was not required for the catalytic cycloaddition reaction because the Br− counter-ions in the framework can harmoniously activate nearby epoxide interacting with the Lewis acidic site as illustrated in Scheme 6. The cycloaddition reaction between CO2 and epichlorohydrin could be catalyzed by FJI-C10 alone at room temperature under 1 atm CO2 (for 24 h). Unfortunately, the conversion and product selectivity were low, 27.3% and 60.7%, respectively. Upon the increase of the temperature (60 °C), however, the reaction was almost completed (99.7%) with enhanced selectivity (88.2%) within 24 h. It is worthwhile to note that the reaction was completed at a slightly increased temperature without the TBABr cocatalyst.
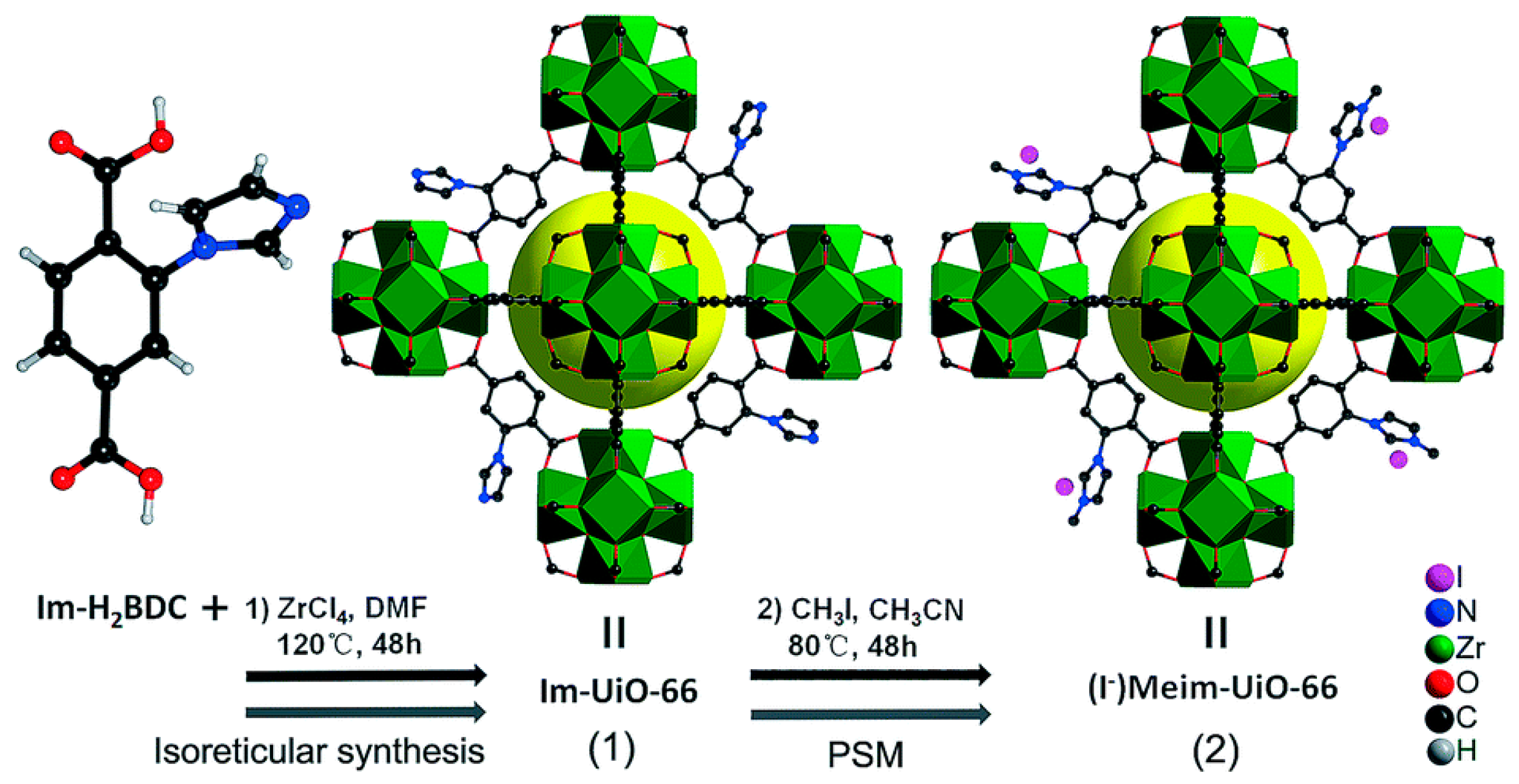
The same group also synthesized a Zr-based MOF with a UiO-66 phase using the same bridging ligand in order to have similar functionalities in the framework as depicted in Figure 15 [36]. In this case, the initially formed Im-UiO-66 could be converted to (I−)Meim-UiO-66 through a post-synthetic modification (PSM) method with an ionization degree of 85%. The (I−)Meim-UiO-66 contained I− nucleophiles near the catalytically active Zr6 clusters. Despite the presence of Lewis basic sites from the unreacted imidazole groups and rich Brønsted acidic sites, such as Zr-OH and Zr-H2O, the cycloaddition activities of (I−)Meim-UiO-66 without TBABr were lower than expected. The conversion of 88.5% for the cycloaddition of epichlorohydrin with CO2 at 100 °C under 1 atm CO2 was observed for the reaction period for 24 h. This result can be partly ascribed to the relatively low BET surface area (328 m2 g−1) of (I−)Meim-UiO-66. Nevertheless, the same reaction conducted at 120 °C under 1 atm CO2 by (I−)Meim-UiO-66 showed a much higher conversion (100%) than the UiO-66 (~0%) and amine-functionalized NH2-UiO-66 (11%) in the absence of TBABr cocatalyst. However, this system still requires a rather high reaction temperature.
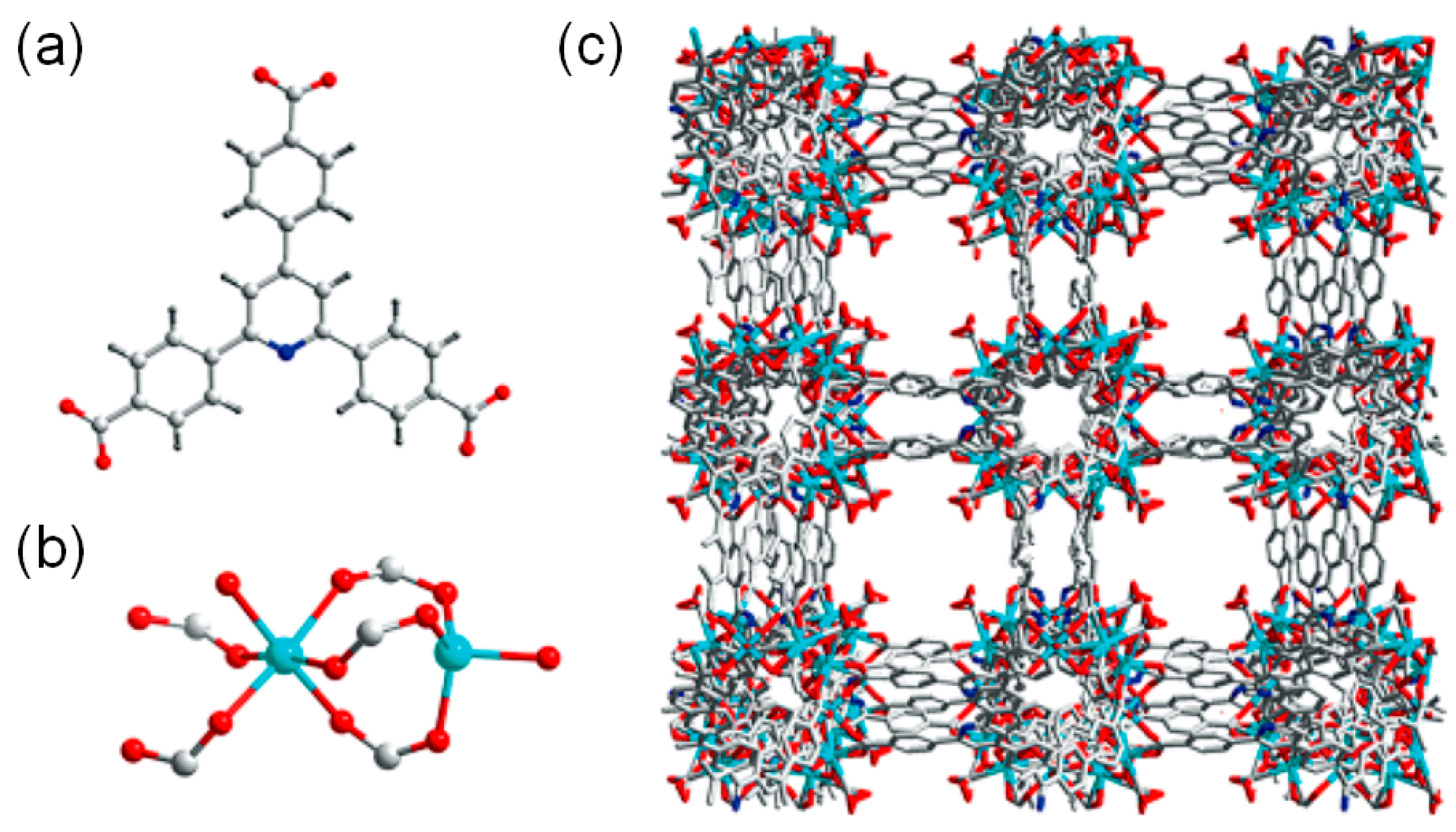
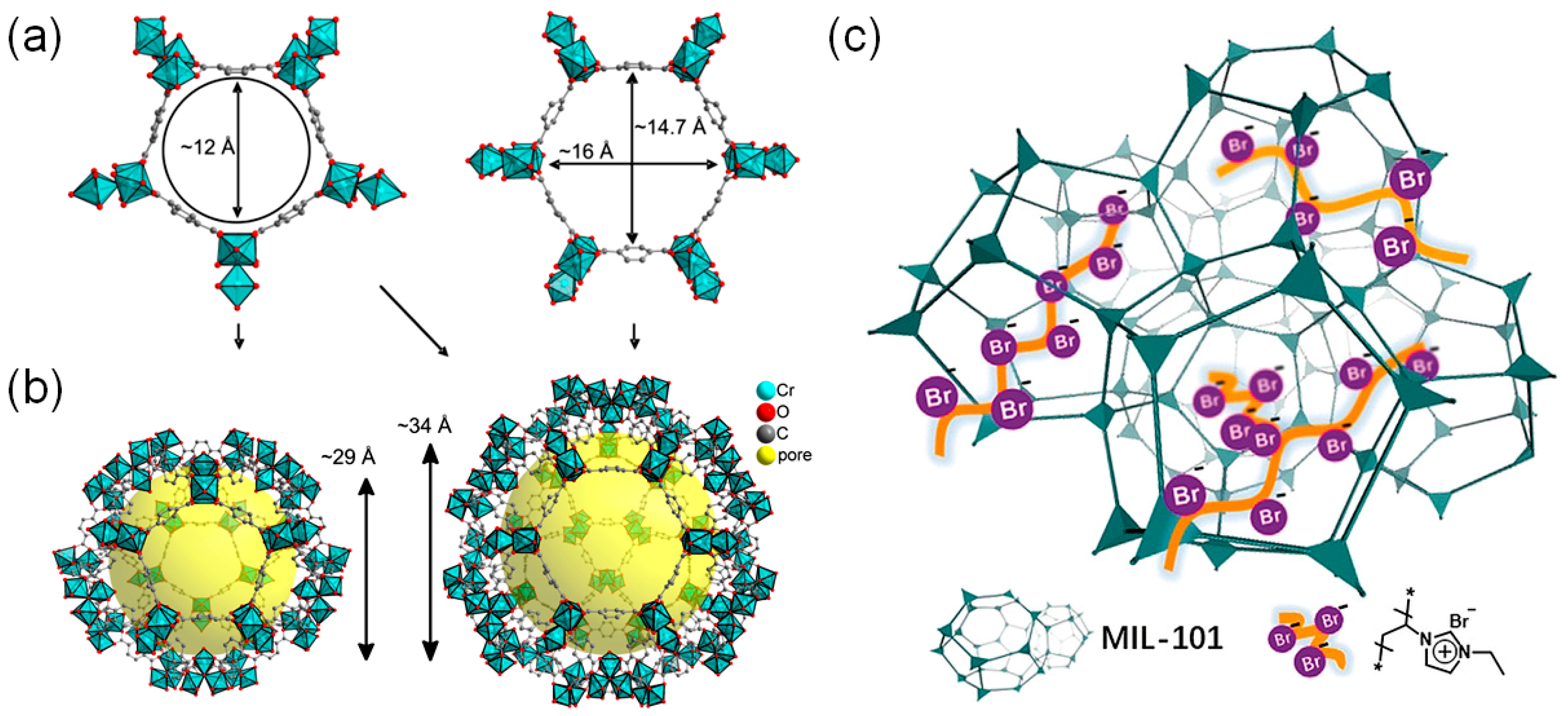
The proximity of the Br− ions and acidic reaction centers may also lead to a new low activation energy pathway. In this context, Ma and coworkers prepared a linear ionic polymer (IP)-encapsulated mesoporous MIL-101(Cr) catalytic system (MIL-101-IP) to realize the presence of cocatalysts near the Cr(III) acidic sites as shown in Scheme 7 [37]. The linear IP could be easily incorporated by a monomer (3-ethyl-1-vinyl-1H-imidazol-3-ium bromide) encapsulation followed by an AIBN-induced radical polymerization. Interestingly, the MIL-101-IP still contained sufficient void spaces as evidenced by the N2 and CO2 gas sorption analysis. The BET surface area of the MIL-101-IP was 2232 m2 g−1. The BET surface area of the original MIL-101 was 2610 m2 g−1. The cycloaddition of CO2 into the propylene oxide at 25 °C under 1 atm CO2 gave almost a complete conversion after 48 h as given in Table 5. In the case of the epichlorohydrin substrate, free IP (3%), MIL-101 (32%) and the physical mixture of IP and MIL-101 (80%) showed smaller conversions compared to the MIL-101-IP (99%) at 50 °C under 1 atm CO2 for 68 h. The respective activation energies were calculated to be 91.6 and 63.6 kJ mol−1 for the physical mixture and MIL-101-IP. Therefore, an energetically more favorable pathway was formed by the MIL-101-IP. It is worth noting that a simple mixing of the mesoporous MIL-101 and Br− ion-containing IP also gave a much higher conversion compared to the low reactivities of each component.
4. Conclusions and Prospects
We briefly surveyed the recent progress in the cycloaddition reaction between the epoxides and CO2 catalyzed by MOFs under mild reaction conditions as a means of efficient sequestering of CO2 and converting it to value-added chemicals. We found that the efficient MOF-based catalytic systems that were fully operable under mild conditions can be divided into three main categories (also see Table 6):
- (i)
- MOFs with rich Lewis/Brønsted acid sites or well-defined Lewis acidic reaction centers,
- (ii)
- MOFs with cooperative bifunctional acid-base catalytic sites, and
- (iii)
- MOFs with directly incorporated Br− nucleophiles inside channels.
Both the large pore dimensions and surface areas of the MOFs are also important factors for the mild cycloaddition reaction. Although homogeneous catalysts have also been investigated for past decades, most of them require high temperature and pressure for the cycloaddition reaction of CO2 to epoxides [15,39,40]. Al-salen- and Co-salen-based homogeneous catalysts together with TBABr or Et2NSiMe3 cocatalysts showed exceptionally good activities under mild reaction conditions [15,22,23,40]. In our opinion, there must be more MOF catalytic systems operable under mild conditions because there are many other MOF systems catalyzing this reaction but only tested under relatively high temperatures and gas pressure within short reaction periods [18,19,20,21,41]. Notwithstanding, most MOF catalytic systems showing good cycloaddition activities under mild reaction conditions summarized in Table 6 still require rather long reaction periods. Continuous efforts to reduce the reaction periods under the same conditions will provide much more effective catalytic systems. Furthermore, large epoxide substrates still show slower reaction kinetics due to the diffusion problem. Newly designed MOF systems with sufficiently large pores will give a solution to this diffusion issue. We also envision that efficient MOF-based catalytic systems without TBABr cocatalyst will open up new avenues for this important reaction. Thus, there will be plenty of opportunity for developing more efficient and cost-effective recyclable MOF catalytic systems for the cycloaddition of epoxides with CO2.
