Heck Reaction—State of the Art


Department of Chemistry, Baburaoji Gholap College, Sangvi, Pune 411027, India
Catalysts 2017, 7(9), 267; https://doi.org/10.3390/catal7090267
Submission received: 20 August 2017
/
Revised: 5 September 2017
/
Accepted: 6 September 2017
/
Published: 11 September 2017
(This article belongs to the Special Issue Catalyzed Mizoroki–Heck Reaction or C–H activation)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
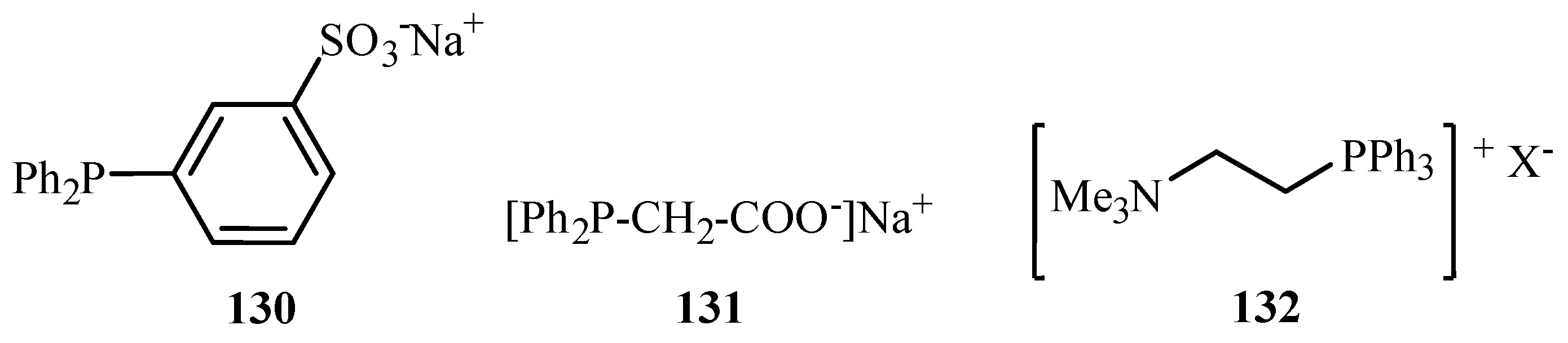{kind=link}
{kind=link}
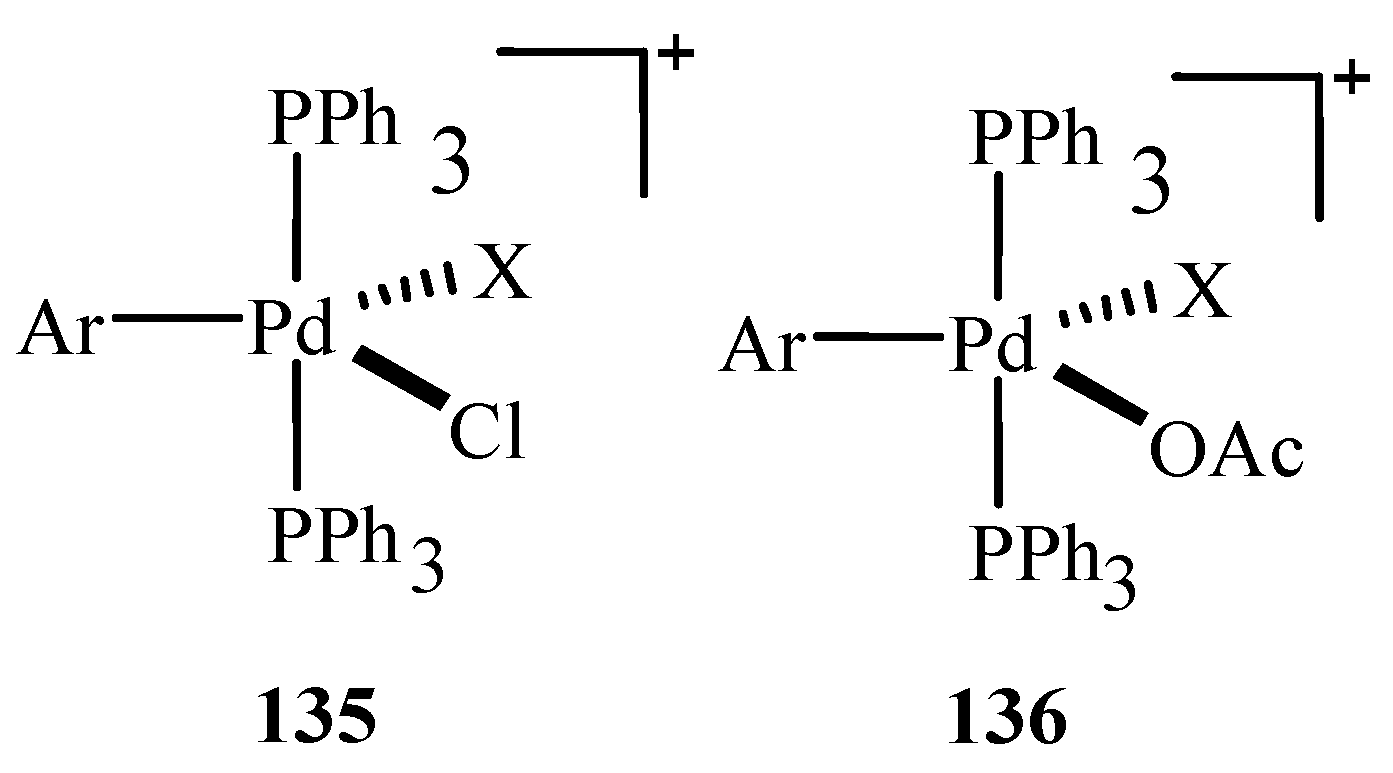{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The Heck reaction is one of the most studied coupling reactions and is recognized with the Nobel Prize in Chemistry. Thousands of articles, hundreds of reviews and a number of books have been published on this topic. All reviews are written exhaustively describing the various aspects of Heck reaction and refer to the work done hitherto. Looking at the quantum of the monographs published, and the reviews based on them, we found a necessity to summarize all reviews on Heck reaction about catalysts, ligands, suggested mechanisms, conditions, methodologies and the compounds formed via Heck reaction in one review and generate a resource of information. One can find almost all the catalysts used so far for Heck reaction in this review.
1. Introduction
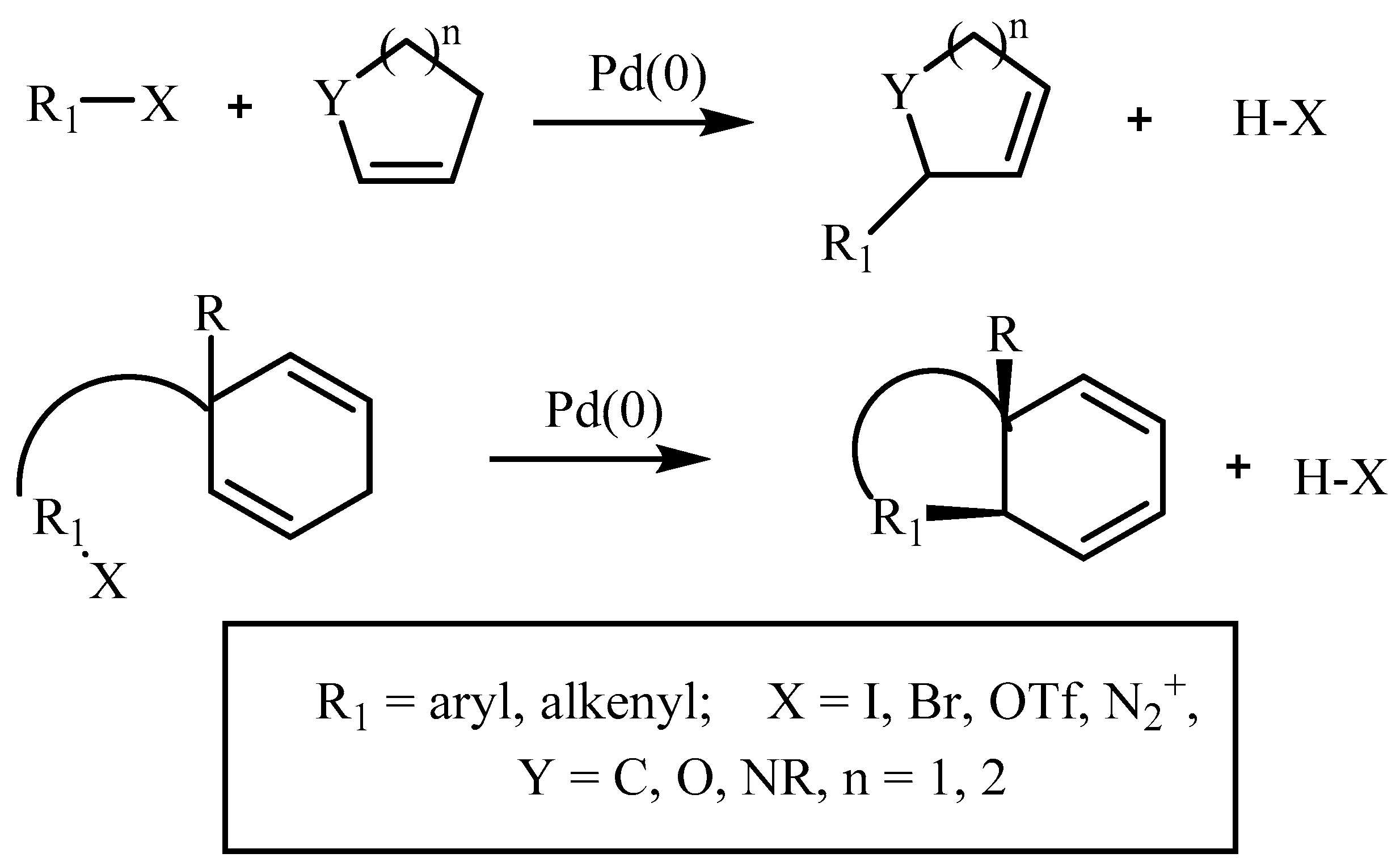
The new era of research started after the introduction of coupling reactions, i.e., carbon–carbon bond forming reactions like Heck [1], Suzuki [2], Sonogoshira [3], Negishi [4], Kumada [5], Stille [6], Tsuji-Trost [7], etc. These reactions have played an enormously decisive and important role in shaping chemical synthesis and have revolutionized the way one thinks about synthetic organic chemistry. The Heck reaction (Equation (1)) is used extensively in many syntheses, including agrochemical, fine chemicals, pharmaceutical, etc. The reaction was introduced by Mizoroki [8] and Heck [9] independently more than four decades ago. It has drawn much attention due to high efficiency and simplicity. Heck methodology is attractive from a synthetic point of view because of its high chemoselectivity and mild reaction conditions along with low toxicity and cost of the reagent if, specifically, the catalyst is recycled.
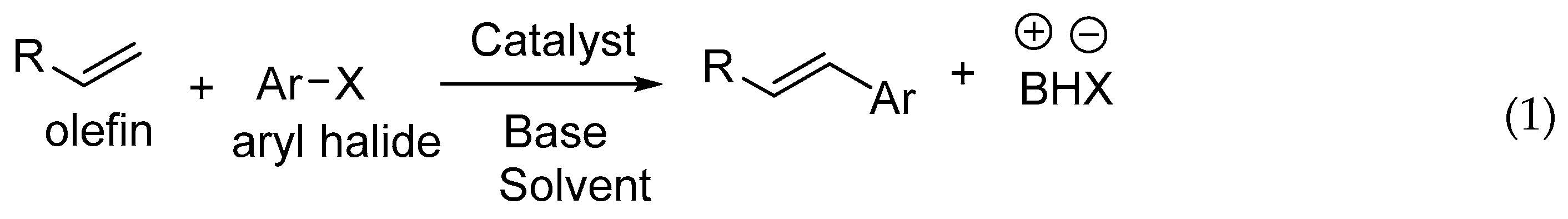
The Heck reaction is described as a vinylation or arylation of olefins where a large variety of olefins can be used, like derivatives of acrylates, styrenes or intramolecular double bonds. The aryl halide variants developed in addition to typical aryl bromides and iodides are aromatic triflates, aroyl chlorides, aryl sulfonyl chlorides, aromatic diazonium salts, aroyl anhydrides, aryl chlorides and arylsilanols. The catalyst is the essential part of a reaction where a variety of metals along with a huge range of ligands is studied. Significant progress for the preparation and characterization of variety of ligands and catalysts has been made for avoiding protection and deprotection procedures, therefore allowing for syntheses to be carried out in fewer steps. Development, novel catalytic properties and extensive mechanistic studies are summarized in several reviews based on seminal work of many researchers and reviewers as described in following sections. Palladium is usually the preferred metal as it tolerates a wide variety of functional groups and it has a remarkable ability to assemble C-C bonds between appropriately functionalized substrates. Most palladium based methodologies proceed with stereo- and regioselectivity and with excellent yields. Generally, the less crowded structure is preferred during Heck reaction, and often favours a trans product. Few mechanisms are also supported by the discussion on the regioselectivity and stereoselectivity of Heck reaction.
Sometimes compounds such as TBAB (Tetra butyl ammonium bromide) are added in the reaction mixture along with organic or inorganic bases needed for the sequestration of acid generated. Typical solvents for the Heck reaction are dipolar aprotic solvents like DMF (dimethyl formamide) and NMP (N-methyl-2-pyrrolidone), however, the reaction is also performed in many other different solvents and in fact there are large numbers of reviews dedicated to the use of various solvents. Besides these, many manuscripts describe the reaction in the absence of one of the component (other than substrates) such as ligand free, organic solvent free and so on. Recent publications also describe how to recover used catalyst specially using aqueous medium which implies the potential for greener approach for organic reactions.
There are a large number of excellent reviews available on Heck reaction and related topics based on applications, mechanism, quest for high turn over numbers (TONs), asymmetric synthesis, separation techniques, etc. In addition to this, there are a few manuscripts that give a general overview about Heck reaction, some of them are detailed and some are not so descriptive. Many reviews discuss various catalysis techniques with certain aspects where along with the Heck reaction, other coupling reactions are also discussed, however for the present review, only the Heck related chemistry is considered. It should be noted that for this review only the reviews and few related articles covering Heck chemistry are considered. Consideration of book articles, although informative, is beyond the scope of this review.
2. Reviews on Catalyst Development
The majority of the work on Heck reaction has been focused on the catalyst development. There are numerous reviews based on this and hence are further categorized for better understanding.
2.1. Overviews
When the mechanism was not fully explored, the reaction and its possible mechanistic pathways are reviewed by one of its pioneers, R.F. Heck [10,11,12] by focusing on possibilities of the intermediate formed during Heck reaction by considering various examples for its support. These reviews mention the working reaction conditions explored for the reaction and focuses on the requirements of the substrates, bases, solvents and the conditions to be used. It was observed that the reaction is usually regioselective and stereospecific and is tolerant of almost every functional group. Data have proved that the direction of addition of the organopalladium complex to the olefin depends upon the steric and electronic influence of the substituent present. The direction of addition is most often dominated by steric effects where the organic group is seen to be added to the least substituted carbon of the double band.
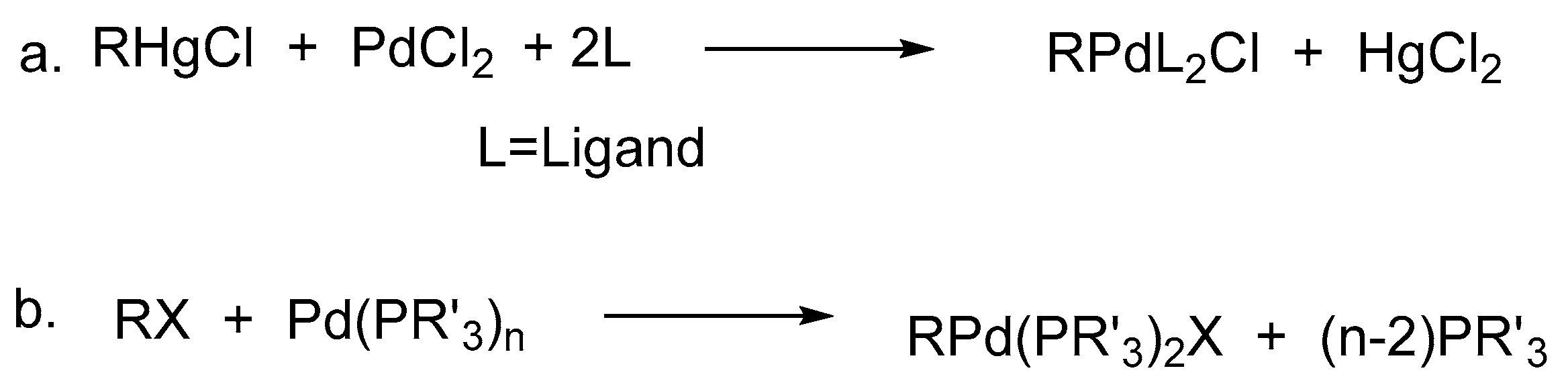
Initially, the intermediate mono-organopalladium(II) species, RPdL2X, were prepared by exchange reactions of mercurials in general, with palladium(II) salts (Scheme 1a), however it suffered from the disadvantage that, many such main group organometallics were not easily accessible and moreover they were needed to be used in stoichiometric amounts in the synthesis of organic compounds. Hence, the finely divided palladium metal or palladium(0) phosphine complexes were used for such reactions (Scheme 1b).
Effects of different triaryl phosphines have also been studied. It is mentioned that, generally the reaction does not require anhydrous or anaerobic conditions although it is advisable to limit access of oxygen when arylphosphines are used as a component of the catalyst. Aryl, heterocyclic, benzyl, or vinyl halides are commonly employed often with bromides and iodides in comparison to halides with an easily eliminated beta-hydrogen atom (i.e., alkyl derivatives) since they form only olefins by elimination under the normal reaction conditions. Generally used bases are secondary or tertiary amine, sodium or potassium acetate, carbonate, or bicarbonates. The catalyst is commonly palladium acetate, although palladium chloride or preformed triarylphosphine palladium complexes, as well as palladium on charcoal, have been used.
Triarylphosphine or a secondary amine is required when organic bromides are used although a reactant, product, or solvent may serve as the ligand for reactions involving organic iodides. Solvents such as acetonitrile, dimethylformamide, hexamethylphosphoramide, N-methylpyrrolidinone, and methanol have been used, but are often not necessary. The procedure is applicable to a very wide range of reactants and yields are generally good to excellent. Temperature used is in the range of 50 to 160 °C, where the reaction proceeds homogeneously. When nucleophilic secondary amines are used as co-reactants with most vinylic halides, a variation occurs that often produces tertiary allylic amines as major products. A comparison of Heck reaction with similar types of reaction where the organic halide is replaced by other reagents such as organometallics, diazonium salts, or aromatic hydrocarbons has also been discussed.
De Meijere and Meyer [13] have reviewed mainly the Heck couplings with various oligohaloarenes along with an overall discussion on development in mechanism and catalysts until 1994. The review stresses upon the careful choice of substrates and skilful tailoring of reaction conditions leading to impressive sequences.
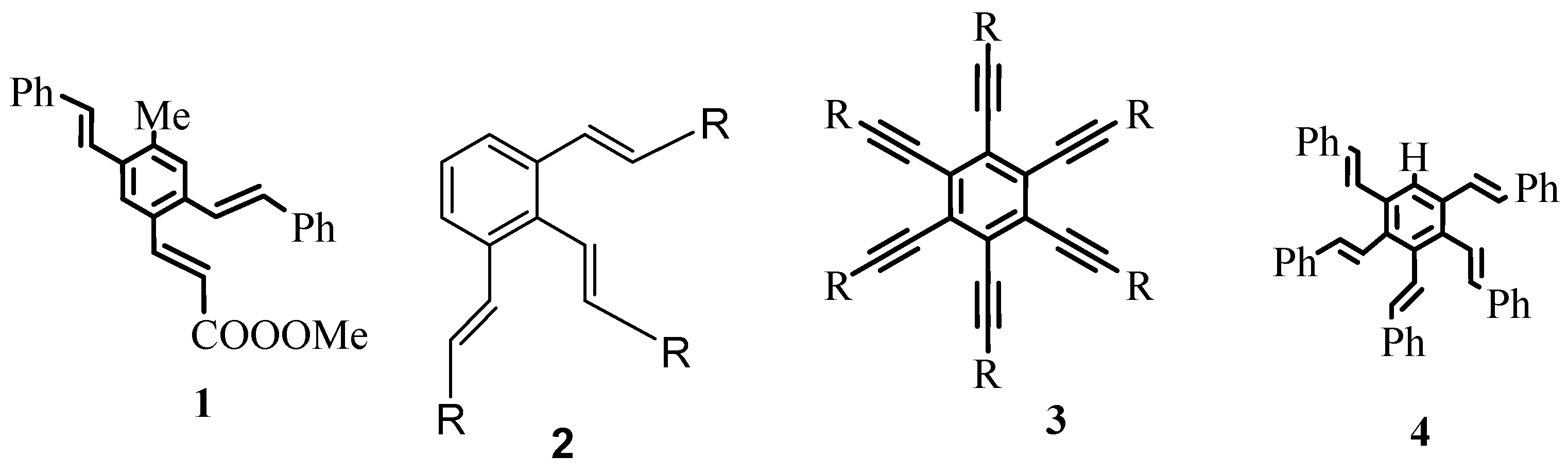
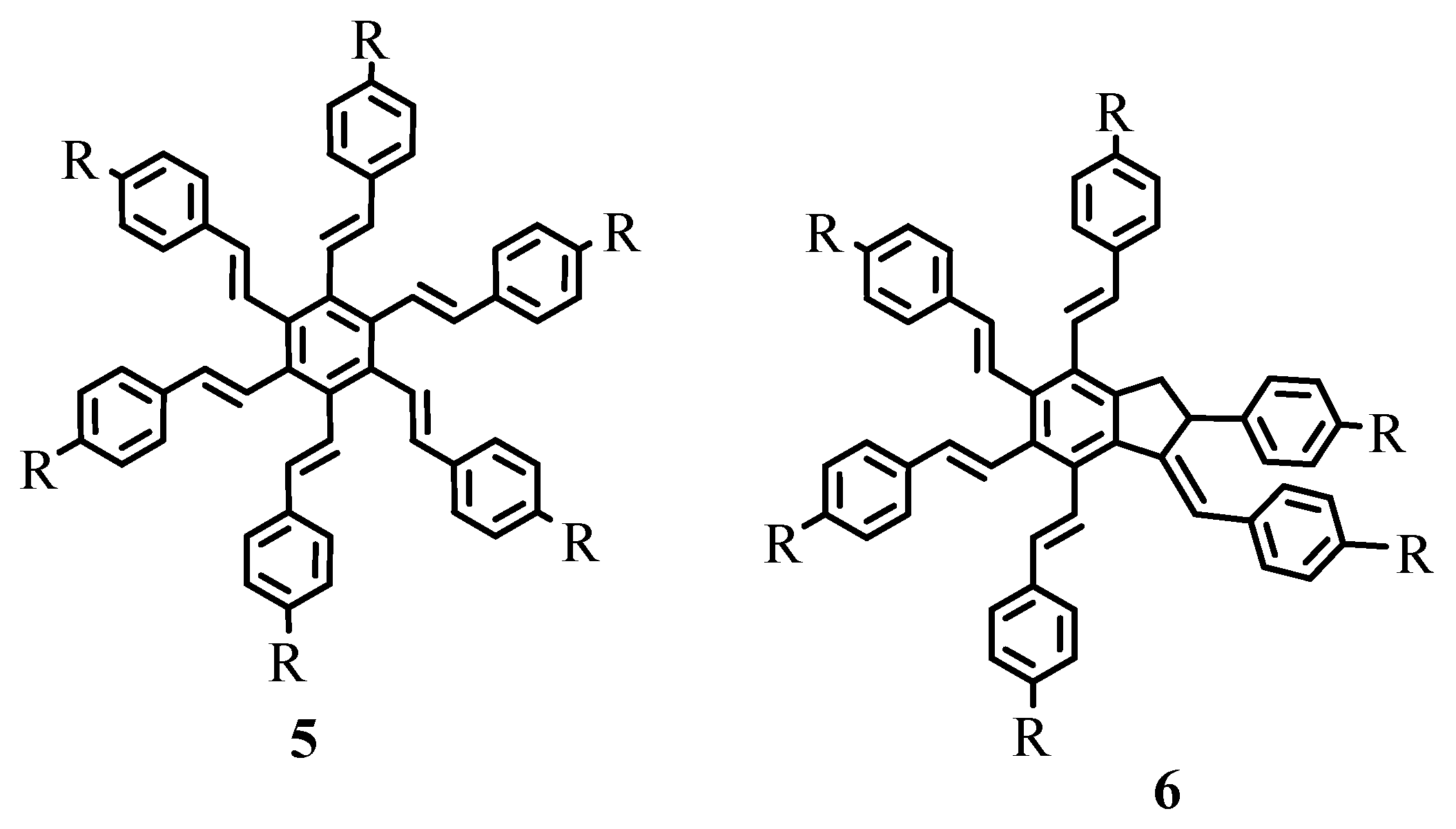
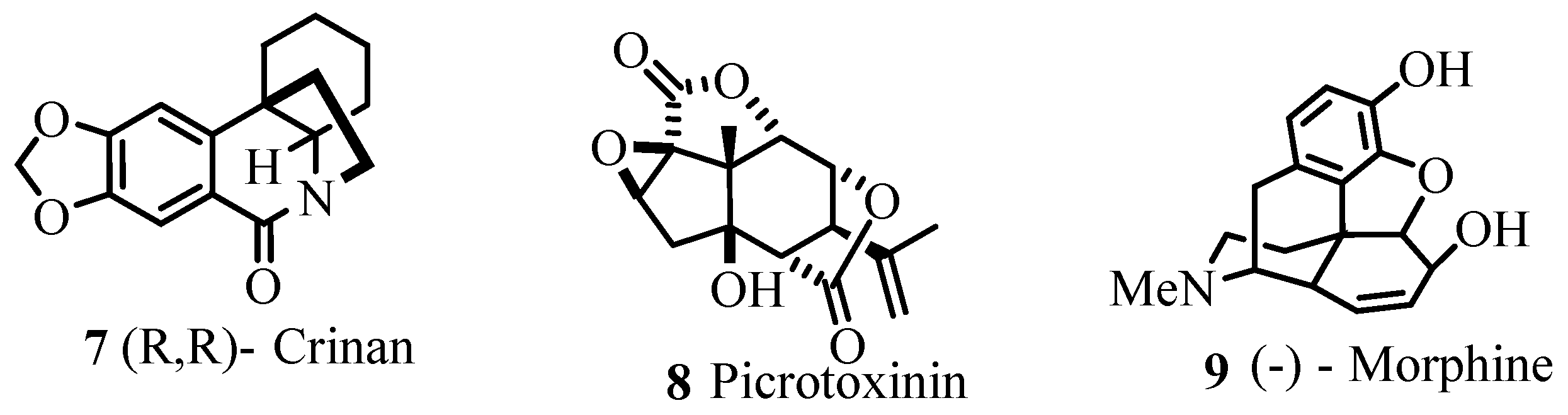
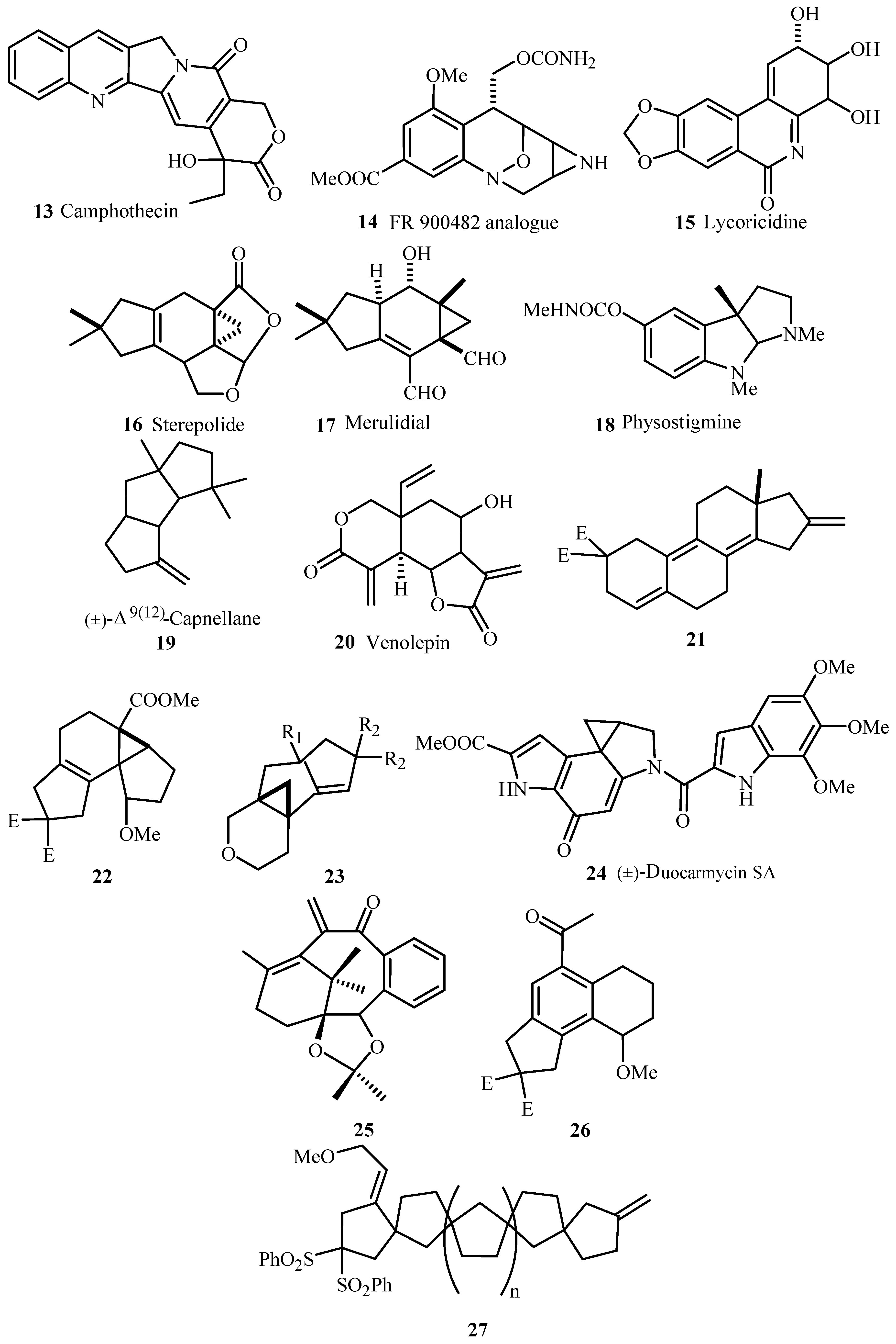
As exemplified in the review, the Heck reaction, together with other mechanistically related palladium catalysed transformations with arene, alkene and alkyne derivatives, opens the door to a tremendous variety of elegant and highly convergent routes to structurally complex molecules. The reaction is not hindered by heteroatoms such as oxygen, nitrogen, sulphur and phosphorus in most of the cases. With Heck reaction, a range of chemoselective and regioselective monocouplings of highly functionalized substrates with unsymmetrical and multisubstituted reaction partners could be achieved. A number of examples are given that demonstrate the cascade reactions in which three, four, five, and even eight new C-C bonds are formed to yield oligofunctional and oligocyclic products like 1–6 (Figure 1) with impressive molecular complexity. Cases are presented to establish the reactivity for enantioselective construction of complex natural products with quaternary stereocenters as exemplified by the synthesis of (R,R)-crinan 7, picrotoxinin 8, and morphine 9 (Figure 2).
