Plasma Oxidation of H2S over Non-stoichiometric LaxMnO3 Perovskite Catalysts in a Dielectric Barrier Discharge Reactor


Abstract
:
1. Introduction
2. Results and Discussions
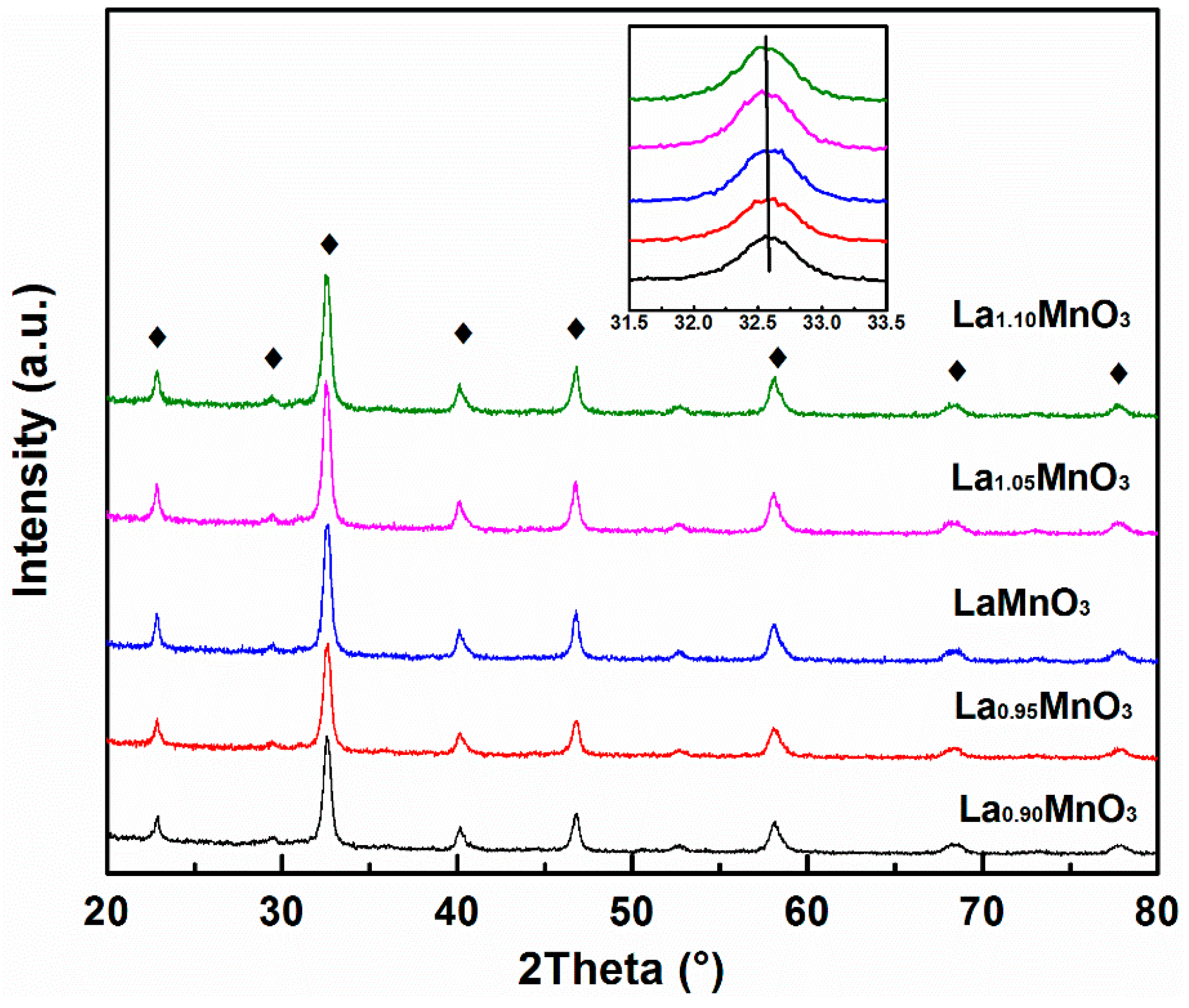
2.1. Physicochemical Properties of the Catalysts
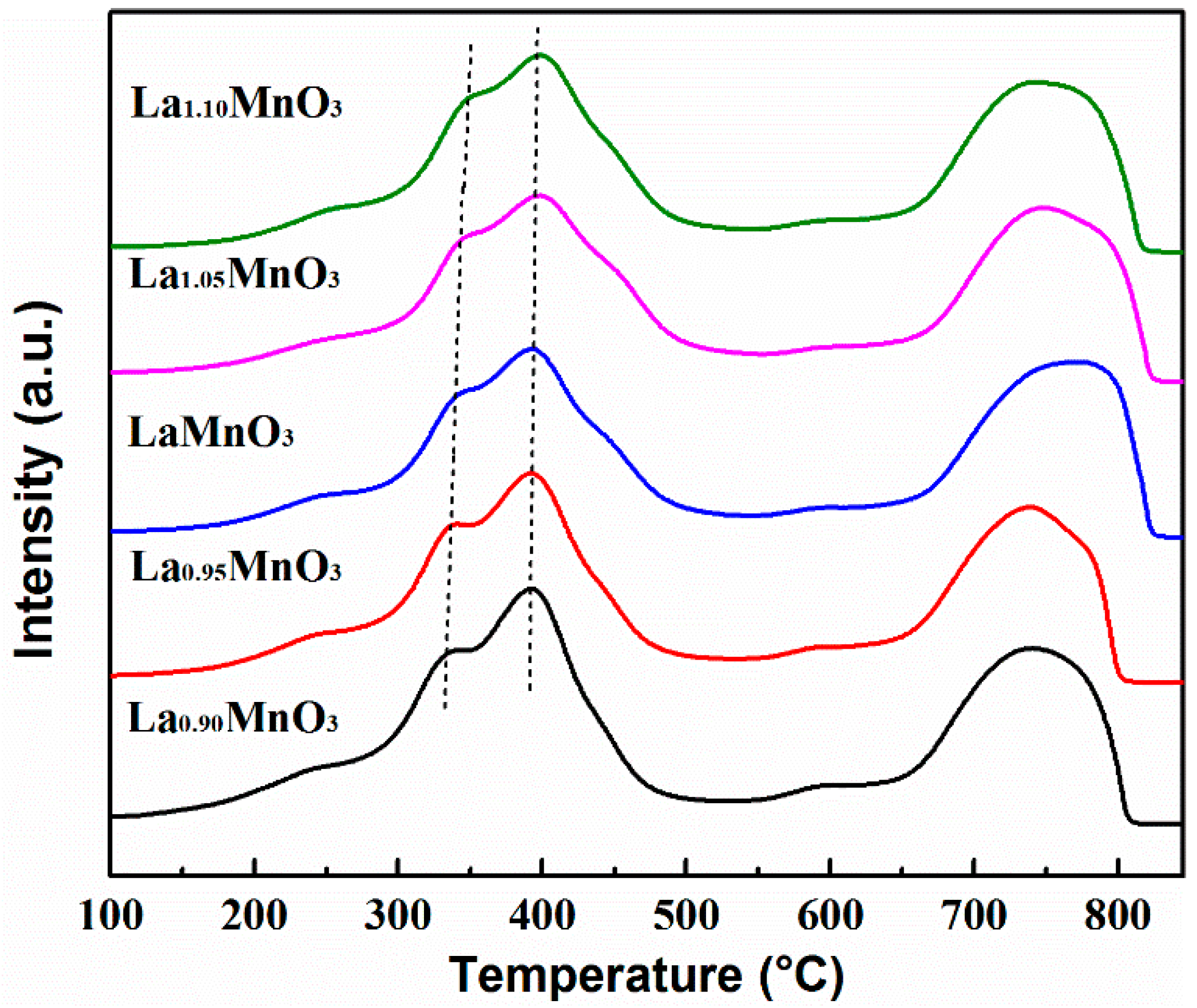
2.2. Redox Properties of the Catalysts
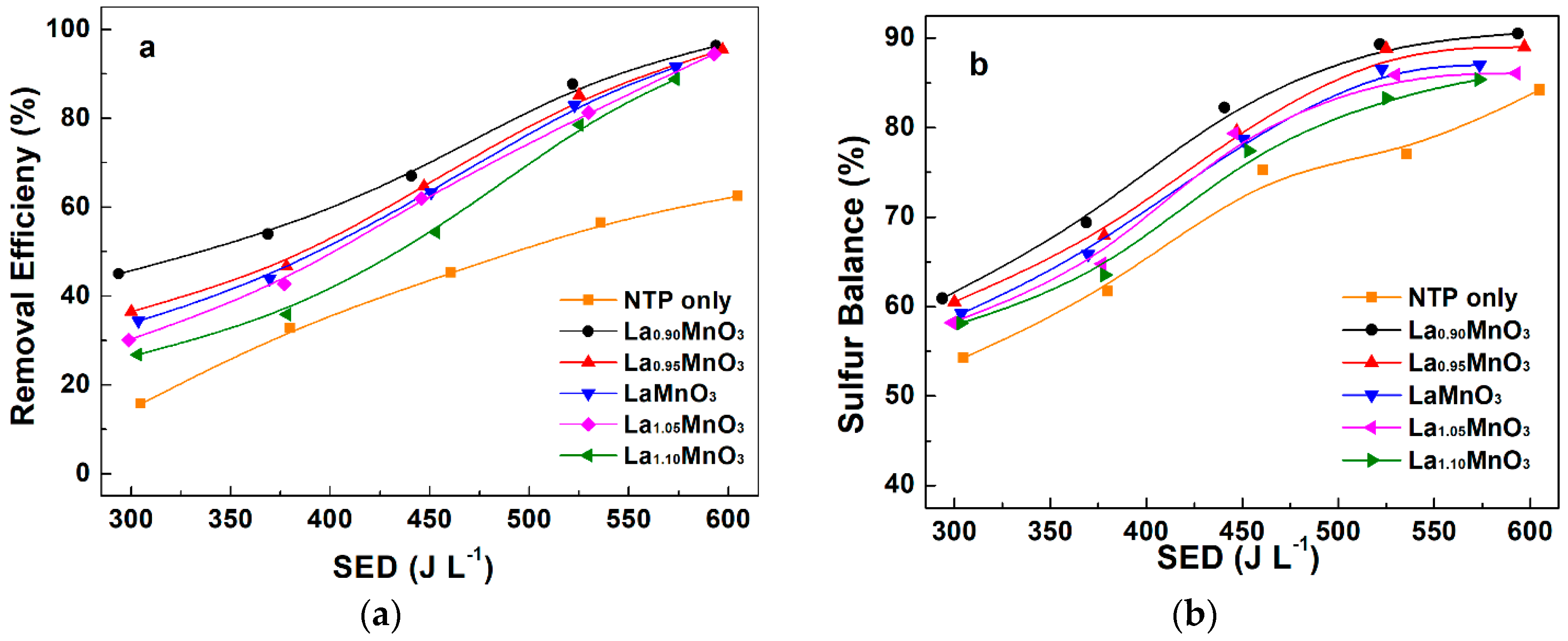
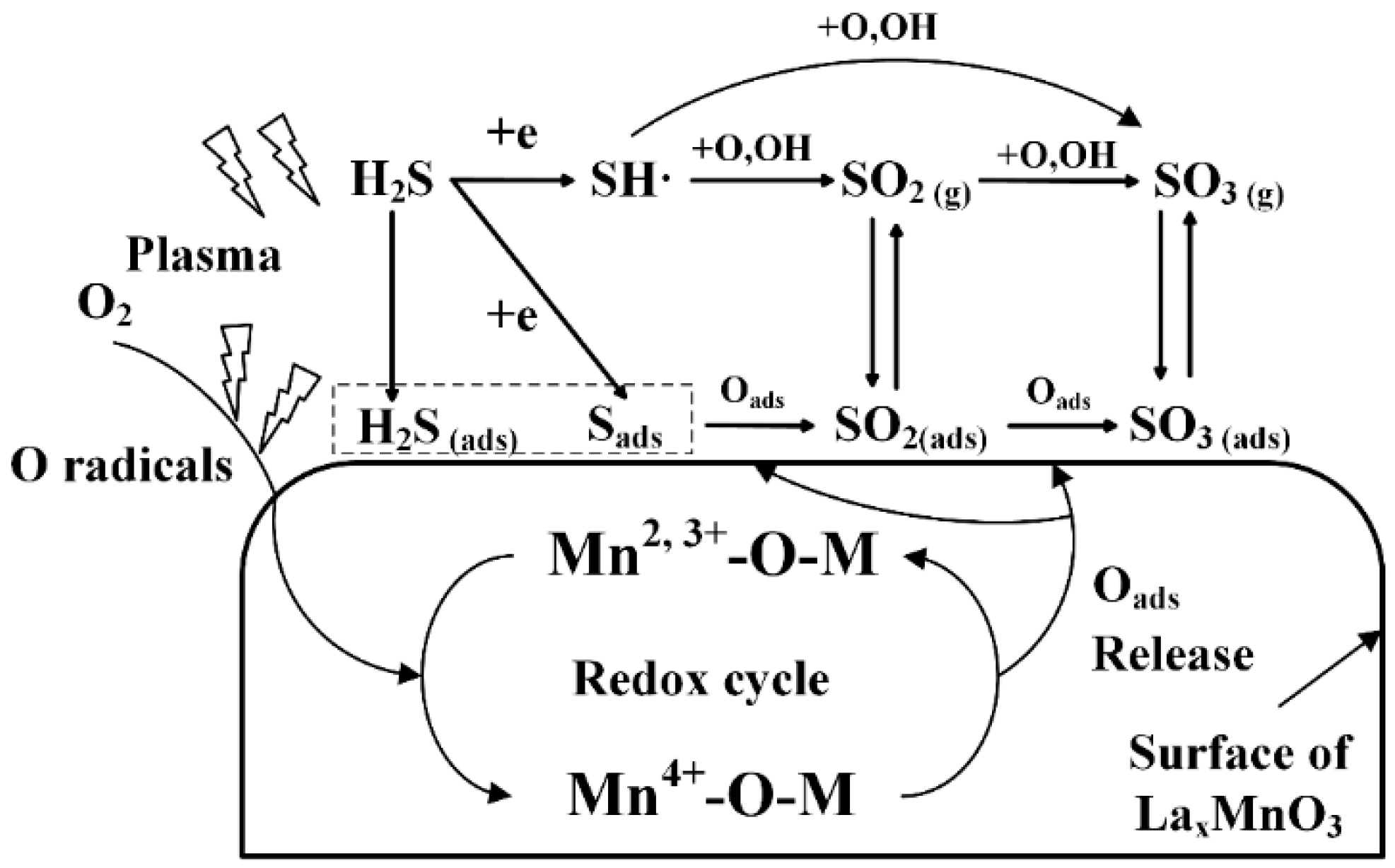
2.3. Plasma-Catalytic Oxidation of H2S
3. Experimental Setup
3.1. Catalyst Preparation
3.2. Catalyst Characterizations
3.3. Experimental Set-Up
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bogner, J.E.; Chanton, J.P.; Blake, D.; Abichou, T.; Powelson, D. Effectiveness of a florida landfill biocover for reduction of CH4 and nmhc emissions. Environ. Sci. Technol. 2010, 44, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.E.; Parsons, S.A.; Stuetz, R.M. Developments in odour control and waste gas treatment biotechnology: A review. Biotechnol. Adv. 2001, 19, 35–63. [Google Scholar] [CrossRef]
- Schlegelmilch, M.; Streese, J.; Stegmann, R. Odour management and treatment technologies: An overview. Waste Manag. 2005, 25, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; De Schryver, P.; De Gusseme, B.; De Muynck, W.; Boon, N.; Verstraete, W. Chemical and biological technologies for hydrogen sulfide emission control in sewer systems: A review. Water Res. 2008, 42, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hoff, S.J. Mitigating odors from agricultural facilities: A review of literature concerning biofilters. Appl. Eng. Agric. 2009, 25, 751–766. [Google Scholar] [CrossRef]