Acknowledgments
This work was supported by the Basic Science Research Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2018R1D1A1B07043017). This work was supported by the Hankuk University of Foreign Studies Research Fund of 2018.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sumida, K.; Rogow, D.L.; Mason, J.A.; McDonald, T.M.; Bloch, E.D.; Herm, Z.R.; Bae, T.-H.; Long, J.R. Carbon Dioxide Capture in Metal-Organic Frameworks. Chem. Rev. 2012, 112, 724–781. [Google Scholar] [CrossRef] [PubMed]
- D’Alessandro, D.M.; Smit, B.; Long, J.R. Carbon Dioxide Capture: Prospects for New Materials. Angew. Chem. Int. Ed. 2010, 49, 6058–6082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-R.; Ma, Y.; McCarthy, M.C.; Sculley, J.; Yu, J.; Jeong, H.-K.; Balbuena, P.B.; Zhou, H.-C. Carbon dioxide capture-related gas adsorption and separation in metal-organic frameworks. Coord. Chem. Rev. 2011, 255, 1791–1823. [Google Scholar] [CrossRef]
- Lu, A.-H.; Hao, G.-P. Porous materials for carbon dioxide capture. Annu. Rep. Prog. Chem. Sect. A Inorg. Chem. 2013, 109, 484–503. [Google Scholar] [CrossRef]
- Liu, J.; Thallapally, P.K.; McGrail, B.P.; Brown, D.R.; Liu, J. Progress in adsorption-based CO2 capture by metal-organic frameworks. Chem. Soc. Rev. 2012, 41, 2308–2322. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; D’Alessandro, D.M. Tuning the functional sites in metal-organic frameworks to modulate CO2 heats of adsorption. CrystEngComm 2015, 17, 706–718. [Google Scholar] [CrossRef]
- Kim, Y.; Huh, S. Pore engineering of metal-organic frameworks: Introduction of chemically accessible Lewis basic sites inside MOF channels. CrystEngComm 2016, 18, 3524–3550. [Google Scholar] [CrossRef]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The Chemistry and Applications of Metal-Organic Frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef]
- Datta, S.J.; Khumnoon, C.; Lee, Z.H.; Moon, W.K.; Docao, S.; Nguyen, T.H.; Hwang, I.C.; Moon, D.; Oleynikov, P.; Terasaki, O.; et al. CO2 capture from humid flue gases and humid atmosphere using a microporous coppersilicate. Science 2015, 350, 302–306. [Google Scholar] [CrossRef]
- Würfel, P. Problems of the Energy Economy. In Physics of Solar Cells; Wiley-VCH: Weinheim, Germany, 2009; pp. 1–10. [Google Scholar]
- Zornoza, B.; Martinez-Joaristi, A.; Serra-Crespo, P.; Tellez, C.; Coronas, J.; Gascon, J.; Kapteijn, F. Functionalized flexible MOFs as fillers in mixed matrix membranes for highly selective separation of CO2 from CH4 at elevated pressures. Chem. Commun. 2011, 47, 9522–9524. [Google Scholar] [CrossRef]
- Hu, Y.; Dong, X.; Nan, J.; Jin, W.; Ren, X.; Xu, N.; Lee, Y.M. Metal-organic framework membranes fabricated via reactive seeding. Chem. Commun. 2011, 47, 737–739. [Google Scholar] [CrossRef] [PubMed]
- Rodenas, T.; Luz, I.; Prieto, G.; Seoane, B.; Miro, H.; Corma, A.; Kapteijn, F.; Llabrés i Xamena, F.X.; Gascon, J. Metal-organic framework nanosheets in polymer composite materials for gas separation. Nat. Mater. 2015, 14, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Aresta, M.; Dibenedetto, A. Utilisation of CO2 as a chemical feedstock: Opportunities and challenges. Dalton Trans. 2007, 2975–2992. [Google Scholar] [CrossRef] [PubMed]
- Sakakura, T.; Kohno, K. The synthesis of organic carbonates from carbon dioxide. Chem. Commun. 2009, 1312–1330. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.; Qi, C.; Wu, W.; Jiang, H. Recent advances in organic synthesis with CO2 as C1 synthon. Curr. Opin. Green Sustain. Chem. 2017, 3, 22–27. [Google Scholar] [CrossRef]
- Yang, Z.-Z.; He, L.-N.; Dou, X.-Y.; Chanfreau, S. Dimethyl carbonate synthesis catalyzed by DABCO-derived basic ionic liquids via transesterification of ethylene carbonate with methanol. Tetrahedron Lett. 2010, 51, 2931–2934. [Google Scholar] [CrossRef]
- Beyzavi, M.H.; Stephenson, C.J.; Liu, Y.; Karagiaridi, O.; Hupp, J.T.; Farha, O.K. Metal-organic framework-based catalysts: Chemical fixation of CO2 with epoxides leading to cyclic organic carbonates. Front. Energy Res. 2015, 2, 63. [Google Scholar] [CrossRef]
- Maina, J.W.; Pozo-Gonzalo, C.; Kong, L.; Schütz, J.; Hill, M.; Dumée, L.F. Metal organic framework based catalysts for CO2 conversion. Mater. Horiz. 2017, 4, 345–361. [Google Scholar] [CrossRef]
- Zou, B.; Hu, C. Halogen-free processes for organic carbonate synthesis from CO2. Curr. Opin. Green Sustain. Chem. 2017, 3, 11–16. [Google Scholar] [CrossRef]
- He, H.; Perman, J.A.; Zhu, G.; Ma, S. Metal-Organic Frameworks for CO2 Chemical Transformations. Small 2016, 12, 6309–6324. [Google Scholar] [CrossRef]