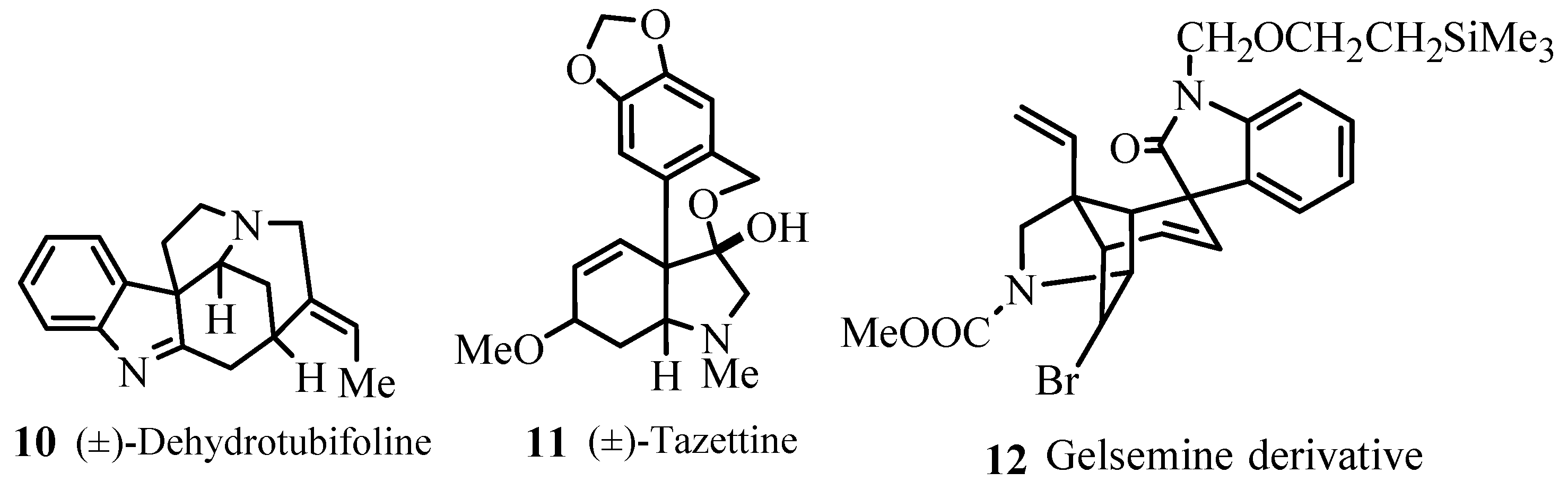
Several examples for syntheses of natural products 10–27 (Figure 3) have been provided that involve different types of Heck transformations as one of the important key steps like,
- Heck reactions with intermolecular asymmetric induction where compounds with ee (enantiomeric excess) up to 99%.
- Multiple component reactions and domino coupling reactions performed with several alkenes and various haloarenes.
- Pd-catalysed heteroannelations.
- Intramolecular Heck reactions.
- Palladium catalysed additions to triple bonds and the coupling products derived from norbornene and dicyclopentadiene that serve as precursors for diverse polycyclic aromatic compound.
Thus, the review gives the expanse of Heck reaction which is one of the important synthetic methods available to organic chemists.
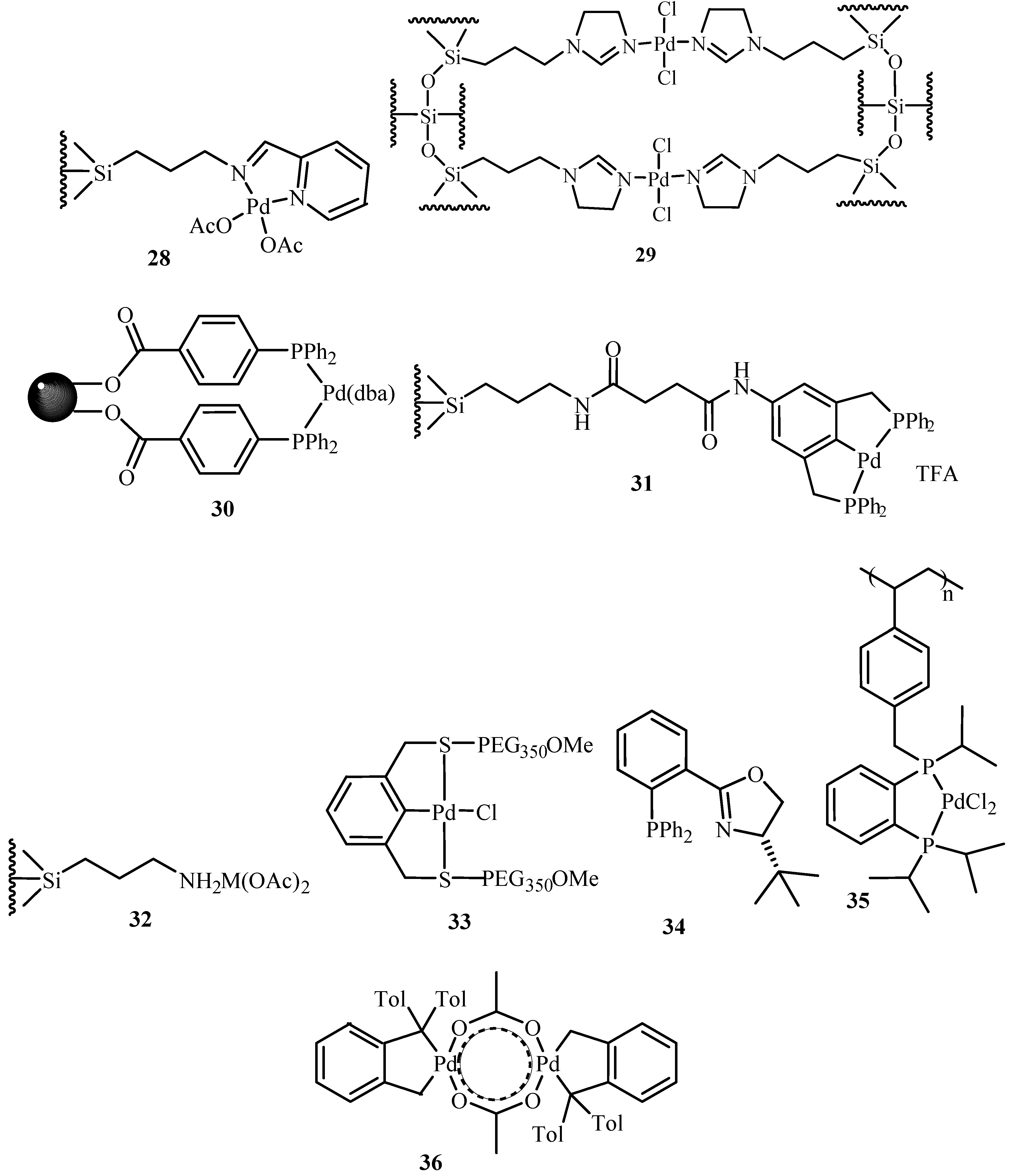
Kobetić and Biliškov [14] have summarized the basics of Heck reaction mechanism, various compositions of applicable catalyst, their developments and applications of Heck reaction until then. Sequential development of both homogeneous and heterogeneous catalysis has been discussed giving examples. Use of palladium with carbon, metal oxides and their salts, silica, porous glass tube, glass beads and guanidinium phosphane clay, zeolites and molecular sieves, MCM 41, polymer, dendrimer, etc., are elaborated with their reaction conditions and yield. Many of these catalysts shows good activity and few of them have proved to be good in recycle study as well. The review also takes an account of substrates, solvents and reaction conditions used. There are few examples of cascade and multiple coupling, synthesis of natural and biologically active compounds and enantioselective Heck-type reactions. Catalysts 28–36 (Figure 4) are seen to give yields in the range of 48 to 93%. Out of all these palladacycles, 36 is the most studied and most reviewed catalyst. In the review, the use of a phase transition catalyst (quaternary ammonium salts), and the solid base that accelerates the Heck reaction are considered to be big breakthrough discoveries.
A review by Sahu and Sapkale [15] discusses the mechanism of palladium catalysed coupling reactions, details about palladium metal, its reactivity and use for reaction like Heck, Suzuki, Negishi, Hiyama, Stille, Fukuyama, Sonogoshira, Buchwald Hartwig and their variants.
The reactivity of palladium is explained on the basis of few points such as
- Electronegativity of palladium develops a polarised Pd-X bond leading to relatively strong Pd-H and Pd-C bonds.
- Its variable oxidation states such as Pd(0), Pd(II) and sometimes Pd(IV) are essential for processes such as oxidative addition, transmetalation and reductive elimination.
- Oxidation states such as Pd(I), Pd(III) and Pd(IV) are also known, where Pd(IV) species are essential in C-H activation mechanisms.
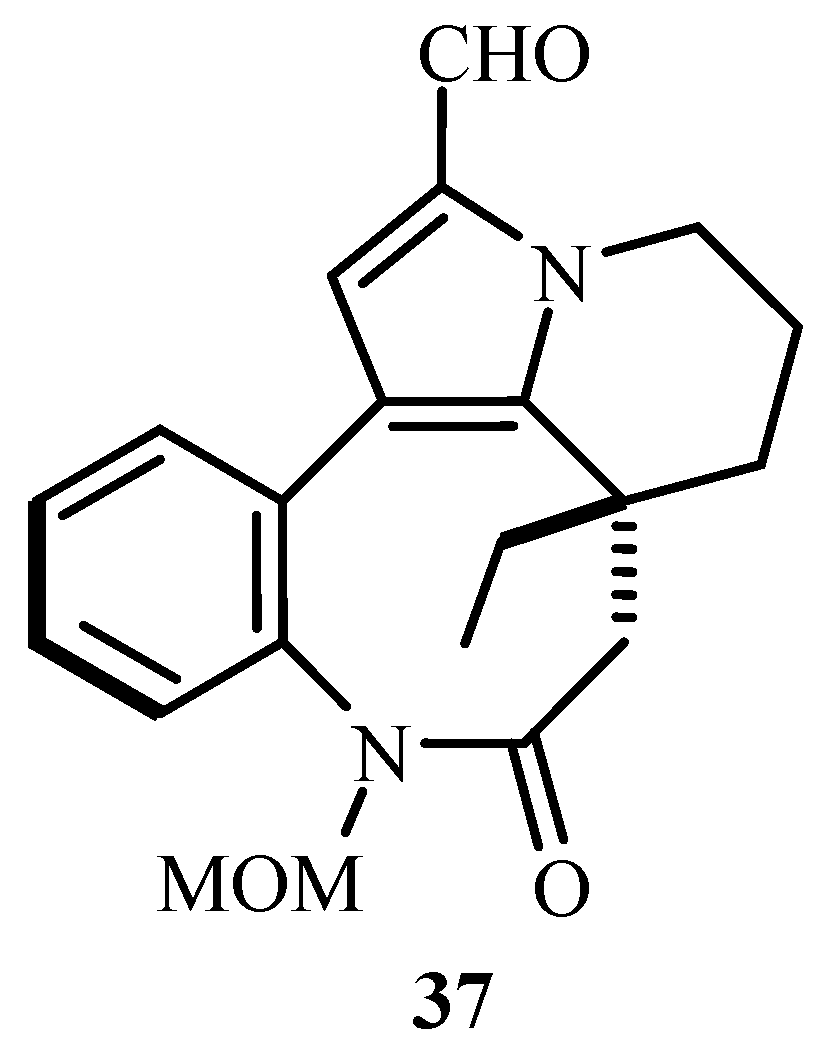
Although not in detail, references for Heck reaction in ionic liquid and for Amino-Heck are given along with the mention of application of Heck reaction in the synthesis of an alkaloid Rhazinal 37 (Figure 5), an antimitotic agent obtained from the stem extract of Kopsia teoi and a promising starting point for anticancer agent. MOM-Rhazinal (MOM = Methoxymethyl ether) derivative is made from the derivative of pyrrol where intramolecular Heck reaction takes place in presence on Pd(OAc)2.
2.2. Catalysts Variants
Every research in catalysis mostly proceeds with the aim of improving the overall catalytic activity for a particular reaction. A progressive development targeting the increase in activity has been discussed thoroughly for Heck reaction as well. Researchers work hard to find cheaper ligands, catalyst system with higher activity so as to minimize the load of palladium and ligand and to find suitable systems for the processing of aryl chlorides too.
2.2.1. Overall Progress
Beletskaya and Cheprakov [16] have published a critical and profound review on Heck reaction that systematically gives details of the work done up to 2000. It is still one of the most comprehensive reviews on Heck reactions.
A number of examples are presented to elaborate the use of various catalysts and ligands for Heck reactions like palladacycles, pincers, carbene complexes, etc. The review supports the mechanism involving Pd(0)/Pd(II) cycle by giving many examples, however the possibility of a mechanism involving Pd(IV)/Pd(II) cycle for few ligands has also been considered. For the reaction of styrene and aryl halide with a mechanism involving a Pd(0)/Pd(II) cycle, regiochemistry of the addition to styrene depends on the solvent and the nature of the anionic ligand. At higher solvent polarity and the weaker coordination ability of ligand there is a greater contribution of a truly cationic form of palladium complex, and the higher relative yield of 1,1-diphenylethylene instead of stilbene.
The review elaborates on the necessity of the stability of a catalyst, particularly for recyclable catalytic systems. Use of bidentate phosphines ligands are the better option for this, where use of excess ligand is not necessary to make stable complexes, and a ratio of 1:1 leading to (L-L)Pd complex is effective for more stable catalyst and thus leading to higher TONs.
The problem of catalyst deactivation, various solvent systems such as aqueous, molten salts, fluorous, supercritical, subcritical fluids, Heck reaction under pressure and microwave conditions are also discussed. It has been seen that high pressure has a beneficial effect on Heck reactions where oxidative addition and migratory insertions steps of the Heck cycles have a negative activation volume and thus are likely to be accelerated by pressure. However, the PdH elimination can be retarded by high pressure, leading to a change of the product distribution while potentially extending the lifetime of palladium catalyst. For microwave assisted Heck reaction, very fast heating by means of microwaves lead to shortening of the reaction time, while the yields and selectivity do not greatly differ when compared with the same reactions carried out using conventional heating.
Many examples of recyclable (phase-separation) catalysis of liquid-liquid and solid-liquid systems are discussed in the review. Heck chemistry with less usual leaving groups like diazonium salts, thallium(III) and lead(IV) derivatives and acid chlorides and anhydrides are given that provides an alternative leaving groups so as to have more reactive substrates and milder procedures. A section on reactions using metals other than palladium like Cu, Ni, Co, Rh, Ir is present in review that says ‘though none of them can rival palladium in synthetic versatility, some features may complement Heck chemistry to provide either cheaper catalysts or catalysts capable of effective processing of some specific substrates’.
A number of simple N-heterocyclic carbene (NHC) palladium-based complexes have emerged as effective catalysts for a variety of cross-coupling reactions and have been proved to be excellent substitutes for phosphines ligands in homogeneous catalysis in a wide range of catalytic processes. Their efficiency is not limited to their binding ability to any transition metal as they also bind to main group elements such as beryllium, sulphur, and iodine. Because of their such specific coordination chemistry, they both stabilize and activate metal centres in quite different key catalytic steps of organic syntheses.
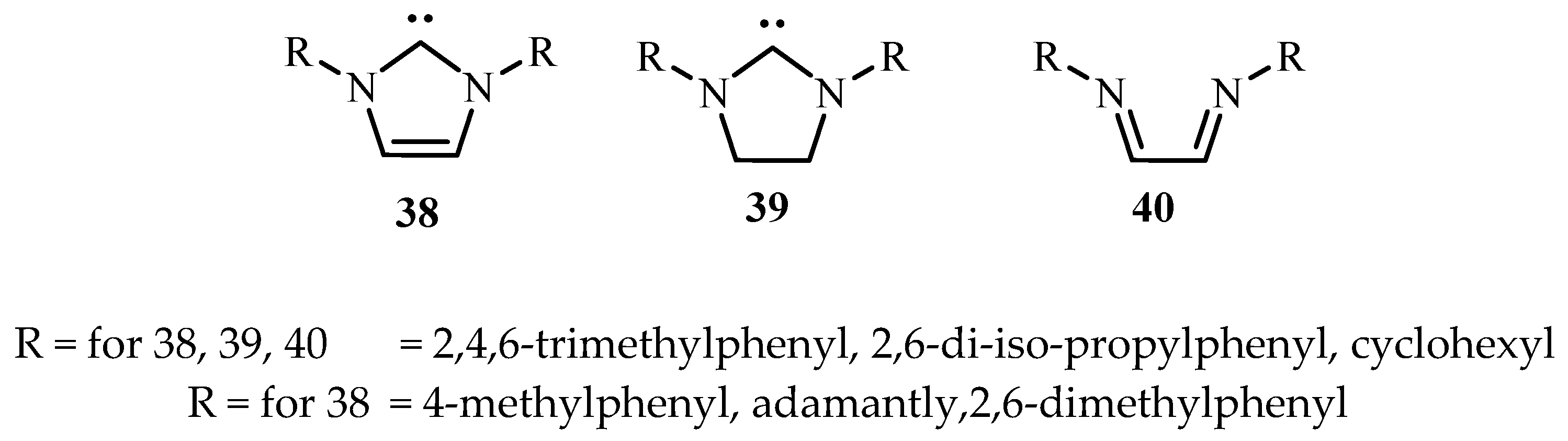
Hillier et al. [17] have reviewed the catalytic cross-coupling reactions mediated by palladium with nucleophilic NHC as ancillary ligands of diazabutadienes 38–40 (Figure 6) mostly based on their own work.
On application of these catalysts to Heck reactions, excellent yields except few were seen. In all cases, the trans products were selectively obtained. However, no activity was observed for aryl chloride substrates. It was also observed that the reactions involving the less reactive aryl halides require bulky electron-donating phosphines. Even excess phosphine and higher Pd loading is required as they are prone to decomposition under Heck conditions increasing the cost of large-scale processes.
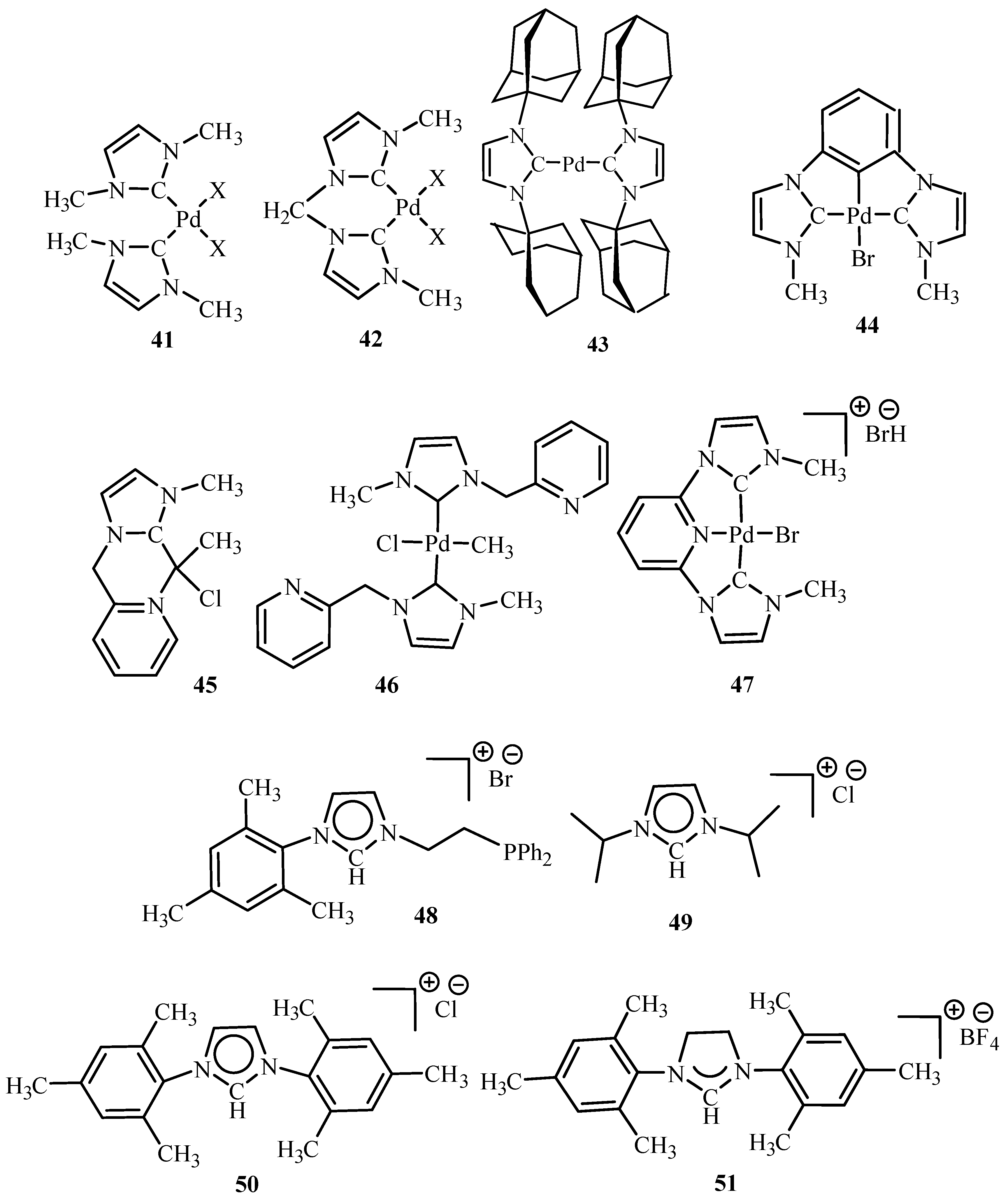
Herrmann [18] has reviewed N-heterocyclic carbenes as ligands where ligands and metal complexes 41–51 (Figure 7) are found to be active towards Heck reaction giving TONs up to 1.7 × 106. These ligands are preferred mainly for the reason that they not only bind to transition metal but also to main group elements. They are robust and found to have high thermal and hydrolytic durability, easy accessibility and require lower amount of loading. They could be derivatized in future to have water-soluble catalysts (two-phase catalysis), immobilization, and in chiral modifications. However, it was observed that it is necessary to have bulky NHC ligands for successful reactions with catalysts made of these ligands.
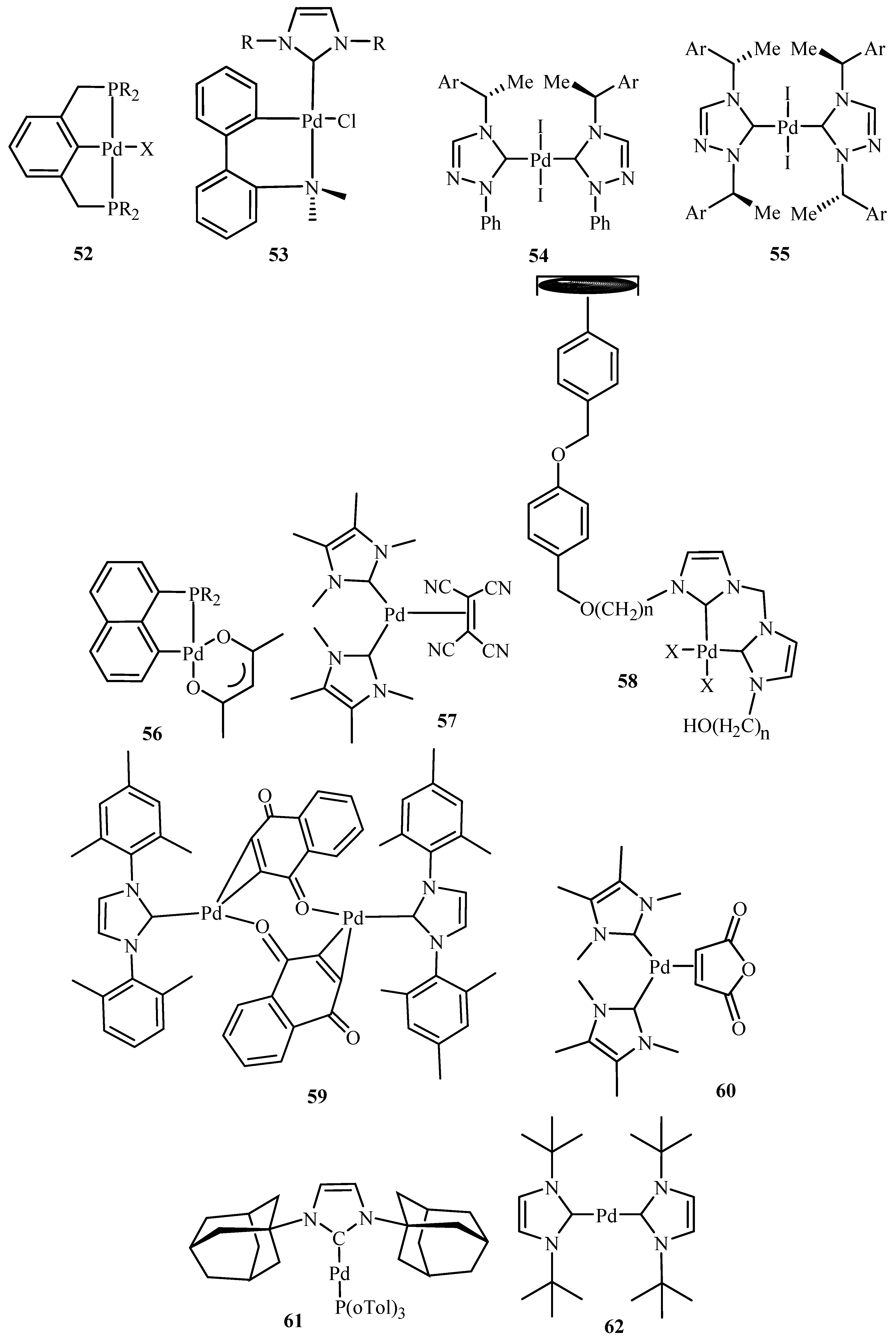
Herrmann et al. [19] presents an eloquent summary about catalytic applications of palladium complexes with phosphorus ligands containing a metallated sp3-carbon centre (palladacycles) 36, 52 and with N-heterocyclic carbene ligands 41–46 and 53–62 (Figure 8) for C-C and C-N coupling reactions of aryl halides. The activity of 36 was found to get increased to TONs of up to 4 × 104 after addition of tetrabutyl ammonium bromide in the reaction of aryl chlorides like 4-chloroacetophenone with n-butyl acrylate under standard conditions. Recycling of this catalyst could be achieved by using non-aqueous ionic liquids (NAILs) as media.
A number of evidences are given for mechanistic discussions and about their role in the catalytic cycle. The catalytic activity of these complexes strongly depends on the steric bulk of the NHC ligand. Structural versatility is a great advantage of N-heterocyclic carbenes where chirality, functionalization, immobilisation and chelate effects can be achieved by easy means. Few complexes contain both NHC and phosphine ligands leading to combine the advantages of stability of bis(carbene) complexes with the good activity of phosphine complexes in C-C coupling reactions.
