Urea Activation by an External Brønsted Acid: Breaking Self-Association and Tuning Catalytic Performance
by
 , ,
, ,
Isaac G. Sonsona
1,2 ,
,
Eugenia Marqués-López
1 ,
,
Marleen Häring
2,
David Díaz Díaz
2,3,* and
Raquel P. Herrera
1,* 1
Laboratorio de Organocatálisis Asimétrica, Departamento de Química Orgánica, Instituto de Síntesis Química y Catálisis Homogénea (ISQCH) CSIC-Universidad de Zaragoza, C/ Pedro Cerbuna 12, 50009 Zaragoza, Spain
2
Institut für Organische Chemie, Universität Regensburg Universitätsstr. 31, 93053 Regensburg, Germany
3
Institute of Advanced Chemistry of Catalonia-Spanish National Research Council (IQAC-CSIC),
Jordi Girona 18-26, 08034 Barcelona, Spain
*
Authors to whom correspondence should be addressed.
Catalysts 2018, 8(8), 305; https://doi.org/10.3390/catal8080305
Submission received: 8 July 2018
/
Revised: 24 July 2018
/
Accepted: 25 July 2018
/
Published: 28 July 2018
(This article belongs to the Special Issue Hydrogen Bonding Activation)
Abstract
:In this work, we hypothesize that Brønsted acids can activate urea-based catalysts by diminishing its self-assembly tendency. As a proof of concept, we used the asymmetric Friedel–Crafts alkylation of indoles with nitroalkenes as a benchmark reaction. The resulting 3-substituted indole derivatives were obtained with better results due to cooperative effects of the chiral urea and a Brønsted acid additive. Such synergy has been rationalized in terms of disassembly of the supramolecular catalyst aggregates, affording a more acidic and rigid catalytic complex.
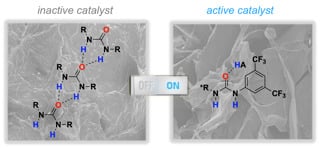
1. Introduction
Asymmetric organocatalysis has become a powerful synthetic strategy over the last decade, being crucial for the development of new catalytic processes [1,2,3,4,5,6,7,8,9,10,11,12,13]. Among different families of organocatalysts [14], those acting by hydrogen bond interactions represent a noteworthy part of the overall developed organocatalysts [15,16,17]. One of the main organocatalytic structures covering this large group are (thio)urea derivatives [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]. After the pioneering work of Curran’s group on the use of urea as an organocatalyst [35,36], a number of considerable efforts have been devoted to the design and synthesis of new (thio)urea structures as suitable catalysts for a series of important enantioselective processes [18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34]. It is remarkable that the works reported by Etter’s group, where they were able to get co-crystal complexes with a urea and a variety of hydrogen bond acceptors, were the real starting point for the development of this field [37,38,39,40].
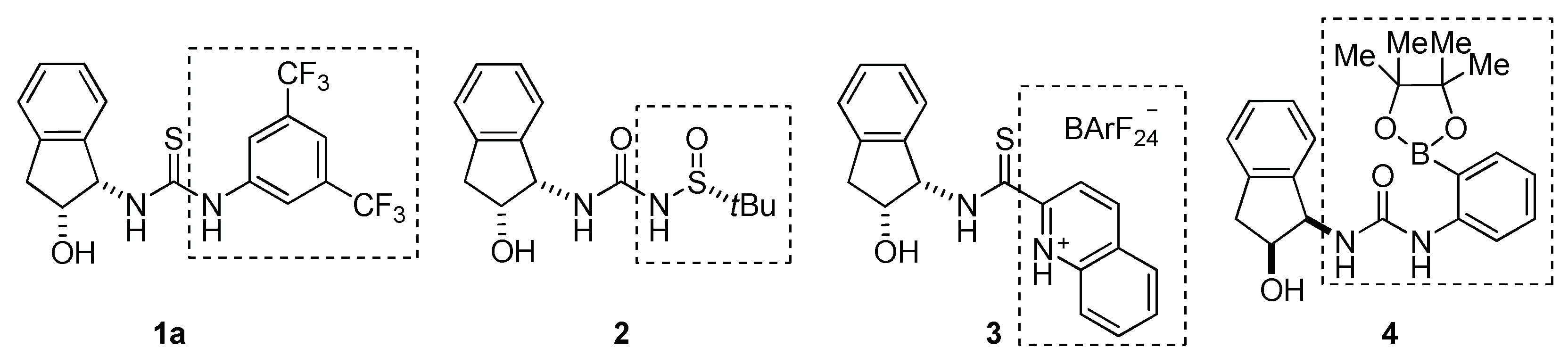
Most of the works in this field have so far focused on the development of more reactive catalytic systems in order to overcome disadvantages exhibited by organocatalysis [41], such as the necessity of high catalyst loadings and ensuing low turnover rates [42]. In this sense, progress has been made on modification of the (thio)ureas skeleton in order to increase their acidity. Within this context, more efficient catalysts were prepared, on introducing internal acidic elements (Figure 1). For instance, taking our catalyst 1a [43,44,45,46,47,48,49,50,51] as a model, subsequent analogues 2 [52], 3 [53] and 4 [54] were designed and have been proved to increase catalytic activity. Using this approach, the authors changed the most acidic moiety of the model catalyst (i.e., 3,5-bis-CF3-phenyl group) for different acidic moieties, while retaining the aminoindanol structure (Figure 1) [55,56].
In this respect, Sigman and co-workers demonstrated that both the reaction rate and the enantioselectivity could be correlated to catalyst acidity and it could be efficiently used for the tuning and design of new catalyst structures for hydrogen-bond-catalyzed enantioselective reactions [57].
Among the most appealing organocatalyzed reactions, the Friedel-Crafts alkylation reaction has received great attention and is a very efficient strategy for C–C bond formation [58,59,60,61]. Based on our previous experience using (thio)urea catalysts [43,44,45,46,47,48,49,50,51,62,63,64,65,66], we reported a pioneering concept regarding the cooperative effect between a Brønsted acid additive and a chiral thiourea organocatalyst [45]. In this work, we hypothesize that similar beneficial effects could take place with urea organocatalysts in combination with a Brønsted acid, which could activate the urea by diminishing significantly its self-association tendency and, therefore, increasing its acidity.
2. Results and Discussion
2.1. Cooperative Effect in the Mixture (Urea Catalyst + Brønsted Acid)
We have previously studied both (thio)urea-catalyzed Friedel-Crafts-type alkylation reactions (Figure 2) [43,44,45,46,47,48,49,50,51]. However, the poorer results obtained with the ureas in comparison with thiourea analogues made us discard this family of structures in successive studies. The unfavorable results found with ureas have been explained based on the most acidic N-H protons in the thioureas [67] and their less tendency to self-aggregate due to lower electron density on the sulfur atom [68,69,70]. Hence, it is not surprising to find more thiourea-based organocatalysts than ureas reported in the literature [16].
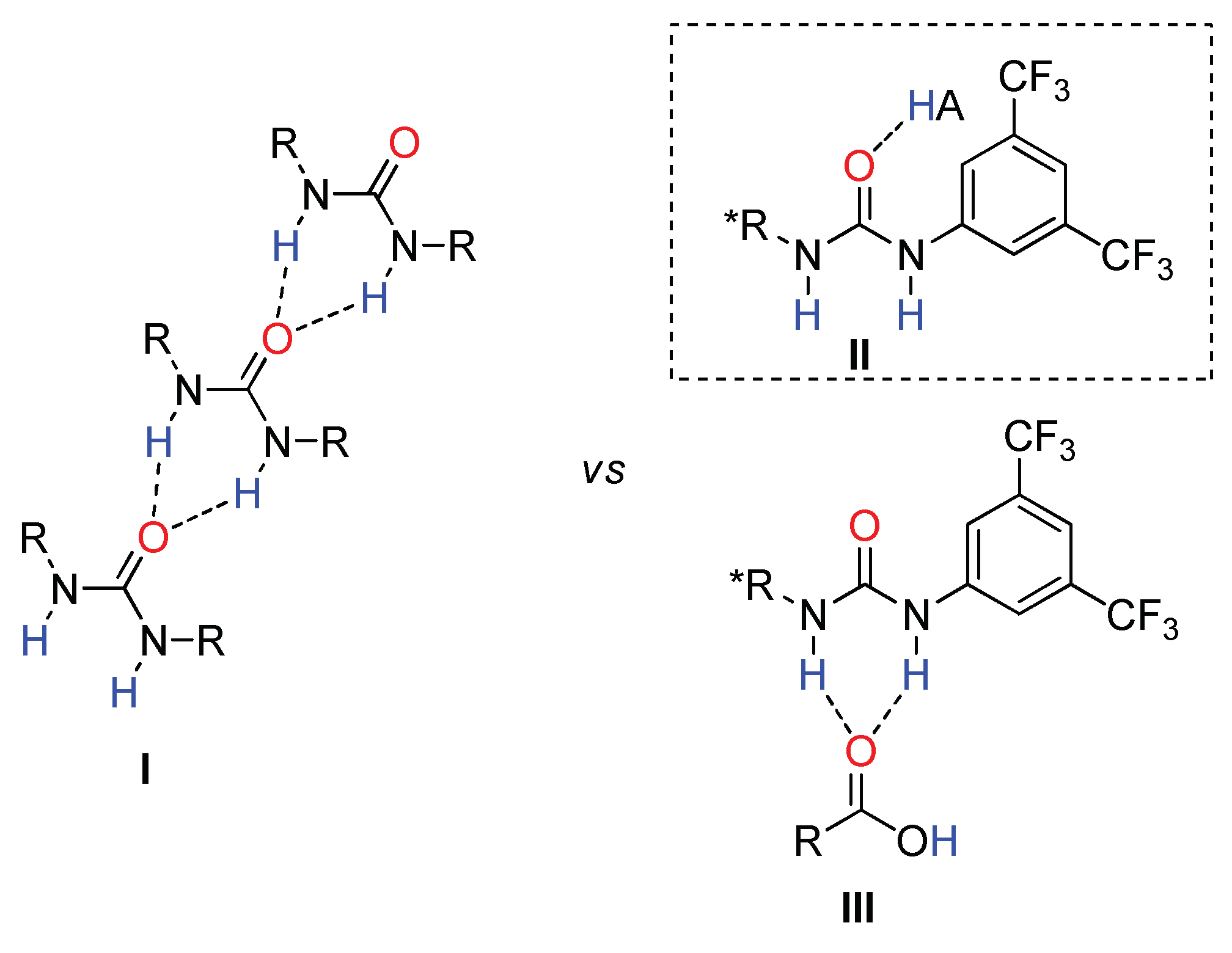
As a matter of fact, self-association of ureas has been identified as the major drawback for hydrogen-bonding organocatalysis (Figure 3, I) [71,72]. Our previous results obtained with thioureas motivated us to investigate whether the addition of a Brønsted acid hampers the hydrogen-bonding pattern of the urea, allowing an equilibrium with free molecules in solution and/or smaller and soluble aggregates. In addition, molecules of “free” urea could be activated by a stronger proton donor (II), disrupting the interactions between neighboring urea molecules and, thus, increasing their efficiency as a hydrogen-bonding organocatalyst (Figure 3). In principle, the activation mode III should also be considered, since it was proposed by others when using thiourea catalysts and carboxylic acids [73,74,75,76,77,78].
To this aim, we explored in detail the Friedel-Crafts alkylation of indole 6a with nitrostyrene 7a using urea catalyst 1b, both in the absence and in the presence of an external Brønsted acid (Scheme 1).
We were delighted to observe a positive effect when using 1b in the presence of acid (±)-mandelic acid (9a). Interestingly, the enantiomer S (product 8aa) obtained when using complex 1b (±)-mandelic acid (9a) was the same when using only urea 1b.
At this point, we decided to explore the different parameters that could influence this activation mode. Thus, we first studied the effect of the solvent, since polarity might play an important role in governing the interaction between catalysts and reagents (Table 1).
A lack of reactivity in polar solvents, with high complexing character, was observed even when the solubility of the urea was certainly higher in those solvents (Table 1, entries 7–12). Thus, the rupture or weakening of the hydrogen interactions between the catalyst and the reagents seems to be higher in these solvents, hampering the reaction. In sharp contrast, good results were achieved on using less coordinating solvents, such as toluene and xylene (Table 1, entries 4–6). Finally, CHCl3 provided the best results (Table 1, entry 2) and, we hence selected this solvent for further studies. It is remarkable that, in all cases, better results were obtained in terms of enantioselectivity and reactivity using catalyst 1b in the presence of mandelic acid (±)-9a (e.g. entries 2, 4, 6, 8 vs. entries 1, 3, 5, 7). Interestingly, the same enantiomeric product S was obtained with thiourea 1a and urea 1b, as well as with a mixture of urea 1b and Brønsted acid (±)-9a. These results suggest that urea catalyst, and not the Brønsted acid, drives the enantioselectivity of the process.