- North, M.; Pasquale, R. Mechanism of Cyclic Carbonate Synthesis from Epoxides and CO2. Angew. Chem. Int. Ed. 2009, 48, 2946–2948. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, J.; North, M.; Pasquale, R. Synthesis of Cyclic Carbonates from Atmospheric Pressure Carbon Dioxide Using Exceptionally Active Aluminium(salen) Complexes as Catalysts. Eur. J. Inorg. Chem. 2007, 3323–3326. [Google Scholar] [CrossRef]
- Chui, S.S.-Y.; Lo, S.M.-F.; Charmant, J.P.H.; Orpen, A.G.; Williams, I.D. A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n. Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef]
- Ji, X.-H.; Zhu, N.-N.; Ma, J.-G.; Cheng, P. Conversion of CO2 into cyclic carbonates by a Co(II) metal-organic framework and the improvement of catalytic activity via nanocrystallization. Dalton Trans. 2018, 47, 1768–1771. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.-Y.; Chen, Y.; Niu, Y.; Williams, K.; Cash, L.; Perez, P.J.; Wojtas, L.; Cai, J.; Chen, Y.-S.; Ma, S. Crystal Engineering of an nbo Topology Metal-Organic Framework for Chemical Fixation of CO2 under Ambient Conditions. Angew. Chem. Int. Ed. 2014, 53, 2615–2619. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, S.-M.; Han, Z.-B.; Ding, M.; Yuan, D.-Q.; Jiang, H.-L. Exceptionally Robust In-Based Metal-Organic Framework for Highly Efficient Carbon Dioxide Capture and Conversion. Inorg. Chem. 2016, 55, 3558–3565. [Google Scholar] [CrossRef] [PubMed]
- Beyzavi, M.H.; Klet, R.C.; Tussupbayev, S.; Borycz, J.; Vermeulen, N.A.; Cramer, C.J.; Stoddart, J.F.; Hupp, J.T.; Farha, O.K. A Hafnium-Based Metal-Organic Framework as an Efficient and Multifunctional Catalyst for Facile CO2 Fixation and Regioselective and Enantioretentive Epoxide Activation. J. Am. Chem. Soc. 2014, 136, 15861–15864. [Google Scholar] [CrossRef]
- Wang, H.-H.; Hou, L.; Li, Y.-Z.; Jiang, C.-Y.; Wang, Y.-Y.; Zhu, Z. Porous MOF with Highly Efficient Selectivity and Chemical Conversion for CO2. ACS Appl. Mater. Interfaces 2017, 9, 17969–17976. [Google Scholar] [CrossRef]
- Li, X.-Y.; Ma, L.-N.; Liu, Y.; Hou, L.; Wang, Y.-Y.; Zhu, Z. Honeycomb Metal-Organic Framework with Lewis Acidic and Basic Bifunctional Sites: Selective Adsorption and CO2 Catalytic Fixation. ACS Appl. Mater. Interfaces 2018, 10, 10965–10973. [Google Scholar] [CrossRef]
- Cao, C.-S.; Shi, Y.; Xu, H.; Zhao, B. A multifunctional MOF as a recyclable catalyst for the fixation of CO2 with aziridines or epoxides and as a luminescent probe of Cr(VI). Dalton Trans. 2018, 47, 4545–4553. [Google Scholar] [CrossRef]
- Kumar, S.; Verma, G.; Gao, W.-Y.; Niu, Z.; Wojtas, L.; Ma, S. Anionic Metal-Organic Framework for Selective Dye Removal and CO2 Fixation. Eur. J. Inorg. Chem. 2016, 4373–4377. [Google Scholar] [CrossRef]
- Babu, R.; Kathalikkattil, A.C.; Roshan, R.; Tharun, J.; Kim, D.-W.; Park, D.-W. Dual-porous metal organic framework for room temperature CO2 fixation via cyclic carbonate synthesis. Green Chem. 2016, 18, 232–242. [Google Scholar] [CrossRef]
- Li, P.-Z.; Wang, X.-J.; Liu, J.; Lim, J.S.; Zou, R.; Zhao, Y. A Triazole-Containing Metal-Organic Framework as a Highly Effective and Substrate Size-Dependent Catalyst for CO2 Conversion. J. Am. Chem. Soc. 2016, 138, 2142–2145. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Xie, Y.-Q.; Wang, X.-S.; Wang, Q.; Liu, T.-T.; Huang, Y.-B.; Cao, R. An imidazolium-functionalized mesoporous cationic metal-organic framework for cooperative CO2 fixation into cyclic carbonate. Chem. Commun. 2018, 54, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Chen, R.-P.; Wang, X.-Y.; Liu, T.-T.; Wang, X.-S.; Huang, Y.-B.; Cao, R. Postsynthetic ionization of an imidazole-containing metal-organic framework for the cycloaddition of carbon dioxide and epoxides. Chem. Sci. 2017, 8, 1570–1575. [Google Scholar] [CrossRef] [PubMed]
- Aguila, B.; Sun, Q.; Wang, X.; O’Rourke, E.; Al-Enizi, A.M.; Nafady, A.; Ma, S. Lower Activation Energy for Catalytic Reactions through Host-Guest Cooperation within Metal-Organic Frameworks. Angew. Chem. Int. Ed. 2018, 57, 10107–10111. [Google Scholar] [CrossRef] [PubMed]
- Jeazet, H.B.T.; Koschine, T.; Staudt, C.; Raetzke, K.; Janiak, C. Correlation of Gas Permeability in a Metal-Organic Framework MIL-101(Cr)–Polysulfone Mixed-Matrix Membrane with Free Volume Measurements by Positron Annihilation Lifetime Spectroscopy (PALS). Membranes 2013, 3, 331–353. [Google Scholar] [CrossRef] [Green Version]
- Dibenedetto, A.; Angelini, A.; Stufano, P. Use of carbon dioxide as feedstock for chemicals and fuels: Homogeneous and heterogeneous catalysis. J. Chem. Technol. Biotechnol. 2014, 89, 334–353. [Google Scholar] [CrossRef]
- Lu, X.-B.; Darensbourg, D.J. Cobalt catalysts for the coupling of CO2 and epoxides to provide polycarbonates and cyclic carbonates. Chem. Soc. Rev. 2012, 41, 1462–1484. [Google Scholar] [CrossRef]
- Noh, J.; Kim, Y.; Park, H.; Lee, J.; Yoon, M.; Park, M.H.; Kim, Y.; Kim, M. Functional group effects on a metal-organic framework catalyst for CO2 cycloaddition. J. Ind. Eng. Chem. 2018, 64, 478–483. [Google Scholar] [CrossRef]
Scheme 1.
A proposed catalytic cycle for the cycloaddition of CO2 with epoxide in the presence of Lewis acid catalyst and TBABr cocatalyst [22].
Scheme 1.
A proposed catalytic cycle for the cycloaddition of CO2 with epoxide in the presence of Lewis acid catalyst and TBABr cocatalyst [22].