- Vandenbroucke, A.M.; Morent, R.; De Geyter, N.; Leys, C. Non-thermal plasmas for non-catalytic and catalytic VOC abatement. J. Hazard. Mater. 2011, 195, 30–54. [Google Scholar] [CrossRef] [PubMed]
- Van Durme, J.; Dewulf, J.; Leys, C.; Van Langenhove, H. Combining non-thermal plasma with heterogeneous catalysis in waste gas treatment: A review. Appl. Catal. B Environ. 2008, 78, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.B.; Feilberg, A.; Beukes, J.A. Abating odour nuisance from pig production units by the use of a non-thermal plasma system. In Proceedings of the International conference on environmental odour monitoring and control (NOSE), Florence, Italy, 22–24 September 2010; pp. 351–356. [Google Scholar]
- Kuwahara, T.; Okubo, M.; Kuroki, T.; Kametaka, H.; Yamamoto, T. Odor removal characteristics of a laminated film-electrode packed-bed nonthermal plasma reactor. Sensors 2011, 11, 5529–5542. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.Y.; Chen, L.; Huang, Q.X.; Yang, L.Q.; Du, C.M.; Li, X.D.; Yan, J.H. Decomposition of ammonia and hydrogen sulfide in simulated sludge drying waste gas by a novel non-thermal plasma. Chemosphere 2014, 117, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.B.; Feilberg, A.; Beukes, J.A. Use of non-thermal plasma and UV-light for removal of odour from sludge treatment. Water Sci. Technol. 2012, 66, 1656. [Google Scholar] [CrossRef] [PubMed]
- Dobslaw, D.; Ortlinghaus, O.; Dobslaw, C. A combined process of non-thermal plasma and a low-cost mineral adsorber for VOC removal and odor abatement in emissions of organic waste treatment plants. J. Environ. Chem. Eng. 2018, 6, 2281–2289. [Google Scholar] [CrossRef]