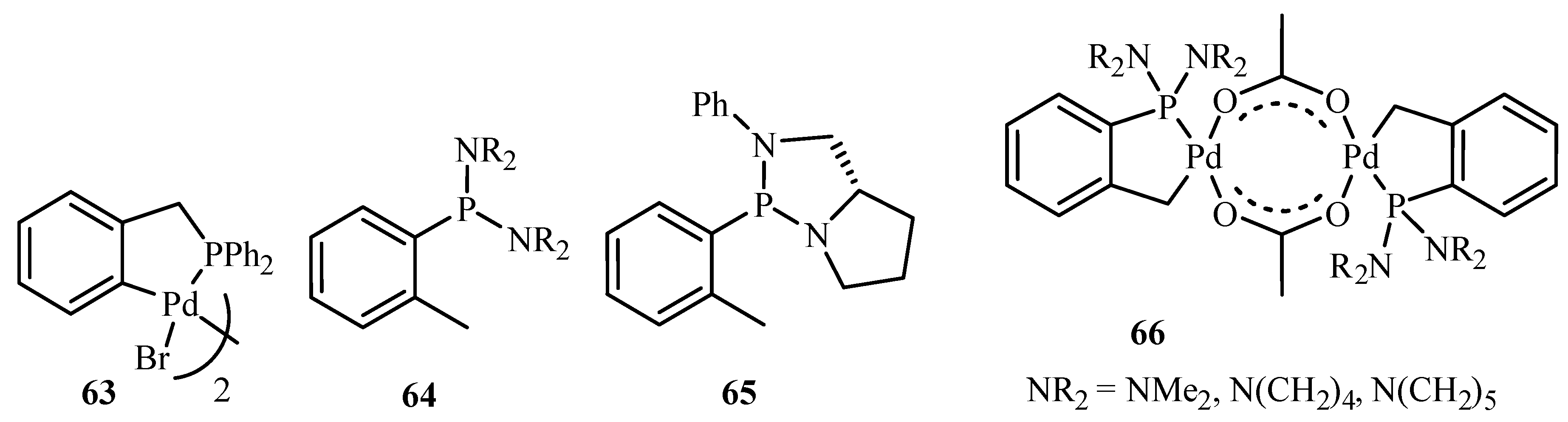
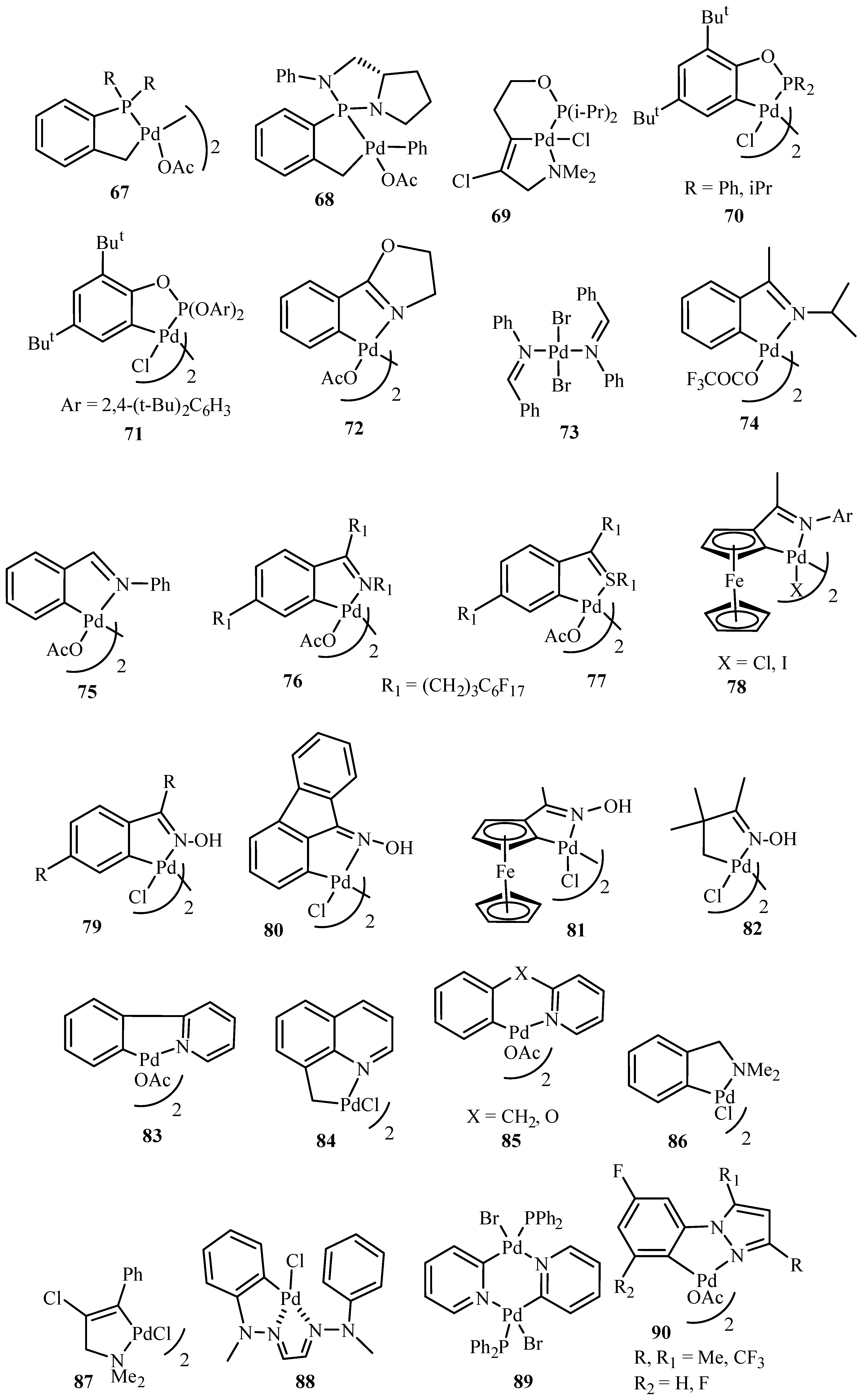
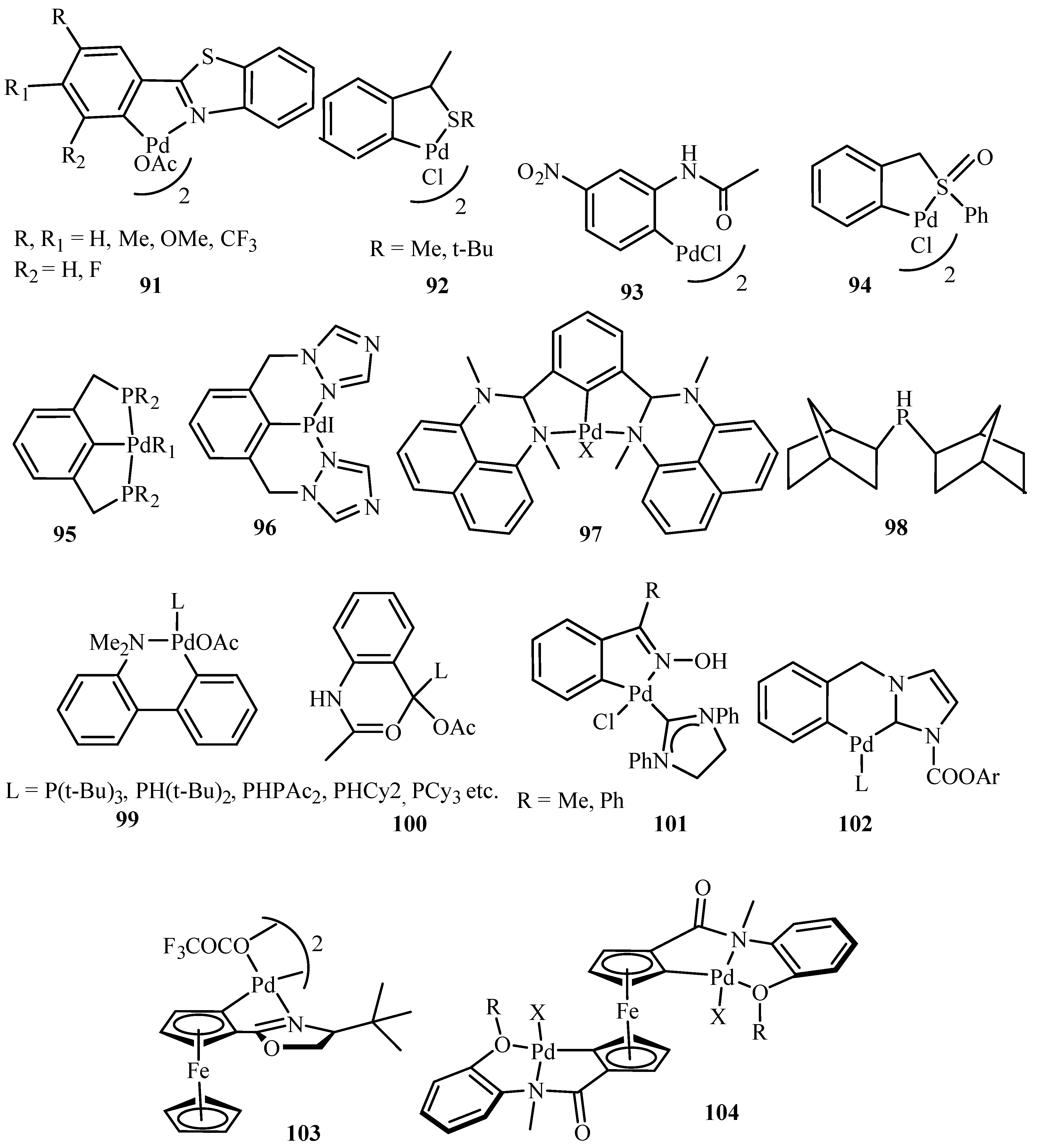
Beletskaya and Cheprakov [20] have given a critical survey on the application of palladacycles as catalysts for cross-coupling and similar reactions where the advantages and limitations of palladacycle catalysts are discussed. The advantages being the slow release of Pd(0) that helps to suppress unwanted processes of nucleation and growth of large inactive Pd metal particles. Bulky ligands with electron-rich phosphines or heterocyclic carbenes are essential and should be used for desired selectivity. However, the phosphine-free chemistry has limited scope. These ligands can be combined with palladacycles into hybrid catalysts, which retain the advantages of both. Such complexes are usually not more active than non-palladacyclic complexes with the same ligands. A review of various palladacycles with subclass of phosphine-free catalysts such as phosphine-derived palladacycles 36, 56, 63–68, phosphite palladacycles 69–71, CN-palladacycles including imine palladacycles 72–78, oxime palladacycles 79–82 and some miscellaneous CN- 83–91, CS- and CO-palladacycles 92–94, pincer palladacycles 95–97, hybrid palladacyclic catalysts 98–102 and couple of palladacycles as structurally defined catalysts 103, 104 (Figure 9) is taken. It was seen that in most of the cases palladacycles serve as a source of highly active zero-valent palladium species.
Pincer catalysts are bis-chelated palladacycles of XCX (X = P, N, S) type. They are extremely stable. It was observed that using pincer catalysts, electron rich phosphine or NHC ligands, the Heck reaction can be carried out with although cheap but otherwise unreactive, chloroarenes. However, based on a survey over the application of palladacycles in catalysis, it was stated the initial promises have not been fulfilled. The catalysts, often announced as outstanding because of very high catalytic activity in several test reactions, very rarely find applications in preparative chemistry. Neither enantioselectivity nor recyclability has been realized. Dozens of palladacycles of all imaginable classes have been studied in various cross-coupling reactions, but none appeared to be the well-defined catalyst, as was thought earlier.
A review by Zafar et al. [21] is about the progress of palladium compounds as a catalyst for Heck-Mizoroki and Suzuki-Miyaura coupling reactions. Some synthesized palladium compounds and their progress in terms of ligand modification and other associated parameters up to early 2014 for Heck-Mizoroki and Suzuki-Miyaura coupling reactions are summarized in it. It was observed that the electron donating ligands such as carbenes, phosphines and nitrogen donor ligands can increase the activity, selectivity and stability of its catalysts. Palladium nanoparticles and palladacycle can also act as precatalysts for Heck and Suzuki coupling reactions. The technological hurdles in using homogeneous catalysts are minimized by putting the catalyst on a polymer support.
2.2.2. Homogeneous Catalysis
Quest for High TON
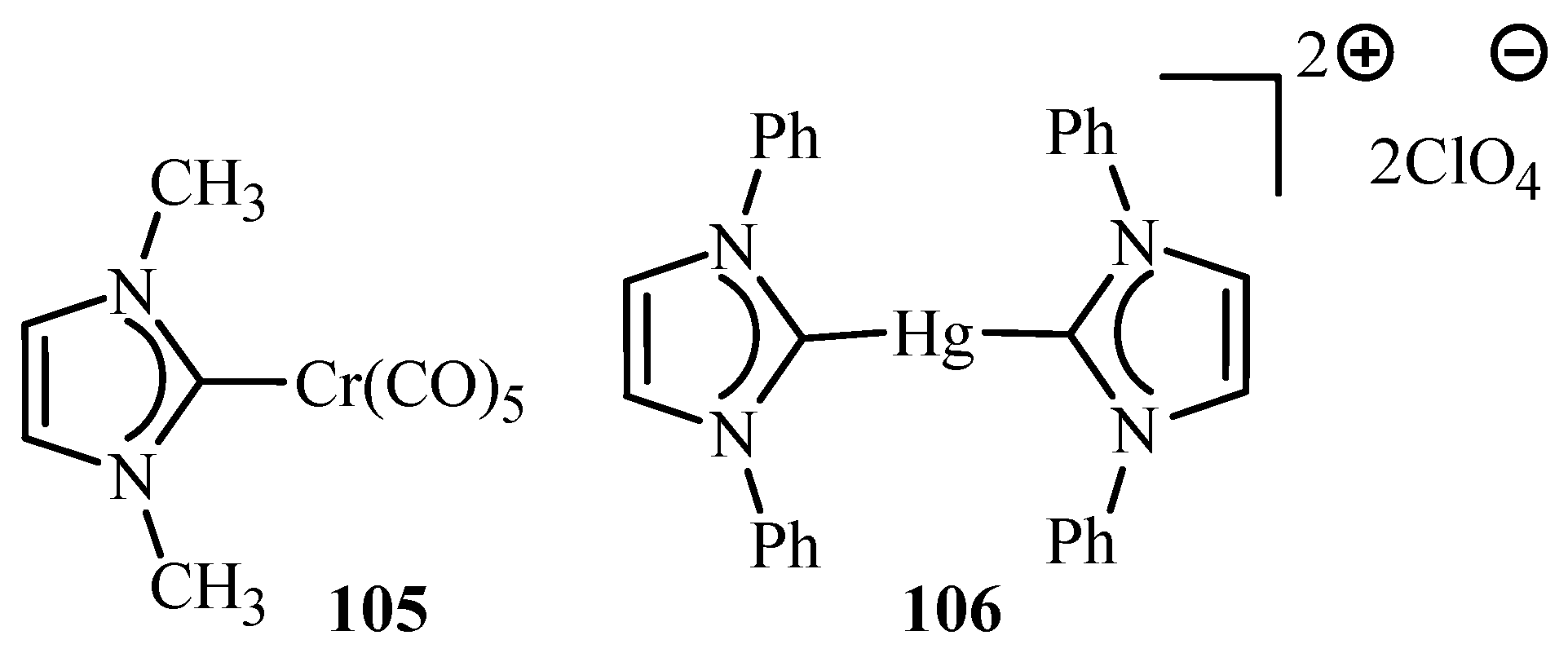
Herrmann et al. [22] have reviewed applications of metal complexes 36, 45, 52, 56, 105 and 106 (Figure 10) in Heck type reactions where detailed information about developments in palladium catalytic systems and their successful approach towards activation of less reactive substrates like aryl chlorides is mentioned. Palladacycles are found to be active against a broad spectrum of reactions and have advantages like active towards more economic aryl halides, high activity at low palladium:ligand ratio (1:1) and improved thermal stability and life-time in solution. The possible mechanisms involving Pd(0)/Pd(II) or Pd(II)/Pd(IV) catalytic cycles has also been reviewed for this class of catalyst. The mechanism of Heck type reactions catalysed by palladacycles is thought to involve active Pd(0) species however the possibility of a Pd(II)/Pd(IV) mechanism, working in competition is also not ruled out. In fact, it is stated that, Pd(II):Pd(IV) could only be a side mechanism which cannot solely be responsible for the high turnovers.
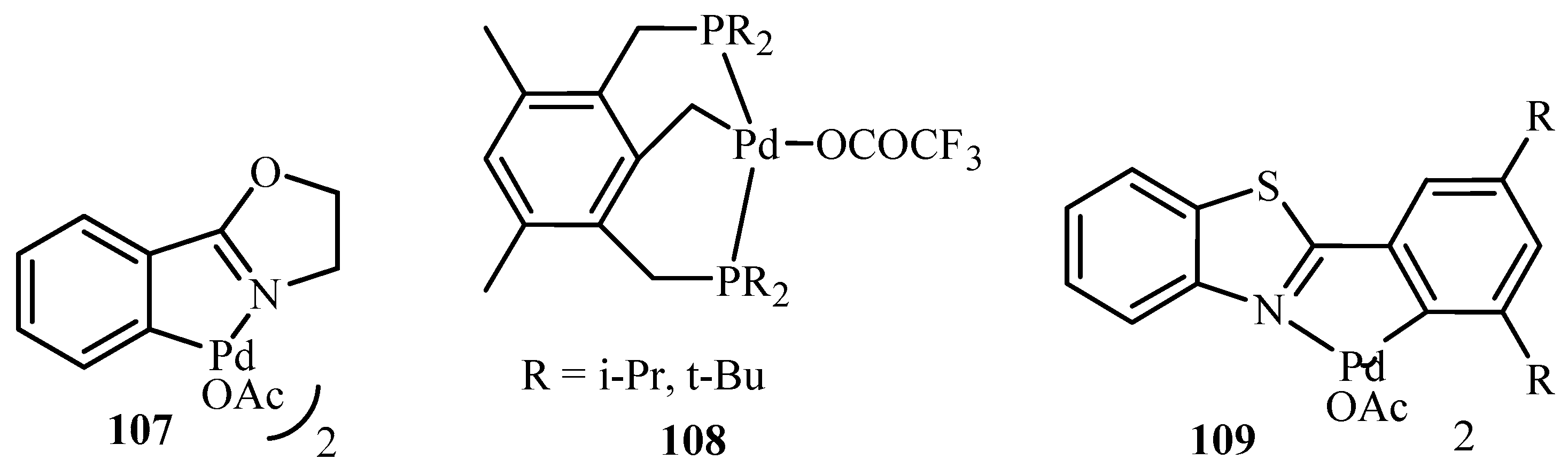
Farina [23] has discussed the problems associated like scope, purity, impurity profile, throughput and time for developing high-turnover catalysts for the cross-coupling and Heck reactions. Numerous examples are given to illustrate the developments in the area of palladacycles and coordinatively unsaturated Pd catalysts featuring bulky phosphanes of high denticity like 107–109 (Figure 11) including 36, 52, 70, 71, 76, 78, 79, 90, 91, 95 and few more similar compounds.
These palladacycles are reviewed from a mechanistic and synthetic standpoint, and compared with more traditional catalysts obtained from conventional mono- and polydentate N and P-based ligands, as well as Pd catalysts without strong ligands, such as Pd colloids or heterogeneous catalysts and polymer supported catalysts. Carbene ligands, though less documented until then were believed to have potential for high-TON research because they are more robust than most phosphines ligands.
Reactions Involving Aryl Chlorides and Bromides
For Heck reactions, aryl bromides and chlorides are the most desirable substrates being cheaper and more readily available; however, not many are used as substrates as they are lower in reactivity due to their stronger C-X bonds and by far having lower TONs by several orders of magnitude. This section describes few reviews that draw attention to future challenges in this area by highlighting advances concerning Heck reactions using aryl bromides and chlorides. A review by Whitecombe and others [24] discuss the development of catalytic systems that can activate unreactive aryl halide towards Heck catalysis. The lesser yields are attributed to the poor reactivity of aryl halides due to their C-X bonds strength as well as the higher temperature used to activate aryl bromides and chlorides resulting in the catalyst decomposition where P-C bond cleavage takes place eventually leading to metal precipitation. This is demonstrated by giving a number of examples using monodentate phosphine ligands, or chelating diphosphine, phosphite, phosphonium salt. However, there are examples of successful reactions taking place under similar conditions with palladacycles, N-heterocyclic carbene complexes and pincers (all previously discussed structures) as they are stable at higher temperature.
The reactivity of ArX is found to be somewhat better if electron withdrawing groups are present in arylhalides; however, reactivity decreases if an electron rich substituent is present typically on aryl halide. The mechanistic study discussed indicates that, the prediction of single mechanism is no longer adequate and there are still unknown factors at play in Heck mechanism. The review exhaustively elaborates the mechanism involving classical Pd(0)/Pd(II) cycles and also Pd(II)/Pd(IV) cycle (this is discussed in detail in present review under mechanism section). In addition to these catalytic systems, heterogeneous catalyst and the catalysts using metals other than palladium, like Ni, Cr, Fe and Co, are described where the yield obtained using these metals are reasonable; however, they are used less than Pd. Use of conditions such as molten salts or phosphine free conditions facilitates the product formation at a higher rate.
A review by Littke and Fu [25] describes the progressive developments in the area of palladium-catalysed couplings of aryl chlorides. It was always observed that the palladium-catalysed coupling processes show poor reactivity towards aryl chlorides, although they are more attractive substrates than the corresponding bromides, iodides, and triflates in terms of cost and availability. Traditional palladium triarylphosphane catalysts are effective only for the coupling of certain activated aryl chlorides (for example, heteroaryl chlorides and substrates bearing electron-withdrawing groups), but not for aryl chlorides in general. However, catalysts using bulky, electron-rich phosphine and carbene ligands have proved to be mild and versatile for aryl chloride coupling.
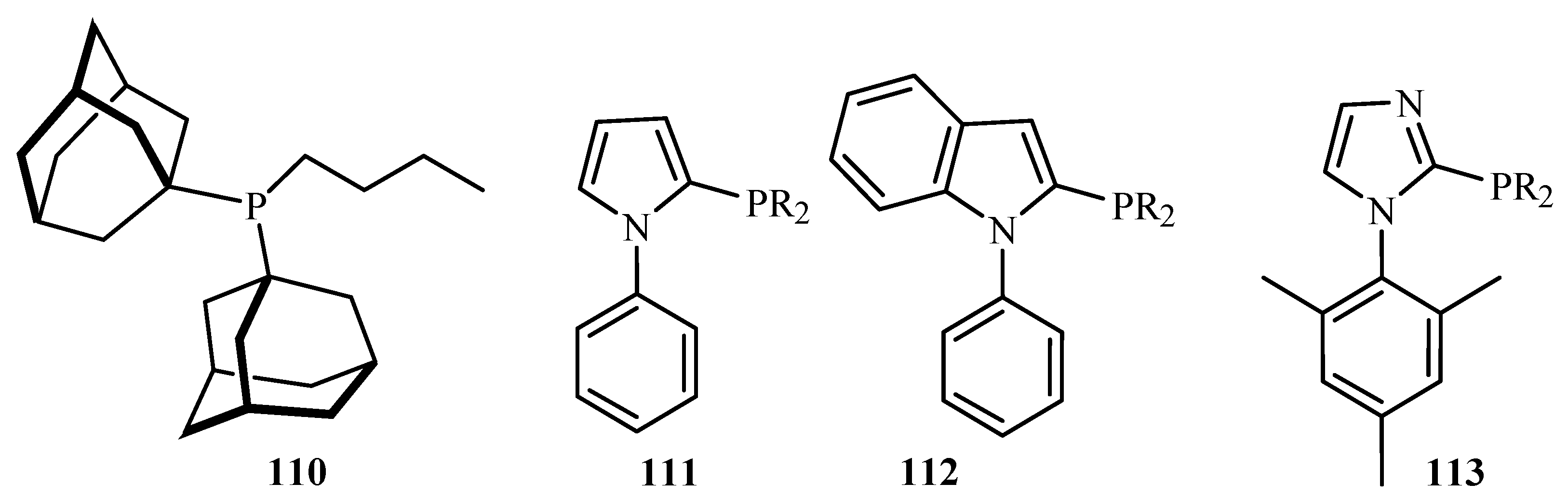
In a similar review by Zapf and Beller [26], ligands and palladacycles 51, 99 and 110 to 115 (Figure 12) are reviewed for their activity towards the C-C and C-N coupling reactions of aryl halides, especially aryl chlorides. An important advantage of this class of ligands is their significantly increased stability towards air and moisture. Due to their basicity and steric bulkiness, they constitute excellent ligands for palladium-catalysed coupling reactions. It was found that the palladacycles with high ligand–palladium ratios are suitable for aryl-X activation reactions at elevated temperatures. However, specially designed basic and sterically demanding phosphines show superior performance under milder reaction conditions. In addition, when monocarbene palladium(0) quinone complexes were tested for the Heck reaction using tetra-n-butylammonium bromide as an ionic liquid, a reasonable to good activity for both electron deficient and electron rich aryl chlorides was obtained.
2.2.3. Heterogeneous Catalysis
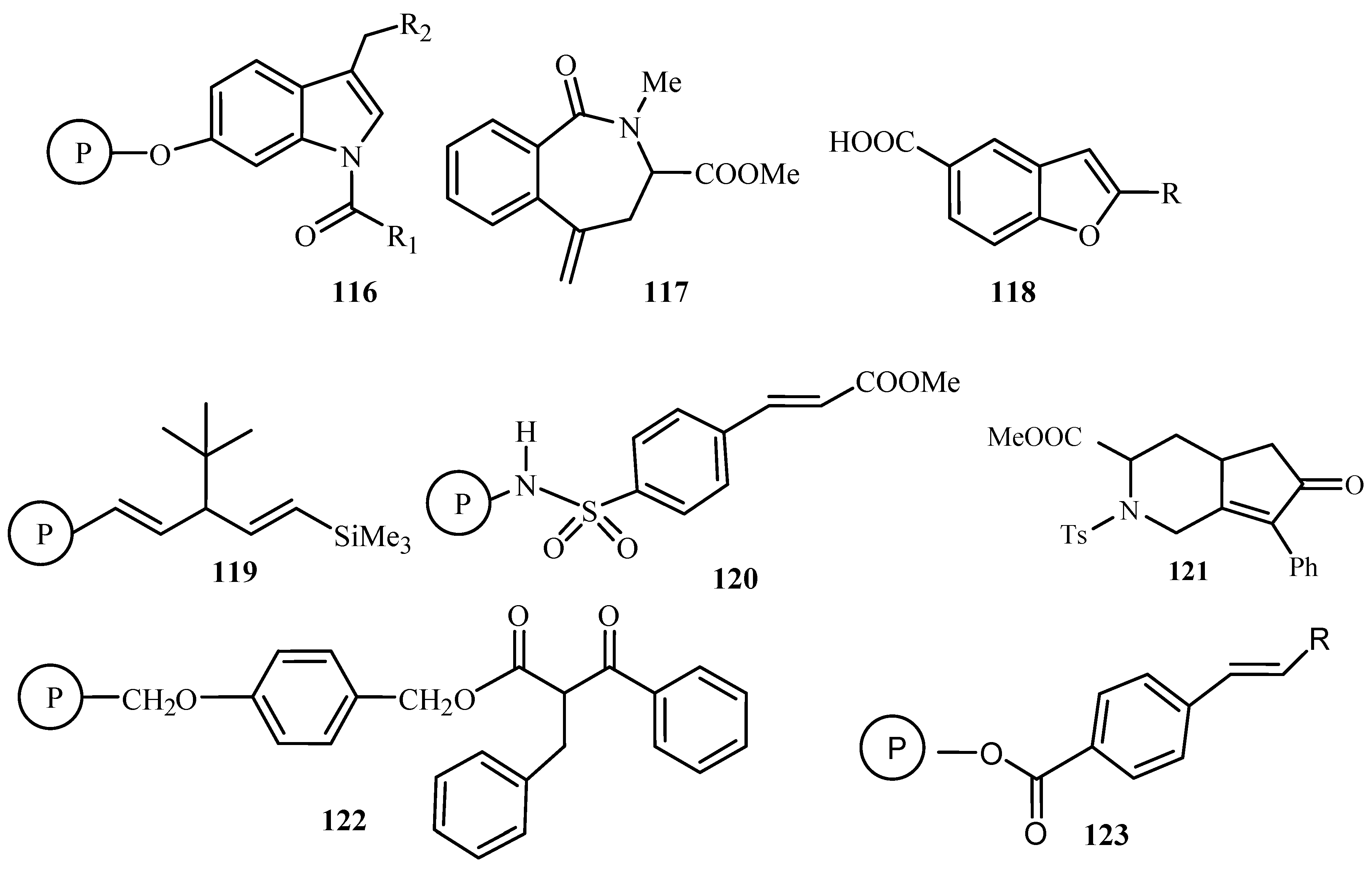
Carbon—carbon bond coupling reactions—Suzuki, Heck and Stille—using catalyst on solid support has been reviewed by Franzén [27]. Metal-catalysed coupling reactions are reportedly efficient and reliable methods for the introduction of new carbon-carbon bonds onto molecules attached to a solid support. A concise summary of the use of these reactions, in the field of solid phase organic synthesis resulting in small organic molecule libraries is presented. This involves the palladium-catalysed intramolecular Heck reaction for the solid-phase synthesis of indole analogues 116, cyclic tetrapeptide derivative via macrocyclization 117, 2-substituted benzofuran carboxylic acids 118, phenyl acetylene oligomers 119, α,β-unsaturated methyl ester phenyl Sulfonide 120, fused bicyclic amino acid derivatives 121, β-keto esters 122 and in the generation of 1,2-disubstituted olefin libraries 123 (Figure 13).
A review by Biffis et al. [28] articulately describes the application of palladium metal catalysts to Heck reaction. The major advantages of supported palladium metal are the simplification of the work-up procedure and the possibility of facile recovery of the precious metal. Initially, the review gives a brief outline of the historical development of heterogeneous catalysis as applied to the Heck reaction followed by the discussion on both supported metal catalysts and stabilized colloidal palladium catalysts. Heterogeneous catalysts supported over different kinds of supports (carbon, inorganic oxides, molecular sieves, polymeric materials, etc.) are reviewed with particular attention to the metal leaching and the nature of catalysis. Ample of examples are given to suggest the presence of leaching, precautions to be taken to prevent leaching or use of methods like re-capture of the leached palladium species with some support. The data provide convincing evidences suggesting the catalytic cycle is sustained mainly by soluble species leached out from the starting solid material after using palladium metal catalysts, either supported or colloidal. The advantage of using heterogeneous catalysis is that it does not necessarily require ligand; the reaction temperature can be high enough for speeding up of the reaction and more of all, the recycling of catalyst is possible although not to a large extent due to extensive metal leaching, metal phase restructuring, structural damage of the support, fouling by carbonaceous deposits, etc., nevertheless systematic tailoring of support can overcome these problems.