Scheme 2.
Preparation of dimethyl carbonate from cyclic carbonates through transesterification with methanol [17].
Scheme 2.
Preparation of dimethyl carbonate from cyclic carbonates through transesterification with methanol [17].

Scheme 3.
(a) The chemical structure of salenH2 ligand in (salen)Al-O-Al(salen) catalyst. (b) Proposed catalytic cycle for the cycloaddition of CO2 with epoxide catalyzed by the dinuclear (salen)Al-O-Al(salen) complex in the presence of TBABr cocatalyst. Adapted with permission from [22]. Copyright 2009, Wiley-VCH Verlag GmbH.
Scheme 3.
(a) The chemical structure of salenH2 ligand in (salen)Al-O-Al(salen) catalyst. (b) Proposed catalytic cycle for the cycloaddition of CO2 with epoxide catalyzed by the dinuclear (salen)Al-O-Al(salen) complex in the presence of TBABr cocatalyst. Adapted with permission from [22]. Copyright 2009, Wiley-VCH Verlag GmbH.

Figure 1.
(a) Chemical structure of thiazolidine 2,4-dicarboxylic acid (H2L). (b) 3D framework of solvent-free Co-MOF with 1D channels viewed along the c-axis. Color codes: Co (cyan), C (gray), N (blue), O (red), and S (yellow). Hydrogen atoms are omitted for clarity. (c) SEM image of Co-MOF. Adapted with permission from [25]. Copyright 2018, The Royal Society of Chemistry.
Figure 1.
(a) Chemical structure of thiazolidine 2,4-dicarboxylic acid (H2L). (b) 3D framework of solvent-free Co-MOF with 1D channels viewed along the c-axis. Color codes: Co (cyan), C (gray), N (blue), O (red), and S (yellow). Hydrogen atoms are omitted for clarity. (c) SEM image of Co-MOF. Adapted with permission from [25]. Copyright 2018, The Royal Society of Chemistry.