- Dobslaw, D.; Schulz, A.; Helbich, S.; Dobslaw, C.; Engesser, K.-H. VOC removal and odor abatement by a low-cost plasma enhanced biotrickling filter process. J. Environ. Chem. Eng. 2017, 5, 5501–5511. [Google Scholar] [CrossRef]
- Maxime, G.; Amine, A.A.; Abdelkrim, B.; Dominique, W. Removal of gas-phase ammonia and hydrogen sulfide using photocatalysis, nonthermal plasma, and combined plasma and photocatalysis at pilot scale. Environ. Sci. Pollut. Res. 2014, 21, 13127–13137. [Google Scholar] [CrossRef] [PubMed]
- Ragazzi, M.; Tosi, P.; Rada, E.C.; Torretta, V.; Schiavon, M. Effluents from MBT plants: Plasma techniques for the treatment of VOCs. Waste Manag. 2014, 34, 2400–2406. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, T.; Ichihashi, E.; Nishida, N.; Machida, T.; Uchida, Y.; Hayashi, Y.; Morito, Y.; Fujishima, A. Field performance test of an air-cleaner with photocatalysis-plasma synergistic reactors for practical and long-term use. Molecules 2014, 19, 17424–17434. [Google Scholar] [CrossRef] [PubMed]
- Almarcha, D.; Almarcha, M.; Jimenez-Coloma, E.; Vidal, L.; Puigcercós, M.; Barrutiabengoa, I. Treatment efficiency by means of a nonthermal plasma combined with heterogeneous catalysis of odoriferous volatile organic compounds emissions from the thermal drying of landfill leachates. J. Eng. 2014, 2014, 831584. [Google Scholar] [CrossRef]
- Dinh, M.T.N.; Giraudon, J.M.; Lamonier, J.F.; Vandenbroucke, A.; De Geyter, N.; Leys, C.; Morent, R. Plasma-catalysis of low tce concentration in air using LaMnO3+δ as catalyst. Appl. Catal. B Environ. 2014, 147, 904–911. [Google Scholar] [CrossRef]