Furthermore, we extended the exploration of this catalytic system using different Brønsted acids. Scheme 2 shows the results obtained with representative derivatives of acetic acid as the structural core of the mandelic acid (±)-9a.
The results shown in Scheme 2 revealed a cooperative effect between urea 1b and all external acids used, although without a clear correlation with the pKa of the acid (Scheme S1). (R)- and (S)-mandelic acid 9a were tested in order to explore the match and mismatch effect. In both cases, the S enantiomer and the same results were obtained, suggesting that chirality is induced only by catalyst 1b. With this experiment, we discarded activation mode III (Figure 3), since in such case the chirality of the acid would be expected to influence the absolute configuration of the final products. In contrast, these results are in agreement with the activation mode II (Figure 3), where Brønsted acid could be activating urea 1b. Remarkably, (S)-9c and (±)-9d also presented similar results than (±)-9a. However, further studies were done using mandelic acid since, being the cheaper acid, its structure seems to be one of the most influential on urea self-aggregation, providing good results in terms of enantioselectivity and reactivity.
Table 2 summarizes the effect of temperature on the behavior of the complex 1b·(±)-mandelic acid (9a).
The results demonstrated that the effect of Brønsted acid on the urea-based catalysis is maintained even at lower temperatures (15 and −25 °C), although a gradual drop in the reaction yield was observed. On the other hand, it seems that the effect of the external Brønsted acid decreases at a lower temperature, where presumably the formation of urea aggregates prevails and increases the insolubility of the acid.
At this point, and since our goal was to test this alternative approach to improve the efficiency of the urea catalyst, we explored the substrate scope at room temperature (Scheme 3).
In order to compare the results obtained with 1b·HA and the sole catalyst 1b, the reactions were stopped after three days. The results shown in Scheme 3 indicate that the working hypothesis was valid using different substituted nitroalkenes 7a–i and indoles 6a–d, affording the corresponding adducts 8 with higher yields and enantioselectivities compared to reactions in the absence of an external acid. The absolute configuration of adducts 8 was assigned by comparison of their optical rotation with those reported in the literature for the same compounds.
2.2. Effect of Brønsted Acid on Urea Aggregation and Mechanistic Hypothesis
The foregoing results suggest two important facts to be considered when adding an external acid to the reaction medium: first, the plausible rupture of self-assembled ureas by the acid; and second, the activation of the “free” urea through coordination by the acid. The fragmentation of urea aggregates, affording a higher concentration of “free” urea (or smaller soluble aggregates) in solution, could explain the enhancement of the reaction yield, but not necessarily an increase of the ee. However, a concurrent activation of the urea with the disassembly process could positively influence both the yield and the enantioselectivity of the process, as shown in Scheme 2.
With this in mind and in order to gain additional insight on the coordination mode between the Brønsted acid and the urea, we tested some additional key catalysts and additives (Table 3).
Additional important aspects can also be concluded from the results disclosed in Table 3. For instance, the presence of an OH group in the skeleton of 1b with cis configuration seems to be crucial for both the enantioselectivity and the reactivity of the process (compare entry 2 against entries 7 and 8), which is in good agreement with our previous observations with thioureas [45]. When additives 9h, 9i or MeOH were used (entries 4–6), the results were similar to those obtained with urea 1b alone (entry 1) in terms of selectivity, although a slightly better yield was obtained with the additives. This supports the scenario in which the additive could break urea interactions, generating more “free” urea, although in the absence of an additional activation, since only the OH of the carboxylic acid seems to be responsible for the activation of the urea catalyst. The effect observed with catalysts 1c and 1d was only evident in the reactivity of the process, since both catalysts alone afforded racemic products (entries 7 and 8). With these results, we were also able to discard the possible mode of activation in which both urea and acid simultaneously activate nitrostyrene, as previously proposed by other authors [79], since in this case the yield should be higher than that achieved with the use of acid (±)-9a alone (Scheme 1, 30%).
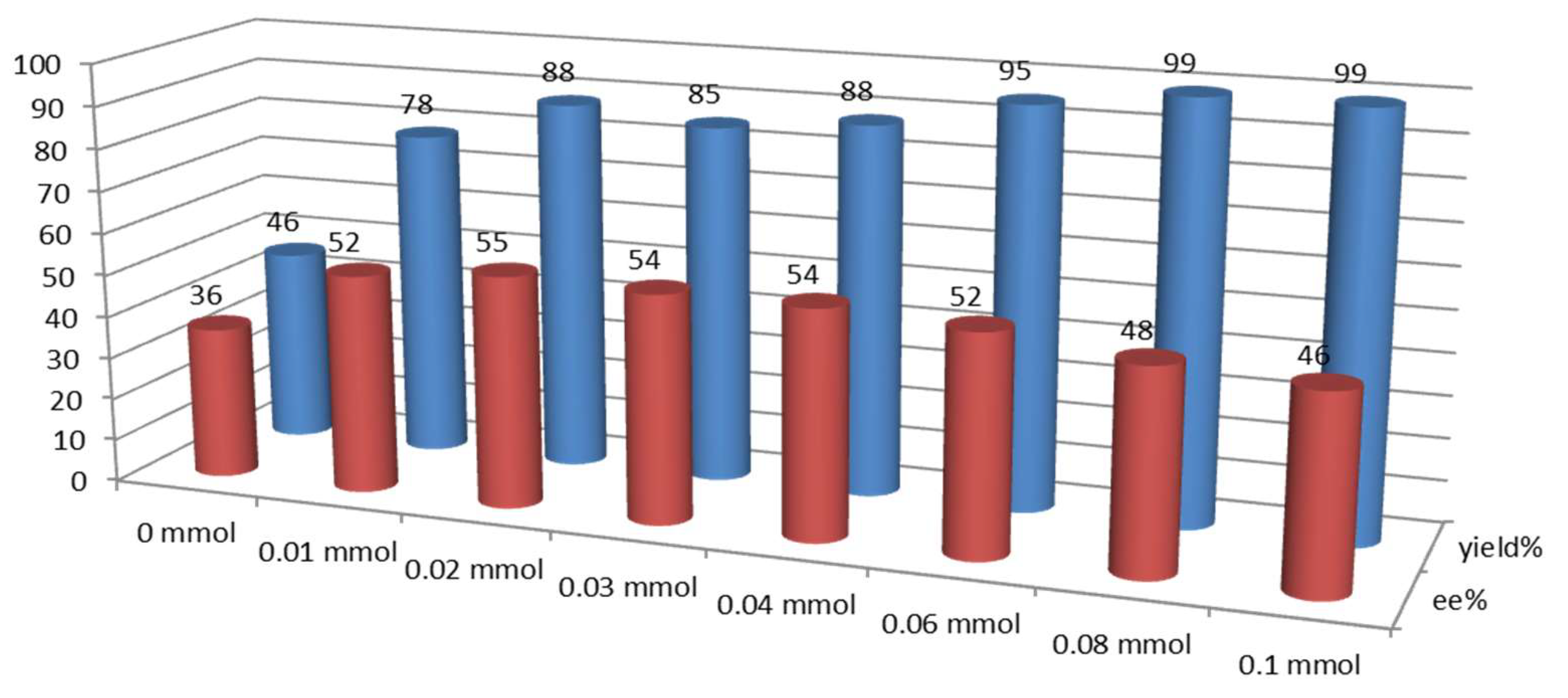
Next, we analyzed the effect on both ee and reaction yield using progressive amounts of (±)-9a with respect to urea 1b (0.02 mmol) (Figure 4).
The results indicated that ca. 20–40 mol% of acid is optimal for the activation effect. Higher amounts of acid further increased the yield, but caused a slightly decrease in the ee of the product, because the activation of the reaction through the acid alone could also take place, giving rise to a racemic product.
Since NMR techniques can provide important information on self-assembly processes in solution and in order to support the fragmentation of urea aggregates in the presence of acid, different 1H-NMR experiments were performed. We thus studied the variation in the signals of the urea catalyst 1b after progressive addition of acid (±)-9a in CDCl3 (Table 4 and Figures S1–S8).
Among these NMR spectra, we only observed one species of the urea due to the fact that the chemical exchange between the self-assembled and non-self-assembled species is rapid on the NMR timescale. Additionally, with the addition of acid, urea signals show sharper peaks in comparison with broader peaks found when it is mainly aggregated (see supporting information Figure S1–S8). This is because when urea is aggregated, the major part of the signals belongs to the NMR-silent (entry 1, ratio integrals ureas:standard) [80]. In contrast, when urea self-association is progressively broken with the addition of acid, the integrals of the urea increase in relation to the standard (ratio integrals ureas:standard). It was also observed that the NHa signal of urea for a given concentration (0.02 mmol/0.5 mL CHCl3), when the equilibrium is shifted in favor of the aggregates (entry 1), is sensitive to the presence of acid and is progressively highfield shifted with the addition of acid. This is in agreement with observations made by other authors in different self-assembly systems [81,82]. These facts could relate to the presence of more free molecules of urea or smaller aggregates in solution and, therefore, support the disruption of urea self-assembly with the presence of acid. Other signals of the urea are also affected by the presence of acid, as shown in Table 4. When 5 equivalents of acid are added (entry 8), the signals of the urea catalysts move in the opposite direction (to downfield). This could be in agreement with the equilibrium shifted to a predominant activation of the urea molecules, and therefore, with the ensuing acidification of the signals. A major variation in urea signals is observed with the addition of 0.02 mmol of acid, as in the catalytic reactions, a plateau is more or less reached after that, and increasing the amount of acid does not strongly affect the shifting of the signals. A DOSY NMR experiment [83,84,85] also supports the coordination of the acid to the urea, since it is possible to find two different aggregate species in a given sample (urea·acid 0.02 mmol:0.02 mmol): the urea self-assembled and the urea·acid complex (Figure 5).