Scheme 4.
The chemical structures of (a) 3,3′,5,5′-biphenyltetracarboxylic acid (H4bptc) for MOF-505 and (b) 1,4,7,10-tetrazazcyclododecane-N,N′,N″,N‴-tetra-p-methylbenzoic acid (H4tactmb) for MMCF-2. Adapted with permission from [26]. Copyright 2014, Wiley-VCH Verlag GmbH.
Scheme 4.
The chemical structures of (a) 3,3′,5,5′-biphenyltetracarboxylic acid (H4bptc) for MOF-505 and (b) 1,4,7,10-tetrazazcyclododecane-N,N′,N″,N‴-tetra-p-methylbenzoic acid (H4tactmb) for MMCF-2. Adapted with permission from [26]. Copyright 2014, Wiley-VCH Verlag GmbH.

Figure 2.
The 3D framework structures of (a) MOF-505 and (b) MMCF-2. Hydrogen atoms are omitted for clarity. (c) The comparison of catalytic cycloaddition reactivities for propylene oxide with CO2 catalyzed by Cu(tactmb)-MOF, HKUST-1, MOF-505 and MMCF-2. Adapted with permission from ref 26. Copyright 2014, Wiley-VCH Verlag GmbH.
Figure 2.
The 3D framework structures of (a) MOF-505 and (b) MMCF-2. Hydrogen atoms are omitted for clarity. (c) The comparison of catalytic cycloaddition reactivities for propylene oxide with CO2 catalyzed by Cu(tactmb)-MOF, HKUST-1, MOF-505 and MMCF-2. Adapted with permission from ref 26. Copyright 2014, Wiley-VCH Verlag GmbH.