Recently, the state of the art, benefits, and challenges of coupling microwave heating with heterogeneous Pd/C catalysis are discussed in the review by Cini et al. [29]. Microwave dielectric heating allowed a significant acceleration of the C-C coupling reaction rate, shortening the reaction time from hours to minutes. The deactivation of catalyst was not observed when Pd/C-catalysed Mizoroki-Heck reaction was carried out under microwave heating and the recycling of catalyst was also possible. Palladium supported on macroscopic pattern-vertically-aligned carbon nanotubes (Pd/VA-CNTs (carbon nanotubes)) catalyst exhibits higher activity in comparison to Pd supported on simple activated charcoal under the same reaction conditions.
2.2.4. Industrial Catalysis
A review on ‘Palladium-Catalysed C-C Coupling: Then and Now’ by Barnard [30] focuses on some of the early work in palladium-catalysed C-C bond formation and change in the methodologies during its developments. It talks about how catalyst has been developed from simple Pd compounds (chloride, acetate) with ligands like triphenylphosphine for variety of aryl halides with even sterically restricted partners, up to the development and applicability of stable, bulky and efficient ligands like PCy3, P(tBu)3, biphenyldialkylphosphine, palladacycle, pincer and carbene complexes that can generate highly active species.
The efforts made to develop supported Pd catalysts suitable for recycle, involving both ligandless and anchored ligand systems, the quest for achieving improved conditions allowing high turnover numbers for aryl bromides and reaction of less reactive aryl chlorides are also discussed. It also mentions about two important reviews, i.e., by Corbet and Mignani [31] on the range of patented cross-coupling technologies and by Yin and Liebscher [32] on C-C coupling by heterogeneous Pd catalysts.
2.2.5. Nano Catalysis
Narayanan [33] has highlighted some of the advances in the application of noble metal nanoparticles as catalysts for Suzuki and Heck reactions. Metal nanoparticles suspended in colloidal solutions and those adsorbed onto bulk supports are attractive catalysts for a wide variety of organic and inorganic reactions, compared to bulk catalysts as they have a high surface-to-volume ratio and very active surface atoms. Important aspects such as shape dependence on the catalytic activity, novel types of supported metal nanoparticles as nanocatalysts and the use of bi-metallic, tri-metallic and multi-metallic nanoparticles as catalysts for the Suzuki and Heck cross-coupling reactions are considered.
A review by Cai et al. [34] provides a summary of bimetallic nanomaterial-catalysed organic transformations and expresses the potential for such bimetallic nanoparticle catalysis to have significant reaction scope, especially with palladium such as magnetically separable “quasi-homogeneous” Pd-Ni nanoalloys, Au-Pd particles confined in silica nanorattles, carbon-supported bimetallic Pd-M (M = Ag, Ni, and Cu) nanoparticles, etc. It was observed that the carbon-supported bimetallic nanoparticles Pd-Cu/C prepared by γ-irradiation at room temperature exhibits high catalytic efficiency in the Suzuki- and Heck-type coupling reactions.
Baboo [35] has reviewed the multimetallic nanomaterial based catalysis for the reactions like oxidation, hydrogenation, coupling reactions, viz. Heck, Suzuki and Sonogoshira, Hydrodechlorination, amidation, reductive amination and hydrogenolysis. Although it is known that nanomaterial based catalysts can easily be separated and reused with same catalytic activities, researchers have paid more attention to the use of multimetallic nano catalysts that show excellent performance than their monometallic nano catalysts. It is mentioned that, despite the great success of bimetallic nanomaterials in terms of their application to oxidation, hydrogenation, and coupling reactions, they have not yet found a wider application in the reactions for the synthesis of complex molecule. The review talks about the use of Pd-based bimetallic nanomaterials and magnetically separable “quasi-homogeneous” Pd-Ni nanoalloys for coupling reactions.
Recently, Labulo et al. [36] have reviewed the CNTs as efficacious supports for palladium-catalysed carbon–carbon cross-coupling reactions. Such catalysts have shown superior catalytic performance and better recyclability for these reactions as they impart stability to the palladium catalyst. The wide variety of surface functionalization techniques for CNTs that improve their properties as catalyst supports, as well as the methods available for loading the catalyst nanoparticles onto the CNTs with a particular focus on the effect of the solvent, base and catalyst loading has been discussed in detail in this review. It was observed that the yield is largely affected by the choice of solvent and base employed for the catalytic reaction. An improved yield could be achieved with para- and meta-substituted aryl halides and not much improvement is seen with those substituted at the ortho-position. Although this catalyst possesses excellent catalytic activities compared with commercial Pd/C catalysts, it suffers from the problem of leaching.
2.3. Array of Heck Type Reactions
This section deals with the Heck reaction performed with some modifications. Reviews are arranged in the way they fit best into one of the category however there are few with interlinks.
2.3.1. Dehydrogenative/Oxidative Heck Reaction
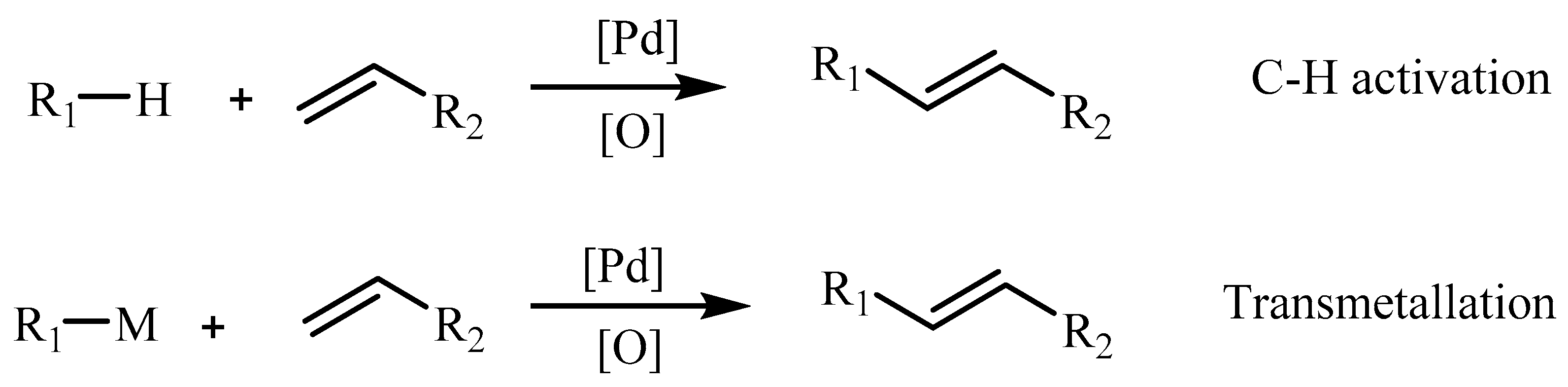
When Ar-H is used at the place of Ar-X in Heck reaction it is called as dehydrogenative Heck reaction also known as Oxidative Heck reaction or Fujiwara-Moritani [37] reaction or simply Fujiwara reaction sometimes. The fact is this version of the Mizoroki–Heck reaction was the first catalytic Heck reaction to be discovered in 1968, where the palladium(II)-catalysed arylation of olefins from phenylmercuric chloride, using catalytic amounts of copper(II) chloride assisted by oxygen for the regeneration of palladium(II) was carried out. This reaction is closely related to the classical Mizoroki–Heck reaction, where instead of initiation by oxidative addition process, it follows transmetallation step or a C-H activation step (Scheme 2).
This is one of the important reactions with respect to atom economy principle since it directly uses the Ar-H compounds thus eliminating additional synthetic step in many total syntheses for halogenations, i.e., in other words, C-H activation of arenes eliminates the need for halogen. The best example of this reaction is the synthesis of benzofuran and dihydrobenzofuran. These structures are important components of numerous biologically active compounds and are preferred to be prepared by dehydrogenative Heck reaction.
Gligorich and Sigman [38] presented a review on advancements and challenges of palladium(II) catalysed oxidation reactions with molecular oxygen as the sole oxidant, where reviewers have discussed some oxidative Heck reactions. The review paper highlights some of the developments until then in direct molecular oxygen-coupled Pd(II)-catalysed oxidation reactions. Although there are reports with positive results for the development of more efficient ligand-modulated oxidative Heck reactions that can be performed under an air atmosphere at room temperature, the asymmetric oxidative Heck reaction still suffers from many limitations and requires further studies.
Karimi et al. [39] have reviewed similar types of reactions describing the oxidative Heck reactions of organometallic compounds such as organomercuric acetates, organoboronic acids, organofluorosilicates and arylstannanes. A number of successful examples are given for the intermolecular, intramolecular, nonsymmetrical, asymmetrical oxidative Heck reaction in presence or in absence of air, with ligand-free or ligand based catalysts formed from palladium, polymer supported palladium(II), transition metal and organometallic catalysts other than palladium along with the examples of asymmetric reactions, intermolecular and intramolecular oxidative Heck reactions via C-H activation. However, the substrate scope of these transformations is still extremely limited and there is much room for the development of newer methods that working well under more convenient reaction conditions.
A review by Su and Jiao [40] reveals the development in the area of palladium catalysed oxidative Heck reactions of alkenes with organometallic compounds, which are effective arylating or akenylating agents. It was observed that the organometallic compounds specially derived from Group III to Group VII like organoboronic. organothallium, organosilicon, organotin, organotin, organophosphorus, organoantimony, organobismuth, organotellurium, hypervalent iodonium salts or simply arenes are efficient substrates for the oxidative Heck reaction. They are found to be remarkably stable and easy to prepare. In addition, various metals other than Pd, including Ru, Rh, Ir and Ni, though less explored for oxidative Heck reaction, are also discussed. The yields of product obtained using them are reasonable up to 72%. The mechanism based on many experimental studies has been reported where the evidence of detection of single charged cationic palladium (II) complexes are given.
A review by Le Bras and Muzart [41] highlights the same subject particularly with respect to its progress of procedures. Numerous results of stoichiometric as well as catalytic palladium mediated arylations of various arenes and heteroarenes are presented. Most of these reactions use an excess of either the arene or the alkene, often with a relatively high Pd catalyst loading and require a terminal oxidant other than molecular oxygen. This becomes an expensive issue and can be a challenge for applications on large scales and even their compatibility with the atom economy principle. Thus, there is wide scope for researchers to work in this area.
Yet another review by Le Bras and Muzart [42] covers the palladium-catalysed annelations of internal alkynes through reactions leading to the loss of only two hydrogens from the substrates. This process is explained with the mechanism that involves (i) dual C-H bond activation; (ii) both C-H and N-H bond activation; (iii) successive amino (or oxy) palladation and C-H bond activation; or (iv) C-H bond activation followed by a Heck-type process. Though not much work has been done in this area, such sustainable processes will become valuable tools for the synthesis of diverse carbocycles and heterocycles.
A review by Lee [43] highlights the use of the oxidative Heck reaction (also referred to as oxidative boron Heck when boron is used in transmetallation reaction (Scheme 2)) in enantioselective Heck-type couplings. This technique overcomes several limitations of the traditional Pd(0)-catalysed Heck coupling and has subsequently allowed for intermolecular couplings of challenging systems such as cyclic enones, acyclic alkenes, and even site selectively on remote alkenes. This has also enabled enantioselective intermolecular couplings of more challenging systems such as desymmetrisation of quaternary centres, cyclic enones, acyclic alkenes and even site selectively on remote alkenes via a redox–relay coupling. A number of examples have been cited along with probable mechanisms for its justification.
2.3.2. Reductive Heck Reaction
The term reductive Heck or reductive arylation is used when the intermediate forms the conjugate addition product in palladium-catalysed Mizoroki-Heck reactions instead of giving substitution product via β-hydride elimination (Scheme 3). Reductive Heck product is a regularly observed side product and the extent of its formation in the reaction varies greatly with base, temperature, substrate and solvent.
Xiaomei et al. [44] have reviewed the progress in reductive Heck reaction, where unsaturated halides react with olefinic compounds at the similar Heck conditions to form the addition products. Discussion on catalytic systems, mechanism, applications and limitation in organic chemistry is given.
2.3.3. Intramolecular Heck Reaction
Intramolecular Heck reaction is generally more efficient than the intermolecular Heck reactions for many reasons like, in the intermolecular Heck reactions, only mono- and disubstituted olefins can participate, while in the intramolecular case tri- and tetrasubstiuted olefins can readily get inserted; regiocontrol of olefin addition is difficult in the intermolecular for electronically neutral olefins whereas the regioselectivity in intramolecular process is governed by ring-size of the newly formed cycle and is generally directed by steric considerations giving highly regioselective couplings. In addition, construction of cyclic compounds containing an endo- or exo-cyclic double bond can efficiently be brought about by using the intramolecular Heck reaction.
A review by Guiry and Kiely [45] summarizes the development of the intramolecular Heck chemistry and the methodology used for the construction of carbocycles and heterocycles along with the mechanism. The review is mainly focused on the optimization of palladium catalysts derived from various diphosphine and phoshinamine ligands for the preparation of a variety of cyclic products like cis-decalins, hydrindans, indolizidines, diquinanes and the synthesis of quaternary carbon centres. A number of examples of the application of intramolecular Heck cyclization as the key step in the preparation of many complex natural product syntheses are given. Some of their own work on ligand synthesis used for such reactions is also discussed.
A review by Oestreich [46] summarizes the exciting development of the enantioselective intermolecular Heck reaction (particularly the Heck-Matsuda [47] reaction that uses arene diazonium salts as an alternative to aryl halides and triflates) of acyclic alkenes and the synthetic utility of enantioselective intermolecular couplings of cyclic alkenes. Many such examples that give very high enantioselectivity for intermolecular Heck reactions are cited.
2.3.4. Asymmetric Heck Reaction
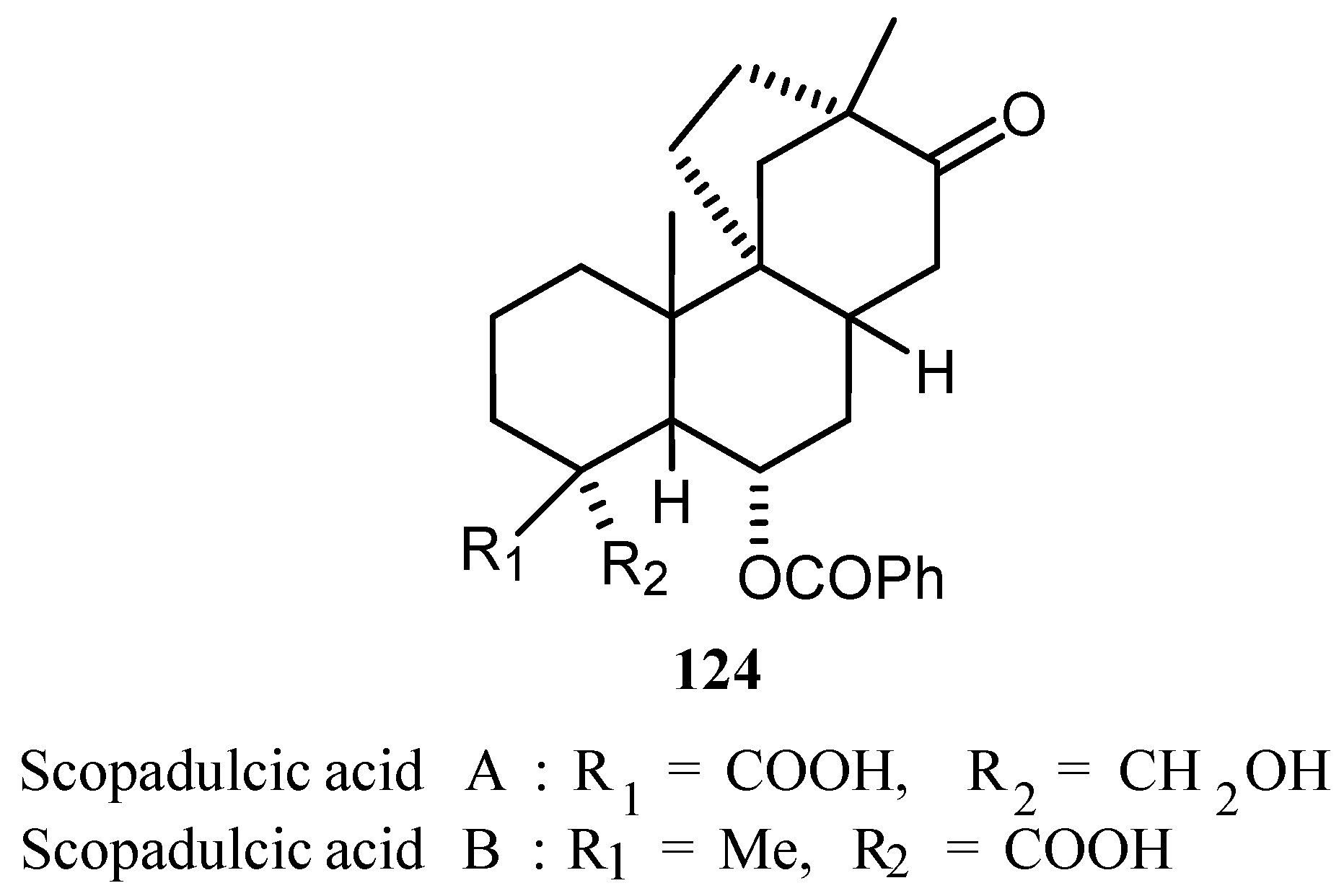
A breakthrough in Heck reaction took place when in 1989 Shibasaki [48] and Overman [49] independently reported the first examples of asymmetric Heck reactions. They used chiral ligands BINAP and (R,R)-DIOP for such reactions, respectively. A classic example of the intramolecular Heck reaction in action is Overman’s synthesis of (−)-scopadulcic acid A 124 (Figure 14), where a tandem double Heck cyclisation (6-exo-trig followed by a 5-exo-trig) rapidly accesses the carbon skeleton found in the natural product.
From 2003, the catalytic asymmetric variant of Heck reaction emerged as a reliable method for enantioselective carbon-carbon bond formation. Dounay and Overman [50] reviewed the application of catalytic asymmetric Heck cyclization in natural product total synthesis for the formation of tertiary and quaternary stereocenters by considering synthesis of some terpenoids (like ernolepin, Oppositol and Prepinnaterpene, Desmethyl-2-methoxycalamenene, Capnellenols, Δ9(12)-Capnellene, Kaurene and Abietic Acid, Retinoids); Alkaloids (like Lentiginosine and Gephyrotoxin 209D, 5-Epiindolizidine 167B and 5E,9Z-indolizidine, 223AB. Physostigmine and Physovenine, Quadrigemine C and Psycholeine, Idiospermuline, Spirotryprostatin, Eptazocine); Polyketides (like Halenaquinone and Halenaquinol, Xestoquinone, Wortmannin). A brief discussion on understanding of the mechanisms of catalytic reactions is provided necessary for the rational development of efficient asymmetric processes.
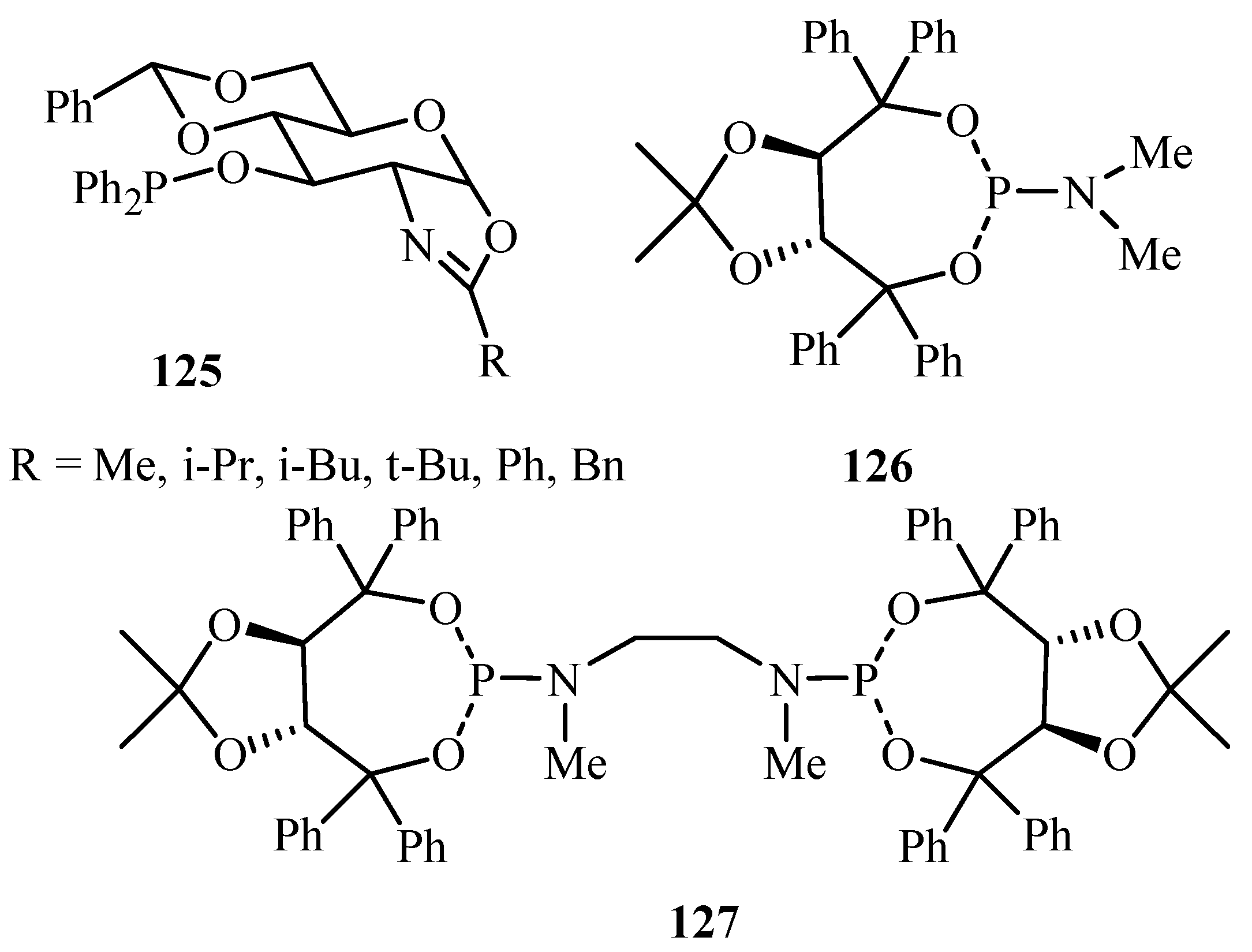
A review by Diéguez [51] covers reports on the ligands derived from carbohydrates for asymmetric catalysis until 2003. High enantioselectivities have been observed by using bidentate ligands, usually diphosphines and phosphine-oxazoline ligands. They also have excellent control on selectivity, based on the properties of the ligand. Carbohydrate ligands have proved to be some of the most versatile ligands for enantioselective catalysis. Examples of numerous carbohydrate based ligands are discussed in the review, however, for Pd-catalysed Heck reaction, only the following kinds of carbohydrate derivative ligands 125–127 (Figure 15) have been seen to be efficiently applied.
McCartney and Guiry [52] comprehensively reviewed the asymmetric inter- and intramolecular Heck (Scheme 4) and related reactions since their original development until 2011 with respect to substrate scope, reactivity, regio- and enantioselectivity. The formation of products is supported by predicting mechanism. The classification is based on the nature of ligands in terms of their denticity, chirality and nature of donor atoms involved so as to understand the continued development of ligand architectural design and their application. The review addresses the significant improvements in reaction times, a disadvantage of Heck reactions performed in classical way, use of microwave-assisted protocols and ligand design. The asymmetric Fujiwara-Moritani and oxidative boron Heck-type reactions and the recent additions to Heck type processes, are also discussed.
2.3.5. Heck Reactions Involving Heteroatom
A review by Daves and Hallberg [53] highlights typical heteroatom-substituted olefins to organopalladium reagents, where a new carbon-carbon bond is formed via 1,2-addition of an organopalladium species to the often strongly polarized carbon-carbon double bond of a heteroatom substituted olefin. These types of reactions afford versatile and efficient synthetic routes to a wide variety of compounds. Heck and other similar types of reactions involving intermediate palladium complex undergoing 1,2-additions have been discussed. Compounds with heteroatoms O, N, S, P are considered to explore their reactivity towards such addition reaction. A number of examples with cyclic enol ethers, glycals, and cyclic enol ethers derived from carbohydrates, acyclic enol ethers like thioenol ethers, enol carboxylates, enamides and enamines, vinylsilanes and vinylphosphonates are given. Product formation is explained based on well accepted mechanism for Heck reaction involving Pd(0) complex. Increased understanding of the pathways by which these reactions occur, that include delineation of the factors determining reaction regio- and stereochemistry, and control of competing modes of σ-organopalladium adduct decomposition to products, make possible the utilization of these reactions in the synthesis of complex structures.
Bedford [54] has reviewed the use of palladacyclic catalysts in C-C and C-heteroatom bond-forming reactions. Palladacycles are air and moisture stable inexpensive catalysts, can easily be handled and stored, and act as clean sources of low-coordinate palladium(0). They as pre-catalysts play a significant role in a range of C-C and C-heteroatom bond forming reactions. Activity of the catalysts comprised of P-C palladacyclic, Palladium P,C,P-pincer complexes, L-C palladacyclic, L,L,C- and L,C,L-pincer based (where L = N, S) have been compared for the C-C and C-heteroatom bond-forming reactions including discussion on the likely nature of the true active catalysts produced in situ. These catalysts show the TON ranging from a few thousand up to a few million. Consideration to the Aryl chlorides as substrates and the mechanism involving palladacycle for such reaction is thoroughly discussed. It was found that the complexes 36, 63 and 70 show reasonable activity with some, usually less electronically challenging substrates. The possibility of recyclability of a few catalysts for Heck and other reactions has been discussed. It was predicted previously that when the catalysts are immobilized on solid supports they could work as recyclable catalyst. Polystyrene-immobilized catalyst 74 shows comparable activity to homogeneous analogues in the Heck coupling of iodobenzene with styrene. However, in recycle study the filtrate after reaction and not the recovered catalyst that showed the activity comparable to that obtained in the first run. This activity was concluded to be due to formation of nanoparticulate palladium which is stabilized by an ammonium salt.
2.4. Methodology
Various attempts have been made by researchers to improve the catalyst activity, lowering the cost of catalyst; hence different methodologies are adopted using non-conventional techniques for Heck reactions. Reviews based on these methodologies are described here.
2.4.1. For Improvement in Activity
Use of tetraalkylammonium salts to improve the yields in Heck reactions particularly is considered to be a remarkable contribution in catalysis. The combination of such salts (phase-transfer catalysts) and insoluble bases accelerates the rate of reaction to great extent even at lower reaction temperatures and is commonly known as Jeffery conditions. A review of these salts in Heck type reactions is taken by Jeffery [55]. It was seen that, under appropriate conditions, tetraalkylammonium hydrogensulfate can just be as efficient as tetraalkylammonium chloride or bromide for facilitating Heck-type reactions. Thus, an appropriate selection of the catalyst system (Pd/Base/QX) can allow this type of reactions to be efficiently realized at will, in a strictly anhydrous medium or in a water-organic solvent mixture or in water alone.
2.4.2. For Lowering the Cost
Tucker and de Vries [56] describe their own efforts in the area of palladium- and nickel-catalysed aromatic substitution carbon-carbon bond formation reactions in the review titled ‘homogeneous catalysis for the production of fine chemicals’. The main focus of this review is on low cost and low waste production methods. A number of examples are discussed to prove methodology for lowering production cost such as reducing the amount of catalyst, eliminating use of ligands or use of cheaper phosphite or phosphoramidite ligands, carrying out reactions at lower temperatures, replacing palladium by nickel, replacing aryl bromides or iodides with the cheaper chlorides and simplified work-up. For waste free production methods, use of aromatic anhydrides as aryl donor is suggested. Methods for recycling of palladium in ligand-free Heck and Suzuki reactions is described that involves treatment of palladium black, precipitating at the end of the reaction, with a small excess of I2 prior to its re-use in the next run.
Typically, the Heck reactions are catalysed by palladium, a precious metal. However, in many cases low cost transition metals are found to play a similar role which is reviewed by Wang and Yang [57]. This review focuses on low-cost transition metal catalysed Heck-type reactions. The cited examples indicate that some low-cost transition metals like Ni, Co, Cu, Fe are active for Heck-type reactions. Ni is found to give best performance among them; in fact, sometimes the results with Ni are comparable to that with Pd. Co and Cu exhibit outstanding activity to alkyl electrophile involved Heck- type reactions. This is attributed to their abilities to generate alkyl radical. It was also observed in a few cases that under certain conditions these low-cost transition metals may show unique catalytic properties, which are absent in Pd-mediated systems. Two mechanisms have been predicted for Heck-type reactions using these transition metals, cationic mechanism and radical mechanism based on the reacting species. Phenyl or benzyl halides predominantly take the cationic mechanism, whereas alkyl halides usually follow the radical mechanism, especially in Co- and Cu- catalysed systems because Co and Cu have good capacity of producing radical from alkyl halides. However, the investigations on low-cost transition metals catalysed Heck-type reactions highlight the extension of the substrate scope and draw a little attention to the development of catalysts, ligands and solvents. Hence, more study is needed in this area.
2.4.3. Non-Conventional Methodologies
Beletskaya et al. [58] have published a critical overview on unconventional methodologies for transition-metal catalysed Heck reaction highlighting the efforts and interest in developing more efficient processes according to the new requirements of chemistry. A vast array of non-conventional methodologies is described considering different parameters involved in the reaction like substrates, catalytic system, solvent, reaction conditions, or work-up. However, it is also stated that, although large numbers of interesting methodologies are available from an academic point of view, they are useless from a practical point of view. Hence, there is a need for reconsideration of a few points while performing Heck reactions at the industrial level such as:
- Most of the methodologies deal with the simpler reactions between the reactive substrates like aryl iodides or activated aryl bromides and acrylates in contrast to the more desirable, but less reactive aryl chlorides or other olefinic substrates such as electron-rich olefins.
- More attention should be paid at the workup and separation of by-products formed during achieving a high regio- and stereoselective products.
- Whenever possible, commercially available starting materials, reagents, ligands, catalysts, or solvents must be used and proper time should be given to experimental work needed to prepare the different reaction components.
- The catalyst must be recyclable and/or display high TONs.