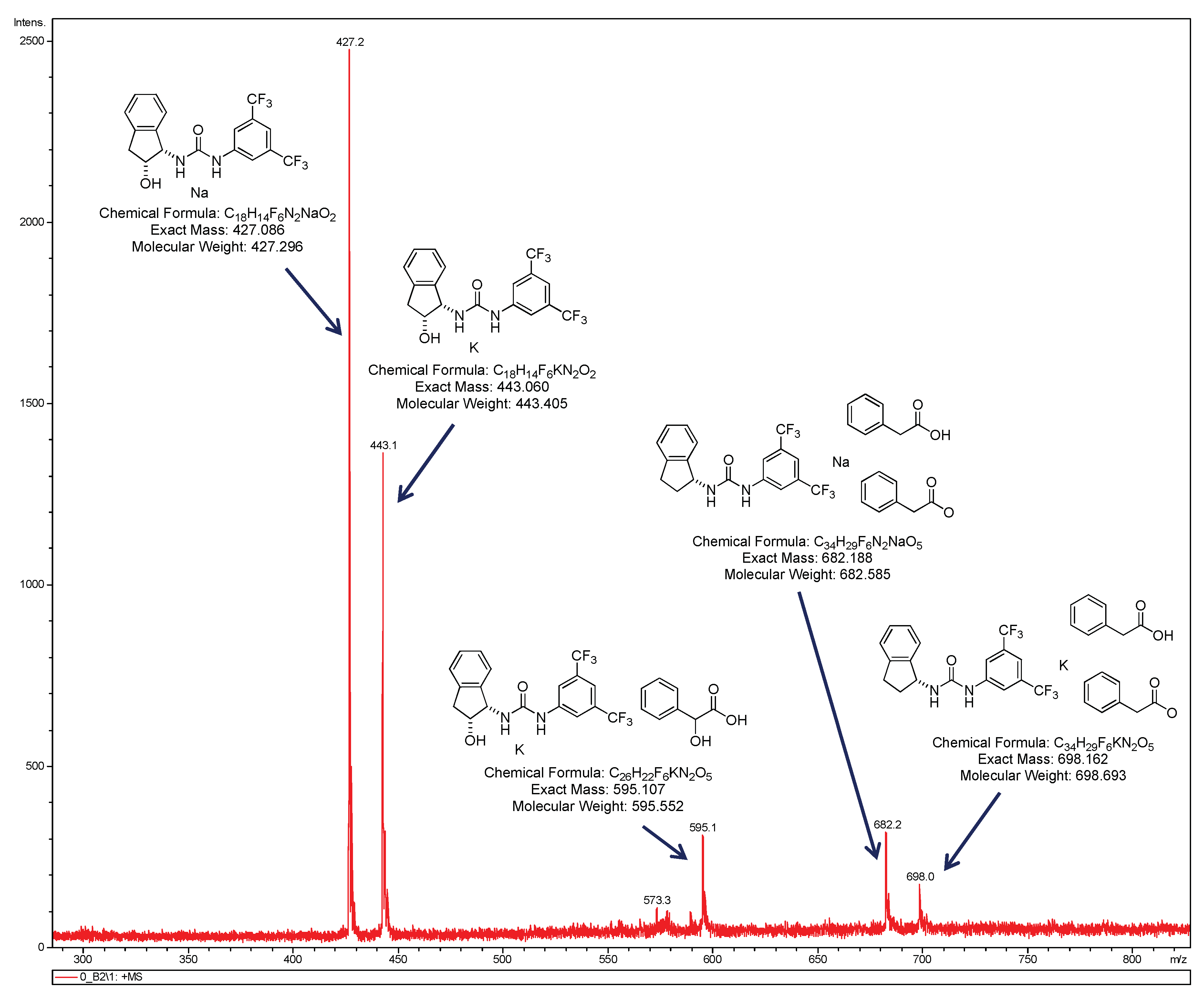
We also performed a mass spectrum of the mixture urea 1b·acid (±)-9a (0.02 mmol:0.02 mmol) in 0.5 mL of CHCl3 (Figure 6), since ESI-MS has been incorporated as an important technique for mechanistic studies of organic reactions [86]. In the cationic ESI spectra obtained directly from solution to the gas phase, we found different species, and among them the remarkable mass peak of the active complex proposed in this manuscript (catalyst 1b + mandelic acid (±)-9a) (M + K = 595.1). Other superior species related to the previous one have also been found in the mixture. Interestingly, we could not observe mass from the union of two or more ureas, although we cannot discard the presence of this complex in the mixture of reaction. It could be due to the impact of this technique that they split into free molecules of urea.
Based on these evidences and the results of our previous investigations [45,46], TSI is proposed as the plausible mode of activation between Brønsted acid, urea 1b and the reagents (Figure 7).
1b·HA complex could be identified as the most reactive species due to the increased acidity of NH in urea 1b after synergic coordination with Brønsted acid. Furthermore, improvement in enantioselectivity could be attributed to the formation of a more rigid assembly (on combination of both structures after breaking urea self-association) in the transition state. This mode of activation supports the fact that the observed enantiomer is a result of the chirality of the organocatalyst used. Further computational calculations could shed light on this singular mode of activation.
2.3. Effect of Brønsted Acids on the Stability of Urea Aggregates
When the reaction was performed in CH2Cl2 or CHCl3 using urea 1b alone, the formation of dense gel-like aggregates was observed, which was in agreement with our previous studies about the self-assembly behavior of these ureas in solution [47]. The viscosity of the mixture was visually decreased upon the addition of acid 9a, which could theoretically be in agreement with a weakening of the self-assembled network, spontaneously generated by the urea catalyst and, hence, with our mechanistic assumptions from the catalytic experiments. This hypothesis was demonstrated by analyzing the thermal stability, flow properties and morphological features of the supramolecular gel made of urea 1b in the absence or presence of various acid additives at different molar ratios urea 1b:acid (i.e. 0.1, 0.3, 0.9). Two solvents (i.e. chloroform—solvent used in the reactions—and toluene) were used in this study for comparative purposes. In general, gels were transparent in appearance, although some systems prepared in chloroform were slightly translucent (Figure 8). Most studies in this section were conducted with gels prepared within approximately 30 min of the corresponding critical gelation concentrations (CGC) of 1b, which were previously established in 7 g/L (chloroform) and 3 g/L (toluene) [47]. The detrimental effect of the acid additives was considerably more evident for the gels in chloroform than in toluene. Therefore, and for the sake of clarity, the most relevant data refereed to the gels in chloroform are included in this section, whereas most data derived from the systems in toluene are provided in Supporting Information (Table S1). The first sign of the weakening effect that the acid additive has on the physical gel network formed by self-assembled urea molecules was a partial gel-to-sol phase transition, visible to the naked eye (Figure 8B–E). In general, the amount of expelled fluid usually increased with increasing amounts of acid.
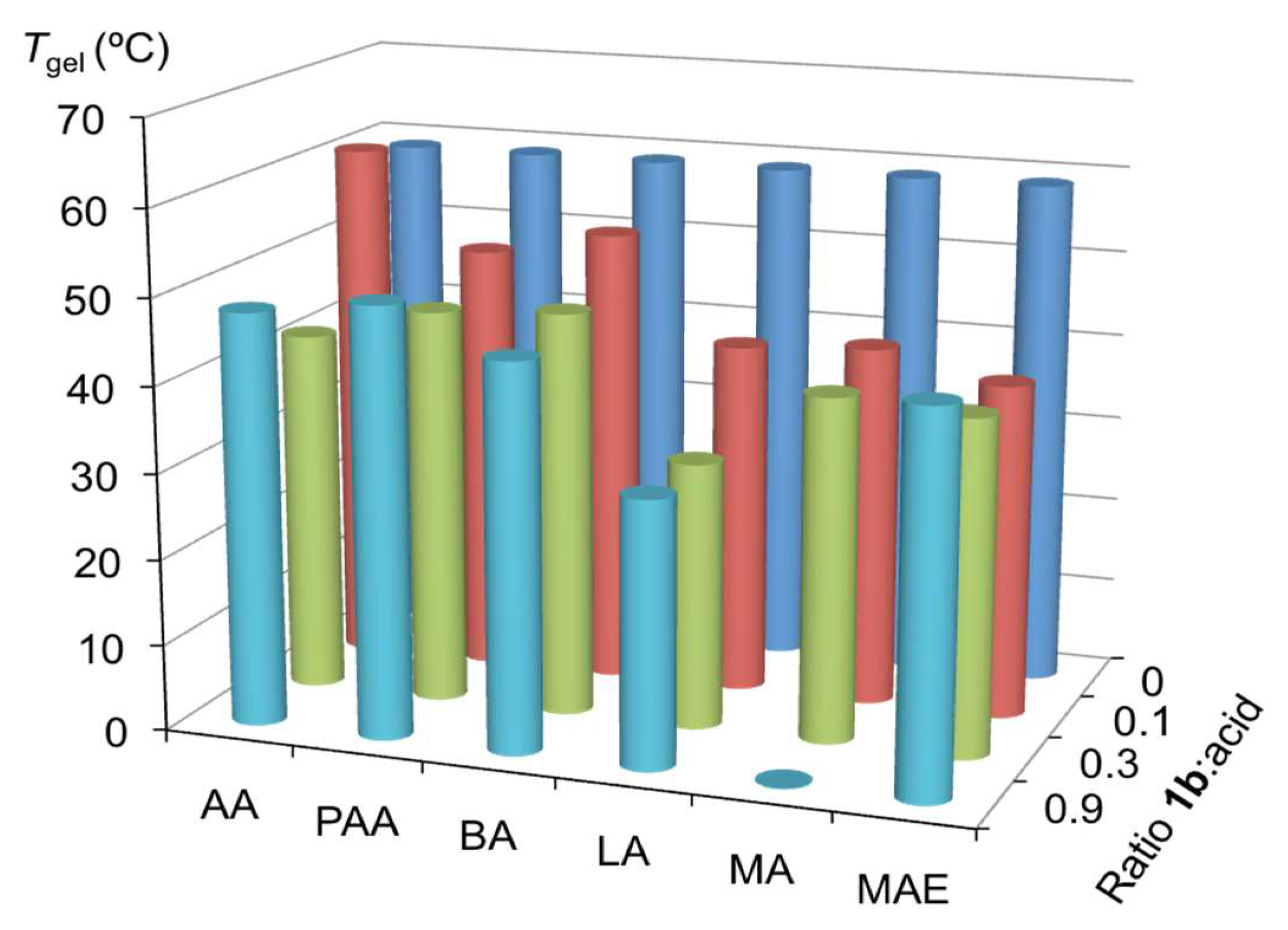
This was also accompanied by a gradual decrease of the Tgel of the remaining gel phase (Figure 9 and Table S1) in comparison with the Tgel of the pristine gel made of 1b alone (~60 °C). A preliminary design of experiments with chiral Brønsted acids (e.g. mandelic acid) showed no significant differences on the above-mentioned effects when using either the racemic mixture of any of the corresponding enantiomers (Table S1). Moreover, gelation kinetics of the doped gels was also notably reduced to several hours in most cases, compared to pristine gels (Table S1).
In terms of the effect of different acids, the most remarkable outcome was observed with mandelic acid, which prevented gelation at molar ratio (1b:acid) = 0.9. Taking experiments with mandelic acid and benzoic acid as representative examples, molar ratios (1b:acid) of 0.6 and 0.9 gave similar results (Table S1). Moreover, experiments using the corresponding methyl ester caused minor disruption in the gel, suggesting the critical role of the free carboxylic group in the disassembly of the supramolecular aggregates. These results are in good agreement with the favorable effect observed in reactions using 1b and mandelic acid in equimolar amounts. In general, other acids such as acetic, phenylacetic, benzoic and lactic acids also caused destabilization of the gel network, albeit to a lesser extent. The results suggest that the acidity of the carboxylic group (e.g., mandelic acid is the most acidic among these examples) might play a more important role than structural features.
The decrease in thermostability of supramolecular urea aggregates upon incorporation of the acid was also accompanied by a detriment of their mechanical properties, as evidenced by oscillatory rheological measurements of selected gels prepared in the absence and presence of the acid. For instance, dynamic frequency sweep (DFS) measurements of gels based on 1b in chloroform showed that the incorporation of small amounts of mandelic acid (i.e. molar ratio 1b:acid = 0.1) caused a drop in the storage modulus G’ of ~1.8 times compared to the undoped gel (i.e. G’ (undoped) ~30493 Pa vs. G’ (doped) ~17096 Pa) (Figure 10A). In addition, the loss factor or tan δ, which represents mechanical damping or internal friction of the viscoelastic system, increased from ~0.180 (undoped) to ~0.239 (doped). This indicates that the material becomes more dissipative in the presence of the acid. Both undoped and doped gels were very brittle in nature, as shown by dynamic strain sweep (DSS) measurements (i.e. gel-to-sol transition occurred between 9–10% strain for all samples) (Figure 10B). Dynamic time sweep (DTS) plots showing constant moduli values over time within the linear regime are provided in Supporting Information (Figure S45A). In concordance with the minor effect that acids showed on the thermostability of toluene gels, additional rheological experiments also showed a lower influence on the mechanical strength of the gels prepared in toluene (Figure S45B–D).
Finally, the observed disrupting effect of the acids on the supramolecular aggregates of 1b could also be related to changes in the microstructure of the xerogels obtained by freeze-drying the corresponding gels. An entangled microfibrillar network over a dense and continuous structure was observed for updoped xerogels (Figure 11), whereas the incorporation of 10 mol% of mandelic acid (with respect to 1b) induced the formation of laminar and smoother structures (Figure 11A vs. Figure 11B). The discontinuous appearance of the doped xerogel could explain a less-effective immobilization of solvent molecules, which is in agreement with the observed lower Tgel and mechanical strength of the bulk material. In contrast, FE-SEM images of the corresponding xerogels showed little effect on the microstructure in comparison with the undoped network (Figure 11C vs. Figure 11D). It is worth mentioning that the harmful effect of acids on the stability of supramolecular gel networks was not exclusive to urea 1b.
