Biocatalytic Approach to Chiral β-Nitroalcohols by Enantioselective Alcohol Dehydrogenase-Mediated Reduction of α-Nitroketones

 , ,
, ,
Abstract
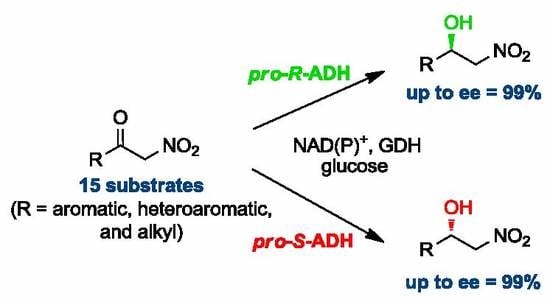
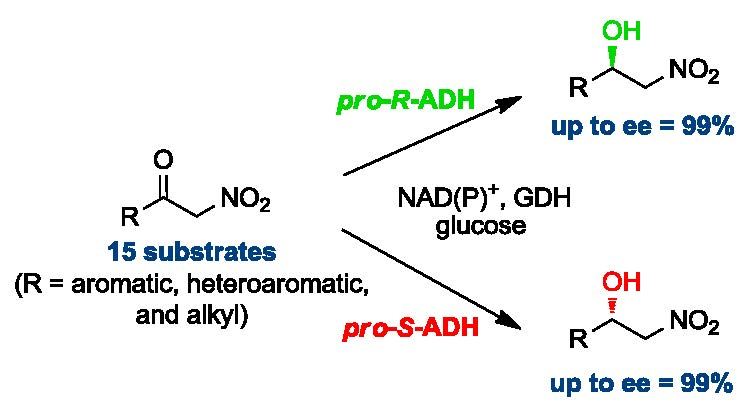
:
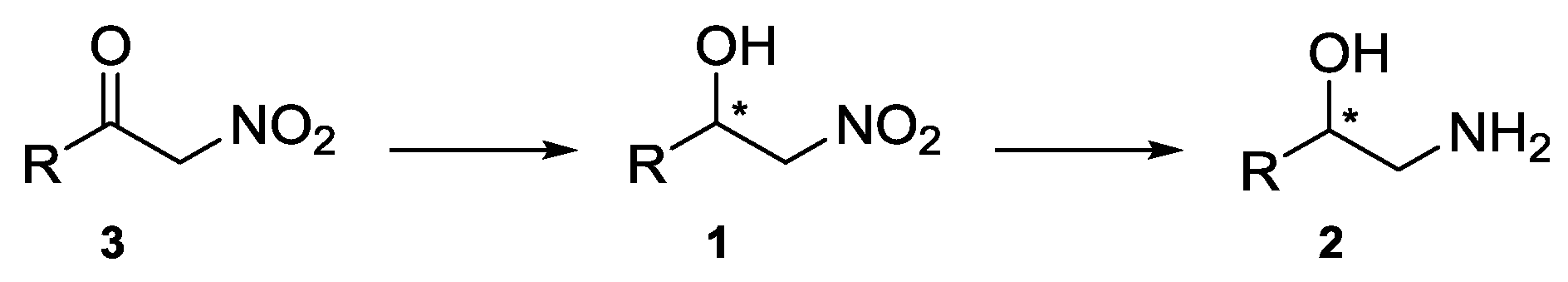
1. Introduction
2. Results and Discussion
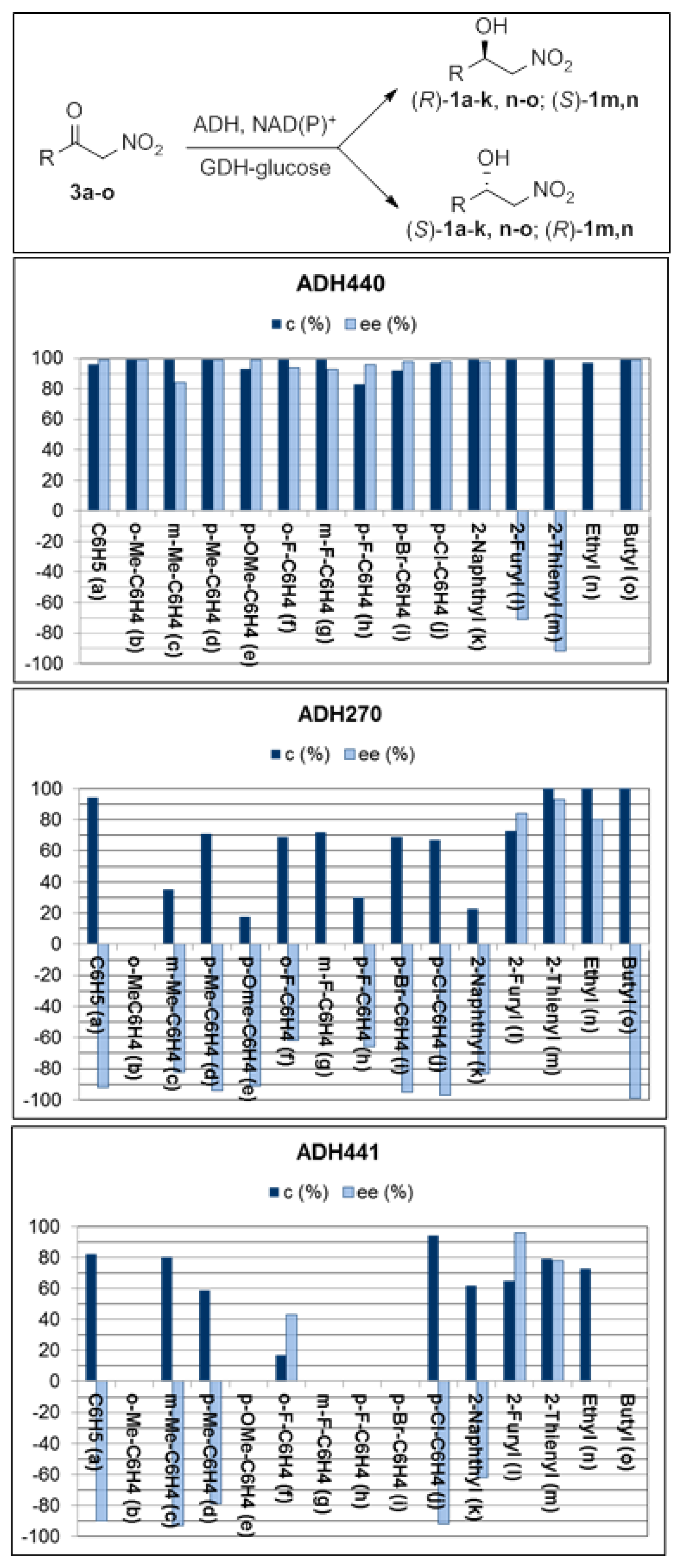
2.1. Synthesis of Nitroketones 3 and Biocatalysed Reduction to Derivatives 1
2.2. Bioreductions of Nitroketones in Biphasic Medium
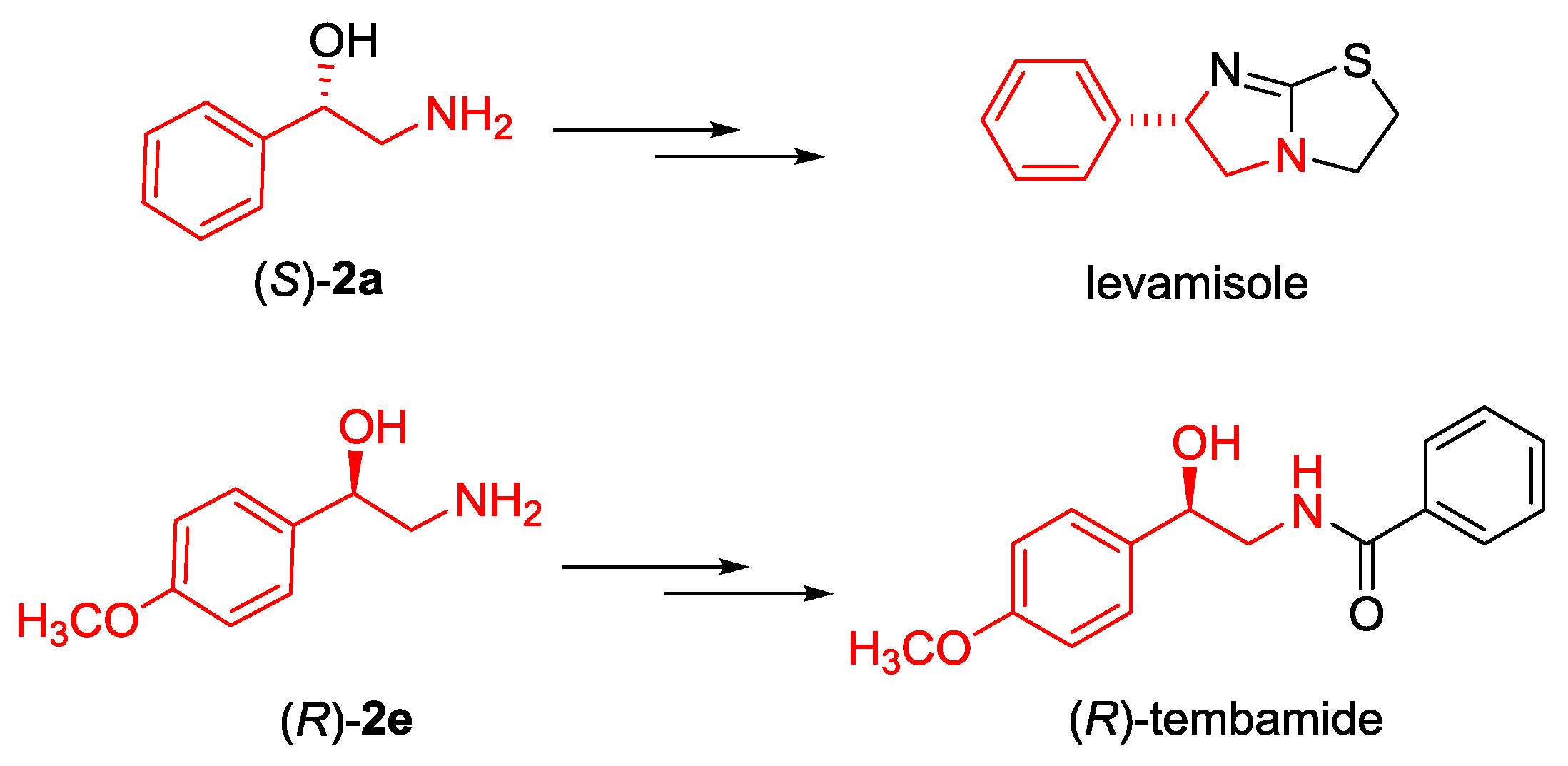
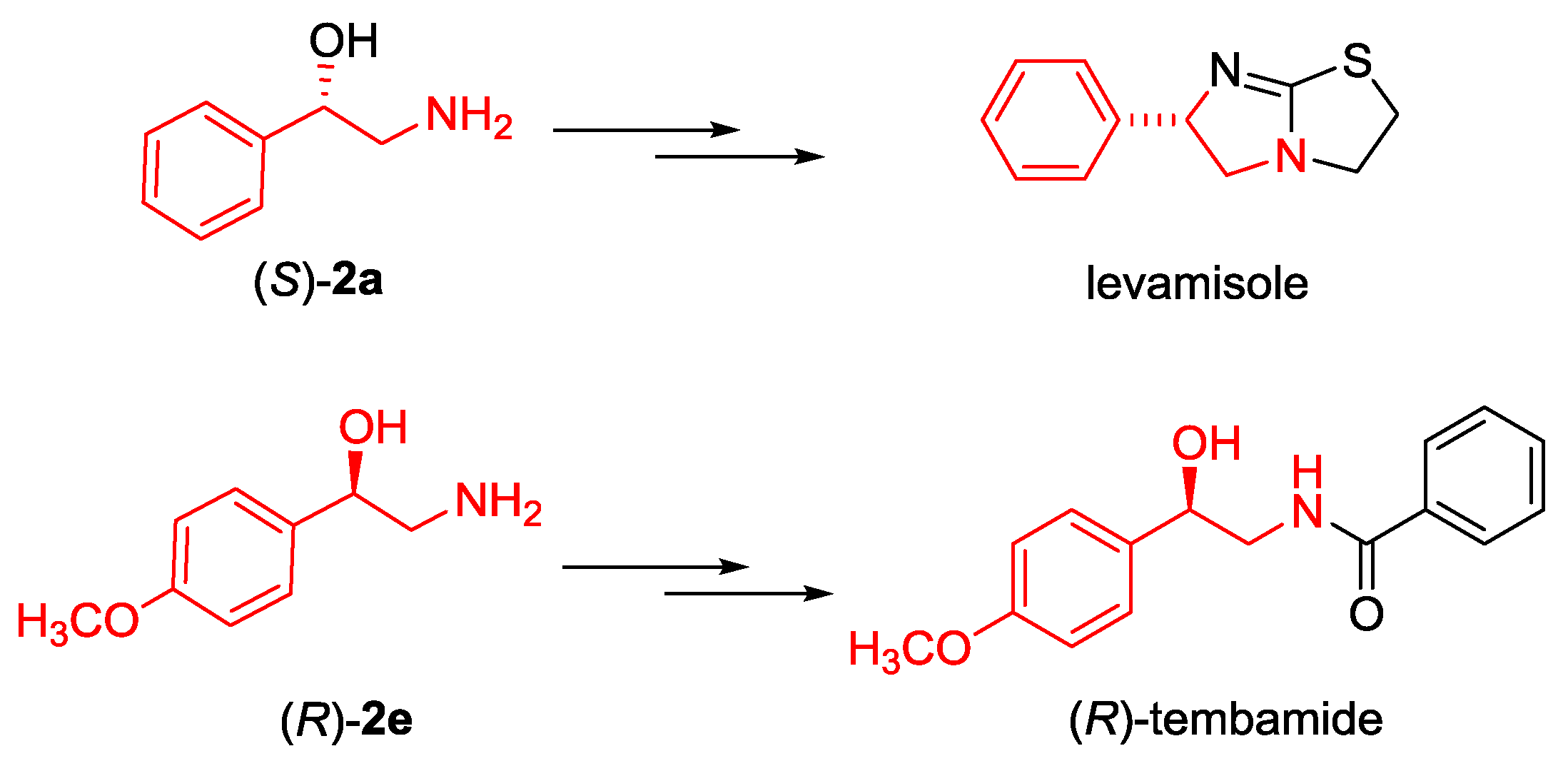
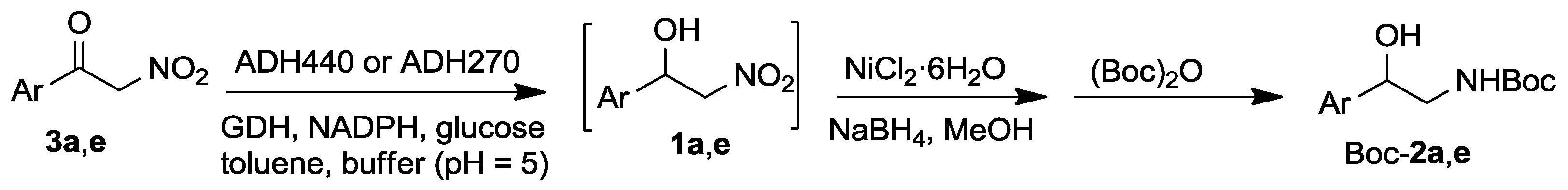
2.3. Synthesis of Boc-protected β-Aminoalcohols 2
3. Materials and Methods
3.1. Sources of Enzymes
3.2. General Procedure for the ADH-Mediated Reduction of α-Nitroketones 1a–o (Screening)
3.3. General Procedure for the Reduction of Nitroketone 3a in a Biphasic System Mediated by ADH440 and ADH270.
3.4. General Procedure for the Conversion α-Nitroketones 3a and 3e into Boc Protected Amino Alcohols 2a and 2e
3.4.1. (S)-Tert-butyl (2-hydroxy-2-phenylethyl) carbamate ((S)-2a)
3.4.2. (R)-Tert-butyl (2-hydroxy-2-(4-methoxyphenyl)ethyl) carbamate ((R)-2e)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luzzio, F.A. The Henry reaction: recent examples. Tetrahedron 2001, 57, 915–945. [Google Scholar] [CrossRef]
- Klingler, F.D. Asymmetric hydrogenation of prochiral amino ketones to amino alcohols for pharmaceutical use. Acc. Chem. Res. 2007, 40, 1367–1376. [Google Scholar] [CrossRef] [PubMed]
- Takasu, T.; Ukai, M.; Sato, S.; Matsui, T.; Nagase, I.; Maruyama, T.; Sasamata, M.; Miyata, K.; Uchida, H.; Yamaguchi, O. Effect of (R)-2-(2-aminothiazol-4-yl)-4-{2-[(2-hydroxy-2-phenylethyl)amino]ethyl} acetanilide (YM178), a novel selective beta(3)-adrenoceptor agonist, on bladder function. J. Pharmacol. Exp. Ther. 2007, 321, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Hicks, A.; McCafferty, G.P.; Riedel, E.; Aiyar, N.; Pullen, M.; Evans, C.; Luce, T.D.; Coatney, R.W.; Rivera, G.C.; Westfall, T.D.; et al. GW427353 (solabegron), a novel, selective beta(3)-adrenergic receptor agonist, evokes bladder relaxation and increases micturition reflex threshold in the dog. J. Pharmacol. Exp. Ther. 2007, 323, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Blay, G.; Hernández-Olmos, V.; Pedro, J.P. Synthesis of (S)-(+)-sotalol and (R)-(−)-isoproterenol via a catalytic enantioselective Henry reaction. Tetrahedron 2010, 21, 578–581. [Google Scholar] [CrossRef]
- Alvarez-Casao, Y.; Marques-Lopez, E.; Herrera, R.P. Organocatalytic enantioselective Henry reactions. Symmetry 2011, 3, 220–245. [Google Scholar] [CrossRef] [Green Version]
- Palomo, C.; Oiarbide, M.; Laso, A. Recent advances in the catalytic asymmetric nitroaldol (Henry) reaction. Eur. J. Org. Chem. 2007, 2561–2574, 2561–2574. [Google Scholar] [CrossRef]
- Marcelli, T.; van der Haas, R.N.S.; van Maarseveen, J.H.; Hiemstra, H. Asymmetric organocatalytic Henry reaction. Angew. Chem. Int. Ed. 2006, 45, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Shibazaki, M.; Kumagaya, N.; Nitabara, T. Preparation of Optically Active Anti-1,2-Nitroalcohols by Stereoselective Nitroaldol Reaction. Jpn. Kokai Tokkyo Koho JP 2009114071 A, 28 May 2009. [Google Scholar]
- Shibazaki, M.; Matsunaga, S.; Handa, S. Preparation of Optically Active Anti-1,2-Nitroalcohols by Stereoselective Nitroaldol Reaction with Palladium Lanthanum-Schiff Base Complex Catalysts. Jpn. Kokai Tokkyo Koho JP 2009108012 A, 21 May 2009. [Google Scholar]
- Ooi, T.; Uraguchi, D. Chiral Tetraaminophosphonium salts, catalyst for asymmetric synthesis and method for producing chiral beta Nitroalcohol. US 20090131716 A1, 21 May 2009. [Google Scholar]