Figure 3.
The chemical structures of (a) 1,2,4-benzenetricarboxylic acid (H3btc) and (b) its derivative formed with piperazine (H2L). (c) 1D channel structure of In-MOF. (d) 3D framework of In-MOF shown along the b-axis. Hydrogen atoms are omitted for clarity. Adapted with permission from [27]. Copyright 2016, American Chemical Society.
Figure 3.
The chemical structures of (a) 1,2,4-benzenetricarboxylic acid (H3btc) and (b) its derivative formed with piperazine (H2L). (c) 1D channel structure of In-MOF. (d) 3D framework of In-MOF shown along the b-axis. Hydrogen atoms are omitted for clarity. Adapted with permission from [27]. Copyright 2016, American Chemical Society.

Figure 4.
Pore structure of Hf-NU-1000. The 1,3,6,8-tetrakis(p-benzoic acid)pyrene bridging linker and Hf6 node are also shown. Hydrogen atoms are omitted for clarity. Adapted with permission from [28]. Copyright 2014, American Chemical Society.
Figure 4.
Pore structure of Hf-NU-1000. The 1,3,6,8-tetrakis(p-benzoic acid)pyrene bridging linker and Hf6 node are also shown. Hydrogen atoms are omitted for clarity. Adapted with permission from [28]. Copyright 2014, American Chemical Society.

Figure 5.
(a) The chemical structure of 5-(4-(tetrazol-5-yl)phenyl)isophthalic acid (H2tzpa), (b) coordination environment of the trinuclear Co(II) cluster, and (c) the structure of ribbon-like SBU in the Co-MOF. Adapted with permission from [29]. Copyright 2017, American Chemical Society.
Figure 5.
(a) The chemical structure of 5-(4-(tetrazol-5-yl)phenyl)isophthalic acid (H2tzpa), (b) coordination environment of the trinuclear Co(II) cluster, and (c) the structure of ribbon-like SBU in the Co-MOF. Adapted with permission from [29]. Copyright 2017, American Chemical Society.

Figure 6.
3D framework structure of the Co-MOF indicating 1D channels along the c-axis. Adapted with permission from [29]. Copyright 2017, American Chemical Society.
Figure 6.
3D framework structure of the Co-MOF indicating 1D channels along the c-axis. Adapted with permission from [29]. Copyright 2017, American Chemical Society.

Figure 7.
(a) Chemical structure of N,N′-bis(isophthalic acid)-oxalamide (H4BDPO), (b) coordination environment of the Ba2+ ions in Ba-MOF, (c) its tubular pore structure, and (d) hexagonally oriented 1D tubes shown along the c-axis. Adapted with permission from [30]. Copyright 2018, American Chemical Society.
Figure 7.
(a) Chemical structure of N,N′-bis(isophthalic acid)-oxalamide (H4BDPO), (b) coordination environment of the Ba2+ ions in Ba-MOF, (c) its tubular pore structure, and (d) hexagonally oriented 1D tubes shown along the c-axis. Adapted with permission from [30]. Copyright 2018, American Chemical Society.

Figure 8.
(a) The chemical structure of 1,5-bis(5-tetrazole)-3-oxapentane (H2btz) linker. (b) Coordination environment of Zn(II) ion. (c) A 3D view of Zn-btz-MOF. Hydrogen atoms are omitted for clarity. Adapted with permission from [31]. Copyright 2018, The Royal Society of Chemistry.
Figure 8.
(a) The chemical structure of 1,5-bis(5-tetrazole)-3-oxapentane (H2btz) linker. (b) Coordination environment of Zn(II) ion. (c) A 3D view of Zn-btz-MOF. Hydrogen atoms are omitted for clarity. Adapted with permission from [31]. Copyright 2018, The Royal Society of Chemistry.

Figure 9.
A proposed catalytic cycle for the cycloaddition of aziridines with CO2 into oxazolidinones catalyzed by Zn-btz-MOF in the presence of TBABr [31].
Figure 9.
A proposed catalytic cycle for the cycloaddition of aziridines with CO2 into oxazolidinones catalyzed by Zn-btz-MOF in the presence of TBABr [31].

Figure 10.
(a) The chemical structure of 4,4′,4′′-(pyridine-2,4,6-triyl)tribenzoic acid (H3PTB). (b) Coordination environment of Zn(II) ions in Zn-MOF. (c) 3D packing diagram of Zn-MOF showing 1D square channels. Hydrogen atoms are omitted for clarity. Adapted with permission from [32]. Copyright 2016, Wiley-VCH Verlag GmbH.
Figure 10.
(a) The chemical structure of 4,4′,4′′-(pyridine-2,4,6-triyl)tribenzoic acid (H3PTB). (b) Coordination environment of Zn(II) ions in Zn-MOF. (c) 3D packing diagram of Zn-MOF showing 1D square channels. Hydrogen atoms are omitted for clarity. Adapted with permission from [32]. Copyright 2016, Wiley-VCH Verlag GmbH.

Scheme 5.
Schematic representation of the preparation of cyclic organic carbonates from epoxides and CO2. Adapted with permission from [33]. Copyright 2016, The Royal Society of Chemistry.
Scheme 5.
Schematic representation of the preparation of cyclic organic carbonates from epoxides and CO2. Adapted with permission from [33]. Copyright 2016, The Royal Society of Chemistry.

Figure 11.
Proposed catalytic cycle for the cycloaddition of CO2 and propylene oxide catalyzed by UMCM-1-NH2/TBABr. Adapted with permission from [33]. Copyright 2016, The Royal Society of Chemistry.
Figure 11.
Proposed catalytic cycle for the cycloaddition of CO2 and propylene oxide catalyzed by UMCM-1-NH2/TBABr. Adapted with permission from [33]. Copyright 2016, The Royal Society of Chemistry.