3. Materials and Methods
3.1. Experimental Details
All commercially available solvents and reagents were used as received. CHCl3 was filtered through basic alumina prior to use to avoid the presence of trace amounts of acid. 1H-NMR spectra were recorded at 300 MHz. CDCl3 was used as a deuterated solvent. Chiral urea catalysts were obtained following literature procedures: 1b [47], 1c [47], 1d [47] and 1e [87]. 1H and 13C NMR spectra for compounds 8aa [43], 8ab [88], 8ac [88], 8ad [88], 8ae [88], 8af [89], 8ag [90], 8ah [88], 8ai [88], 8ba [43], 8ca [47] and 8ba [53] are consistent with values previously reported in the literature.
3.2. General Procedure for the 1b·HA-Catalyzed Friedel–Crafts Alkylation Reaction
To a mixture of urea 1b (20 mol%), (±)-mandelic acid 9a (20 mol%), and nitroalkene 7a–i (0.1 mmol) at 25 °C in a test tube CHCl3 (filtered through basic alumina, 0.5 mL) was added and then indole 6a–d (0.15 mmol). After 3 days, the residue was purified by liquid chromatography (SiO2; hexane/EtOAc, 8:2) to obtain the desired adducts 8. Yields and enantioselectivities are reported in Scheme 3. Spectral and analytical data for compounds 8 are in agreement with those previously reported in the literature.
(S)-3-(2-Nitro-1-phenylethyl)-1H-indole (8aa) [43]. Following the general procedure, compound 8aa was obtained after 3 days of reaction at room temperature in 88% yield. The ee of the product was determined to be 55% by HPLC using a Daicel Chiralpak IA column (n-hexane/i-PrOH = 90:10, flow rate 1 mL·min−1, λ = 254.0 nm): τmajor = 15.3 min; τminor = 14.0 min. [α]D24 = +10.7 (c 0.40, CH2Cl2, 55% ee) {lit. [53], [α]D24 + 25.3 (c 0.53, CH2Cl2) for (S)-8aa, 88% ee}.
(S)-3-(1-(4-Chlorophenyl)-2-nitroethyl)-1H-indole (8ab) [88]. Following the general procedure, compound 8ab was obtained after 3 days of reaction at room temperature in 94% yield. The ee of the product was determined to be 55% by HPLC using a Daicel Chiralpak IA column (n-hexane/i-PrOH = 90:10, flow rate 1 mL·min−1, λ = 254.0 nm): τmajor = 20.2 min; τminor = 16.8 min. [α]D24 = +1.5 (c 0.53, CH2Cl2, 55% ee). {lit. [88], [α]D20 + 7.5 (c 1.2, CH2Cl2), for (S)-8ab, 82% ee}.
(S)-3-(1-(4-Bromophenyl)-2-nitroethyl)-1H-indole (8ac) [88]. Following the general procedure, compound 8ac was obtained after 3 days of reaction at room temperature in 95% yield. The ee of the product was determined to be 55% by HPLC using a Daicel Chiralpak IA column (n-hexane/i-PrOH = 90:10, flow rate 1 mL·min−1, λ = 254.0 nm): τmajor = 21.2 min; τminor = 17.6 min. [α]D24 = −4.2 (c 0.47, CH2Cl2, 55% ee) {lit. [88], [α]D20 − 1.7 (c 1.0, CH2Cl2), for (S)-8ac, 90% ee}.
(S)-3-(2-Nitro-1-p-tolylethyl)-1H-indole (8ad) [88]. Following the general procedure, compound 8ad was obtained after 3 days of reaction at room temperature in 80% yield. The ee of the product was determined to be 54% by HPLC using a Daicel Chiralpak IA column (n-hexane/i-PrOH = 90:10, flow rate 1 mL·min−1, λ = 254.0 nm): τmajor = 14.4 min; τminor = 13.0 min. [α]D25 = +5.1 (c 0.43, CH2Cl2, 54% ee) {lit. [88], [α]D20 + 16.4 (c 0.9, CH2Cl2), for (S)-8ad, 81% ee}.
(S)-3-(1-(4-methoxyphenyl)-2-nitroethyl-1H-indole (8ae) [88]. Following the general procedure, compound 8ae was obtained after 3 days of reaction at room temperature in 62% yield. The ee of the product was determined to be 48% by HPLC using a Daicel Chiralpak IB column (n-hexane/i-PrOH = 80:20, flow rate 1 mL·min−1, λ = 254.0 nm): τmajor = 22.4 min; τminor = 24.3 min. [α]D25 = +14.4 (c 0.19, CH2Cl2, 48% ee) {lit. [88], [α]D20 + 26.4 (c 1.1, CH2Cl2), for (S)-8ae, 81% ee}.
(R)-3-(1-(2,4-dichlorophenyl)-2-nitroethyl)-1H-indole (8af) [89]. Following the general procedure, compound 8af was obtained after 3 days of reaction at room temperature in 97% yield. The ee of the product was determined to be 53% by HPLC using a Daicel Chiralpak IA column (n-hexane/i-PrOH = 90:10, flow rate 1 mL·min−1, λ = 254.0 nm): τmajor = 14.1 min; τminor = 12.7 min. [α]D25 = +32.5 (c 0.27, CH2Cl2, 53% ee).
(R)-3-(1-(2-Bromophenyl)-2-nitroethyl)-1H-indole (8ag) [90]. Following the general procedure, compound 8ag was obtained after 3 days of reaction at room temperature in 94% yield. The ee of the product was determined to be 54% by HPLC using a Daicel Chiralpak IB column (n-hexane/i-PrOH = 80:20, flow rate 1 mL·min−1, λ = 254.0 nm): τmajor = 26.7 min; τminor = 18.4 min. [α]D25 = +56.4 (c 0.20, CH2Cl2, 54% ee) {lit. [90], [α]D22 − 99.9 (c 1.3, CH2Cl2), for (S)-8ag, 96% ee}.
(S)-3-(2-nitro-1-(thiophen-2-yl)ethyl)-1H-indole (8ah) [88]. Following the general procedure, compound 8ah was obtained after 3 days of reaction at room temperature in 68% yield. The ee of the product was determined to be 52% by HPLC using a Daicel Chiralpak IA column (n-hexane/i-PrOH = 90:10, flow rate 1 mL·min−1, λ = 254.0 nm): τmajor = 19.1 min; τminor = 17.2 min. [α]D24 = +10.6 (c 0.46, CH2Cl2, 52% ee) {lit. [88], [α]D20 + 24.3 (c 1.0, CH2Cl2), for (S)-8ah, 82% ee}.
(S)-3-(1-(furan-2-yl)-2-nitroethyl)-1H-indole (8ai) [88]. Following the general procedure, compound 8ai was obtained after 3 days of reaction at room temperature in 58% yield. The ee of the product was determined to be 50% by HPLC using a Daicel Chiralpak IB column (n-hexane/i-PrOH = 80:20, flow rate 1 mL·min−1, λ = 254.0 nm): τmajor = 16.7 min; τminor = 12.6 min. [α]D25 = −16.4 (c 1.24, CH2Cl2, 50% ee) {lit. [88], [α]D20 − 78 (c 1.0, CH2Cl2, for (S)-8ai, 78% ee}.
(S)-5-chloro-3-(2-nitro-1-phenylethyl)-1H-indole (8ba) [43]. Following the general procedure, compound 8ba was obtained after 3 days of reaction at room temperature in 36% yield. The ee of the product was determined to be 50% by HPLC using a Daicel Chiralpak IA column (n-hexane/i-PrOH = 90:10, flow rate 1 mL·min−1, λ = 254.0 nm): τmajor = 13.3 min; τminor = 12.1 min. [α]D25 = −14.7 (c 0.83, CH2Cl2, 50% ee) {lit. [91], [α]D20 − 27.8 (c 1.1, CH2Cl2, for (S)-8ba, 90% ee}.
(S)-5-fluoro-3-(2-nitro-1-phenylethyl)-1H-indole (8ca) [47]. Following the general procedure, compound 8ca was obtained after 3 days of reaction at room temperature in 43% yield. The ee of the product was determined to be 53% by HPLC using a Daicel Chiralpak IA column (n-hexane/i-PrOH = 90:10, flow rate 1 mL·min−1, λ = 254.0 nm): τmajor = 13.7 min; τminor = 12.2 min. [α]D25 = +6.3 (c 0.92, CHCl3, 53% ee) {lit. [47], [α]D26 + 11 (c 0.38, CHCl3, for (S)-8ca, 86% ee}.
(S)-5-methoxi-3-(2-nitro-1-phenylethyl)-1H-indole (8da) [53]. Following the general procedure, compound 8da was obtained after 3 days of reaction at room temperature in 98% yield. The ee of the product was determined to be 55% by HPLC using a Daicel Chiralpak IA column (n-hexane/i-PrOH = 90:10, flow rate 1 mL·min−1, λ = 254.0 nm): τmajor = 20.4 min; τminor = 17.1 min. [α]D23 = −15.5 (c 0.34, CH2Cl2, 55% ee) {lit. [53], [α]D20 − 28.4 (c 1.0, CH2Cl2, for (S)-8da, 98% ee}.
3.3. Evaluation of the Stability of Urea Aggregates
Thermoreversible gels (1 mL volume) were prepared at desired concentration by the gentle heating-cooling procedure, as previously described [47]. Unless stated otherwise, acid additives (from a stock solution) were mixed with the urea and the volume was then adjusted with pure solvent. The heating-cooling cycle was then applied and the formed gels were considered to be stable if they did not exhibit gravitational flow upon turning the vial upside-down. See Supporting Information for additional details.