- Nuns, N.; Beaurain, A.; Dinh, M.T.N.; Vandenbroucke, A.; De Geyter, N.; Morent, R.; Leys, C.; Giraudon, J.M.; Lamonier, J.F. A combined tof-sims and Xps study for the elucidation of the role of water in the performances of a post-plasma process using LaMnO3+δ as catalyst in the total oxidation of trichloroethylene. Appl. Surf. Sci. 2014, 320, 154–160. [Google Scholar] [CrossRef]
- Shi, C.; Zhang, Z.S.; Crocker, M.; Xu, L.; Wang, C.Y.; Au, C.T.; Zhu, A.M. Non-thermal plasma-assisted NOx storage and reduction on a LaMn0.9Fe0.1O3 perovskite catalyst. Catal. Today 2013, 211, 96–103. [Google Scholar] [CrossRef]
- Hueso, J.; Cotrino, J.; Caballero, A.; Espinos, J.; Gonzalezelipe, A. Plasma catalysis with perovskite-type catalysts for the removal of NO and CH4 from combustion exhausts. J. Catal. 2007, 247, 288–297. [Google Scholar] [CrossRef]
- Hueso, J.L.; Caballero, A.; Cotrino, J.; González-Elipe, A.R. Plasma catalysis over lanthanum substituted perovskites. Catal. Commun. 2007, 8, 1739–1742. [Google Scholar] [CrossRef]
- Vandenbroucke, A.M.; Nguyen Dinh, M.T.; Nuns, N.; Giraudon, J.M.; De Geyter, N.; Leys, C.; Lamonier, J.F.; Morent, R. Combination of non-thermal plasma and Pd/LaMnO3 for dilute trichloroethylene abatement. Chem. Eng. J. 2016, 283, 668–675. [Google Scholar] [CrossRef]
- Choi, J.J.; Billinge, S.J. Perovskites at the nanoscale: From fundamentals to applications. Nanoscale 2016, 8, 6206–6208. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-H.; Choi, Y.J.; Jeon, E.C.; Sunwoo, Y. Characterization of malodorous sulfur compounds in landfill gas. Atmos. Environ. 2005, 39, 1103–1112. [Google Scholar] [CrossRef]