Figure 12.
(a) The chemical structure of octatopic “clicked” ligand H8L1. (b) Connectivity between L1 and eight paddlewheel Cu2 units. (c) Cu2 clusters connected by isophthalate moieties from Cu-MOF generating 2D lamellar structure with periodically arranged open metal sites. (d) 3D framework of Cu-MOF. Hydrogen atoms are omitted for clarity. (e) (4,3,4)-network of Cu-MOF. Adapted with permission from [34]. Copyright 2016, American Chemical Society.
Figure 12.
(a) The chemical structure of octatopic “clicked” ligand H8L1. (b) Connectivity between L1 and eight paddlewheel Cu2 units. (c) Cu2 clusters connected by isophthalate moieties from Cu-MOF generating 2D lamellar structure with periodically arranged open metal sites. (d) 3D framework of Cu-MOF. Hydrogen atoms are omitted for clarity. (e) (4,3,4)-network of Cu-MOF. Adapted with permission from [34]. Copyright 2016, American Chemical Society.

Figure 13.
Comparison of the reaction yields of various cyclic carbonates obtained from the cycloaddition of CO2 with epoxide substrates catalyzed by Cu-MOF (black) and HKUST-1 (grey). The reaction was conducted using epoxide (20 mmol) with CO2 (1 atm) at room temperature with TBABr (0.65 g, 10 mol %) for 48 h. Adapted with permission from [34]. Copyright 2016, American Chemical Society.
Figure 13.
Comparison of the reaction yields of various cyclic carbonates obtained from the cycloaddition of CO2 with epoxide substrates catalyzed by Cu-MOF (black) and HKUST-1 (grey). The reaction was conducted using epoxide (20 mmol) with CO2 (1 atm) at room temperature with TBABr (0.65 g, 10 mol %) for 48 h. Adapted with permission from [34]. Copyright 2016, American Chemical Society.

Figure 14.
Schematic illustration of the self-assembly of (Br−)Etim-BDCs, BDCs, and Cr3O clusters to generate cationic framework (FJI-C10). Coordinated aqua ligands are represented as dashed circles. Hydrogen atoms are omitted for clarity. Adapted with permission from [35]. Copyright 2018, The Royal Society of Chemistry.
Figure 14.
Schematic illustration of the self-assembly of (Br−)Etim-BDCs, BDCs, and Cr3O clusters to generate cationic framework (FJI-C10). Coordinated aqua ligands are represented as dashed circles. Hydrogen atoms are omitted for clarity. Adapted with permission from [35]. Copyright 2018, The Royal Society of Chemistry.

Scheme 6.
Proposed mechanism for the cycloaddition reaction of CO2 with epoxides catalyzed by FJI-C10. The hashed bonds indicate weak coordination interactions. Adapted with permission from [35]. Copyright 2018, The Royal Society of Chemistry.
Scheme 6.
Proposed mechanism for the cycloaddition reaction of CO2 with epoxides catalyzed by FJI-C10. The hashed bonds indicate weak coordination interactions. Adapted with permission from [35]. Copyright 2018, The Royal Society of Chemistry.

Figure 15.
Preparations of Im-UiO-66 and (I−)Meim-UiO-66 via conventional reticular chemistry and by a post-synthetic modification method, respectively. Green polyhedra indicate Zr6 clusters. Yellow balls indicate micropores. Adapted with permission from [36]. Copyright 2017, The Royal Society of Chemistry.
Figure 15.
Preparations of Im-UiO-66 and (I−)Meim-UiO-66 via conventional reticular chemistry and by a post-synthetic modification method, respectively. Green polyhedra indicate Zr6 clusters. Yellow balls indicate micropores. Adapted with permission from [36]. Copyright 2017, The Royal Society of Chemistry.