Unless indicated otherwise, gels were equilibrated for at least overnight at room temperature before thermal analysis. Tgel values were determined at desired times after gelation using a calibrated thermoblock, which was heated using an electric heating plate equipped with a temperature control couple at ~0.4 °C/min [47]. The temperature at which the bulk gel started to break was defined as Tgel. Each measurement was randomly made at least twice and the average value was reported with an estimated error of ±2 °C. Reported Tgel values correspond to the gel phase that supported the inversion of the vial, which enabled separation first of any liquid phase generated by the acid additive.
Flow properties of our compounds were studied by oscillatory rheology using an AR 2000 Advanced rheometer (TA Instruments, Regensburg, Germany) equipped with a Julabo C cooling system (Regensburg, Germany). A 1000 mm gap setting and a torque of 5 × 10−4 N/m at 25 °C were employed for the measurements in a plain-plate (20 mm, stainless steel). The following experiments were performed for each sample, using 2 mL total gel volume: (a) Dynamic strain sweep (DSS) = variation of storage modulus G′ and loss modulus G″ with strain from 0.01 to 100%; (b) dynamic frequency sweep (DFS) = variation of G′ and G″ with frequency from 0.1 to 10 Hz at 0.1% strain; (c) dynamic time sweep (DTS) = variation of G′ and G″ with time keeping the strain and frequency values constant and within the linear viscoelastic regime (i.e., strain = 0.1% strain; frequency = 1 Hz). Mechanical inertial effects of the measuring head were determined by the software package. In addition, fixed rest time after sample loading and pre-shearing to equilibrium at different shear rates were routinely made in order to minimize prehistory effects.
Field Emission Scanning Electron Microscopy (FESEM) images of xerogels were recorded with a Zeiss Merlin, Field Emission Scanning Electron Microscope (Zaragoza, Spain) operated at an accelerating voltage of 10 kV. For visualization, samples were prepared by freeze-drying the corresponding gels. The obtained fibrous solids were placed on top of tin plates and shielded with Pt (40 mA during 30–60 s; film thickness = 5–10 nm). Images were taken at the University of Zaragoza (Servicio General de Apoyo a la Investigación-SAI)
4. Conclusions
The results obtained in the Friedel-Crafts alkylation reaction suggest that the synergic effect between a Brønsted acid and a urea catalyst is higher than the effect promoted by each species separately. The experiments are also in agreement first, with the critical role played by the free carboxylic group in the disassembly of the supramolecular urea aggregates and secondly, in the activation of the “free” urea through coordination by the acid. This mechanism was demonstrated by analyzing the thermal and mechanical stability, as well as morphological properties of the supramolecular gel aggregates in the absence and presence of acid additives at different molar ratios. In terms of the effect of different acids, the most remarkable outcome (i.e., decrease of gel stability and significant changes in morphology) was observed with mandelic acid, which prevented the gelation at molar ratio of 0.9. In contrast, experiments using the corresponding methyl ester caused minor disruption in the gel, in good agreement with the observations made during the catalytic studies.
The observed behavior is studied for the first time in the literature and could open a new interesting research line related to the effect of acid on urea-catalyzed reactions. Moreover, this contribution could become an important starting point for further investigations, since this study suggests that this phenomenon could be extended to other molecules with similar acidity and/or hydrogen-bonding ability. This possibility is currently being investigated in our laboratories.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4344/8/8/305/s1, Scheme S1: pKa values in waters, Figures S1–S8: Effect of the acid in the 1H NMR signals of the urea catalysts (500 MHz), Figures S9–S44: HPLC chromatograms, Figure S45: Gel measurements, Table S1: Effects of the addition of different additives on the aspect, gelation time and Tgel with respect to the pristine gels prepared in CHCl3 and in toluene.
Author Contributions
Conceptualization, E.M.-L., D.D.D. and R.P.H.; Methodology, Software, Validation, Formal Analysis, Investigation, Resources, Data Curation, all authors; Writing-Original Draft Preparation, D.D.D. and R.P.H.; Writing-Review & Editing, all authors; Visualization, E.M.-L., D.D.D. and R.P.H.; Supervision, E.M.-L., D.D.D. and R.P.H.; Project Administration, E.M.-L., D.D.D. and R.P.H.; Funding Acquisition, E.M.-L., D.D.D. and R.P.H.
Funding
This research was funded by Ministerio de Economía, Industria y Competitividad (CTQ2017-88091-P), Universidad de Zaragoza (JIUZ-2017-CIE-05) and Gobierno de Aragón-Fondo Social Europe (Research Groups E-07_17R).
Acknowledgments
I. G. S. thanks the Obra Social de Ibercaja-CAI for mobility aid. D. D. D. thanks the Deutsche Forschungsgemeinschaft (DFG) for the Heisenberg Professorship Award.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Berkessel, A.; Gröger, H. Asymmetric Organocatalysis; Whiley-VHC: Weinheim, Germany, 2004. [Google Scholar]
- Acc. Chem. Res. 2004, 37 (8). Special Issue on Organocatalysis; Houk, K.N.; List, B. (Eds.) American Chemical Society: Washington, DC, USA, 2004. [Google Scholar]
- Enantioselective Organocatalysis; Dalko, P.I. (Ed.) Whiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Chem. Rev. 2007, 107 (12). Special Issue on Organocatalysis; List, B. (Ed.) American Chemical Society: Washington, DC, USA, 2007. [Google Scholar]
- Recent Developments in Asymmetric Organocatalysis; Pellissier, H. (Ed.) RSC Publishing: Cambridge, UK, 2010. [Google Scholar]
- Asymmetric Organocatalysis; List, B. (Ed.) Springer: Heidelberg, Germany, 2010. [Google Scholar]
- Current Organic Chemistry, Volume 15, Number 13; Herrera, R.P. (Ed.) Bentham Science: Soest, The Netherlands, 2011. [Google Scholar]
- Science of Synthesis Asymmetric Organocatalysis; List, B.; Maruoka, K. (Eds.) Thieme: Sttutgart, Germany, 2012. [Google Scholar]
- Comprehensive Enantioselective Organocatalysis: Catalysts, Reactions, and Application; Dalko, P.I. (Ed.) Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- De Figueiredo, R.M.; Christmann, M. Organocatalytic Synthesis of Drugs and Bioactive Natural Products. Eur. J. Org. Chem. 2007, 2007, 2575–2600. [Google Scholar] [CrossRef]
- Marqués-López, E.; Herrera, R.P.; Christmann, M. Asymmetric organocatalysis in total synthesis—A trial by fire. Nat. Prod. Rep. 2010, 27, 1138–1167. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; Cabrera, S. Applications of asymmetric organocatalysis in medicinal chemistry. Chem. Soc. Rev. 2013, 42, 774–793. [Google Scholar] [CrossRef] [PubMed]
- Marqués-López, E.; Herrera, R.P. Comprehensive Enantioselective Organocatalysis; Dalko, P., Ed.; Wiley-VCH: Weinheim, Germany, 2013; pp. 1359–1383. [Google Scholar]
- Dalko, P.I.; Mosan, L. In the Golden Age of Organocatalysis. Angew. Chem. Int. Ed. 2004, 43, 5138–5175. [Google Scholar] [CrossRef] [PubMed]
- Berkessel, A. Organocatalysis by Hydrogen Bonding Networks in Organocatalysis; Reetz, M.T., List, B., Jaroch, H., Weinmann, H., Eds.; Springer: Berlin, Germany, 2008; Volume 2, pp. 281–297. [Google Scholar]
- Hydrogen Bonding in Organic Synthesis; Pihko, P.M. (Ed.) Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Kerstin, E.-E.; Berkessel, A. Noncovalent organocatalysis based on hydrogen bonding: elucidation of reaction paths by computational methods. Top. Curr. Chem. 2010, 291, 1–27. [Google Scholar] [CrossRef]