- Zhu, X.; Gao, X.; Yu, X.; Zheng, C.; Tu, X. Catalyst screening for acetone removal in a single-stage plasma-catalysis system. Catal. Today 2015, 256, 108–114. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, C.; Zhan, W.; Guo, Y.; Guo, Y.; Lu, G.; Baylet, A.; Giroir-Fendler, A. Catalytic oxidation of vinyl chloride emission over LaMnO3 and LaB0.2Mn0.8O3 (B = Co, Ni, Fe) catalysts. Appl. Catal. B Environ. 2013, 129, 509–516. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, S.; Cai, Y.; Gao, X.; Zhou, J.; Zheng, C.; Tu, X. Post-plasma catalytic removal of methanol over Mn-Ce catalysts in an atmospheric dielectric barrier discharge. Appl. Catal. B Environ. 2016, 183, 124–132. [Google Scholar] [CrossRef]
- Hou, Y.-C.; Ding, M.-W.; Liu, S.-K.; Wu, S.-K.; Lin, Y.-C. Ni-substituted LaMnO3 perovskites for ethanol oxidation. RSC Adv. 2014, 4, 5329. [Google Scholar] [CrossRef]
- Shen, M.Q.; Zhao, Z.; Chen, J.H.; Su, Y.G.; Wang, J.; Wang, X.Q. Effects of calcium substitute in LaMnO3 perovskites for NO catalytic oxidation. J. Rare Earth. 2013, 31, 119–123. [Google Scholar] [CrossRef]
- Quiroz, J.; Giraudon, J.-M.; Gervasini, A.; Dujardin, C.; Lancelot, C.; Trentesaux, M.; Lamonier, J.-F. Total oxidation of formaldehyde over MnOx-CeO2 catalysts: The effect of acid treatment. ACS Catal. 2015, 5, 2260–2269. [Google Scholar] [CrossRef]
- Continetti, R.E.; Balko, B.A.; Lee, Y.T. Photodissociation of H2S and the HS radical at 193.3 nm. Chem. Phys. Lett. 1991, 182, 400–405. [Google Scholar] [CrossRef]