- Baxter, C.E.J. Preparation of (1R*,2S*)-1-Phenyl-2-Nitroalcohols from Benzaldehyde and the Corresponding Nitroalkane in the Presence of Amine Catalysts. US 5750802 A, 12 May 1998. [Google Scholar]
- Kodama, K.; Sugawara, K.; Hirose, T. Synthesis of chiral 1,3-diamines derived from cis-2-benzamidocyclohexanecarboxylic acid and their application in the Cu-catalyzed enantioselective Henry reaction. Chem. Eur. J. 2011, 17, 13584–13592. [Google Scholar] [CrossRef] [PubMed]
- Chinnaraja, E.; Arunachalam, R.; Subramanian, P.S. Enantio- and diastereoselective synthesis of beta-nitroalcohol via Henry reaction catalyzed by Cu(II), Ni(II), Zn(II) complexes of chiral BINIM ligands. ChemistrySelect 2016, 1, 5331–5338. [Google Scholar] [CrossRef]
- Cwiek, R.; Niedziejko, P.; Kaluzia, Z. Synthesis of tunable diamine ligands with spiro indane-2,2-pyrrolidine backbone and their applications in enantioselective Henry reaction. J. Org. Chem. 2014, 79, 1222–1234. [Google Scholar] [CrossRef] [PubMed]
- Angelin, M.; Vongvilai, P.; Fischer, A.; Ramström, O. Crystallization-driven asymmetric synthesis of pyridine-beta-nitroalcohols via discovery-oriented self-resolution of a dynamic system. Eur. J. Org. Chem. 2010, 2010, 6315–6318. [Google Scholar] [CrossRef]
- Milner, S.E.; Moody, T.S.; Maguire, A.R. Biocatalytic approaches to the Henry (nitroaldol) reaction. Eur. J. Org. Chem. 2012, 2012, 3059–3067. [Google Scholar] [CrossRef]
- Purkarthofer, T.; Gruber, K.; Gruber-Khadjawi, M.; Waich, K.; Skranc, W.; Mink, D.; Griengl, H. A biocatalytic henry reaction—the hydroxynitrile lyase from Hevea brasiliensis also catalyzes nitroaldol. Angew. Chem. Int. Ed. 2006, 45, 3454–3456. [Google Scholar] [CrossRef] [PubMed]