- Schreiner, P.R. Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 2003, 32, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, Y. Recognition and activation by ureas and thioureas: stereoselective reactions using ureas and thioureas as hydrogen-bonding donors. Org. Biomol. Chem. 2005, 3, 4299–4306. [Google Scholar] [CrossRef] [PubMed]
- Connon, S.J. Organocatalysis Mediated by (Thio)urea Derivatives. Chem. Eur. J. 2006, 12, 5418–5429. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric Catalysis by Chiral Hydrogen-Bond Donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef] [PubMed]
- Doyle, A.G.; Jacobsen, E.N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef] [PubMed]
- Miyabe, H.; Takemoto, Y. Discovery and Application of Asymmetric Reaction by Multi-Functional Thioureas. Bull. Chem. Soc. Jpn. 2008, 81, 785–795. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Schreiner, P.R. (Thio)urea organocatalysis—What can be learnt from anion recognition? Chem. Soc. Rev. 2009, 38, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Marqués-López, E.; Herrera, R.P. El renacer de un nuevo campo: La Organocatálisis Asimétrica. Tioureas como organocatalizadores. An. Quím. 2009, 105, 5–12. (In Spanish) [Google Scholar]
- Takemoto, Y. Development of Chiral Thiourea Catalysts and Its Application to Asymmetric Catalytic Reactions. Chem. Pharm. Bull. 2010, 58, 593–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohtome, Y.; Nagasawa, K. The Design of Chiral Double Hydrogen Bonding Networks and Their Applications to Catalytic Asymmetric Carbon-Carbon and Carbon-Oxygen Bond-Forming Reactions. Synlett 2010, 1–22. [Google Scholar] [CrossRef]
- Sohtome, Y.; Nagasawa, K. Dynamic asymmetric organocatalysis: Cooperative effects of weak interactions and conformational flexibility in asymmetric organocatalysts. Chem. Commun. 2012, 48, 7777–7789. [Google Scholar] [CrossRef] [PubMed]
- Marqués-López, E.; Herrera, R.P. New Strategies in Chemical Synthesis and Catalysis; Pignataro, B., Ed.; Wiley-VCH: Weinheim, Germany, 2012; pp. 175–199. [Google Scholar]
- Serdyuk, O.V.; Heckel, C.M.; Tsogoeva, S.B. Bifunctional primary amine-thioureas in asymmetric organocatalysis. Org. Biomol. Chem. 2013, 11, 7051–7071. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Shi, X. Cinchonine and thiourea. Chem. Commun. 2013, 49, 8583–8585. [Google Scholar] [CrossRef] [PubMed]
- Narayanaperumal, S.; Rivera, D.G.; Silva, R.C.; Paixão, M.W. Terpene-Derived Bifunctional Thioureas in Asymmetric Organocatalysis. ChemCatChem. 2013, 5, 2756–2773. [Google Scholar] [CrossRef]
- Zhang, Z.; Bao, Z.; Xing, H. N,N′-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea: a privileged motif for catalyst development. Org. Biomol. Chem. 2014, 12, 3151–3162. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, C.-J. Recent advances in asymmetric organocatalysis mediated by bifunctional amine–thioureas bearing multiple hydrogen-bonding donors. Chem. Commun. 2015, 51, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Curran, D.P.; Kuo, L.H. Altering the Stereochemistry of Allylation Reactions of Cyclic. alpha.-Sulfinyl Radicals with Diarylureas. J. Org. Chem. 1994, 59, 3259–3261. [Google Scholar] [CrossRef]
- Curran, D.P.; Kuo, L.H. Acceleration of a dipolar Claisen rearrangement by Hydrogen bonding to a soluble diaryl^urea. Tetrahedron Lett. 1995, 36, 6647–6650. [Google Scholar] [CrossRef]
- Etter, M.C.; Panunto, T.W. 1,3-Bis(m-nitrophenyl)urea: An exceptionally good complexing agent for proton acceptors. J. Am. Chem. Soc. 1988, 110, 5896–5897. [Google Scholar] [CrossRef]
- Etter, M.C.; Urbañczyk-Lipkowska, Z.; Zia-Ebrahimi, M.; Panunto, T.W. Hydrogen bond-directed cocrystallization and molecular recognition properties of diarylureas. J. Am. Chem. Soc. 1990, 112, 8415–8426. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Kelly, T.R.; Kim, M.H. Relative Binding Affinity of Carboxylate and Its Isosteres: Nitro, Phosphate, Phosphonate, Sulfonate, and. delta.-Lactone. J. Am. Chem. Soc. 1994, 116, 7072–7080. [Google Scholar] [CrossRef]
- Auvil, T.J.; Schafer, A.G.; Mattson, A.E. Design Strategies for Enhanced Hydrogen-Bond Donor Catalysts. Eur. J. Org. Chem. 2014, 2633–2646. [Google Scholar] [CrossRef]
- Giacalone, F.; Gruttadauria, M.; Agrigento, P.; Noto, R. Low-loading asymmetric organocatalysis. Chem. Soc. Rev. 2012, 41, 2406–2447. [Google Scholar] [CrossRef] [PubMed]
- Herrera, R.P.; Sgarzani, V.; Bernardi, L.; Ricci, A. Catalytic enantioselective Friedel-Crafts alkylation of indoles with nitroalkenes by using a simple thiourea organocatalyst. Angew. Chem. Int. Ed. 2005, 44, 6734–6737. [Google Scholar] [CrossRef] [PubMed]
- Herrera, R.P.; Monge, D.; Martín-Zamora, E.; Fernández, R.; Lassaletta, J.M. Organocatalytic Conjugate Addition of Formaldehyde N,N-Dialkylhydrazones to β,γ-Unsaturated α-Keto Esters. Org. Lett. 2007, 9, 3303–3306. [Google Scholar] [CrossRef] [PubMed]
- Marqués-López, E.; Alcaine, A.; Tejero, T.; Herrera, R.P. Enhanced Efficiency of Thiourea Catalysts by External Brønsted Acids in the Friedel—Crafts Alkylation of Indoles. Eur. J. Org. Chem. 2011, 3700–3705. [Google Scholar] [CrossRef] [Green Version]
- Roca-López, D.; Marqués-López, E.; Alcaine, A.; Merino, P.; Herrera, R.P. A Friedel–Crafts alkylation mechanism using an aminoindanol-derived thiourea catalyst. Org. Biomol. Chem. 2014, 12, 4503–4510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schön, E.-M.; Marqués-López, E.; Herrera, R.P.; Alemán, C.; Díaz, D.D. Exploiting Molecular Self-Assembly: From Urea-Based Organocatalysts to Multifunctional Supramolecular Gels. Chem. Eur. J. 2014, 20, 10720–10731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juste-Navarro, V.; Marqués-López, E.; Herrera, R.P. Thiourea-Catalyzed Addition of Indoles to Aliphatic β,γ-Unsaturated α-Ketoesters. Asian J. Org. Chem. 2015, 4, 884–889. [Google Scholar] [CrossRef]
- Schiller, J.; Pérez-Ruiz, R.; Sampedro, D.; Marqués-López, E.; Herrera, R.P.; Díaz, D.D. Fluoride Anion Recognition by a Multifunctional Urea Derivative: An Experimental and Theoretical Study. Sensors 2016, 16, 658. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, M.C.; Herrera, R.P. Hydrogen Bonding Networks in Chiral Thiourea Organocatalysts: Evidence on the Importance of the Aminoindanol Moiety. Cryst. Growth Des. 2016, 16, 5091–5099. [Google Scholar] [CrossRef] [Green Version]
- Izaga, A.; Herrera, R.P.; Gimeno, M.C. Gold(I)-Mediated Thiourea Organocatalyst Activation: A Synergic Effect for Asymmetric Catalysis. ChemCatChem 2017, 9, 1313–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robak, M.T.; Trincado, M.; Ellman, J.A. Enantioselective Aza-Henry Reaction with an N-Sulfinyl Urea Organocatalyst. J. Am. Chem. Soc. 2007, 129, 15110–15111. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, M.; Seidel, D. Catalytic Enantioselective Additions of Indoles to Nitroalkenes. J. Am. Chem. Soc. 2008, 130, 16464–16465. [Google Scholar] [CrossRef] [PubMed]
- So, S.; Burkett, J.A.; Mattson, A.E. Internal Lewis Acid Assisted Hydrogen Bond Donor Catalysis. Org. Lett. 2011, 13, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Sonsona, I.G.; Marqués-López, E.; Herrera, R.P. The aminoindanol core as a key scaffold in bifunctional organocatalysts. Beilstein J. Org. Chem. 2016, 12, 505–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Kass, S.R. Enantioselective Friedel–Crafts Alkylation between Nitroalkenes and Indoles Catalyzed by Charge Activated Thiourea Organocatalysts. J. Org. Chem. 2017, 82, 13288–13296. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.H.; Sigman, M.S. Systematically Probing the Effect of Catalyst Acidity in a Hydrogen-Bond-Catalyzed Enantioselective Reaction. Angew. Chem. Int. Ed. 2007, 46, 4748–4750. [Google Scholar] [CrossRef] [PubMed]