- Liang, W.-J.; Fang, H.-P.; Li, J.; Zheng, F.; Li, J.-X.; Jin, Y.-Q. Performance of non-thermal DBD plasma reactor during the removal of hydrogen sulfide. J. Electrostat. 2011, 69, 206–213. [Google Scholar] [CrossRef]
- Aerts, R.; Martens, T.; Bogaerts, A. Influence of vibrational states on CO2 splitting by dielectric barrier discharges. J. Phys. Chem. C 2012, 116, 23257–23273. [Google Scholar] [CrossRef]
- Zheng, C.; Zhu, X.; Gao, X.; Liu, L.; Chang, Q.; Luo, Z.; Cen, K. Experimental study of acetone removal by packed-bed dielectric barrier discharge reactor. J. Ind. Eng. Chem. 2014, 20, 2761–2768. [Google Scholar] [CrossRef]
- Aerts, R.; Tu, X.; De Bie, C.; Whitehead, J.C.; Bogaerts, A. An investigation into the dominant reactions for ethylene destruction in non-thermal atmospheric plasmas. Plasma Process. Polym. 2012, 9, 994–1000. [Google Scholar] [CrossRef]
- Zhu, X.; Gao, X.; Qin, R.; Zeng, Y.; Qu, R.; Zheng, C.; Tu, X. Plasma-catalytic removal of formaldehyde over Cu-Ce catalysts in a dielectric barrier discharge reactor. Appl. Catal. B Environ. 2015, 170–171, 293–300. [Google Scholar] [CrossRef]
- Holzer, F.; Kopinke, F.D.; Roland, U. Influence of ferroelectric materials and catalysts on the performance of non-thermal plasma (NTP) for the removal of air pollutants. Plasma Chem. Plasma Process. 2005, 25, 595–611. [Google Scholar] [CrossRef]
- Jarrige, J.; Vervisch, P. Decomposition of gaseous sulfide compounds in air by pulsed corona discharge. Plasma Chem. Plasma Process. 2007, 27, 241–255. [Google Scholar] [CrossRef]