- Heterogeneous catalysis or a use of ligandless catalysts, recoverable ligands or stabilized nanoparticles are preferred for better recovery and lower cost provided metal leaching is prevented.
- An ideal recyclable catalyst is the one in which filtration of the reaction mixture produces a catalytically active solid and an inactive filtrate.
- Use of high temperature with some stable catalysts may prove to be unfavourable for the selectivity of the product.
- The expensive equipment or reaction medium utilized in some methodologies cannot compensate for the little or no improvement observed in many cases with respect to the conventional methodologies; in addition, the application of these methodologies is normally restricted to a small scale.
- Reproducibility, atom-economy, low-cost, scalable and practical procedures, are needed to extend the methodologies from the academic laboratory to the industrial plant.
Further research must be undertaken in order to clarify the reaction mechanisms involved in the different processes, which remain unclear in most cases; it is crucial to have a better knowledge of the nature and properties of the real catalytic species in order to improve any given reaction.
2.5. Selectivity
Under the section Asymmetric Heck reaction (Section 2, Section 3 and Section 4) reviews dealing with the generation of asymmetric centre by means of Heck reactions were summarized whereas under this section the progress of ligands and catalysts for the development of various regioselective and enantioselective transformations via Heck reactions are discussed.
A review by Tietze et al. [59] on ‘enantioselective palladium catalysed transformation’ discusses many other important organic reactions including Heck reaction where enantioselectivity in intramolecular and intermolecular Heck reaction is discussed with lot of examples collected right from its first example of such kind by Shibasaki [48] and Overman [49]. A number of examples with new improved ligands for development of various enantioselective transformations has been discussed aiming at higher (preferably over 95%) ee values. The review asserts that there is no field where enantioselective Pd catalysis cannot be employed. However, the disadvantage of such catalysis is the high price of Pd and the usually small turnover numbers, making the processes too expensive for industrial use. Nevertheless, novel chiral Pd catalysts resulting in high turnover number can be synthesized to overcome this issue in addition to a broad range of enantioselective transformations suitable for the chemical industry.
Almost at the same time, Shibasaki et al. [60] reviewed a similar topic particularly aiming at the asymmetric Heck reaction. Since a variety of carbocyclic, heterocyclic and spirocyclic systems can be constructed, the asymmetric Heck reaction becomes a powerful method for the synthesis of both tertiary and quaternary chiral carbon centres, with an enantiomeric excess often in the range of 80% to 99%. The scope of the reaction with respect to the product alkene isomerization was limited due to regioselectivity, and was predicted to be solved by development of new generation of ligands dissociating more rapidly from the products, thus improving both enantio- and regiocontrol.
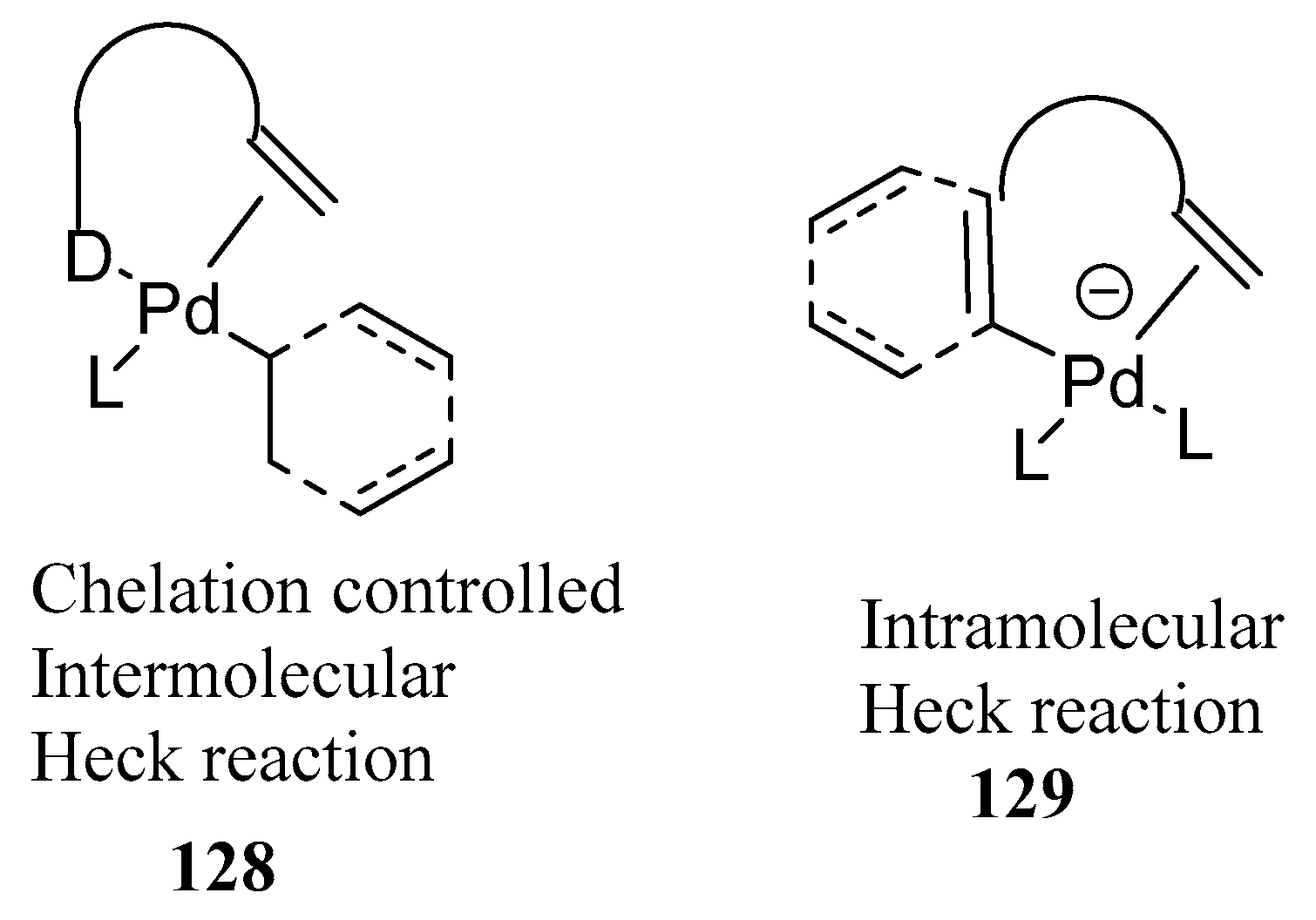
Oestreich [61] describes the evolution of inter- as well as intramolecular Heck reactions from regio- to diastereo- and finally to enantioselective transformations with a special reference to heteroatom-directed Heck reactions in his review. The concept of “Chelation Control” 128, 129 (Figure 16) controls regio- and stereoselectivity with the aid of attractive interactions between substrate and reagent/catalyst is discussed.
Several examples of removable catalyst-directing groups developed for the preferential regioselective intermolecular arylation of alkenes are given such as amino-directed intermolecular Heck reaction of vinyl ethers. Mono- versus bidentate phosphines influence a regiochemical switch based on the bite angle and the mechanistic rationale for inverted regioselectivity is also discussed. Review covers the syntheses of stereodefined, multi-arylated alkenes, the diastereoselective construction of tertiary and quaternary carbon centres, and also the combination of substrate with catalyst-control in an enantioselective transformation. Many examples have been discussed to show the neighbouring-group effects playing important role in Heck chemistry and expect few more discoveries in this field. The example of a substrate-controlled enantioselective reaction illustrates that the enantioselection is sometimes discriminated not only by the chiral reagent but also by a suitably located donor.
2.6. Aqueous Media
Nowadays, use of water has become increasingly popular for fine synthetic chemistry in industry for the reasons: water is non-toxic, nonflammable, inexpensive, and environmentally friendly solvent and there by use of organic solvent can be avoided. In addition, one of the major drawbacks of homogeneous metal catalysis lies in the separation of the reaction product from the catalyst and requires costly procedures. The concept of transition metal catalysis in water is used where the catalyst is easily recovered by separation of the aqueous and organic phase if a biphasic system is used. Its popularity has been increased since the development of the Ruhrchemie–Rhone Poulenc process using a modified water-soluble rhodium complex in the hydroformylation methodology. However, the limitations of aqueous phase catalysis are:
- Stability of substrate or product in water.
- Partial solubility of substrate in the aqueous phase to avoid mass transfer limitation.
- Necessity of preparation of water soluble ligands or dispersing agents to maintain catalyst in aqueous phase.
- Challenges for future developments in this area to develop catalysts with scope and activity comparable to the best organic-phase catalyst systems.
Nevertheless, there is still strong interest in developing efficient and recoverable catalysts for use in pharmaceutical and other fine chemical synthetic processes, as can be seen from following reviews.
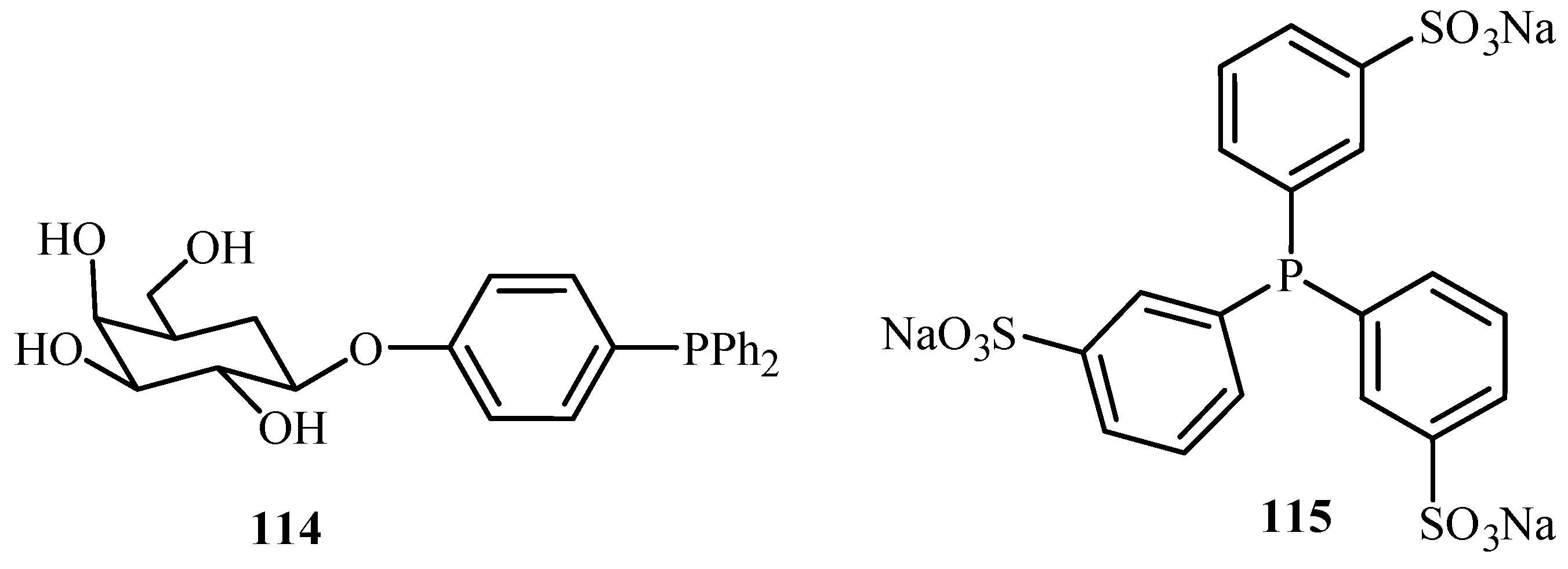
Genet and Savignac [62] have reviewed the palladium cross-coupling reactions carried out in aqueous medium. Reactions like Heck, Sonogashira, Tsuji-Trost, Suzuki, Stille as well as protecting group chemistry in aqueous media are discussed in the review. Although palladium is known to be unstable in aqueous medium, there are reports of the excellent compatibility of water-soluble palladium catalysts with water- soluble phosphines such as TPPTS (3,3′,3′′-Phosphanetriyltris(benzenesulfonic acid) trisodium salt) 115, TPPMS (Sodium Diphenylphosphinobenzene-3-sulfonate) 130 and salts of acid or amines 131, 132 (Figure 17), offering new opportunities for such reactions at mild conditions and with new selectivity.
It was seen that the careful selection of reaction conditions, co-solvents and catalysts, is very important for long life catalyst and new selectivities. The advantages of the two-phase aqueous system, i.e., easy separation of the products and recycling the expensive palladium can be obtained by using palladium catalysed reactions with water-soluble phosphines that has increased the potential of modern palladium catalysis.
Li [63] has reviewed many organic reactions in aqueous media where water serves as a medium for various palladium-catalysed reactions of aryl halides with acrylic acid or acrylonitrile to give the corresponding coupling products in high yields. In addition, reactions at superheated and microwave heating conditions with bulky phosphine ligands along with arenediazonium salts instead of aryl halides in the Heck-type reaction are mentioned. The reason for a high yield of coupling product for reaction involving the use of Pd(OAc)2 with water-soluble ligand, TPPTS, and formation of complex mixture was attributed to high dielectric constant of water. Examples of many transition metals other than palladium in water have been cited.
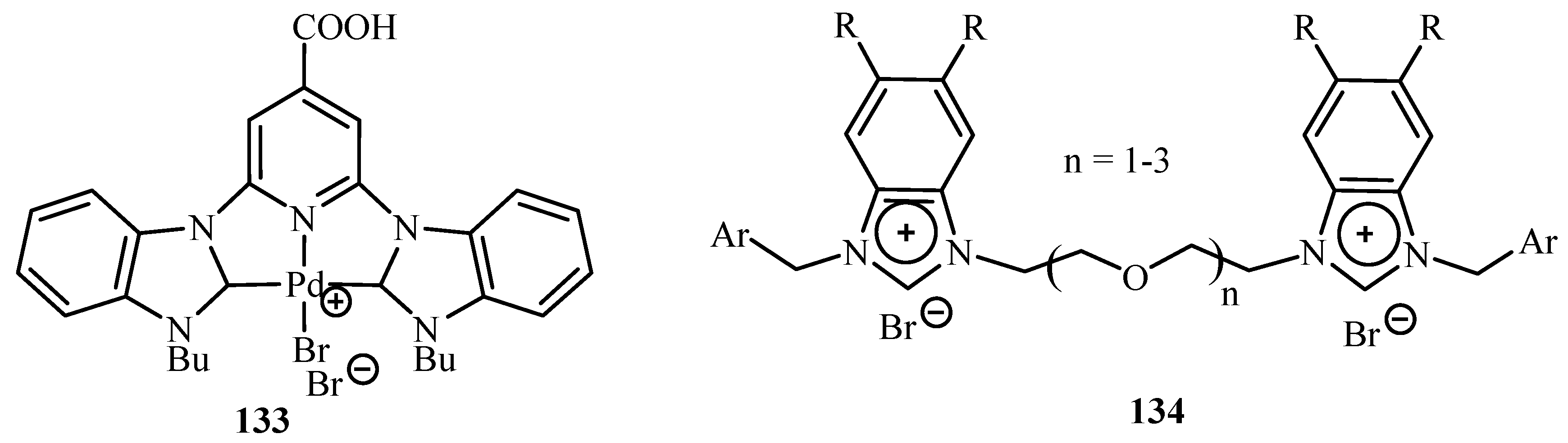
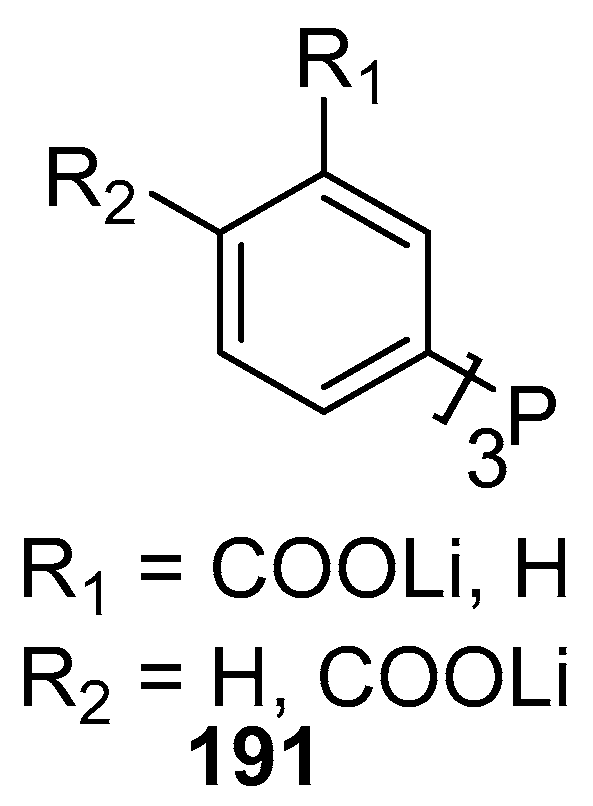
Velazquez and Verpoort [64] have reviewed the reports on the use of N-heterocyclic carbene transition metal complexes for catalysis in water. The typical phosphine and amine-type ligands can thus be displaced by this type of catalysis because of their higher stability and reactivity. Palladium complex 133 and a complex of ligand 134 (Figure 18) are used for catalysing the Heck reaction successfully in water.
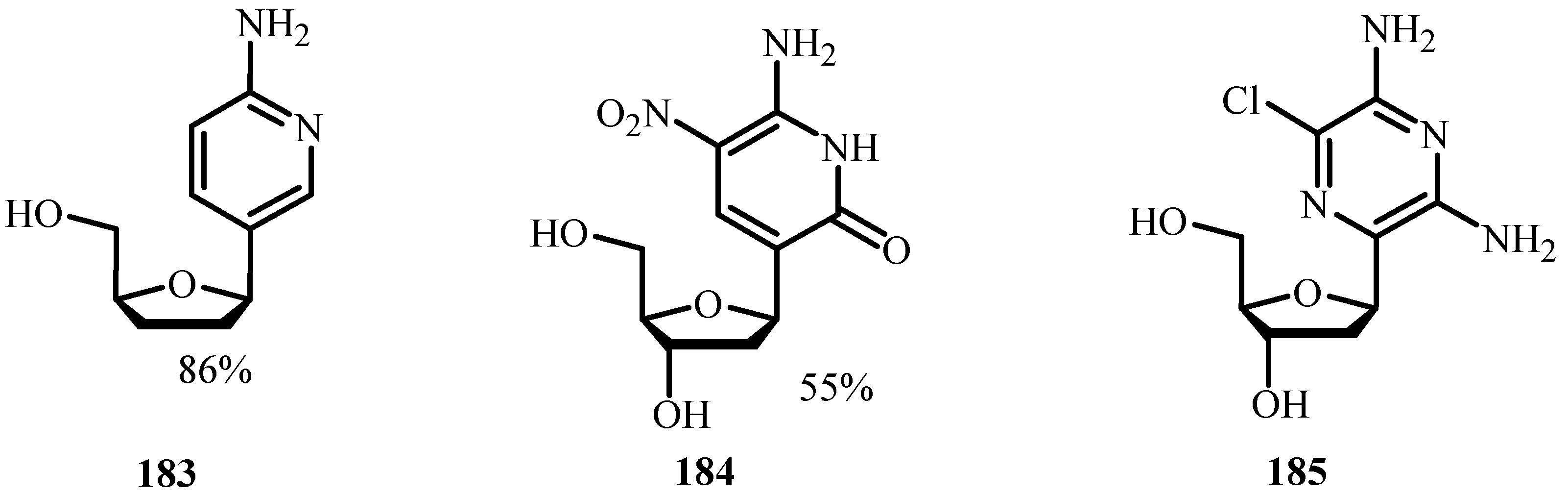
A review by Hervé and Len [65] is based on both Heck and Sonogashira cross-coupling of nucleosides following two important aspects of the green chemistry, i.e., use of aqueous medium and no protection/deprotection steps. It focuses on the study of C5-modified pyrimidines and C7-deaza or C8-modified purines where these chemical modifications have been developed using palladium cross-coupling reactions. The review encompasses variations of the starting materials, alkene and alkyne, nature of the solvent, palladium source and ligand at either room temperature or higher temperature. Heck cross-couplings were performed using Na2PdCl4 (80 mol %) and Pd(OAc)2 (5–10 mol %) in the presence of TPPTS as ligand in a mixture of CH3CN/H2O and in sole water. Using these procedures, yields up to 98% have been reported.
3. Reviews on Mechanism
The mechanism of Heck reaction is discussed and reviewed by many researchers since 1972 when for the first time it was given in complete detail by Heck and Nolley [9]. There are articles that argue the case for having a Pd(0)/Pd(II) mechanism while others favour the Pd(II)/Pd(IV) mechanism based on evidential data. Few discuss the specific intermediate formed during a particular mechanism while others demonstrate the presence of different intermediate. This section deals with reviews on such reviews on mechanism of Heck reaction.
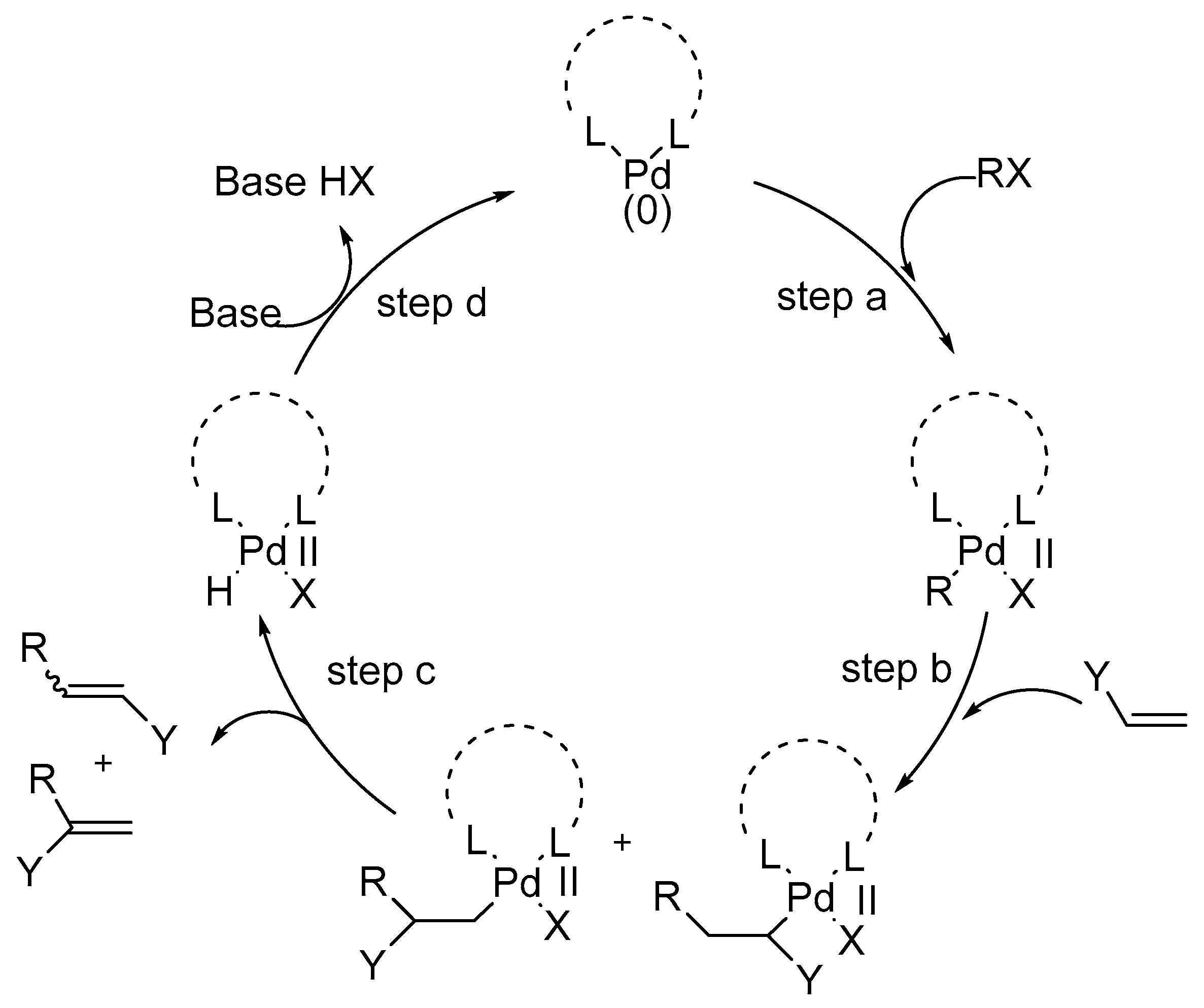
A review by Cabri and Candiani [66] explains the basis of a common mechanistic hypothesis for the coordination-insertion process of unsaturated systems on palladium(I1) complexes based on the results obtained until then by several research groups on the Heck reaction. All of these results are explained by the commonly accepted mechanism based on Pd(0)/Pd(II) cycle (Scheme 5).
This mechanistic model gives a better understanding of the scope and limitations of the Heck reaction. The steps involved in the mechanism of Heck reaction are:
- step (a)
- Oxidative addition
- step (b)
- Coordination-insertion
- step (c)
- β-Hydride elimination-dissociation
- step (d)
- Recycling of the L2Pd(0)
Several factors like leaving groups, neutral ligands, additives (like bases, salts, etc.), and olefin substituents play important roles in overall reaction course via coordination-insertion process as shown in Scheme 6.
It is essential that the insertion process requires a coplanar assembly of the metal, ethylene, and the hydride and hence the insertion process is stereoselective and occurs in a syn manner. In addition, the energy barrier for the generation of the reactive configuration in a tetracoordinated complex is low with respect to a pentacoordinated one. Therefore, pentacoordinated species are possibly not involved in the coordination process. The β-hydride elimination is also stereoselective and occurs in a syn manner. Its efficiency is related to the dissociation of the olefin from the palladium(I1)-hydride complex. It was also observed that, the presence of a base is necessary in order to transform the L2Pd(H)X into the starting L2Pd(0) complex and complete the catalytic cycle.
The mechanism is also supported by the discussion on the regioselectivity and stereoselectivity of Heck reaction. It was observed that the preferential formation of the branched products in the arylation of heterosubstituted olefins like enol ethers, enol amides, vinyl acetate, allyl alcohols, and homoallyl alcohols was independent of the substituents on the aromatic ring, the reaction temperature, and the solvent and is related only to the coordination-insertion pathway, though the stereoselectivity was dependent on the added base. In the intramolecular asymmetric Heck reaction, sometimes the enantioselectivity was related to the geometry of the substrate olefin.