- Marqués-López, E.; Diez-Martinez, A.; Merino, P.; Herrera, R.P. The Role of the Indole in Important Organocatalytic Enantioselective Friedel-Crafts Alkylation Reactions. Curr. Org. Chem. 2009, 13, 1585–1609. [Google Scholar] [CrossRef]
- You, S.-L.; Cai, Q.; Zeng, M. Chiral Brønsted acid catalyzed Friedel–Crafts alkylation reactions. Chem. Soc. Rev. 2009, 38, 2190–2210. [Google Scholar] [CrossRef] [PubMed]
- Terrasson, V.; de Figueiredo, R.M.; Campagne, J.M. Organocatalyzed Asymmetric Friedel–Crafts Reactions. Eur. J. Org. Chem. 2010, 2635–2655. [Google Scholar] [CrossRef]
- Zeng, M.; You, S.-L. Asymmetric Friedel-Crafts Alkylation of Indoles: The Control of Enantio- and Regioselectivity. Synlett 2010, 1289–1301. [Google Scholar] [CrossRef]
- Dessole, G.; Herrera, R.P.; Ricci, A. H-Bonding Organocatalysed Friedel-Crafts Alkylation of Aromatic and Heteroaromatic Systems with Nitroolefins. Synlett 2004, 2374–2378. [Google Scholar] [CrossRef]
- Pettersen, D.; Herrera, R.P.; Bernardi, L.; Fini, F.; Sgarzani, V.; Fernández, R.; Lassaletta, J.M.; Ricci, A. A Broadened Scope for the Use of Hydrazones as Neutral Nucleophiles in the Presence of H-Bonding Organocatalysts. Synlett 2006, 239–242. [Google Scholar] [CrossRef]
- Bernardi, L.; Fini, F.; Herrera, R.P.; Ricci, A.; Sgarzani, V. Enantioselective aza-Henry reaction using cinchona organocatalysts. Tetrahedron 2006, 62, 375–380. [Google Scholar] [CrossRef]
- Alcaine, A.; Marqués-López, E.; Merino, P.; Tejero, T.; Herrera, R.P. Thiourea catalyzed organocatalytic enantioselective Michael addition of diphenyl phosphite to nitroalkenes. Org. Biomol. Chem. 2011, 9, 2777–2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auria-Luna, F.; Marqués-López, E.; Mohammadi, S.; Heiran, R.; Herrera, R.P. New Organocatalytic Asymmetric Synthesis of Highly Substituted Chiral 2-Oxospiro-[indole-3,4′-(1′,4′-dihydropyridine)] Derivatives. Molecules 2015, 20, 15807–15826. [Google Scholar] [CrossRef] [PubMed]
- Bordwell, F.G.; Algrim, D.J.; Harrelson, J.A., Jr. The relative ease of removing a proton, a hydrogen atom, or an electron from carboxamides versus thiocarboxamides. J. Am. Chem. Soc. 1988, 110, 5903–5904. [Google Scholar] [CrossRef]
- This behavior can be also rationalized in terms of ab initio methods, which support differences in energy for the N-H···S compared with N-H···O interaction in the chain dimers, being weaker in thioureas than in ureas (Ref. 69 and 70).
- Masunov, A.; Dannenberg, J.J. Theoretical Study of Urea. I. Monomers and Dimers. J. Phys. Chem. A 1999, 103, 178–184. [Google Scholar] [CrossRef]
- Masunov, A.; Dannenberg, J.J. Theoretical Study of Urea and Thiourea. 2. Chains and Ribbons. J. Phys. Chem. B 2000, 104, 806–810. [Google Scholar] [CrossRef]
- Berkessel, A.; Cleemann, F.; Mukherjee, S.; Müller, T.N.; Lex, J. Highly Efficient Dynamic Kinetic Resolution of Azlactones by Urea-Based Bifunctional Organocatalysts. Angew. Chem. Int. Ed. 2005, 44, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Scheerder, J.; Engbersen, J.F.J.; Casnati, A.; Ungaro, R.; Reinhoudt, D.N. Complexation of Halide Anions and Tricarboxylate Anions by Neutral Urea-Derivatized p-tert-Butylcalix[6]arenes. J. Org. Chem. 1995, 60, 6448–6454. [Google Scholar] [CrossRef]
- Weil, T.; Kotke, M.; Kleiner, C.M.; Schreiner, P.R. Cooperative Brønsted Acid-Type Organocatalysis: Alcoholysis of Styrene Oxides. Org. Lett. 2008, 10, 1513–1516. [Google Scholar] [CrossRef] [PubMed]
- Companyó, X.; Valero, G.; Crovetto, L.; Moyano, A.; Rios, R. Highly Enantio- and Diastereoselective Organocatalytic Desymmetrization of Prochiral Cyclohexanones by Simple Direct Aldol Reaction Catalyzed by Proline. Chem. Eur. J. 2009, 15, 6564–6568. [Google Scholar] [CrossRef] [PubMed]
- Reis, Ö.; Eymur, S.; Reis, B.; Demir, A.S. Direct enantioselective aldol reactions catalyzed by a proline–thiourea host–guest complex. Chem. Commun. 2009, 1088–1090. [Google Scholar] [CrossRef] [PubMed]
- El-Hamdouni, N.; Companyó, X.; Rios, R.; Moyano, A. Substrate-Dependent Nonlinear Effects in Proline–Thiourea-Catalyzed Aldol Reactions: Unraveling the Role of the Thiourea Co-Catalyst. Chem. Eur. J. 2010, 16, 1142–1148. [Google Scholar] [CrossRef] [PubMed]
- Klausen, R.S.; Jacobsen, E.N. Weak Brønsted Acid-Thiourea Co-catalysis: Enantioselective, Catalytic Protio-Pictet-Spengler Reactions. Org. Lett. 2009, 11, 887–890. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Zuend, S.J.; Woll, M.G.; Tao, Y.; Jacobsen, E.N. Asymmetric Cooperative Catalysis of Strong Brønsted Acid–Promoted Reactions Using Chiral Ureas. Science 2010, 327, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-L.; Shi, M. Chiral Thiourea-Phosphine Organocatalysts in the Asymmetric Aza-Morita–Baylis–Hillman Reaction. Adv. Synth. Catal. 2007, 349, 2129–2135. [Google Scholar] [CrossRef]
- Escuder, B.; LLusar, M.; Miravet, J.F. Insight on the NMR Study of Supramolecular Gels and Its Application to Monitor Molecular Recognition on Self-Assembled Fibers. J. Org. Chem. 2006, 71, 7747–7752. [Google Scholar] [CrossRef] [PubMed]
- Schoonbeek, F.S.; van Esch, J.H.; Hulst, R.; Kellogg, R.M.; Feringa, B.L. Geminal Bis-ureas as Gelators for Organic Solvents: Gelation Properties and Structural Studies in Solution and in the Gel State. Chem. Eur. J. 2000, 6, 2633–2643. [Google Scholar] [CrossRef]
- Jung, J.H.; Shinkai, S.; Shimizu, T. Spectral Characterization of Self-Assemblies of Aldopyranoside Amphiphilic Gelators: What is the Essential Structural Difference Between Simple Amphiphiles and Bolaamphiphiles? Chem. Eur. J. 2002, 8, 2684–2690. [Google Scholar] [CrossRef]
- Morris, K.F.; Johnson, C.S., Jr. Diffusion-ordered two-dimensional nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 1992, 114, 3139–3141. [Google Scholar] [CrossRef]
- Price, W.S. Pulsed-field gradient nuclear magnetic resonance as a tool for studying translational iffusion: Part 1. Basic theory. Conc. Magn. Reson. 1997, 9, 299–336. [Google Scholar] [CrossRef]
- Cohen, Y.; Avram, L.; Frish, L. Diffusion NMR Spectroscopy in Supramolecular and Combinatorial Chemistry: An Old Parameter—New Insights. Angew. Chem. Int. Ed. 2005, 44, 520–554. [Google Scholar] [CrossRef] [PubMed]
- Reactive Intermediates; Santos, L.S. (Ed.) Wiley-VCH: Weinheim, Germany, 2010. [Google Scholar]
- Liu, X.-G.; Jiang, J.-J.; Shia, M. Development of axially chiral bis(arylthiourea)-based organocatalysts and their application in the enantioselective Henry reaction. Tetrahedron: Asymmetry 2007, 18, 2773–2781. [Google Scholar] [CrossRef]
- Jia, Y.-X.; Zhu, S.-F.; Yang, Y.; Zhou, Q.-L. Asymmetric Friedel-Crafts Alkylations of Indoles with Nitroalkenes Catalyzed by Zn(II)-Bisoxazoline Complexes. J. Org. Chem. 2006, 71, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Meshram, H.M.; Rao, N.N.; Kumar, G.S. Boric Acid–Mediated Mild and Efficient Friedel–Crafts Alkylation of Indoles with Nitro Styrenes. Synth. Commun. 2010, 40, 3496–3500. [Google Scholar] [CrossRef]
- Wu, J.; Li, X.; Wu, F.; Wan, B. A New Type of Bis(sulfonamide)-Diamine Ligand for a Cu(OTf)2-Catalyzed Asymmetric Friedel-Crafts Alkylation Reaction of Indoles with Nitroalkenes. Org. Lett. 2011, 13, 4834–4837. [Google Scholar] [CrossRef] [PubMed]
- Itoh, J.; Fuchibe, K.; Akiyama, T. Chiral Phosphoric Acid Catalyzed Enantioselective Friedel–Crafts Alkylation of Indoles with Nitroalkenes: Cooperative Effect of 3 Å Molecular Sieves. Angew. Chem. Int. Ed. 2008, 47, 4016–4018. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Comparative variation of acidity in (thio)urea structures.