- Chang, M.B.; Balbach, J.H.; Rood, M.J.; Kushner, M.J. Removal of SO2 from gas streams using a dielectric barrier discharge and combined plasma photolysis. J. Appl. Phys. 1991, 69, 4409–4417. [Google Scholar] [CrossRef]
- Hyun Ha, K.; Wu, C.; Takashima, K.; Mizuno, A. The influence of reaction conditions on SO2 oxidation in a discharge plasma reactor. IEEE Trans. Ind. Appl. 1999, 3, 1478–1482. [Google Scholar]
- Neyts, E.C. Plasma-surface interactions in plasma catalysis. Plasma Chem. Plasma Process. 2015, 36, 185–212. [Google Scholar] [CrossRef]
- Tu, X.; Whitehead, J.C. Plasma-catalytic dry reforming of methane in an atmospheric dielectric barrier discharge: Understanding the synergistic effect at low temperature. Appl. Catal. B Environ. 2012, 125, 439–448. [Google Scholar] [CrossRef]
- Hosseini, S.A.; Salari, D.; Niaei, A.; Oskoui, S.A. Physical-chemical property and activity evaluation of LaB0.5Co0.5O3 (B = Cr, Mn, Cu) and LaMnxCo1−xO3 (x = 0.1, 0.25, 0.5) nano perovskites in VOC combustion. J. Ind. Eng. Chem. 2013, 19, 1903–1909. [Google Scholar] [CrossRef]
- Chen, J.; Shen, M.; Wang, X.; Qi, G.; Wang, J.; Li, W. The influence of nonstoichiometry on LaMnO3 perovskite for catalytic NO oxidation. Appl. Catal. B Environ. 2013, 134–135, 251–257. [Google Scholar] [CrossRef]
- Dai, Y.; Wang, X.Y.; Li, D.; Dai, Q.G. Catalytic combustion of chlorobenzene over Mn-Ce-La-O mixed oxide catalysts. J. Hazard. Mater. 2011, 188, 132–139. [Google Scholar]
- Zhang, Z.; Jiang, Z.; Shangguan, W. Low-temperature catalysis for VOCs removal in technology and application: A state-of-the-art review. Catal. Today 2016, 264, 270–278. [Google Scholar] [CrossRef]