- Gruber-Khadjawi, M.; Purkarthofer, T.; Skranc, W.; Griengl, H. A biocatalytic Henry reaction—The hydroxynitrile lyase from Hevea brasiliensis also catalyzes nitroaldol reactions. Adv. Synth. Catal. 2007, 349, 1445–1450. [Google Scholar] [CrossRef]
- Yuryev, R.; Purkarthofer, T.; Gruber, M.; Griengl, H.; Liese, A. Kinetic studies of the asymmetric Henry reaction catalyzed by hydroxynitrile lyase from Hevea brasiliensis. Biocatal. Biotransform. 2010, 28, 348–356. [Google Scholar] [CrossRef]
- Fuhshuku, K.; Asano, Y. Synthesis of (R)-beta-nitro alcohols catalyzed by R-selective hydroxynitrile lyase from Arabidopsis thaliana in the aqueous-organic biphasic system. J. Biotechnol. 2011, 153, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Bekerle-Bogner, M.; Gruber-Khadjawi, M.; Wiltsche, H.; Wiedner, R.; Schwab, H.; Steiner, K. (R)-Selective nitroaldol reaction catalyzed by metal-dependent bacterial hydroxynitrile lyase. ChemCatChem 2016, 8, 2214–2216. [Google Scholar] [CrossRef]
- Fujisawa, T.; Hayashi, H.; Kishioka, Y. Enantioselective Synthesis of Optically Pure Amino Alcohol Derivatives by Yeast Reduction. Chem. Lett. 1987, 16, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Brenelli, E.; de Carvalho, M.; Marques, M.; Moran, P.J.S.; Rodrigues, J.A.R.; Sorrilha, A.E.P.M. Enantioselective synthesis of (R)-(−)-phenylethanolamines using Baker’s yeast reduction of some substituted methyl phenyl ketones. Indian J. Chem. 1992, 31B, 821–823. [Google Scholar]
- Wallner, S.R.; Lavandera, I.; Mayer, S.F.; Öhrlein, R.; Hafner, A.; Edegger, K.; Faber, K.; Kroutil, W. Stereoselective anti-Prelog reduction of ketones by whole cells of Comamonas testosteroni in a substrate-coupled approach. J. Mol. Catal. B. 2008, 55, 126–129. [Google Scholar] [CrossRef]
- Venkataraman, S.; Chadha, A. Enantio- & chemo-selective preparation of enantiomerically enriched aliphatic nitro alcohols using Candida parapsilosis ATCC 7330. RSC Adv. 2015, 5, 73807–73813. [Google Scholar] [CrossRef]