Figure 2.
Thiourea and urea catalysts previously tested in Friedel-Crafts-type alkylation reactions.
Figure 2.
Thiourea and urea catalysts previously tested in Friedel-Crafts-type alkylation reactions.

Figure 3.
Mechanistic hypothesis.

Scheme 1.
Exploring the urea-catalyzed Friedel-Crafts-type alkylation reaction in the presence of a Brønsted acid.
Scheme 1.
Exploring the urea-catalyzed Friedel-Crafts-type alkylation reaction in the presence of a Brønsted acid.

Scheme 2.
Screening of acids using urea catalyst 1b.

Scheme 3.
Scope of the Friedel-Crafts alkylation reaction. The purple values are obtained in the presence of 1b·(±)-9a and the black values are obtained in the absence of acid (±)-9a. The yields are obtained after isolation by column chromatography and the ee is determined by chiral HPLC analysis.
Scheme 3.
Scope of the Friedel-Crafts alkylation reaction. The purple values are obtained in the presence of 1b·(±)-9a and the black values are obtained in the absence of acid (±)-9a. The yields are obtained after isolation by column chromatography and the ee is determined by chiral HPLC analysis.

Figure 4.
Effect of the addition of acid (±)-9a using 20 mol% of urea 1b.

Figure 5.
DOSY NMR experiment in CDCl3 (300 MHz).

Figure 6.
Cationic ESI-MS spectra experiment in CHCl3.

Figure 7.
Proposed transition states for the [urea⋅acid]-catalyzed Friedel-Crafts reaction.

Figure 8.
Representative photographs of undoped gels and gels doped with different additives under different conditions: (A) Undoped gel made of 1b in CHCl3 (c = 7 g/L; 1 mL); (B–D) doped gels made of 1b in CHCl3 (c = 7 g/L; 1 mL) and mandelic acid at different molar ratios 1b:acid: (B) 0.9, (C) 0.3, (D) 0.1; (E) doped gel made of 1b in CHCl3 (c = 7 g/L; 1 mL) and benzoic acid (molar ratios 1b:acid = 0.9); (F) doped gel made of 1b in CHCl3 (c = 7 g/L; 1 mL) and acetic acid (molar ratio 1b:acid = 0.9); (G) undoped gel made of 1b in toluene (c = 3 g/L; 1 mL); (H) doped gel made of 1b in toluene (c = 3 g/L; 1 mL) and mandelic acid (molar ratio 1b:acid = 0.9); (I) doped gel made of 1b in toluene (c = 3 g/L; 1 mL) and acetic acid (molar ratio 1b:acid = 0.9).
Figure 8.
Representative photographs of undoped gels and gels doped with different additives under different conditions: (A) Undoped gel made of 1b in CHCl3 (c = 7 g/L; 1 mL); (B–D) doped gels made of 1b in CHCl3 (c = 7 g/L; 1 mL) and mandelic acid at different molar ratios 1b:acid: (B) 0.9, (C) 0.3, (D) 0.1; (E) doped gel made of 1b in CHCl3 (c = 7 g/L; 1 mL) and benzoic acid (molar ratios 1b:acid = 0.9); (F) doped gel made of 1b in CHCl3 (c = 7 g/L; 1 mL) and acetic acid (molar ratio 1b:acid = 0.9); (G) undoped gel made of 1b in toluene (c = 3 g/L; 1 mL); (H) doped gel made of 1b in toluene (c = 3 g/L; 1 mL) and mandelic acid (molar ratio 1b:acid = 0.9); (I) doped gel made of 1b in toluene (c = 3 g/L; 1 mL) and acetic acid (molar ratio 1b:acid = 0.9).

Figure 9.
Tgel of the gel made of 1b (c = 7 g/L) in CHCl3 and the effect caused by the incorporation of selected acids at different ratios (molar ratio 1b:acid = 0.1, 0.3, 0.9). Abbreviations: AA = acetic acid; PAA = phenylacetic acid; BA = benzoic acid; LA = lactic acid; MA = mandelic acid; MAE = mandelic acid methyl ester. The average values of at least two independent measurements are shown. Estimated error ±2 °C.
Figure 9.
Tgel of the gel made of 1b (c = 7 g/L) in CHCl3 and the effect caused by the incorporation of selected acids at different ratios (molar ratio 1b:acid = 0.1, 0.3, 0.9). Abbreviations: AA = acetic acid; PAA = phenylacetic acid; BA = benzoic acid; LA = lactic acid; MA = mandelic acid; MAE = mandelic acid methyl ester. The average values of at least two independent measurements are shown. Estimated error ±2 °C.

Figure 10.
(A) DFS and (B) DSS measurements of undoped gel made of 1b in CHCl3 (c = 14 g/L), and doped gel made of 1b (c = 14 g/L) and mandelic acid (molar ratio 1b:acid = 0.1) in CHCl3.
Figure 10.
(A) DFS and (B) DSS measurements of undoped gel made of 1b in CHCl3 (c = 14 g/L), and doped gel made of 1b (c = 14 g/L) and mandelic acid (molar ratio 1b:acid = 0.1) in CHCl3.

Figure 11.
Representative FE-SEM images of xerogels derived from (A) pristine gel made of 1b (c = 7 g/L) in CHCl3, (B) doped gel made of 1b (c = 7 g/L) and mandelic acid (molar ratio 1b:acid = 0.1) in CHCl3, (C) pristine gel made of 1b (c = 3 g/L) in toluene, (D) doped gel made of 1b (c = 3 g/L) and mandelic acid (molar ratio 1b:acid = 0.1) in toluene.
Figure 11.
Representative FE-SEM images of xerogels derived from (A) pristine gel made of 1b (c = 7 g/L) in CHCl3, (B) doped gel made of 1b (c = 7 g/L) and mandelic acid (molar ratio 1b:acid = 0.1) in CHCl3, (C) pristine gel made of 1b (c = 3 g/L) in toluene, (D) doped gel made of 1b (c = 3 g/L) and mandelic acid (molar ratio 1b:acid = 0.1) in toluene.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Screening of catalytic complex 1b·(±)-9a in different solvents. a.

| Entry | Solvent | Time (Days) | Yield (%) b | ee (%) c |
|---|---|---|---|---|
| 1 d | CHCl3 | 3 | 46 | 36 |
| 2 | CHCl3 | 3 | 88 | 55 |
| 3 d | Toluene | 3 | 10 | 18 |
| 4 | Toluene | 3 | 38 | 42 |
| 5d | Xylene | 4 | 18 | 28 |
| 6 | Xylene | 4 | 95 | 47 |
| 7 d | CH3CN | 10 | n.r. e | _ |
| 8 | CH3CN | 4 | n.d. f | 8 |
| 9 d | THF | 10 | n.r. e | _ |
| 10 | THF | 10 | n.r. e | _ |
| 11 d | AcOEt | 10 | n.r. e | _ |
| 12 | AcOEt | 10 | n.r. e | _ |
a Unless specified, see Experimental Section; b After isolation by column chromatography; c Determined by chiral HPLC analysis (Chiralpak IA, Hex:iPrOH 90:10, 1 mL/min); d Reaction performed in the absence of mandelic acid; e n.r. = Not reaction observed; f n.d. = Not determined.
Table 2.
Effect of temperature of model reaction. a

| Entry | Acid | T (°C) | Time (Days) | Yield (%) b | ee (%) c |
|---|---|---|---|---|---|
| 1 | - | 15 | 5 | 24 | 46 |
| 2 | 9ad | 15 | 4 | 60 | 62 |
| 3 | 9ae | 15 | 4 | 77 | 60 |
| 4 | 9af | 15 | 4 | 82 | 57 |
| 5 | - | -25 | 5 | 15 | 57 |
| 6 | 9ae | -25 | 5 | 23 | 68 |
a To a mixture of catalyst 1b (20 mol%, 0.02 mmol), (±)-mandelic acid 9a (20 mol%, 0.02 mmol), and nitroalkene 7a (0.1 mmol) at 15 or −25 °C, indole 6a (0.15 mmol) was added; b After isolation by column chromatography; c Determined by chiral HPLC analysis (Chiralpak IA, Hex:iPrOH 90:10, 1 ml/min); d 2 equiv. of acid (0.04 mmol); e 3 equiv. of acid (0.06 mmol); f 4 equiv. of acid (0.08 mmol).
Table 3.
Effect of the nature of the catalytic complex. a

| Entry | Catalyst | Acid | Yield (%) b | ee (%) c |
|---|---|---|---|---|
| 1 | 1b | - | 46 | 36 |
| 2 | 1b | (±)-9a | 88 | 55 |
| 3 | 1b |  | 83 | 48 |
| 4 | 1b |  | 57 | 40 |
| 5 | 1b |  | 55 | 37 |
| 6 | 1b | MeOH | 68 | 40 |
| 7 | 1c | (±)-9a | 18 (13) d | Rac. (Rac.) d |
| 8 | 1d | (±)-9a | 25 (7) d | Rac. (Rac.) d |
a Unless specified, see Experimental Section; b After isolation by column chromatography; c Determined by chiral HPLC analysis (Chiralpak IA, Hex:iPrOH 90:10, 1 ml/min); d values in parenthesis have been obtained in the absence of acid.
Table 4.
Effect of the acid on the 1H NMR signals of the urea catalyst. a

| Entry | (±)-9a (mmol) | δNHa (ppm) | δHb (ppm) | δHc (ppm) | δHd (ppm) | δHd’ (ppm) | Ratio Integrals Urea:Standard |
|---|---|---|---|---|---|---|---|
| 1 | 0 | 7.937 | 5.553 | 4.691 | 3.238 | 2.976 | 0.22:1 |
| 2 | 0.01 | 7.888 | 5.818 | 4.670 | 3.219 | 2.949 | 0.37:1 |
| 3 | 0.02 | 7.852 | 5.971 | 4.635 | 3.177 | 2.923 | 0.62:1 |
| 4 | 0.03 | 7.852 | 6.030 | 4.629 | 3.173 | 2.926 | 0.70:1 |
| 5 | 0.04 | 7.844 | 6.032 | 4.630 | 3.176 | 2.923 | 0.79:1 |
| 6 | 0.06 | 7.845 | 6.034 | 4.625 | 3.167 | 2.920 | 0.78:1 |
| 7 | 0.08 | 7.770 | 5.960 | 4.554 | 3.093 | 2.820 | 0.82:1 |
| 8 | 0.1 | 7.844 | 6.035 | 4.631 | 3.169 | 2.920 | 0.88:1 |
a To a solution of urea 1b (0.02 mmol) in CDCl3 (0.5 mL) and mesitylene (0.0075 mmol), as internal standard, acid (±)-9a is added after each measure of 1H NMR.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sonsona, I.G.; Marqués-López, E.; Häring, M.; Díaz, D.D.; Herrera, R.P. Urea Activation by an External Brønsted Acid: Breaking Self-Association and Tuning Catalytic Performance. Catalysts 2018, 8, 305. https://doi.org/10.3390/catal8080305
AMA Style
Sonsona IG, Marqués-López E, Häring M, Díaz DD, Herrera RP. Urea Activation by an External Brønsted Acid: Breaking Self-Association and Tuning Catalytic Performance. Catalysts. 2018; 8(8):305. https://doi.org/10.3390/catal8080305
Chicago/Turabian StyleSonsona, Isaac G., Eugenia Marqués-López, Marleen Häring, David Díaz Díaz, and Raquel P. Herrera. 2018. "Urea Activation by an External Brønsted Acid: Breaking Self-Association and Tuning Catalytic Performance" Catalysts 8, no. 8: 305. https://doi.org/10.3390/catal8080305
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.