- Yang, Z.; Zheng, C.; Zhang, X.; Zhou, H.; Silva, A.A.; Liu, C.; Snyder, B.; Wang, Y.; Gao, X. Challenge of SO3 removal by wet electrostatic precipitator under simulated flue gas with high SO3 concentration. Fuel 2018, 217, 597–604. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m2·g−1) | 1 Crystalline Size (nm) | Cell Volume (Å3) | Mn4+/(Mn3+ + Mn4+) (%) | Oads/(Oads + Olat) (%) |
|---|---|---|---|---|---|
| La0.90MnO3 | 15.2 | 15.4 | 58.6 | 43.2 | 60.7 |
| La0.95MnO3 | 13.0 | 15.8 | 58.7 | 42.2 | 59.9 |
| LaMnO3 | 12.6 | 16.1 | 58.9 | 41.5 | 58.1 |
| La1.05MnO3 | 13.7 | 15.9 | 233.2 | 40.8 | 57.6 |
| La1.10MnO3 | 14.6 | 15.7 | 234.4 | 38.2 | 56.6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xuan, K.; Zhu, X.; Cai, Y.; Tu, X. Plasma Oxidation of H2S over Non-stoichiometric LaxMnO3 Perovskite Catalysts in a Dielectric Barrier Discharge Reactor. Catalysts 2018, 8, 317. https://doi.org/10.3390/catal8080317
Xuan K, Zhu X, Cai Y, Tu X. Plasma Oxidation of H2S over Non-stoichiometric LaxMnO3 Perovskite Catalysts in a Dielectric Barrier Discharge Reactor. Catalysts. 2018; 8(8):317. https://doi.org/10.3390/catal8080317
Chicago/Turabian StyleXuan, Kejie, Xinbo Zhu, Yuxiang Cai, and Xin Tu. 2018. "Plasma Oxidation of H2S over Non-stoichiometric LaxMnO3 Perovskite Catalysts in a Dielectric Barrier Discharge Reactor" Catalysts 8, no. 8: 317. https://doi.org/10.3390/catal8080317
APA StyleXuan, K., Zhu, X., Cai, Y., & Tu, X. (2018). Plasma Oxidation of H2S over Non-stoichiometric LaxMnO3 Perovskite Catalysts in a Dielectric Barrier Discharge Reactor. Catalysts, 8(8), 317. https://doi.org/10.3390/catal8080317