- Albarrán-Velo, J.; González-Martínez, D.; Gotor-Fernández, V. Stereoselective biocatalysis: A mature technology for the asymmetric synthesis of pharmaceutical building blocks. Biocatal. Biotransform. 2017, 36, 102–130. [Google Scholar] [CrossRef]
- Brenna, E.; Crotti, M.; Gatti, F.G.; Monti, D.; Parmeggiani, F.; Pugliese, A.; Tentori, F. Biocatalytic synthesis of chiral cyclic gamma-oxoesters by sequential C−H hydroxylation, alcohol oxidation and alkene reduction. Green Chem. 2017, 19, 5122–5130. [Google Scholar] [CrossRef]
- Brenna, E.; Crotti, M.; Gatti, F.G.; Monti, D.; Parmeggiani, F.; Santangelo, S. Asymmetric bioreduction of beta-acylaminonitroalkenes: Easy access to chiral building blocks with two vicinal nitrogen-containing functional groups. ChemCatChem 2017, 9, 2480–2487. [Google Scholar] [CrossRef]
- Brenna, E.; Cannavale, F.; Crotti, M.; De Vitis, V.; Gatti, F.G.; Migliazza, G.; Molinari, F.; Parmeggiani, F.; Romano, D.; Santangelo, S. Synthesis of enantiomerically enriched 2-hydroxymethylalkanoic acids by oxidative desymmetrisation of achiral 1,3-diols mediated by Acetobacter aceti. ChemCatChem 2016, 8, 3796–3803. [Google Scholar] [CrossRef]
- Brenna, E.; Crotti, M.; Gatti, F.G.; Monti, D.; Parmeggiani, F.; Powell, R.W.; Santangelo, S.; Stewart, J.D. Opposite enantioselectivity in the bioreduction of (Z)-beta-aryl-beta-cyanoacrylates mediated by the tryptophan 116 mutants of Old Yellow Enzyme 1: synthetic approach to (R)- and (S)-beta-aryl-gamma-lactams. Adv. Synth. Catal. 2015, 357, 1849–1860. [Google Scholar] [CrossRef]