Scheme 7.
(a) Two different aperture dimensions of the building blocks of MIL-101, [Cr3(μ3-O)(F,OH)(BDC)3(H2O)2] (BDC = 1,4-benzenedicarboxylate). (b) Mesoporous cages of MIL-101. Hydrogen atoms are omitted for clarity. (c) Schematic illustration of MIL-101-IP with the ionic polymer encapsulated inside the pores. Adapted with permission from [37,38]. Copyrights 2018, Wiley-VCH Verlag GmbH and (2013) MDPI AG.
Scheme 7.
(a) Two different aperture dimensions of the building blocks of MIL-101, [Cr3(μ3-O)(F,OH)(BDC)3(H2O)2] (BDC = 1,4-benzenedicarboxylate). (b) Mesoporous cages of MIL-101. Hydrogen atoms are omitted for clarity. (c) Schematic illustration of MIL-101-IP with the ionic polymer encapsulated inside the pores. Adapted with permission from [37,38]. Copyrights 2018, Wiley-VCH Verlag GmbH and (2013) MDPI AG.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Cyclic carbonate yields and TOFs for various epoxides catalyzed by In-MOF. a Adapted with permission from [27]. Copyright 2016, American Chemical Society.
Table 1.
Cyclic carbonate yields and TOFs for various epoxides catalyzed by In-MOF. a Adapted with permission from [27]. Copyright 2016, American Chemical Society.
| Entry | Epoxides | Products | Yield (%) b | TOF (h−1) |
|---|---|---|---|---|
| 1 |  |  | 77.9 | 7.1 |
| 2 |  |  | 60.1 | 5.4 |
| 3 |  |  | 44.2 | 4.0 |
| 4 |  |  | 31.6 | 2.9 |
a Reaction conditions: epoxide, 20 mmol; TBABr, 1 mmol; In-MOF, 46 μmol; 1 atm of CO2; room temperature; reaction time, 48 h. b Yields were determined by GC.
Table 2.
Cyclic carbonate yields for various epoxides catalyzed by Hf-NU-1000. Adapted with permission from [28]. Copyright 2014, American Chemical Society.
Table 2.
Cyclic carbonate yields for various epoxides catalyzed by Hf-NU-1000. Adapted with permission from [28]. Copyright 2014, American Chemical Society.
| Entry | Catalyst | Epoxides | Products | Temp. (°C) | Press. (atm) | Time (h) | Yield c (%) |
|---|---|---|---|---|---|---|---|
| 1a a | Hf-NU-1000 |  |  | r.t. | 1 | 56 | 100 |
| 1b a | NU-1000 | r.t. | 1 | 56 | 46 | ||
| 1c b | - | r.t. | 1 | 56 | 0 | ||
| 1d a | Hf-NU-1000 | 55 | 1 | 13 | 100 | ||
| 2 a | Hf-NU-1000 |  |  | 55 | 1 | 19 | 100 |
| 3 a | Hf-NU-1000 |  |  | r.t. | 1 | 26 | 100 |
a Reaction conditions: epoxide (0.2 mmol), catalyst (4.0 mol % −OH active site), and TBABr (10 mol %) under CO2 (1 atm gauge pressure). b The same conditions as in footnote [a], but without the catalyst. c Determined by 1H NMR analysis using 1-bromo-3,5-difluorobenzene as the internal standard.
Table 3.
Conversions and TONs for cycloaddition of various epoxides with CO2 into cyclic carbonates by Ba-MOF. a Adapted with permission from [30]. Copyright 2018, American Chemical Society.
Table 3.
Conversions and TONs for cycloaddition of various epoxides with CO2 into cyclic carbonates by Ba-MOF. a Adapted with permission from [30]. Copyright 2018, American Chemical Society.
| Entry | Epoxides | Products | Conversion (%) | TON b |
|---|---|---|---|---|
| 1 |  |  | 98.1 | 196.2 |
| 2 |  |  | 95.7 | 191.4 |
| 3 |  |  | 94.5 | 189.0 |
| 4 |  |  | 90.2 | 180.4 |
| 5 |  |  | 19.8 | 39.6 |
| 6 |  |  | 52.3 | 104.6 |
a Reaction conditions: epoxides (20 mmol), catalyst (0.5% mmol), CO2 (1 atm), TBABr (2% mmol), room temperature, 48 h. b TON is the turnover number (product (mmol)/catalyst (mmol)).
Table 4.
Cyclic carbonate yields for various epoxides catalyzed by Zn-MOF. Adapted with permission from [32]. a Copyright 2016, Wiley-VCH Verlag GmbH.
Table 4.
Cyclic carbonate yields for various epoxides catalyzed by Zn-MOF. Adapted with permission from [32]. a Copyright 2016, Wiley-VCH Verlag GmbH.
| Entry | Epoxides | Products | Yield (%) |
|---|---|---|---|
| 1 |  |  | 92.1 |
| 2 |  |  | 88.6 |
| 3 |  |  | 78.4 |
| 4 |  |  | 69.7 |
| 5 |  |  | 39.2 |
a Reaction conditions: epoxide (25 mmol), Zn-MOF (20 mg), TBABr (0.58 g), room temperature, CO2 (101.3 kPa), 48 h.
Table 5.
Cyclic carbonate yields for various epoxides catalyzed by MIL-101-IP. a Adapted with permission from [37]. Copyright 2018, Wiley-VCH Verlag GmbH.
Table 5.
Cyclic carbonate yields for various epoxides catalyzed by MIL-101-IP. a Adapted with permission from [37]. Copyright 2018, Wiley-VCH Verlag GmbH.
| Entry | Epoxides | Products | Time (h) | Yields (%) |
|---|---|---|---|---|
| 1 |  |  | 48 | 99 |
| 2 |  |  | 48 | 95 |
| 3 |  |  | 96 | 82 |
| 4 |  |  | 96 | 84 |
| 5 |  |  | 72 | 33 |
a Reaction conditions: epoxide (1 g) with MIL-101-IP (50 mg) at 25 °C under 1 atm CO2.
Table 6.
Catalytic activities of MOFs operable under mild reaction conditions. Several high temperature conditions are also included for comparison.
Table 6.
Catalytic activities of MOFs operable under mild reaction conditions. Several high temperature conditions are also included for comparison.
| Types of MOFs | MOF Catalysts | Epoxides | Temp. (°C) | CO2 (atm) | Time (h) | Conversions (%) | Ref. |
|---|---|---|---|---|---|---|---|
| Acid site-rich (with TBABr) | {Co(μ3-L)(H2O)]·0.5H2O}n a |  | 25 | 1 | 48 | 19.3 | 25 |
| | 50 | 1 | 36 | 98.2 | 25 | ||
| [Cu2(Cu-tactmb)(H2O)3 (NO3)2] (MMCF-2) b | | r.t. | 1 | 48 | 95.4 | 26 | |
| In2(OH)(btc)(Hbtc)0.4(L)0.6· 3H2O c | | r.t. | 1 | 48 | 77.9 | 27 | |
| Hf-NU-1000 | | r.t. | 1 | 26 | 100 | 28 | |
| {[Co2(tzpa)(OH)(H2O)2]· DMF}n d | | r.t. | 1 | 48 | 93.8 | 29 | |
| Bifunctional acid–base | [Ba2(BDPO)(H2O)] e | | r.t. | 1 | 48 | 98.1 | 30 |
| (with TBABr) | {[Zn(btz)]·DMF·0.5H2O}n f |  | 30 | 1 | 12 | 39 | 31 |
| | 70 | 1 | 12 | 99 | 31 | ||
| Zn-PTB g | | r.t. | 1 | 48 | 92.1 | 32 | |
| UMCM-1-NH2 | | r.t. | 3.95 | 4 | 78 | 33 | |
| Cu-L1 h | | r.t. | 1 | 48 | 96 | 34 | |
| Incorporated Br− nucleophile | FJI-C10 |  | r.t. | 1 | 24 | 27.3 | 35 |
| (no TBABr) | | 60 | 1 | 24 | 99.7 | 35 | |
| (I−)Meim-UiO-66 | | 100 | 1 | 24 | 88.5 | 36 | |
| MIL-101-IP | | r.t. | 1 | 48 | 99 | 37 |
a L = thiazolidine 2,4-dicarboxylate. b tactmb = (1,4,7,10-tetrazacyclododecane-N,N′,N″,N‴-tetra-p-methylbenzoate). c L = an in-situ generated derivative of btc (btc = 1,2,4-benzenetricarboxylate). d H2tzpa = 5-(4-(tetrazol-5-yl)phenyl)isophthalic acid. e H4BDPO = N,N′-bis(isophthalic acid)-oxalamide. f H2btz = 1,5-bis(5-tetrazole)-3-oxapentane. g PTB = 4,4′,4″-(pyridine-2,4,6-triyl)tribenzoate. h L1 = octatopic “clicked” carboxylate bridging ligand.
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Huh, S. Direct Catalytic Conversion of CO2 to Cyclic Organic Carbonates under Mild Reaction Conditions by Metal—Organic Frameworks. Catalysts 2019, 9, 34. https://doi.org/10.3390/catal9010034
AMA Style
Huh S. Direct Catalytic Conversion of CO2 to Cyclic Organic Carbonates under Mild Reaction Conditions by Metal—Organic Frameworks. Catalysts. 2019; 9(1):34. https://doi.org/10.3390/catal9010034
Chicago/Turabian StyleHuh, Seong. 2019. "Direct Catalytic Conversion of CO2 to Cyclic Organic Carbonates under Mild Reaction Conditions by Metal—Organic Frameworks" Catalysts 9, no. 1: 34. https://doi.org/10.3390/catal9010034
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.