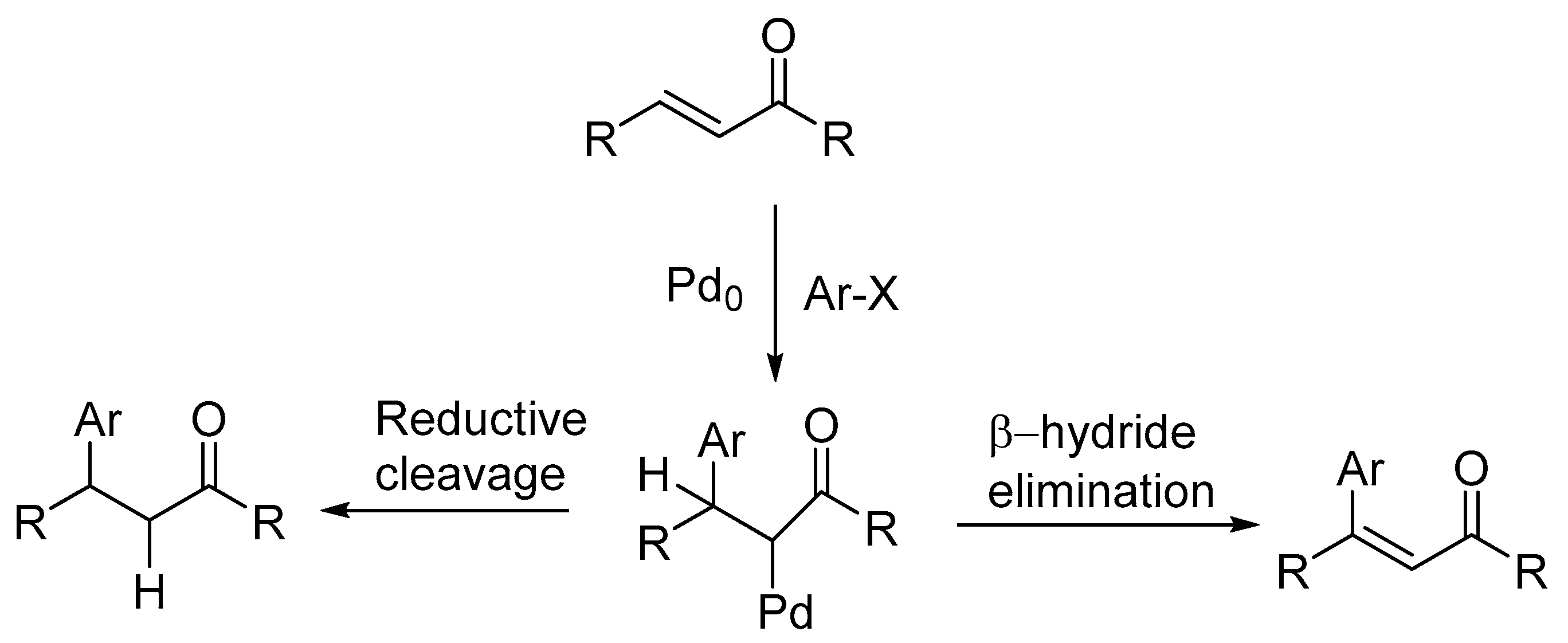
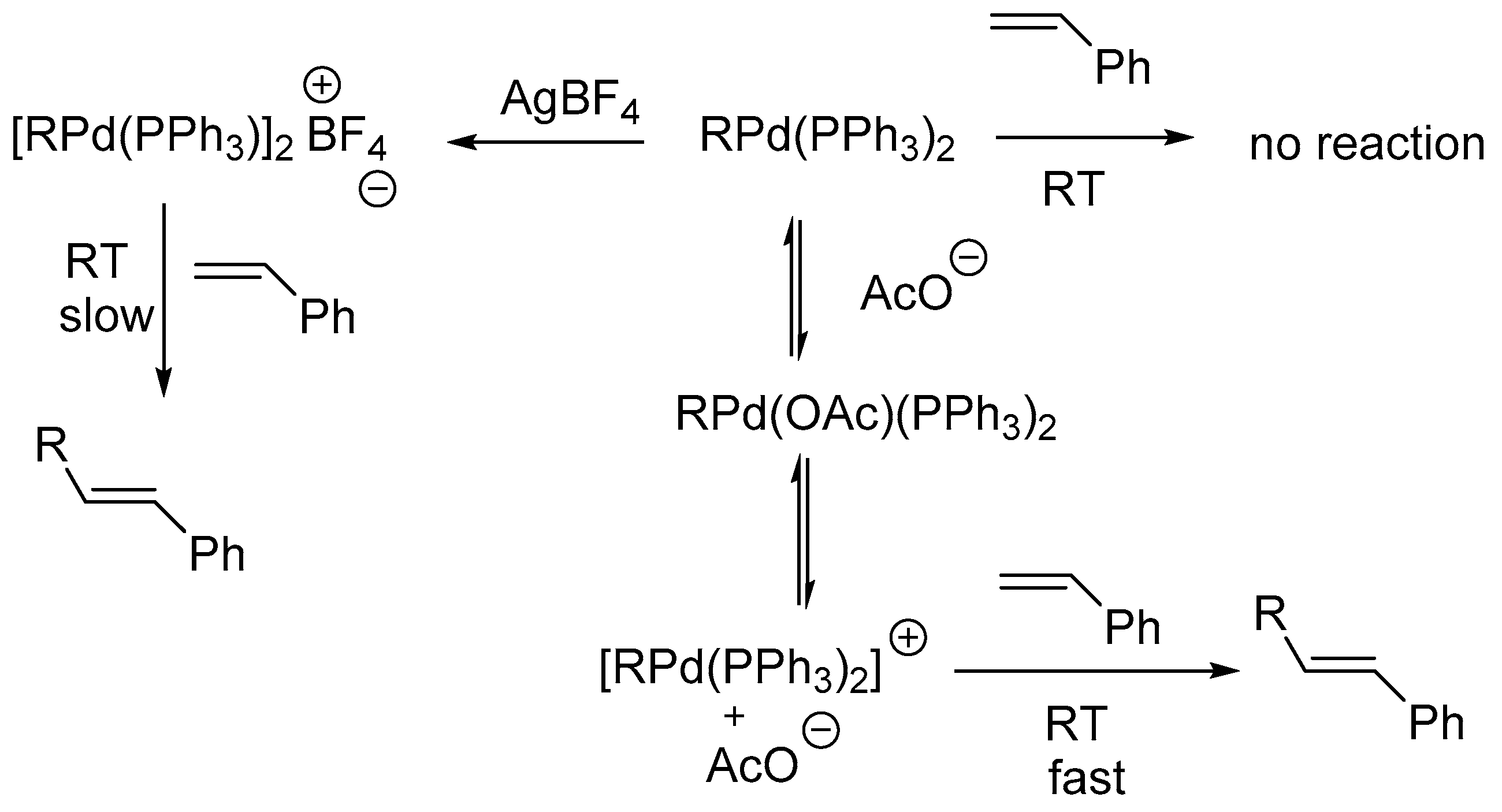
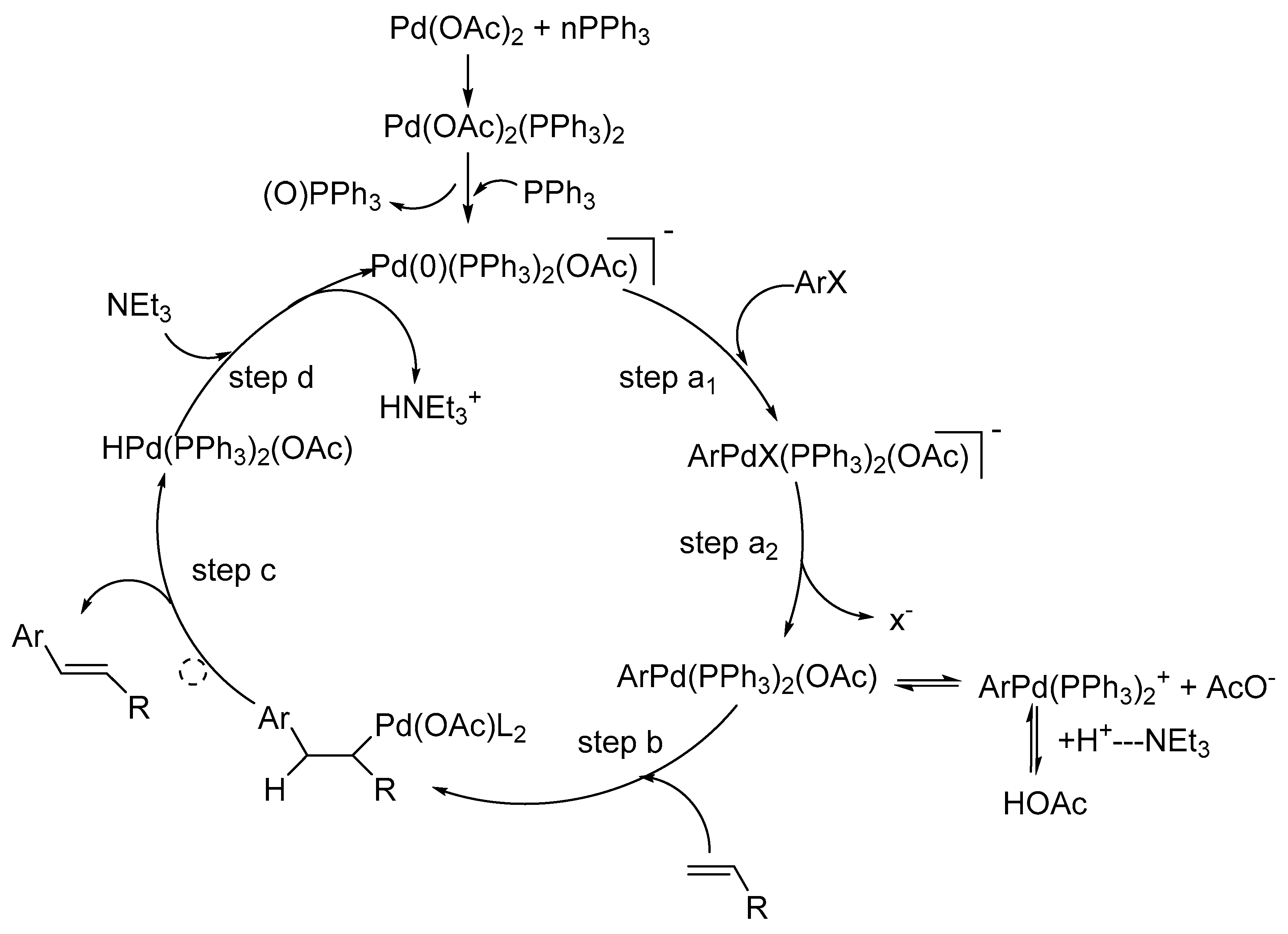
A review by Crisp [67] examines the implications of details on the mechanism of the Heck reaction using both traditional and non-traditional catalytic systems. It is mentioned that, while studying the intermediate formed during oxidative addition of aryl halide to Pd complex, the conventionally thought intermediate ArPdL2X is not produced rather an intermediate ArPd(PPh3)2(OAc) is formed. The formation of this species is also supported by Amator and co-workers [68]. It was observed that the styrene on reaction with preformed PhPd(PPh3)2(OAc) during the carbon–carbon bond forming step of the Heck reaction in DMF at room temperature forms stilbene whereas on reaction with PhPdI(PPh3)2 under similar conditions, stilbene was not formed. Further, when acetate anion was added to a mixture of PhPdI(PPh3)2 and styrene, the formation of stilbene was observed at room temperature. These observations are consistent with the dissociation of acetate ion from PhPd(PPh3)2(OAc) to form an equilibrium mixture containing the cationic complex [PhPd(PPh3)2]+ (Scheme 7).
The observations using various reaction conditions for inter and intramolecular Heck couplings, in presence of different ligands (PPh3/P(o-tolyl)3/Chelating phosphine ligands), bases (organic/inorganic), solvent (polar/nonpolar) and/ or TBAB are cited. In addition to this, the recent modifications to traditional reaction conditions are also reviewed and interpreted that, for intermolecular couplings involving reactive electrophiles (aryl or vinyl iodides) and alkenes containing an electron withdrawing group, a traditional catalyst system such as Pd(OAc)2 with 2–4 equivalents of L [L = PPh3 or P(o-tolyl)3] or PdL2Cl2 or PdL4 along with organic or inorganic base should suffice. Such systems require temperatures in the range 50–100 °C. In order to lower the temperature, the most effective protocol is to add R4NX (X = Cl, Br) and use an aqueous solvent with K2CO3 as the base. For aryl or vinyl triflates with alkenes containing an electron withdrawing group, a traditional catalyst system can also be used. For alkenes not containing an electron withdrawing group, a halide free condition, achieved by either using aryl or vinyl triflates as the electrophile or adding a halide sequestering agent (Ag+) for aryl or vinyl halides, will be advantageous. However, for electrophiles (like aryl bromides with electron donating groups or aryl chlorides) undergoing oxidative addition more slowly, high temperatures (above 120 °C) are usually required and a stable metal-ligand system that does not decompose at higher temperature is essential for prolonged life of the catalyst and hence PPh3 will not be suitable ligand in such cases. For intramolecular Heck cyclizations, the reaction conditions appear to vary depending on the ring size, stereochemistry of the alkene and whether a tertiary or quaternary centre is being formed. The presence of halides does not appear to impede the cyclization at elevated temperatures and can be beneficial for high ee’s in asymmetric Heck couplings. The use of halide free conditions can produce rapid Heck couplings but variable ee’s are seen for asymmetric cyclization.
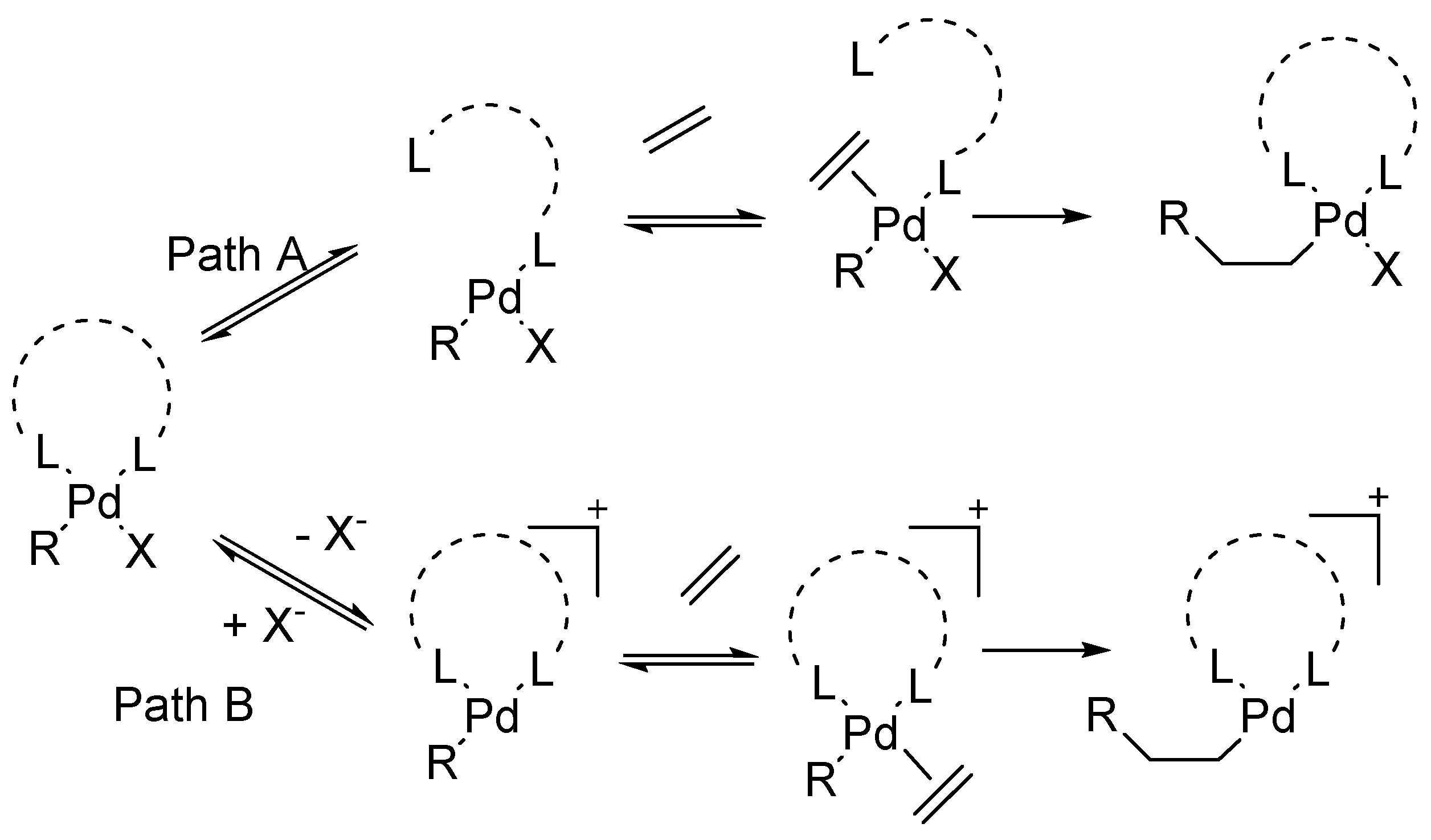
The Amatore group of researchers have invested much effort into investigations of the mechanism of Heck reaction. Kinetic evidences and electrochemical techniques like steady state voltammetry along with spectroscopic techniques like UV (Ultra violet spectroscopy), NMR (Nuclear Magnetic Resonance Spectroscopy) was used for the elucidation of mechanisms of palladium-catalysed reactions [69]. Their work emphasizes the crucial role played by the anions born by the precursors of palladium(0) complexes and rationalize empirical findings dispersed in literature concerning the specificity of palladium catalytic systems. A new adequate catalytic cycle has been proposed for Heck reactions where the fundamental role of chloride or acetate ions brought by palladium(II) complexes, precursors of palladium(0) complexes, is established (Scheme 8).
It is proposed that the new reactive anionic palladium(0) complexes species are formed where palladium(0) is ligated by either chloride ions Pd(0)(PPh3)2Cl− or by acetate ions Pd(0)(PPh3)2(OAc)−. The reactivity of such anionic palladium(0) complexes in oxidative addition to aryl iodides strongly depends on the anion ligated to the palladium(0). The structure of the arylpalladium(II) complexes formed in the oxidative addition also depends on the anion.
The existence of intermediate anionic pentacoordinated arylpalladium(II) complexes ArPdI(Cl)(PPh3)2− 135 and ArPdI(OAc)(PPh3)2− 136 (Figure 19) are also predicted based on evidences. Their stability and reactivity is discussed depending on the presence of chloride or acetate anion. ArPdI(Cl)(PPh3)2− is found to be more stable and it affords trans ArPdI(PPh3)2, whereas, ArPdI(OAc)(PPh3)2− is quite unstable and rapidly affords the stable trans ArPd(OAc)(PPh3)2 complex.
Another review by Amator and Jutand [70] is the resurgence on the mechanism of Heck reaction. It aims at giving the evidences of anionic Pd(0) and Pd(II) intermediates in palladium-catalysed Heck and cross-coupling reactions. It was proved that the anions of PdCl2L2 and Pd(OAc)2, precursors of palladium(0) play a crucial role during the reaction. Thus, Pd(0)L2, postulated as the common catalyst in catalytic systems, is not formed as a main intermediate, instead, previously unsuspected reactive tricoordinated anionic complex species Pd0L2Cl−and Pd0L2(OAc)− are found to be the effective catalysts. These anions also affect the kinetics of the oxidative addition to ArI as well as the structure and reactivity of the arylpalladium(II) complexes produced in this reaction. The pentacoordinated anionic complexes ArPdI(Cl)L2− or ArPdI(OAc)L2− are formed in the reaction.
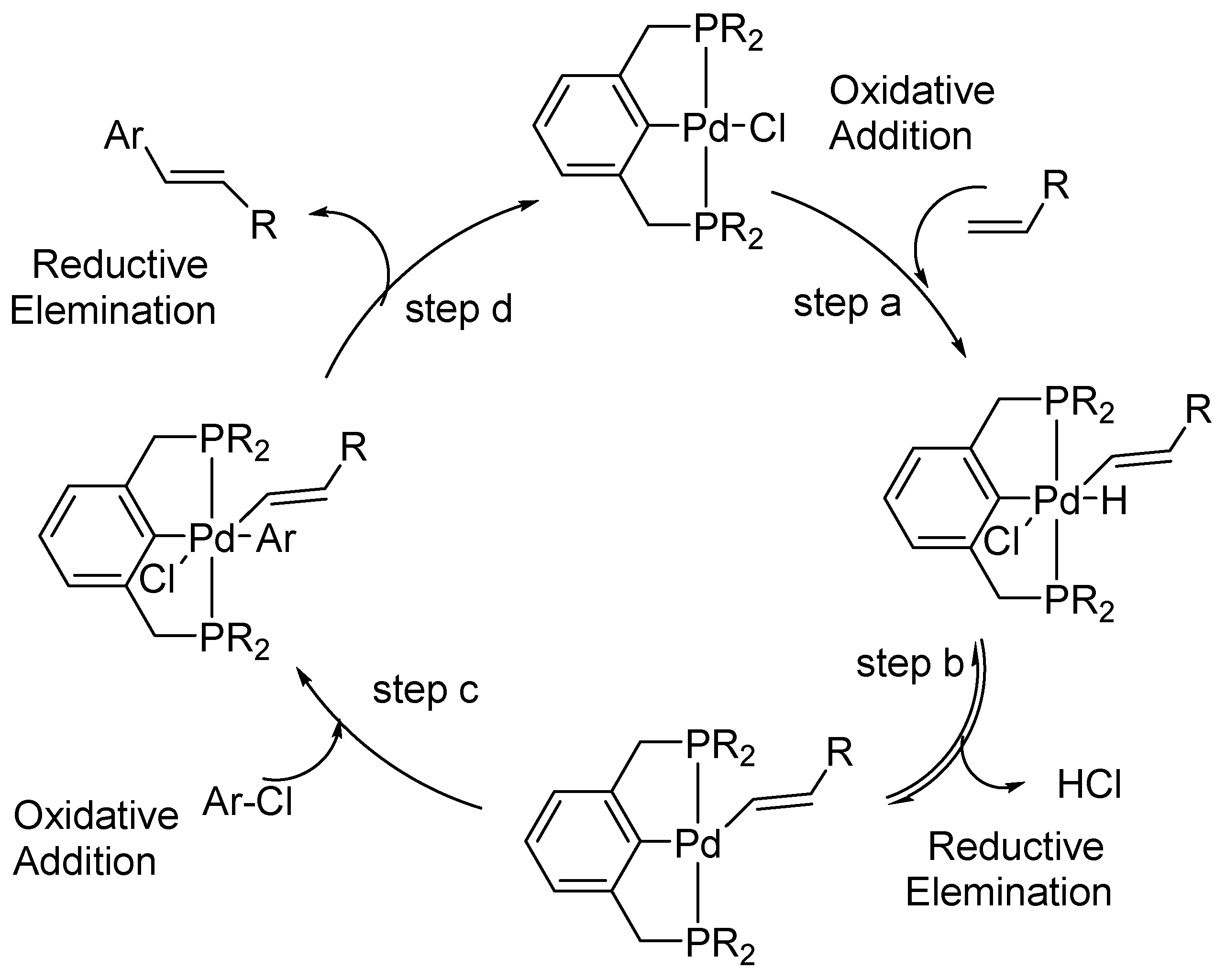
An alternative mechanism via Pd(II)/Pd(IV) cycle (Scheme 9) specially proposed for PCP complexes (pincer palladacycle with Phosphorous as donor ligands) is described in the review by Whitecomb [24]. The initiation of the catalytic cycle takes place via the oxidative addition of a vinyl C-H of CH2 = CHR to the palladium (II) complex (step a). This is followed by the reductive elimination of HCl from the Pd(IV) species (step b) and is a rate determining step to generate Pd(II) species. One more oxidative addition of ArCl (step c) generates another Pd(IV) species collapsing further giving out coupled product via reductive elimination to restore the catalyst (step d). The mechanism is different than a classical Pd(0)/Pd(II) as it involves the successive oxidative addition of both aryl halide and alkene substrates and eliminates the need for the migratory insertion step. Formation of intermediate after a reversible step a, has been supported by NMR studies using deuterium.
Cavell and McGuinness [71] have reviewed the redox process involving N-heterocyclic carbene complexes and associated imidazolium salts in conjunction with Group 10 metals. The mechanistic studies with stoichiometric reaction of the [(tmiy)2Pd(Ar)]+ cation (where Ar = p-nitrophenyl) with butylacrylate was seen to give a complex mixture of products. It was observed that the product distribution was dependent on temperature and other reaction conditions. For example, at −30 °C only the reductive elimination products 2-(4-nitrophenyl)-1,3,4,5-tetramethylimidazolium ion, and traces of the 2-substituted-imidazolium ion were observed. On warming to −20 °C, the Heck coupling product, n-butyl (E)-4-nitrocinnamate, and a further product, 1,3,4,5-tetramethyl imidazolium salt, were observed. At room temperature, the main products were the Heck product and the 1,3,4,5-tetramethylimidazolium salt. This indicates that the raise in temperature leads to the Heck coupling product more rapidly, i.e., the rates of migratory insertion plus β-elimination, as required for Heck catalysis, become more competitive with reductive elimination. When the process is run under catalytic conditions, i.e., base with an excess of p-nitrophenyliodide and butylacrylate at around 120 °C, only the Heck product is observed with high turnover frequencies (TOF) and TON and no products generated from the reductive elimination reaction were observed. This indicates the thermodynamic parameters such as activation barriers and relative exothermicities play a crucial role.
Trzeciak and Ziolkowski [72] have reviewed the catalytic activity of palladium in C-C bond forming processes in respect to Heck and carbonylation reactions. Catalytic systems based on palladium complexes with phosphorus ligands and phosphine-free systems and based on the Pd(0) colloid are discussed by giving a number of examples. These examples reveal that, both monomolecular Pd(0) phosphine complexes and nanosized Pd(0) colloids can act as active catalysts. This review supports the formation of conventional oxidative addition complex PhPdL2X on the basis of evidences. The possibility of typical monomolecular Pd(0) complexes (e.g., [Pd(0)P4], P = phosphorus ligand) undergoing partial decomposition during the catalytic process to form nanosized phosphine-free Pd(0) particles is predicted. Palladium in the form of a colloid was observed to be a good catalyst for Heck and carbonylation reactions, especially when applied in an ammonium salt ([R4N]X) or an ionic liquid (IL) medium. Ammonium salts efficiently contribute to the formation of soluble, monomolecular Pd(0) compounds of probably higher catalytic activity than bigger-sized clusters. The study reveals that carboxylate salts used as bases in the Heck reactions, can act as effective Pd(II) to Pd(0) reducing agents, and therefore they also may initiate the formation of colloidal nanoparticles.
Phan et al. [73] have a critical review on the nature of the active species in palladium catalysed Mizoroki–Heck and Suzuki–Miyaura couplings using homogeneous or heterogeneous catalysis. Regardless of what palladium precatalyst is used, it is critical for researchers to understand the nature of the true catalytic species generally used in their studies because of palladium deactivation via clustering, leaching and redeposition processes. The review summarizes each type of precatalyst of palladium, proposed to be truly active in the various form of catalysts like discrete soluble palladium complexes, solid-supported metal ligand complexes, supported palladium nano- and macroparticles, soluble palladium nanoparticles, soluble ligandfree palladium and palladium-exchanged oxides. A considerable focus is placed on solid precatalysts and on evidence for and against catalysis by solid surfaces vs. soluble species when starting with a range of precatalysts.
Based on various control experiments and tests to assess the homogeneity or heterogeneity of catalyst systems, it was concluded that, the Heck reactions using NC, SC, PC, SCS and PCP palladacycles (catalysts with N, O, P or S donor ligands) operates by Pd(0)/Pd(II) mechanism only and there is no proven example of catalysts operating by a Pd(II)/Pd(IV) catalytic cycle in Heck or Suzuki coupling reactions.
Knowles and Whiting [74] have reviewed advances in the mechanistic perspective of the Heck reaction aiming to review and compare work undertaken on the mechanism until 2007. The data provided give the evidence for a mechanism where the oxidative addition strongly depends on the conditions, particularly whether the reaction is saturated with halide. Eventually, the authors concluded that it is not possible to explain all the phenomena observed for the oxidative addition on the basis of single mechanism only and there may be several mechanisms in operation, either independently, depending on the reaction conditions, or in parallel.
Although the electron rich and chelating nature of the phosphines is required to activate aryl chlorides, it disfavors carbometallation or dissociation. It is also assumed that the rate determining step comes after initial oxidative addition; however, the nature of this step is yet unclear and has a strong dependence on the nature of the species produced in the oxidative addition step.
Kumar and co-workers [75] have reviewed the status regarding in situ generation of palladium chalcogenides phases or Pd(0) protected with organochalcogen fragments when Palladium(II) complexes of organochalcogen ligands (e.g., Pd(II) complex of an (S,C,S) pincer ligand) are used as viable alternatives to complexes of phosphine/carbene ligands for Suzuki−Miyaura and Heck C-C cross coupling reactions. These ligands are thermally stable, easy to handle and air and moisture sensitivity are not impediments with many of them.
Recently, Eremin and Ananikov [76] summarized the studies mentioning the behaviour of a “Cocktail” of catalysts and mechanistic investigations regarding the evolution of transition metal species during catalytic cycles. They have considered the transformations of molecular catalysts, leaching, aggregation and various interconversions of metal complexes, clusters and nanoparticles that occur during catalytic processes termed as “Cocktail”-type systems. These systems are considered from the perspective of the development of a new generation of efficient, selective and re-usable catalysts for synthetic applications. It also attempts to determine ‘‘What are the true active species?” in mechanism specially in coupling reactions like Heck. It has been concluded that there is nothing static in catalysis and catalytic system is always dynamic where ‘‘Cocktail”-type catalytic systems are formed when the transformation of a metal species occurs during the overall catalytic process. It was observed that the “Cocktail” picture can exist not only in homogeneous catalysis in solution but also in a heterogeneous catalytic system.
4. Reviews on Applications
The palladium-catalysed cross-coupling reactions particularly Heck reaction have great significance for both academic and industrial research and in the production of number of compounds on a large industrial scale. Fine chemicals—including pharmaceuticals, agricultural chemicals, and high-tech materials—that benefit society when designed properly in addition to the new reactivities, increased product selectivity and reduced volatile organic consumption, can lead to a great breakthrough in industrial research. Reviews targeting such chemicals are given below.
4.1. Fine Chemical Production
An overview by Blaser et al. [77] describes the industrial application of homogeneous catalysts for the chemical industry. Along with theoretical background of organometallic complexes and homogeneous catalysis, a description of the prerequisites and current problems for their industrial application mainly for manufacture of intermediates for pharmaceuticals and agrochemicals are discussed. Production processes for octyl-4-methoxycinnamate, the most common UVB sunscreen (UVB—Ultra Violet B: These rays penetrate the upper layers of the skin) developed by Dead Sea Company (Scheme 10); Naproxen, a generic analgesic, developed by Albemarle (Scheme 11) and sodium 2-(3,3,3,-trifluoropropyl)- benzenesulfonate, a key intermediate for Novartis’ sulfonylurea herbicide Prosulfuron (Scheme 12) are mentioned, where, Heck reaction is one of the key step in its process, along with their yield and selectivity.
‘The Heck reaction in the production of fine chemicals’ by deVries [78], is an overview mainly focusing the commercial products produced on a scale in excess of 1 ton/year and using Heck or Heck type reaction as one of the steps during synthesis. The synthetic methods for production of a sunscreen agent, 2-ethylhexyl p-methoxy-cinnamate 137; a nonsteroidal anti-inflammatory drug, Naproxen 138; an herbicide, Prosulfuron 139; an antiasthma agent, Singulair 140 and monomers for coatings, divinyltetramethyldisiloxanebisbenzocyclobutene 141 (Figure 20) have been discussed along with the conditions used for their synthesis. Herbicide prosulfuron is synthesized via Matsuda reaction using arenediazonium salts as an alternative to aryl halides and triflates, whereas the production of sunscreen agent 2-ethylhexyl p-methoxy-cinnamate uses Pd/C as catalyst. Naproxen and the monomers are synthesized via bromo derivatives, whereas Heck reaction on allylic alcohol is carried out for the production of antiasthma agent Singulair. The review also talks about the use of metals and ligands as a part of catalyst for industrial production. Although palladium has proved to be the best metal to be used for Heck reaction, from the economic standpoint its cost becomes the biggest hurdle at industrial level.
Similarly, despite the fact that palladacycles, pincers and bulky ligands are robust, they work only on few reactive substrates. Hence ligandless catalysis seems to be attractive for production. In addition, various techniques of recycling of catalyst has been discussed like immobilization of catalysts by attaching ligands to solid support, etc., however, was not found to be useful, because of leaching and reduced activity issues.
A review by Zapf and Beller [79] published in succeeding year gives the importance of several palladium-catalysed reactions like Heck, Suzuki, Sonogashira, telomerization [80] and carbonylation [81], developed and optimized to a stage that enables application on an industrial scale. In addition to Naproxen 138, Prosulfuron 139 and divinyltetramethyldisiloxane-bisbenzocyclobutene 141, use of Heck reaction in the synthesis of L-699,392 142, resveratrol 143, eletriptan 144, cincalcet 145 and montelukast sodium (Salt of Singulair) 146 (Figure 21) by different industries are discussed.