- Lindsay, V.N.G.; Lin, W.; Charette, A.B. Experimental evidence for the all-up reactive conformation of chiral rhodium(II) carboxylate catalysts: enantioselective synthesis of cis-cyclopropane alpha-amino acids. J. Am. Chem. Soc. 2009, 131, 16383–16385. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G.; Anderson, D.H.; Alt, L.L. Mechanism of the hydrolytic cleavage of carbon-carbon bonds. III. Hydrolysis of alpha-nitro and alpha-sulfonyl ketones. J. Am. Chem. Soc. 1955, 77, 527–529. [Google Scholar] [CrossRef]
- Thienpont, D.C.I.; Vanparijs, O.; Raeymaekers, A.; Vandenberk, J.; Demoen, P.; Allewijn, F.; Marsboom, R.; Niemegeers, C.; Schellekens, K.; Janssen, P. Tetramisole (R 8299), a new, potent broad spectrum anthelmintic. Nature 1966, 209, 1084–1086. [Google Scholar] [CrossRef] [PubMed]
- Moertel, C.G.; Fleming, T.T.; Macdonald, J.S.; Haller, D.G.; Laurie, J.A.; Tangen, C.M.; Ungerleider, J.S.; Emerson, W.A.; Tormey, D.C.; Glick, J.H.; et al. Fluorouracil plus levamisole as effective adjuvant therapy after resection of stage III colon carcinoma: a final report. Anal. Int. Med. 1995, 122, 321–326. [Google Scholar] [CrossRef]
- Moertal, G.G.; Fleming, T.R.; Macdonald, J.S. Levamisole and fluorouracil for adjuvant therapy of resected colon-carcinoma. New Engl. J. Med. 1990, 322, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Gwilt, P.; Tempero, M.; Kremer, A.; Connolly, M.; Ding, C. Pharmacokinetics of levamisole in cancer patients treated with 5-fluorouracil. Cancer Chemother. Pharmacol. 2000, 45, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Raeymaekers, A.H.M.; Roevens, L.F.C.; Janssen, P.A.J. The absolute configurations of the optical isomers of the broad spectrum anthelmintic tetramisole. Tetrahedron Lett. 1967, 8, 1467–1470. [Google Scholar] [CrossRef]
- Choudhary, M.K.; Rajkumar Tak, R.; Kureshy, R.I.; Ansari, A.; Khan, N.H.; Abdi, S.H.R.; Bajaj, H.C. Enantioselective aza-Henry reaction for the synthesis of (S)-levamisole using efficient recyclable chiral Cu(II)-amino alcohol derived complexes. J. Mol. Catal. A. 2015, 409, 85–93. [Google Scholar] [CrossRef]
- Roeben, C.; Souto, J.A.; Gonzalez, Y.; Lishchynskyi, A.; Muniz, K. Enantioselective metal-free diamination of styrenes. Angew. Chem., Int. Ed. 2011, 50, 9478–9482. [Google Scholar] [CrossRef] [PubMed]
- Sadhukhan, A.; Sahu, D.; Ganguly, B.; Khan, N.H.; Kureshy, R.I.; Abdi, S.H.R.S.; Bajaj, H.C. Oxazoline-based organocatalyst for enantioselective Strecker reactions: A protocol for the synthesis of levamisole. Chem. Eur. J. 2013, 19, 14224–14232. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Ramesh Khanna, G.B.; Krishnaji, T.; Ramu, R. A new facile chemoenzymatic synthesis of levamisole. Biorg. Med. Chem. Lett. 2005, 15, 613–615. [Google Scholar] [CrossRef] [PubMed]
- Shoeb, A.; Kapil, R.S.; Popli, S.P. Coumarins and alkaloids of Aegle-marmelos. Phytochemistry 1973, 12, 2071–2072. [Google Scholar] [CrossRef]
- Das, A.; Choudhary, M.K.; Kureshy, R.I.; Roy, T.; Khan, N.H.; Abdi, S.H.R.; Bajaj, H.C. Enantioselective Henry and aza-Henry reaction in the synthesis of (R)-tembamide using efficient, recyclable polymeric CuII complexes as catalyst. ChemPlusChem 2014, 79, 1138–1146. [Google Scholar] [CrossRef]
- Russo, A.; Lattanzi, A. Catalytic asymmetric beta-peroxidation of nitroalkenes. Adv. Synth. Catal. 2008, 350, 1991–1995. [Google Scholar] [CrossRef]
- O’Brien, P.; Osborne, S.A.; Parker, D.D. Asymmetric aminohydroxylation of substituted styrenes: applications in the synthesis of enantiomerically enriched arylglycinols and a diamine. J. Chem. Soc. Perkin Trans. 1998, 1, 2519–2526. [Google Scholar] [CrossRef]
- Watanabe, M.; Murata, K.; Ikariya, T. Practical synthesis of optically active amino alcohols via asymmetric transfer hydrogenation of functionalized aromatic ketones. J. Org. Chem. 2002, 67, 1712–1715. [Google Scholar] [CrossRef] [PubMed]