Much later, a review published by Picquet [82] edited by Beller and Blaser presents the state-of-the-art in the industrial use of organometallic or coordination complexes as catalysts for the production of fine chemicals. The review illustrates the great potential of platinum group metal (pgm) complexes as valuable tools in this field for many organic transformations, where these reactions involve platinum group metal complexes as catalysts. Heck reaction for the production of compounds Naproxen 138, Prosulfuron 139, Resveratrol 143, Cinacalcet 145 and Montelukast 146 is discussed. The authors are from the industrial world so were also able to describe the technical challenges encountered in scaling up the reactions from small quantities to production amounts and also tackle issues related to it by taking numerous examples of production processes, pilot plant or bench-scale reactions.
4.2. Pharmaceuticals
A review by Sharma [83] shows the importance of cinnamic acid derivatives (CADs) 147, 148 (Figure 22) for its various biological activities like antioxidant, hepatoprotective, anxiolytic, insect repellent, antidiabetic and anticholesterolemic, etc.
It is mentioned that the various methods for the preparation of cinnamic acid derivatives such as Perkin reaction [84], enzymatic method, Knoevenagel condensation, phosphorous oxychloride method and Claisen–Schmidt condensations has some or the other drawbacks or loopholes like low yield, loss of catalytic activity of enzyme, long duration of reaction time, ozone layer depletion caused by CCl4 and tedious synthetic procedure, etc. Hence, the Heck reaction using various novel supported catalysts is by far the best for the synthesis of cinnamic acid derivatives.
Biajoli et al. [85] have reported the applications of Pd-catalysed C-C cross-coupling reactions for the synthesis of drug components or drug candidates. The review is written in context with two earlier independent reviews by Pfizer researchers Magano and Dunetz on the large-scale applications of transition metal-catalysed coupling reactions for the manufacture of drug components in the pharmaceutical industry [86] and by Torborg and Beller [87] on the applications of Pd-catalysed coupling reactions, both covering the material till 2010. Hence, the period after that until July 2014 is covered in their review.
Cross coupling reactions like Heck, Suzuki, Negishi, Sonogashira, Stille and Kumada are discussed with number of examples. Heck reactions are intensely exploited for inter and intramolecular coupling involving double bonds for the synthesis of a idebenone 149 used for the treatment of Alzheimer’s, Parkinson’s diseases, free radical scavenging and action against some muscular illnesses; ginkgolic acid (13:0) 150, a tyrosinase inhibitor; olopatadine 151 an antihistaminic drug; the vascular endothelial growth factor (VEGF) inhibitor axitinib 152; caffeine-styryl compounds 153 possessing a dual A2A antagonist/MAO-B inhibition properties and with potential application in Parkinson’s disease; styryl analogues of piperidine alkaloids (+)-caulophyllumine B 154 displaying a high anti-cancer activity in vitro; (R)-tolterodine 155, a drug employed in urinary incontinence treatment; ectenascidin 743, the anti-cancer tetrahydroisoquinoline alkaloid 156; bioactive compounds the abamines 157, 158 and naftifine 159 (Figure 23).
Lab scale preparation of three other drugs: cinacalcet hydrochloride 160, alverine 161 and tolpropamine 162 are also discussed using typically Heck-Matsuda reaction (Figure 24). Cinacalcet hydrochloride is a calcimimetic drug commercialized under the trade names Sensipar and Mimpara, and is therapeutically useful for the treatment of secondary hyperthyroidism and also indicated against hypercalcemia in patients with parathyroid carcinoma. Alverine is a smooth muscle relaxant used for the treatment of gastrointestinal disorders such as diverticulitis and irritable bowel syndrome and tolpropamine is an antihistaminic drug used for the treatment of allergies.
4.3. Total Synthesis
The Heck reaction has been used in synthesis of more than 100 different natural products and biologically active compounds. Taxol is the first example where Heck reaction was employed for creating the eight-membered ring during its synthesis. Reviews in this section give glimpses of all such compounds.
Nicolaou et al. have corroborated the role of palladium catalyzed cross-coupling reactions in total synthesis [88]. The review highlights number of selected examples of synthesis using most commonly applied palladium-catalyzed carbon–carbon bond forming reactions including Heck reactions. The involvement of intramolecular Heck reaction in the total synthesis of (±)-dehydrotubifoline 163 with justification based on Jeffery modification [89] for the previously formed unwanted derivative has been discussed. Synthetic routes for (−)-quadrigemine C 164, Δ9(12)-capnellene-3β,8β,10α-triol 165, estrone 166, taxol 167, Δ9(12)-capnellene-3β,8β,10α,14-tetraol 168, (+)-calcidiol 169, Okaramine N 170, α-tocopherol 171 (Figure 25), scopadulcic acid-B 124 and singulair 140 are given. Few examples of zipper polycyclization 172 developed by Trost group [90,91] are also mentioned.
All these processes are impressive since they do not require the preparation of reactive intermediates prior to the carbon–carbon bond forming event since they proceed by activation of stable and readily available starting materials in situ and, therefore, are both more practical and often more efficient in terms of overall yield.
4.4. Targeted Compounds
In addition to all above distinctive natural products, Heck reaction is also used in the production of some particular compounds such as aryl glycosides or heterocycles. A review by Wellington and Benner [92] is about using Heck reaction for the synthesis of aryl glycosides (also termed as C-glycosides or C-nucleosides) though there are many reports of synthesis of such compounds via non-Heck methods. Synthesis of glycosides pyrimidine c-nucleosides, purine c-nucleosides, monocyclic, bicyclic, and tetracyclic C-nucleosides 173–185 (Figure 26) via Heck reaction are discussed.
The review describes the subsequent conversion with respective reagents and conditions of the Heck products corresponding to target molecules and their application. Pd(OAc)2-AsPh3 was observed to be serving well for most of the synthesis although few other palladium-ligand combinations have to be used in some cases. Iodides were found to be more reactive than other halides. Bidentate ligand accelerates the Heck coupling reaction and results in a higher yield of the product.
A review by Zeni and Larock [93] covers a wide range of palladium catalyzed processes involving oxidative addition- reductive elimination chemistry developed to prepare heterocycles, with the emphasis on fundamental processes used to generate the ring systems themselves. A number of examples are given for preparation of heterocycles via intramolecular Heck cyclization of aryl halides, vinylic halides, vinylic and aryl triflates. intermolecular annulation, and via asymmetric Heck cyclization.
5. Reviews on Reuse of Catalyst
Homogeneous catalysts are of great interest for synthesizing fine-chemical, specialty chemical, pharmaceutical products for their advantages of high activity and selectivity. However, the main disadvantages of traditional organic phase reactions employing homogeneous transition metal catalysts are the difficulties associated with separating the catalyst from the product and solvent albeit having less aggressive reaction conditions and increased selectivity. Hence, it is important to discuss the catalyst product separation techniques for heck reactions. This section aims to provide an overview of the current reviews on the separation/recycling methods of homogeneous transition metal catalysts.
5.1. Recapitulation of All Methods
A succinct review by Bhanage and Arai [94] describes all the various catalyst-product separation methodologies developed for Heck reactions until 2001. The review gives information about the various catalyst-product separation methodologies developed for Heck reactions until then and is illustrated with number of examples. The separation techniques are classified into two major categories: heterogeneous catalysts and heterogenized homogeneous catalysts.
Heterogeneous catalysts can easily be removed by physical separation methods. In addition, they are stable at higher temperatures and hence become interesting for the activation of less reactive but less expensive chloroaryls substrates. However, the heterogeneous catalysts have a major drawback of poor selectivity toward Heck coupling products.
Heterogeneous catalysts include
- Conventional supported metal catalysts such as Pd/C, Pd/SiO2, Pd/Al2O3 and Pd/BaSO4 and also nonpalladium based catalysts like Ni/HY zeolite, Ni/Al2O3, Cu/Al2O3 and Co/Al2O3. However most of them end up in leaching of metal and hence making it difficult to do effective recovery and recycling of the active metal.
- Zeolite-encapsulated catalysts include palladium-grafted mesoporous MCM-41, Pd-TMS11, etc. However, depending on zeolite structure the leaching of active Pd species was observed with this type of catalyst as well.
- Colloids–nanoparticles primarily palladium based are reported showing high activity.
- Intercalated metal compound,s e.g., palladium-graphite type catalyst, Pd-chloride- and Cu-nitrate-intercalated montmorillonite K10 clays, etc., are reported to be active for Heck reaction.
The heterogenized metal complex catalysts operate under relatively mild conditions as compared with heterogeneous catalysts, and hence can be applied to the production of pharmaceuticals and fine chemicals. The homogeneous metal complexes are heterogenized using following strategies:
- Modified silica catalysts where metal is adsorbed onto a silica support.
- Polymer-supported catalysts, where Pd anchored to phosphinated polystyrene.
- Biphasic catalysis, another exciting area of environmentally responsible catalysis. This system uses two liquid phases such that the catalyst exists in one phase while reactants and products are present in the other phase. The maximum studied catalyst of this category is sulphonated triphenylphosphine like 115, 130 complexed mainly with palladium although few other metals are also known to react. This catalyst has proven its efficiency for many other reactions too.
- Supported liquid-phase catalysts, where the catalyst-containing phase is dispersed as a thin film on a hydrophilic support like silica and is used in another immiscible solvent leading to the enhancement in reaction rate.
- NAILS also known as molten salts provide a medium in which the catalyst is generally dissolved allowing the product to be easily separated. The catalyst and ionic liquid can then be recycled.
- Perfluorinated solvents uses fluorous phase and perfluorinated ligand based organometallic catalyst and is segregated from reagents and products, either during the process or during the workup.
In addition to all above, recyclable homogeneous complexes (where they are recovered by solvent precipitation), catalysts giving High TON (as even after non-recovery of the catalyst it becomes affordable) and supercritical solvents are discussed.
5.2. Heterogeneous Catalysts
A review by Wall et al. [95] mainly focuses discussion on the Heck reaction followed by review on cinnamic acid synthesis using heterogeneous catalyst, Pd/C in particular. Progress made until then, mechanism and the conditions required for successful Heck reaction are discussed including the factors contributing to electronic control for α- or β-arylation. Limitations to the homogeneous reaction along with possible reasons and need for proceeding with heterogeneous catalysts, although the mechanism for which is not known are mentioned by giving a number of examples. Their efforts in the field of development of methods for the production of cinnamic acids, used as substrates for the synthesis of unnatural amino acids by employing phenylalanine ammonialyase are given.
Heerbeek et al. [96] have reviewed the use of dendrimers as support for recoverable catalysts and reagents. Phosphonated DAB-dendr-[N(CH2PPh2)2]16 dendrimer dimethylpalladium complex 186 (Figure 27) is the first such successful dendritic catalyst to recover through a precipitation procedure where no metallic palladium was observed unlike monomeric catalysts and was recycled however with lesser yields in successive runs.
Solid supports have become valuable tools for catalysts immobilization nowadays for simplified product isolation and catalyst recycling. Horn et al. [97] reviewed such non-covalently solid-phase bound catalysts for organic synthesis. Immobilization of catalysts by non-covalent bonding like hydrogen bridges, or ionic, hydrophobic or fluorous interactions on solid support is found to be an alternative to covalent attachment. Such non-covalent approaches increase the flexibility in the choice of the support material, reaction conditions and work-up strategies compared to covalent attachment. Numerous catalytic reactions employing one of these non-covalent bonding strategies are given along with the examples of Heck reaction with catalyst immobilization by ionic interactions.
The problems, potential and recent advances with general approach in heterogeneously catalyzed Heck reactions especially with supported palladium catalyst such as Pd on activated carbon, oxides, polymers and in zeolites has been reviewed by Kohler et al. [98]. The advantages and limitations for practical applications are considered. A thorough literature for less activated or deactivated compounds is discussed along with the new approaches and strategies for the activation of such compounds by heterogeneous catalysts. Particular attention is given to the relation between homogeneous and heterogeneous catalysis from the mechanistic point of view. It is concluded that palladium species dissolved from the support are proven to be responsible for high activity and selectivity in Heck reactions in supported catalysis. The careful choice of optimum catalyst and reaction conditions can activate even deactivated aryl chlorides with high yields within few hours of reaction time.
Polshettiwar and Molnar [99] have reviewed the Heck coupling with silica supported catalyst and palladium redeposition. Mechanisms for these catalysts considering both less common Pd(II)/Pd(IV) catalytic cycle and the most accepted Pd(0)/Pd(II) cycle are given. A number of examples are cited for the Heck reaction in presence of silica-supported Pd complexes and the structural investigations of functionalized mesoporous silica-supported Pd catalysts. New emerging fields like sol–gel entrapped silica–Pd catalysts, nanosized Pd particles embedded in silica catalysts, colloid palladium layer–silica catalysts, silica/ionic liquid Pd catalysts and silica-supported Pd–TPPTS liquid-phase catalysts are also discussed along with testing of palladium leaching by poisoning test using pyridine and mercury.
Another review on this topic by Molnar [100] gives an account of efficient, selective, and recyclable palladium catalysts in carbon-carbon coupling reactions. The review gives a comprehensive survey, thorough analysis of the available data and discusses critically the questions related to recyclability in general. Publications reporting a stable and well characterized catalyst with high yield and minimum five recycles are cited. It is reported that, both amorphous and ordered silica materials are useful supports for palladium nanoparticles. The combined use of varied ionic liquids and related homogeneous complexes has shown interesting results. The best performance with respect to cumulative TON numbers are reported for catalyst Pd-SBA, 3.9% Pd-HS-Si(HIPE), and 4.1% Pd-grHSSi(HIPE) (HIPE = high internal phase emulsion). Thus, the catalytically active species, Pd particles and immobilized species, or the support material employed may play the decisive role in efficient catalysis.
5.3. Heterogenization of Homogeneous Catalysts
The development of new, highly efficient heterogenized catalysts is an active and important area in fine chemicals production research as it gives the maximum advantages of both homogeneous and heterogeneous catalysis. It offers several significant practical advantages for synthetic chemistry and industrial research. Reviews about various techniques used for heterogenization are given in following subsections.
5.3.1. Ionic Liquid
These days, ionic liquids are proposed as greener alternative to the classical cross-coupling procedure. They have replaced organic solvents in many metal catalyzed reactions and have gained much attention in recent times mainly because they lack the vapour pressure. However, several factors like toxicity, stability, cost, ease of processing, etc., still need to be addressed before this new technology is to be accepted by industry.
A review by Sheldon [101] is one of the finest written reviews on ionic liquids. Discovery of ionic liquids, historical development and progression for various types of reactions in them until 2001 are summarized eloquently. Scientists and industrial researchers are seriously considering ionic liquids as better replacement for toxic and/or hazardous volatile organic compounds and consider that, the use of ionic liquids as novel reaction media may offer a convenient solution to both the solvent emission and the catalyst recycling problem. Ionic liquids also provide a medium for performing clean reactions with minimum waste generation. It was observed that use of ionic liquids (Figure 28) as reaction media for catalytic transformations or, in some cases, as the catalyst itself can have a profound effect on activities and selectivities.
The first example of a Heck coupling in an ionic liquid was reportedly carried out in 1996, where in ionic liquids tetraalkylammonium and tetraalkylphosphonium bromide salts, high yields for Heck reaction were obtained. No leaching of palladium was observed, the product was isolated by distillation from the ionic liquid and the ionic liquid was recycled for two more times. Similarly, ionic liquids like Bu4NBr, bmimPF6 and n-hexylpyridinium PF6 are also reported to give improved activity for Heck reaction. In comparison to pyridinium analogues, the imidazolium ionic liquids were found to have greater catalytic activity when the reactions performed in it. The selectivity of α over β—product for the Heck arylation of electron-rich enol ethers in ionic liquids was found to be >99% in comparison with other solvents generally leading to a mixture of regio isomers owing to competition between cationic and neutral pathways.
Singh et al. [102] reviewed the use of ionic liquids medium for many palladium catalyzed reactions like Heck, Suzuki, Stille, Negishi, Trost–Tsuji and Sonogoshira coupling. A comprehensive literature about the versatility of ionic liquid in conjunction with palladium for these reactions is cited. The studies have shown that the classical transition-metal catalyzed reactions can be performed in ionic liquids. They are supposed to be a medium for clean reactions with minimal waste and efficient product extraction. In addition to the basic ionic liquids discussed by Sheldon [101] ionic liquids specially introduced after 2001 are discussed in detail. Apart from regularly studied ionic liquids, results of few specially studied ionic liquids using palladium catalyst such as functionalized ionic liquids like [bmim][TPPMS] and [bmim][OAc]; N-butyronitrile pyridinium bis(trifluoromethylsulphonyl) imide with PdCl2; palladium acetate immobilized on amorphous silica with the aid of an ionic liquid [Bmim][PF6]; palladium(II) complex from pyrazolylfunctionalized hemilabile NHC; Pd(OAc)2/[PEG-mim][Cl], Pd/C-catalyzed Heck reaction in ionic liquid and Pd(0) nanoparticles (~2 nm diameter), immobilized in [bmim][PF6] are also discussed. Most of them showed reasonable efficiency in recycle studies. Pd-nanoparticles were found to be much more efficient in TBAB than in pyridinium, phosphonium and imidazolium ionic liquids in catalyzing the Heck reactions. Few reports of reactions in ionic liquids using ultrasonic irradiation and microwaves are also mentioned to show better activity.
Bellina and Chiappe [103] have reviewed the progress and challenges for the Heck reaction in ionic liquids. Developments in the application of ionic liquids and related systems like supported ionic liquids; ionic polymers, etc., in the Heck reaction have been reviewed. Merits and achievements of ionic liquids were analysed and discussed considering the possibility of increasing the effectiveness of industrial processes. Numerous examples for homogeneous and heterogeneous conditions used for Heck reactions in ionic liquids are discussed to prove that, the ionic liquids are not only suitable solvents for Heck reaction, but their unique physico-chemical properties have ability to change the course of the reaction, activate and/or stabilize the intermediates or transition states in the reaction mechanisms. It is observed that Heck reactions performed in ionic liquids have higher reaction rates, and may be characterized by a higher control on the regio- and stereoselectivity of the coupling products. Examples with involvement of carbene complex, palladium nanoparticles and palladium anionic complexes have been discussed for the evidence. However, the study of Heck reactions in ionic liquid is still limited to the development of the ionic liquids where only simple reactions such as the coupling of aryl halides with cinnamates or styrene have been investigated. Thus, the use of ionic liquids in Heck reactions involving more complicated substrates that could give important indications about possibility of application of these alternative media on large scale need to be explored further.
Mastrorilli et al. [104] have surveyed the significant developments and the beneficial effect of the ionic liquids in terms of activity, selectivity and recyclability for palladium-catalyzed cross-coupling reactions performed in them. Insights into the reaction mechanisms reveal that, the effect of the ionic liquid on C-C bond forming reactions manifests itself not only in the energy lowering of polar transition states (or intermediates), involved in the catalytic cycles but also, depending on the cases, in the stabilization of palladium nanoparticles, in the synthesis of molecular Pd complexes with the ionic liquid anions or in the enhancement of the chemical reactivity of reactants, etc. The synergistic effect found by using appropriate mixtures of ionic liquids is also discussed.
Santos et al. [105] have reviewed Heck-Mizoroki reactions in ionic liquids, reported to be a possible green alternative to the classical cross-coupling procedure. In this review, the different approaches and achievements on the use of ionic liquids as solvents in Heck-Mizoroki coupling reactions is revisited along with a brief reference to supported ionic liquids. The results show that ionic liquids are able to modify the reaction course and activate and stabilize intermediates or transitions states. However, the examples show that, though there are large numbers of studies in this topic, most of the works involve the coupling of simple aryl halides and olefins and hence we expect more research on the use of ionic liquids in Heck reactions that include more complicated substrates affording environmentally benign chemical processes in the future.
Recently, Limberger et al. [106] have published a review on charge-tagged ligands, where there is an insertion of an ionic side chain into the molecular skeleton of a known ligand. This technique has become a useful protocol for anchoring ligands, and consequently catalysts, in polar and ionic liquid phases. The ionic modification confers a particular solubility profile making catalyst/product recovery possible, and often improving the activity of the catalytic species compared to the parent tag-free analogue. Usability of charge-tagged ligands is increasing and it has become a valuable tool in organometallic catalysis because in this, water can be used as an anchoring medium, thereby combining sustainability and efficiency. It is predicted that this technique can also be used to detect reaction intermediates in organometallic catalysis where the insertion of an ionic tag ensures the charge on the intermediates. Hence, these ligands have been used as ionic probes in mechanistic studies for several catalytic reactions. This review summarizes the selected examples on the use of charge-tagged ligands (CTLs) 187–190 (Figure 29) as immobilising agents in organometallic catalysis and as probes for studying mechanisms through ESI-MS (electrospray ionisation mass spectrometry). In these CTLs, a coordinating imidazole, pyrazole or pyridine group was attached to either C2 or C1 of the imidazolium moiety. These CTLs with palladium salts were found to be active for Heck reaction with the yields ranging from 70 to 99%. Moreover, it could be recycled several times and they can also be used as the solvent and ligand to stabilize the palladium species in Heck reaction.
5.3.2. Catalysis in Multiphase Systems and Supercritical Carbon Dioxide
Bhanage et al. [107] have reviewed the multiphase Heck reactions using various types of catalysts. The multiphase Heck systems are prepared by using different types of catalyst phases, including biphasic catalysis, supported liquid phase catalysts and solvent of supercritical carbon dioxide which replaces the conventional organic solvents. These solvent systems were found to be beneficial towards easy catalyst-product separation and catalyst recycling. Supercritical carbon dioxide can serve as a better solvent system for Heck reaction under homogeneous, heterogeneous and multiphase systems provided the problem of leaching is solved.
A review by Skouta [108] presents an overview of some selected metal-catalysed chemical reactions developed in supercritical carbon dioxide, water, and ionic liquids. Heck reaction was found to give 55% yield of the coupling products in average when was performed in super critical CO2. The water-soluble phosphine compounds m-TPPTC [tris(m-carboxyphenyl) phosphine trilithium salt], and p-TPPTC 191 (Figure 30) in addition to sulphonated phosphines like TPPTS 115, etc., were efficient in organo-aqueous palladium-catalysed Heck reactions, and the excellent results obtained are presumably because of the steric and electronic effects of the carboxylic group in meta-position.
5.3.3. Catalysis in Continuous Flow
Gürsel et al. [109] gives an overview on the separation/recycling methods for homogeneous transition metal catalysts in continuous flow on the lab- and industrial scale by methods like heterogenization, scavenging, using biphasic systems and organic solvent nanofiltration. There are numerous successful demonstrations on the laboratory scale and industrial scale. Examples of Heck reaction using supported ionic liquid phase (SILP) with compressed CO2 as the flowing medium in continuous flow; use of ionic liquid as the recycling reaction medium in an automated microreactor system catalysed by Pd catalyst immobilized in ionic liquid phase; in situ separation of the catalyst with the membrane within the reactor/separator unit, etc., are mentioned. All these methods have the advantage of ease of separation and low energy requirement compared to classical separation methods like distillation.
6. Summary
The Heck reaction has proved to be a remarkably robust and efficient method for carbon–carbon bond formation and remains a flourishing area of research. The enthusiasm and rational optimism of researchers promotes new discoveries resulting in better and efficient catalysts production and providing solutions for old problems. With the use of such reactions, many protection-deprotection procedures during organic synthesis may be reduced so as to reduce the number of steps when designed systematically in addition to the new reactivities, increasing product selectivity and reducing volatile organic consumption. The great functional group tolerance of palladium makes Heck reactions possible on even the most sensitive substrates. In addition, the intramolecular Heck reaction is an incredibly powerful method for the construction of many compounds with quaternary and/or asymmetric carbon centers. The well accepted mechanism for Heck reaction is based on intermediate species involving Pd(0)/Pd(II), however the feasibility of the Pd(II)/Pd(IV) mechanism is not ruled out by many. The Pd(II)/Pd(IV) mechanism is believed to take place when a Pd(0) to Pd(II) conversion is not available clearly or when the Pd(II) species cannot readily undergo β-hydride or reductive elimination. The prospects for application of Heck reaction for many transformations look very good via homogeneous, heterogeneous and even with heterogenized catalysis using organometallic complexes. Some of them are already scaled at an industrial level. Ironically, although a large number of metal-based catalysts are described in the literature promoting Heck reaction for the manufacture of bulk chemicals, when these reactions need to be applied for the synthesis of complex molecules like pharmaceuticals, agrochemicals or fragrances production, Pd(OAc)2, Pd2(dba)3, Pd/C, PdCl2L2, Pd(PPh3)4 and PdCl2(dppf)2 still happen to be catalysts of choice.
Ample study has been made and yet a lot remains to be done. There are several challenges and expectations from the research community that need to be overcome in order to have an economic, robust and reliable industrial process, such as:
- A chase for catalyst precursor and ligands allowing high turnover numbers (TON) and turnover frequencies (TOF), making the process economically attractive, is still a challenge.
- Scaling from laboratory to production level needs a better and a cheaper catalyst precursor and ligands.
- Preparation of air and moisture stable catalyst, since the reactions easily get poisoned by molecular oxygen. In fact, preparation of such a catalyst that works better in aqueous media too.
- Standardizing the reaction conditions that are mild and also allowing lower catalyst loadings.
- Development of new generation of palladacycles promising to be excellent in future applications in industrial processes.
- Understanding of mechanism of heterogeneous catalysis is still rhetoric.
- Increased use of cheaper and easily available starting materials, especially chloroarenes and chloroalkenes, is a niche and much is needed to be done as far as finding newer protocols, along with catalyst and ligand design is concerned.
- Understanding the reasons and thereby successfully prohibiting the precipitation of metal.
- Development of the catalyst where cheaper first-row transition elements are used would truly add to the applicable research.
- Improvements in catalyst efficiency along with catalyst recycling, especially for use in pharmaceutical and other fine chemical synthetic processes.
- Solving the issue of contamination of the product at the end with palladium that needs further purification steps.
Thus, the ever-growing expansion of this coupling reaction is in the phase of new outlook that surely will enable more impressive accomplishments in the future. The useful and practical catalytic systems allowing the Heck reactions involving cheaper transition metals or the easy recovery of metal especially palladium will emerge by further research. This review aims at the stimulation of advance work in these directions.
Conflicts of Interest
The author declares no conflict of interest.