- Soltani, O.; Ariger, M.A.; Vázquez-Villa, H.E.; Carreira, M. Transfer hydrogenation in water: enantioselective, catalytic reduction of alpha-cyano and alpha-nitro substituted acetophenones. Org. Lett. 2010, 12, 2893–2895. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
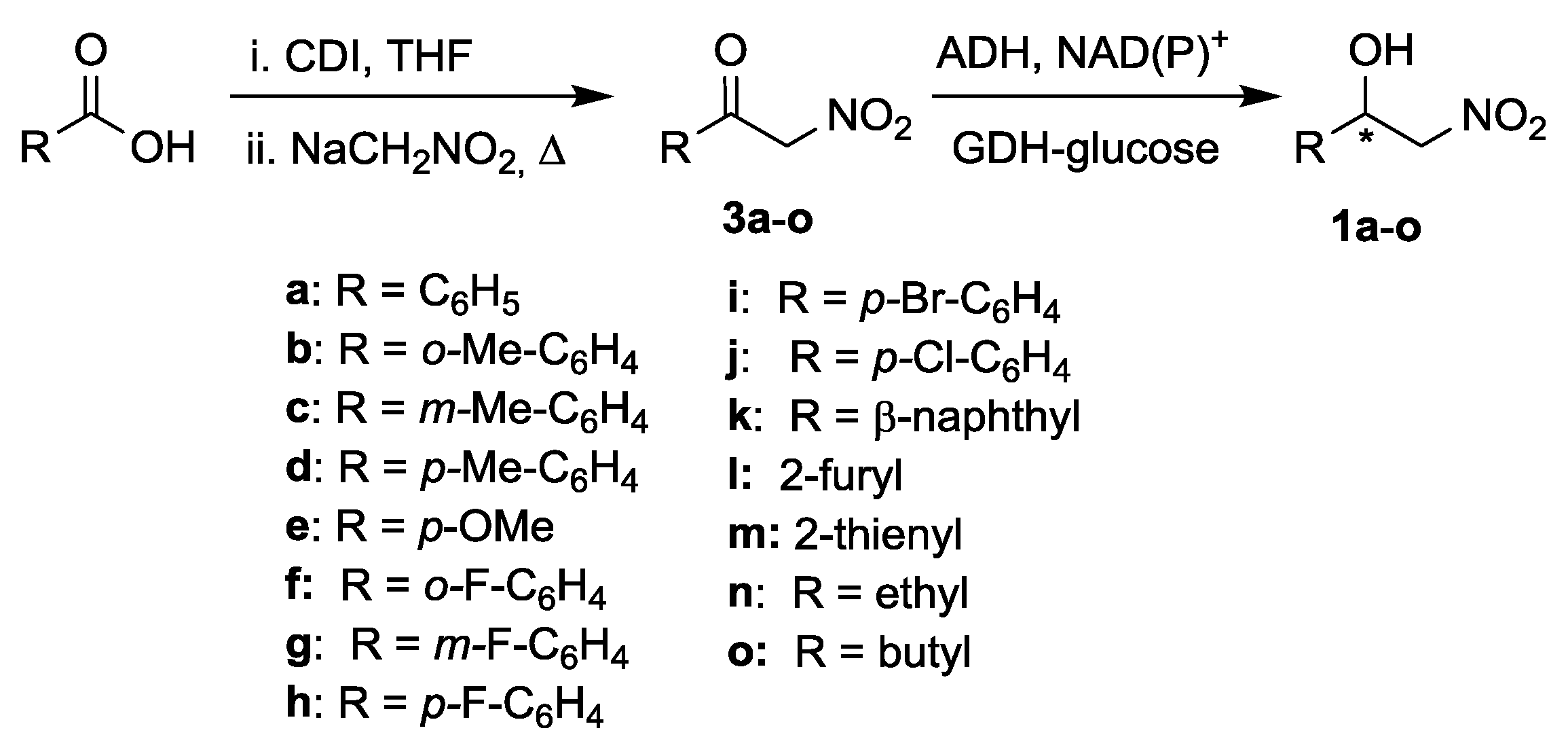{kind=link}
{kind=link}
{kind=link}
| ADH 1 | Organic Solvent | Conversion 2 (%) | Ee 3 (%) |
|---|---|---|---|
| 270 | AcOEt | - | - |
| 440 | AcOEt | 68 | 98 (R) |
| 270 | toluene | 88 | 95 (S) |
| 440 | toluene | 99 | 97 (R) |
| ADH 1 | [Substrate](mg/mL) | Substrate/Enzyme(mg/mg) | Conversion 2(%) |
|---|---|---|---|
| 440 | 1 | 2.0 | 99 |
| 1 | 8.0 | 94 | |
| 2 | 2.0 | 95 | |
| 2 | 8.0 | 91 | |
| 3 | 8.0 | 94 | |
| 3 | 24.0 | 80 | |
| 270 | 1 | 2.0 | 88 |
| 1 | 3.0 | 67 | |
| 2 | 2.0 | 84 | |
| 2 | 3.0 | 74 | |
| 3 | 3.0 | 73 | |
| 3 | 4.0 | 58 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tentori, F.; Brenna, E.; Colombo, D.; Crotti, M.; Gatti, F.G.; Ghezzi, M.C.; Pedrocchi-Fantoni, G. Biocatalytic Approach to Chiral β-Nitroalcohols by Enantioselective Alcohol Dehydrogenase-Mediated Reduction of α-Nitroketones. Catalysts 2018, 8, 308. https://doi.org/10.3390/catal8080308
Tentori F, Brenna E, Colombo D, Crotti M, Gatti FG, Ghezzi MC, Pedrocchi-Fantoni G. Biocatalytic Approach to Chiral β-Nitroalcohols by Enantioselective Alcohol Dehydrogenase-Mediated Reduction of α-Nitroketones. Catalysts. 2018; 8(8):308. https://doi.org/10.3390/catal8080308
Chicago/Turabian StyleTentori, Francesca, Elisabetta Brenna, Danilo Colombo, Michele Crotti, Francesco G. Gatti, Maria Chiara Ghezzi, and Giuseppe Pedrocchi-Fantoni. 2018. "Biocatalytic Approach to Chiral β-Nitroalcohols by Enantioselective Alcohol Dehydrogenase-Mediated Reduction of α-Nitroketones" Catalysts 8, no. 8: 308. https://doi.org/10.3390/catal8080308