References
- Heck, R.F. Acylation, methylation, and carboxyalkylation of olefins by group VIII metal derivatives. J. Am. Chem. Soc. 1968, 90, 5518–5526. [Google Scholar] [CrossRef]
- Miyaura, N.; Yamada, K.; Suzuki, A. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and brompyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar] [CrossRef]
- King, A.O.; Okukado, N.; Negishi, E. Highly general stereo-, regio-, and chemo-selective synthesis of terminal and internal conjugated enynes by the Pd-catalyzed reaction of alkynylzinc reagents with alkenyl halides. J. Chem. Soc. Chem. Commun. 1977, 683–684. [Google Scholar] [CrossRef]
- Tamao, K.; Sumitani, K.; Kumada, M. Selective carbon-carbon bond formation by cross-coupling of Grignard reagents with organic halides. Catalysis by nickel-phosphine complexes. J. Am. Chem. Soc. 1972, 94, 4374–4376. [Google Scholar] [CrossRef]
- Milstein, D.; Stille, J.K. A general, selective, and facile method for ketone synthesis from acid chlorides and organotin compounds catalyzed by palladium. J. Am. Chem. Soc. 1978, 100, 3636–3638. [Google Scholar] [CrossRef]
- Trost, B.M.; Fullerton, T.J. New synthetic reactions. Allylic alkylation. J. Am. Chem. Soc. 1973, 95, 292–294. [Google Scholar] [CrossRef]
- Mizoroki, T.; Mori, K.; Ozaki, A. Arylation of olefin with aryl iodide catalyzed by palladium. Bull. Chem. Soc. Jpn. 1971, 44, 581. [Google Scholar] [CrossRef]
- Heck, R.F.; Nolley, J.P. Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides. J. Org. Chem. 1972, 37, 2320–2322. [Google Scholar] [CrossRef]
- Heck, R.F. New applications of palladium in organic syntheses. Pure Appl. Chem. 1978, 50, 691–701. [Google Scholar] [CrossRef]
- Heck, R.F. Palladium-catalyzed reactions with olefins. Acc. Chem. Res. 1979, 12, 146–151. [Google Scholar] [CrossRef]
- Heck, R.F. Palladium catalysed vinylation of organic halides. Org. React. 1982, 27, 345–390. [Google Scholar] [CrossRef]
- De Meijere, I.A.; Meyer, F.E. Fine feathers make fine birds: The Heck reaction in modern garb. Angew. Chem. Int. Ed. Engl. 1994, 33, 2379–2411. [Google Scholar] [CrossRef]
- Kobetić, R.; Biliškov, N. The Heck reaction—A powerful tool in contemporary organic synthesis. Chem. Ind. 2007, 56, 391–402. [Google Scholar]
- Sahu, M.; Sapkale, P. A review on palladium catalyzed coupling reactions. Int. J. Pharm. Chem. Sci. 2013, 2, 1159–1170. [Google Scholar]
- Beletskaya, I.P.; Cheprakov, A.V. The Heck reaction as a sharpening stone of palladium catalysis. Chem. Rev. 2000, 100, 3009–3066. [Google Scholar] [CrossRef] [PubMed]
- Hillier, A.C.; Grasa, G.A.; Viciu, M.S.; Lee, H.M.; Yang, C.; Nolan, S.P. Catalytic cross-coupling reactions mediated by palladium/nucleophilic carbene systems. J. Organomet. Chem. 2002, 653, 69–82. [Google Scholar] [CrossRef]
- Herrmann, W.A. N-heterocyclic carbenes: A newconcept in organometallic catalysis. Angew. Chem. Int. Ed. 2002, 41, 1290–1309. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Öfele, K.; Preysing, D.V.; Schneider, S.K. Phospha-palladacycles and N-heterocyclic carbene palladium complexes: Efficient catalysts for C-C-coupling reactions. J. Organomet. Chem. 2003, 687, 229–248. [Google Scholar] [CrossRef]
- Beletskaya, I.P.; Cheprakov, A.V. Palladacycles in catalysis—A critical survey. J. Organomet. Chem. 2004, 689, 4055–4088. [Google Scholar] [CrossRef]
- Zafar, M.N.; Mohsin, M.A.; Danish, M.; Nazar, M.F.; Murtaza, S. Palladium catalyzed Heck-Mizoroki and Suzuki–Miyaura coupling reactions. Russ. J. Coord. Chem. 2014, 40, 781–800. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Böhm, V.P.; Reisinger, C.P. Application of palladacycles in Heck type reactions. J. Organomet. Chem. 1999, 576, 23–41. [Google Scholar] [CrossRef]
- Farina, V. High-turnover palladium catalysts in cross-coupling and Heck chemistry: A critical overview. Adv. Synth. Catal. 2004, 346, 1553–1582. [Google Scholar] [CrossRef]
- Whitecombe, N.I.; Hii, K.K.M.; Gibson, S.E. Advances in the Heck chemistry of aryl bromide and chlorides. Tetrahedron 2001, 57, 7449–7476. [Google Scholar] [CrossRef]
- Littke, A.; Fu, G. Palladium-Catalyzed Coupling Reactions of Aryl Chlorides. Angew. Chem. Int. Ed. 2002, 41, 4176–4211. [Google Scholar] [CrossRef]
- Zapf, A.; Beller, M. The development of efficient catalysts for palladium-catalyzed coupling reactions of aryl halides. Chem. Commun. 2005, 28, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Franzén, R. The Suzuki, the Heck, and the Stille reaction—Three versatile methods for the introduction of new C-C bonds on solid support. Can. J. Chem. 2000, 78, 957–962. [Google Scholar] [CrossRef]
- Biffis, A.; Zecca, M.; Basato, M. Palladium metal catalysts in Heck C-C coupling reactions. J. Mol. Catal. A Chem. 2001, 173, 249–274. [Google Scholar] [CrossRef]
- Cini, E.; Petricci, E.; Taddei, M. Pd/C Catalysis under Microwave Dielectric Heating. Catalysts 2017, 7, 89. [Google Scholar] [CrossRef]
- Barnard, C. Palladium-catalysed C-C Coupling: Then and now. Platinum Met. Rev. 2008, 52, 38–45. [Google Scholar] [CrossRef]
- Corbet, J.-P.; Mignani, G. Selected patented cross-coupling reaction technologies. Chem. Rev. 2006, 106, 2651–2710. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Liebscher, J. Carbon-carbon coupling reactions catalyzed by heterogeneous palladium catalysts. Chem. Rev. 2007, 107, 133–173. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, R. Recent advances in noble metal nanocatalysts for Suzuki and Heck cross-coupling reactions. Molecules 2010, 15, 2124–2138. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Wang, D.; Niu, Z.; Li, Y. Progress in organic reactions catalyzed by bimetallic nanomaterials. Chin. J. Catal. 2013, 34, 1964–1974. [Google Scholar] [CrossRef]
- Baboo, R. Multimetallic nanomaterial based catalysis. Int. J. Curr. Res. Acad. Rev. 2015, 3, 153–157. [Google Scholar]
- Labulo, A.H.; Martincigh, B.S.; Omondi, B.; Nyamori, V.O. Advances in carbon nanotubes as efficacious supports for palladium-catalysed carbon-carbon cross-coupling reactions. J. Mater. Sci. 2017, 52, 9225–9248. [Google Scholar] [CrossRef]
- Moritanl, I.; Fujiwara, Y. Aromatic substitution of styrene-palladium chloride complex. Tetrahedron Lett. 1967, 8, 1119–1122. [Google Scholar] [CrossRef]
- Gligorich, K.M.; Sigman, M.S. Recent advancements and challenges of palladiumII-catalyzed oxidation reactions with molecular oxygen as the sole oxidant. Chem. Commun. (Camb.) 2009, 26, 3854–3867. [Google Scholar] [CrossRef] [PubMed]
- Karimi, B.; Behzadnia, H.; Elhamifar, D.; Akhavan, P.F.; Esfahani, F.K.; Zamani, A. Transition-metal-catalyzed oxidative Heck reactions. Synthesis 2010, 1399–1427. [Google Scholar] [CrossRef]
- Su, Y.; Jiao, N. Palladium-catalyzed oxidative Heck reaction. Curr. Org. Chem. 2011, 15, 3362–3388. [Google Scholar] [CrossRef]
- Le Bras, J.; Muzart, J. Intermolecular dehydrogenative Heck reactions. Chem. Rev. 2011, 111, 1170–1214. [Google Scholar] [CrossRef] [PubMed]
- Le Bras, J.; Muzart, J. Dehydrogenative Heck Annelations of Internal Alkynes. Synthesis 2014, 46, 1555–1572. [Google Scholar] [CrossRef]
- Lee, A.L. Enantioselective oxidative boron Heck reactions. Org. Biomol. Chem. 2016, 14, 5357–5366. [Google Scholar] [CrossRef] [PubMed]
- Xiaomei, Y.; Sha, M.; Yanni, D.; Yunhai, T. Progress in reductive Heck reaction. Chin. J. Org. Chem. 2013, 33, 2325–2333. [Google Scholar] [CrossRef]
- Guiry, P.J.; Kiely, D. The Development of the intramolecular asymmetric Heck Reaction. Curr. Org. Chem. 2004, 8, 781–794. [Google Scholar] [CrossRef]
- Oestreich, M. Breaking News on the Enantioselective Intermolecular Heck Reaction. Angew. Chem. Int. Ed. 2014, 53, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Kikukawa, K.; Matsuda, T. Reaction of Diazonium Salts with Transition Metals. I. Arylation of Olefins with Arenediazonium Salts Catalyzed by Zero Valent Palladium. Chem. Lett. 1977, 2, 159–162. [Google Scholar] [CrossRef]
- Sato, Y.; Sodeoka, M.; Shibasaki, M. Catalytic asymmetric carbon-carbon bond formation: Asymmetric synthesis of cis-decalin derivatives by palladium-catalyzed cyclization of prochiral alkenyl iodides. J. Org. Chem. 1989, 54, 4738–4739. [Google Scholar] [CrossRef]
- Carpenter, N.E.; Kucera, D.J.; Overman, L.E. Palladium-catalyzed polyene cyclizations of trienyl triflates. J. Org. Chem. 1989, 54, 5846–5848. [Google Scholar] [CrossRef]
- Dounay, A.B.; Overman, L.E. The asymmetric intramolecular Heck reaction in natural product total synthesis. Chem. Rev. 2003, 103, 2945–2963. [Google Scholar] [CrossRef] [PubMed]
- Diéguez, M.; Pàmies, O.; Claver, C. Ligands derived from carbohydrates for asymmetric catalysis. Chem. Rev. 2004, 104, 3189–3216. [Google Scholar] [CrossRef] [PubMed]
- McCartney, D.; Guiry, P.J. The asymmetric Heck and related reactions. Chem. Soc. Rev. 2011, 40, 5122–5150. [Google Scholar] [CrossRef] [PubMed]
- Daves, G.D.; Hallberg, A. 1,2-Additions to heteroatom-substituted olefins by organopalladium reagents. Chem. Rev. 1989, 89, 1433–1445. [Google Scholar] [CrossRef]
- Bedford, R.B. Palladacyclic catalysts in C-C and C-heteroatom bond-forming reactions. Chem. Commun. 2003, 1787–1796. [Google Scholar] [CrossRef]
- Jeffery, T. On the efficiency of tetraalkylammonium salts in Heck type reactions. Tetrahedron 1996, 52, 10113–10130. [Google Scholar] [CrossRef]
- Tucker, C.E.; de Vries, J.G. Homogeneous catalysis for the production of fine chemicals. Palladium- and nickel-catalysed aromatic carbon-carbon bond formation. Top. Catal. 2002, 19, 111–118. [Google Scholar] [CrossRef]
- Wang, S.S.; Yang, G.Y. Recent developments in low-cost TM-catalyzed Heck-type reactions (TM = transition metal, Ni, Co, Cu, and Fe). Catal. Sci. Technol. 2016, 6, 2862–2876. [Google Scholar] [CrossRef]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Non-conventional methodologies for transition-metal catalysed carbon-carbon coupling: A critical overview. Part 1: The Heck reaction. Tetrahedron 2005, 61, 11771–11835. [Google Scholar] [CrossRef]
- Tietze, L.F.; Ila, H.; Bell, H.P. Enantioselective palladium-catalyzed transformations. Chem. Rev. 2004, 104, 3453–3516. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, M.; Vogl, E.M.; Ohshima, T. Asymmetric Heck reaction. Adv. Synth. Catal. 2004, 346, 1533–1552. [Google Scholar] [CrossRef]
- Oestreich, M. Neighbouring-group effects in Heck reactions. Eur. J. Org. Chem. 2005, 783–792. [Google Scholar] [CrossRef]
- Genet, J.P.; Savignac, M. Recent developments of palladium(0) catalyzed reactions in aqueous medium. J. Organomet. Chem. 1999, 576, 305–317. [Google Scholar] [CrossRef]
- Li, C. Organic reactions in aqueous media with a focus on carbon-carbon bond formations: A decade update. Chem. Rev. 2005, 105, 3095–3165. [Google Scholar] [CrossRef] [PubMed]
- Velazquez, H.D.; Verpoort, F. N-heterocyclic carbene transition metal complexes for catalysis in aqueous media. Chem. Soc. Rev. 2012, 41, 7032–7060. [Google Scholar] [CrossRef] [PubMed]
- Hervé, G.; Len, C. Heck and Sonogashira couplings in aqueous media—Application to unprotected nucleosides and nucleotides. Sustain. Chem. Process. 2015, 3, 3. [Google Scholar] [CrossRef]
- Cabri, W.; Candiani, I. Recent developments and new perspectives in the Heck reaction. Acc. Chem. Res. 1995, 28, 2–7. [Google Scholar] [CrossRef]
- Crisp, G.T. Variations on a theme—Recent developments on the mechanism of the Heck reaction and their implications for synthesis. Chem. Soc. Rev. 1998, 27, 427–436. [Google Scholar] [CrossRef]
- Amatore, C.; Carre, E.; Jutand, A.; M’Barki, M.A.; Meyer, G. Evidence for the Ligation of Palladium(0) Complexes by Acetate Ions: Consequences on the Mechanism of Their Oxidative Addition with Phenyl Iodide and PhPd(OAc)(PPh3)2 as Intermediate in the Heck Reaction. Organometallics 1995, 14, 5605–5614. [Google Scholar] [CrossRef]
- Amatore, C.; Jutand, A. Mechanistic and kinetic studies of palladium catalytic systems. J. Organomet. Chem. 1999, 576, 254–278. [Google Scholar] [CrossRef]
- Amatore, C.; Jutand, A. Anionic Pd(0) and Pd(II) intermediates in palladium-catalyzed Heck and cross-coupling reactions. Acc. Chem. Res. 2000, 33, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Cavell, K.J.; McGuinness, D.S. Redox processes involving hydrocarbylmetal (N-heterocyclic carbene) complexes and associated imidazolium salts: Ramifications for catalysis. Coord. Chem. Rev. 2004, 248, 671–681. [Google Scholar] [CrossRef]
- Trzeciak, A.M.; Zi´ołkowski, J.J. Structural and mechanistic studies of Pd-catalyzed C-C bond formation: The case of carbonylation and Heck reaction. Coord. Chem. Rev. 2005, 249, 2308–2322. [Google Scholar] [CrossRef]
- Phan, N.T.S.; DerSluys, M.V.; Jones, C.W. On the Nature of the Active Species in Palladium Catalyzed Mizoroki–Heck and Suzuki–Miyaura Couplings—Homogeneous or Heterogeneous Catalysis, A Critical Review. Adv. Synth. Catal. 2006, 348, 609–679. [Google Scholar] [CrossRef]
- Knowles, J.P.; Whiting, A. The Heck–Mizoroki cross-coupling reaction: A mechanistic perspective. Org. Biomol. Chem. 2007, 5, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Rao, G.K.; Kumar, S.; Singh, A.K. Formation and Role of Palladium Chalcogenide and Other Species in Suzuki−Miyaura and Heck C-C Coupling Reactions Catalyzed with Palladium(II) Complexes of Organochalcogen Ligands: Realities and Speculations. Organometallics 2014, 33, 2921–2943. [Google Scholar] [CrossRef]
- Eremin, D.B.; Ananikov, V.P. Understanding active species in catalytic transformations: From molecular catalysis to nanoparticles, leaching, “Cocktails” of catalysts and dynamic systems. Coord. Chem. Rev. 2017, 346, 2–19. [Google Scholar] [CrossRef]
- Blaser, H.U.; Indolese, A.; Schnyder, A. Applied homogeneous catalysis by organometallic complexes. Curr. Sci. 2000, 78, 1336–1344. [Google Scholar]
- De Vries, J.G. The Heck reaction in the production of fine chemicals. Can. J. Chem. 2001, 79, 1086–1092. [Google Scholar] [CrossRef]
- Zapf, A.; Beller, M. Fine chemical synthesis with homogeneous palladium catalysts: Examples, status and trends. Top. Catal. 2002, 19, 101–109. [Google Scholar] [CrossRef]
- Olovnikov, A.M. A theory of marginotomy. The incomplete copying of template margin in enzymic synthesis of polynucleotides and biological significance of the phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Zoeller, J.R.; Agreda, V.H.; Cook, S.L.; Lafferty, N.L.; Polichnowski, S.W.; Pond, D.M. Eastman Chemical Company Acetic Anhydride Process. Catal. Today 1992, 13, 73–91. [Google Scholar] [CrossRef]
- Picquet, M. Organometallics as catalysts in the fine chemical industry. Platinum Metals Rev. 2013, 57, 272–280. [Google Scholar] [CrossRef]
- Sharma, P. Cinnamic acid derivatives: A new chapter of various pharmacological activities. J. Chem. Pharm. Res. 2011, 3, 403–423. [Google Scholar]
- Perkin, W.H. On the artificial production of coumarin and formation of its homologues. J. Chem. Soc. 1868, 21, 53–61. [Google Scholar] [CrossRef]
- Biajoli, A.F.P.; Schwalm, C.; Limberger, J.; Claudino, T.S.; Monteiro, A.L. Recent progress in the use of Pd-catalyzed C-C cross-coupling reactions in the synthesis of pharmaceutical compounds. J. Braz. Chem. Soc. 2014, 25, 2186–2214. [Google Scholar] [CrossRef]
- Magano, J.; Dunetz, J.R. Large-scale applications of transition metal-catalyzed couplings for the synthesis of pharmaceuticals. Chem. Rev. 2011, 111, 2177–2250. [Google Scholar] [CrossRef] [PubMed]
- Torborg, C.; Beller, M. Recent applications of palladium-catalyzed coupling reactions in the pharmaceutical, agrochemical, and fine chemical industries. Adv. Synth. Catal. 2009, 351, 3027–3043. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Bulger, P.G.; Sarlah, D. Palladium-catalyzed cross-coupling reactions in total synthesis. Angew. Chem. Int. Ed. 2005, 44, 4442–4489. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, T. Heck-type reactions in water. Tetrahedron Lett. 1994, 35, 3051–3054. [Google Scholar] [CrossRef]
- Trost, B.M.; Shi, Y. A palladium-catalyzed zipper reaction. J. Am. Chem. Soc. 1991, 113, 701–703. [Google Scholar] [CrossRef]
- Trost, B.M. Atom economy—A challenge for organic synthesis: Homogeneous catalysis leads the way. Angew. Chem. Int. Ed. Engl. 1995, 34, 259–281. [Google Scholar] [CrossRef]
- Wellington, K.W.; Benner, S.A. A review: Synthesis of aryl C-glycosides via the Heck coupling reaction. Nucleotides Nucleic Acids 2006, 25, 1309–1333. [Google Scholar] [CrossRef] [PubMed]
- Zeni, G.; Larock, R.C. Synthesis of heterocycles via palladium-catalyzed oxidative addition. Chem. Rev. 2006, 106, 4644–4680. [Google Scholar] [CrossRef] [PubMed]
- Bhanage, B.M.; Arai, M. Catalyst product separation techniques in Heck reaction. Catal. Rev. 2001, 43, 315–344. [Google Scholar] [CrossRef]
- Wall, V.M.; Eisenstadt, A.; Ager, D.J.; Laneman, S.A. The Heck reaction and cinnamic acid synthesis by heterogeneous catalysis. Platinum Met. Rev. 1999, 43, 138–145. [Google Scholar]
- Van Heerbeek, R.; Kamer, P.C.; van Leeuwen, P.W.; Reek, J.N. Dendrimers as support for recoverable catalysts and reagents. Chem. Rev. 2002, 102, 3717–3756. [Google Scholar] [CrossRef] [PubMed]
- Horn, J.; Michalek, F.; Tzschucke, C.C.; Bannwarth, W. Non-covalently solid-phase bound catalysts for organic synthesis. Top. Curr. Chem. 2004, 242, 43–75. [Google Scholar] [CrossRef] [PubMed]
- Köhler, K.; Pröckl, S.S.; Kleist, W. Supported palladium catalysts in Heck coupling reactions—Problems, potential and recent advances. Curr. Org. Chem. 2006, 10, 1585–1601. [Google Scholar] [CrossRef]
- Polshettiwar, V.; Molnár, A. Silica-supported Pd catalysts for Heck coupling reactions. Tetrahedron 2007, 63, 6949–6976. [Google Scholar] [CrossRef]
- Molnar, A. Efficient, selective, and recyclable palladium catalysts in carbon-carbon coupling reactions. Chem. Rev. 2011, 111, 2251–2320. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R. Catalytic reactions in ionic liquids. Chem. Commun. 2001, 2399–2407. [Google Scholar] [CrossRef]
- Singh, R.; Sharma, M.; Mamgain, R.; Rawat, D.S. Ionic liquids: A versatile medium for palladium-catalyzed reactions. J. Braz. Chem. Soc. 2008, 19, 357–379. [Google Scholar] [CrossRef]
- Bellina, F.; Chiappe, C. The Heck reaction in ionic liquids: Progress and challenges. Molecules 2010, 15, 2211–2245. [Google Scholar] [CrossRef] [PubMed]
- Mastrorilli, P.; Monopoli, A.; Dell’Anna, M.M.; Latronico, M.; Cotugno, P.; Nacci, A. Ionic liquids in palladium-catalyzed cross-coupling reactions. Top. Organomet. Chem. 2013, 51, 237–285. [Google Scholar] [CrossRef]
- Santos, C.I.; Barata, J.F.; Faustino, M.A.F.; Lodeiro, C.; Neves, M.G.P. Revisiting Heck–Mizoroki reactions in ionic liquids. RSC Adv. 2013, 3, 19219–19238. [Google Scholar] [CrossRef]
- Limberger, J.; Leal, B.C.; Monteiro, A.L. Charge-tagged ligands: Useful tools for immobilising complexes and detecting reaction species during catalysis. Chem. Sci. 2015, 6, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Bhanage, B.M.; Fujita, S.I.; Arai, M. Heck reactions with various types of palladium complex catalysts: Application of multiphase catalysis and supercritical carbon dioxide. J. Organomet. Chem. 2003, 687, 211–218. [Google Scholar] [CrossRef]
- Skouta, R. Selective chemical reactions in supercritical carbon dioxide, water, and ionic liquids. Green Chem. Lett. Rev. 2009, 2, 121–156. [Google Scholar] [CrossRef]
- Gürsel, I.V.; Noël, T.; Wang, Q.; Hessel, V. Separation/recycling methods of homogeneous transition metal catalysts in continuous flow. Green Chem. 2015, 17, 2012–2026. [Google Scholar] [CrossRef]
Scheme 1.
Initial work by Heck [10] to get an active species (a) RPdL2X and (b) RPd(PR’3)2X.
Scheme 1.
Initial work by Heck [10] to get an active species (a) RPdL2X and (b) RPd(PR’3)2X.
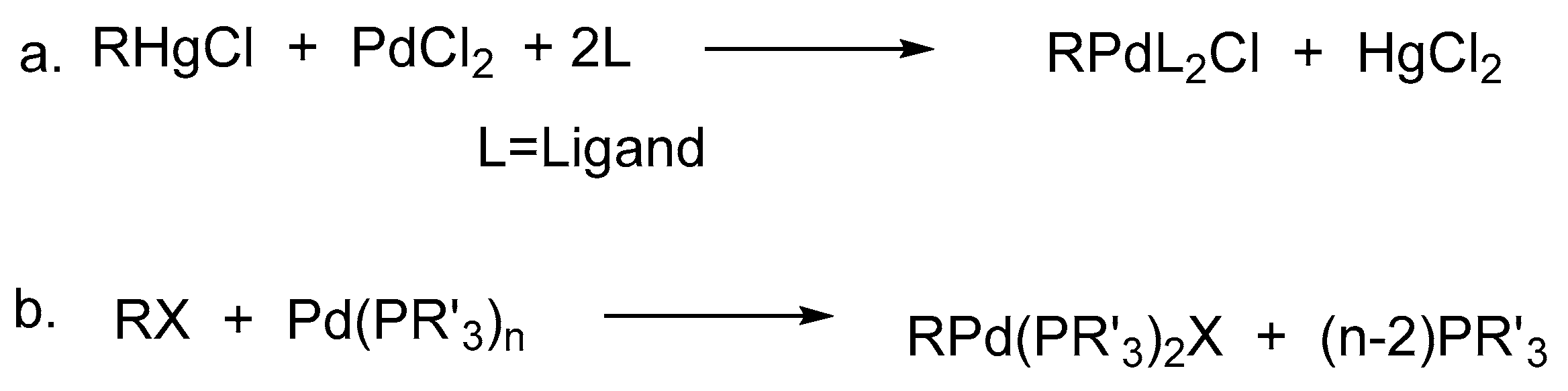
Figure 1.
Oligofunctional and oligocyclic products formed via Heck reaction.
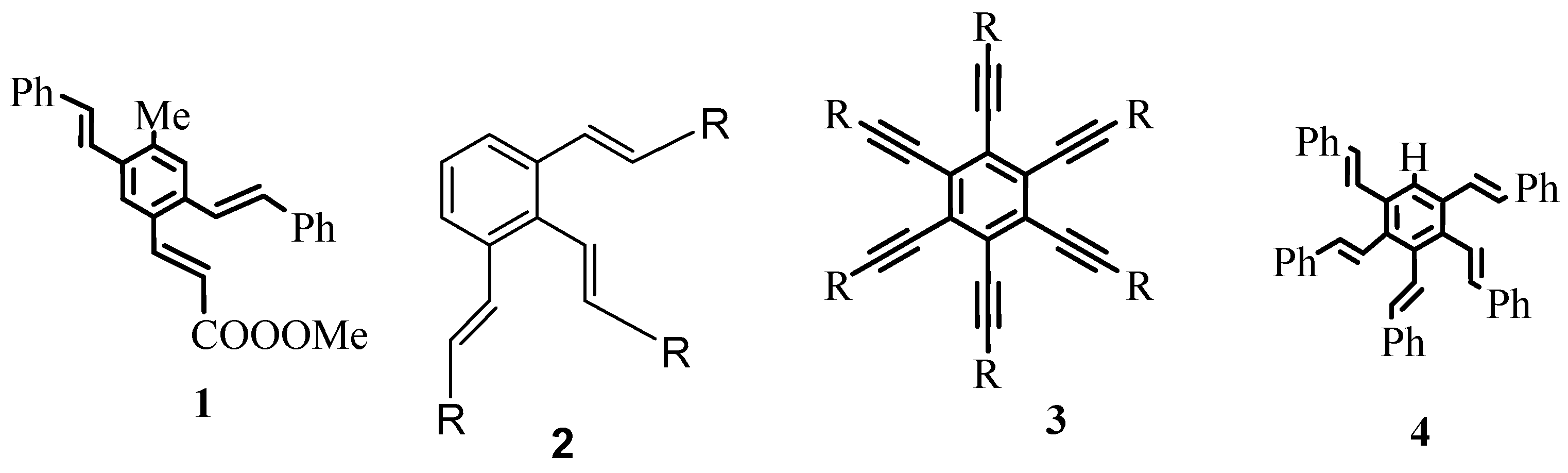
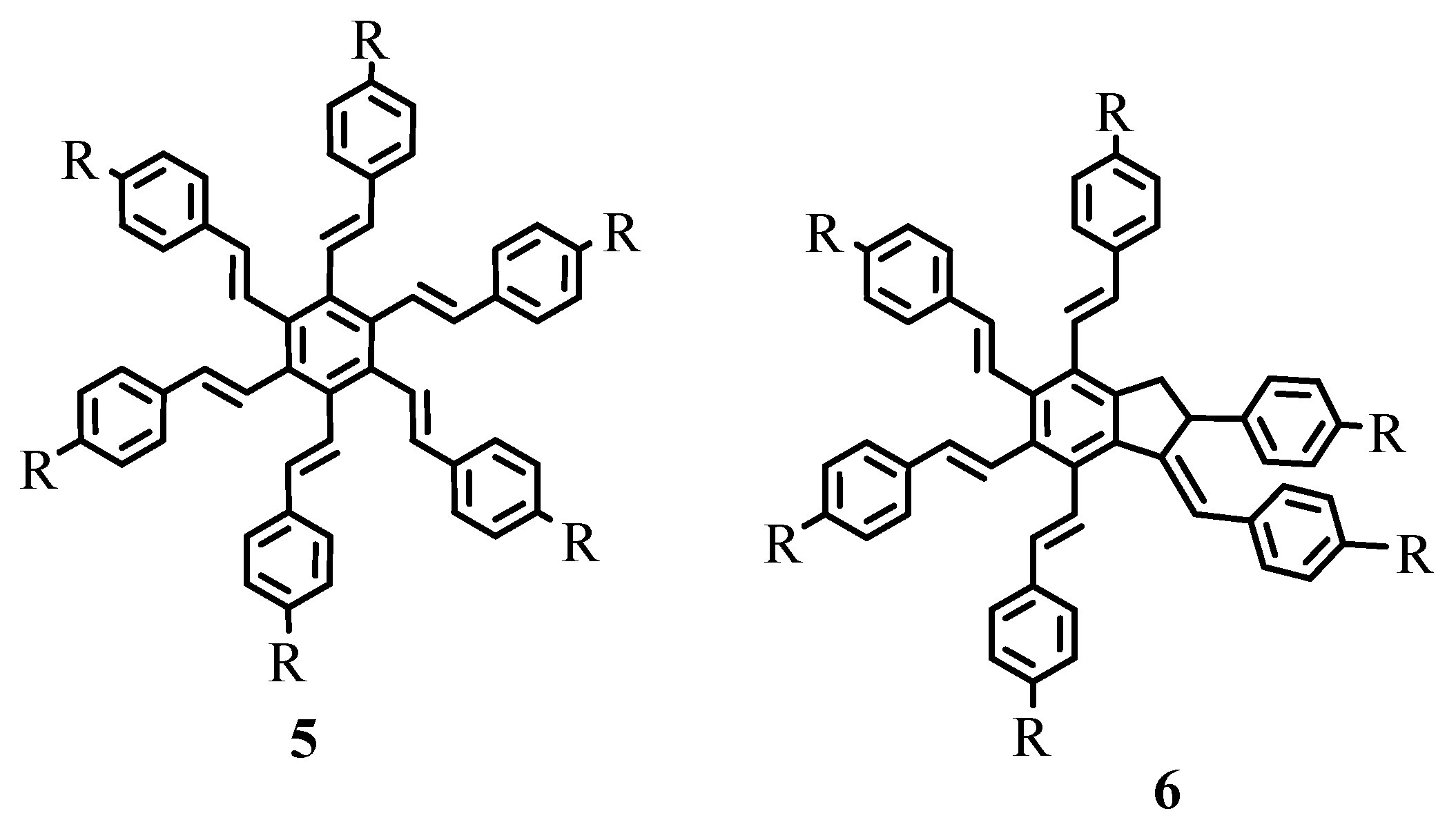
Figure 2.
Enantioselective construction of complex natural products reviewed by de Meijere and Meyer [13].
Figure 2.
Enantioselective construction of complex natural products reviewed by de Meijere and Meyer [13].
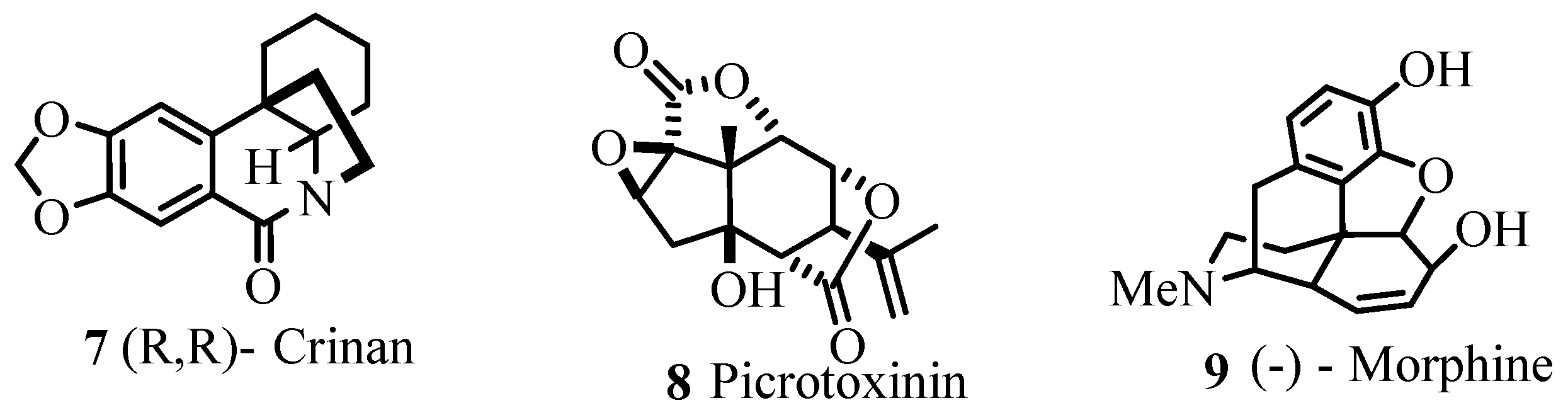
Figure 3.
Natural products involving synthesis via different types of Heck transformations.
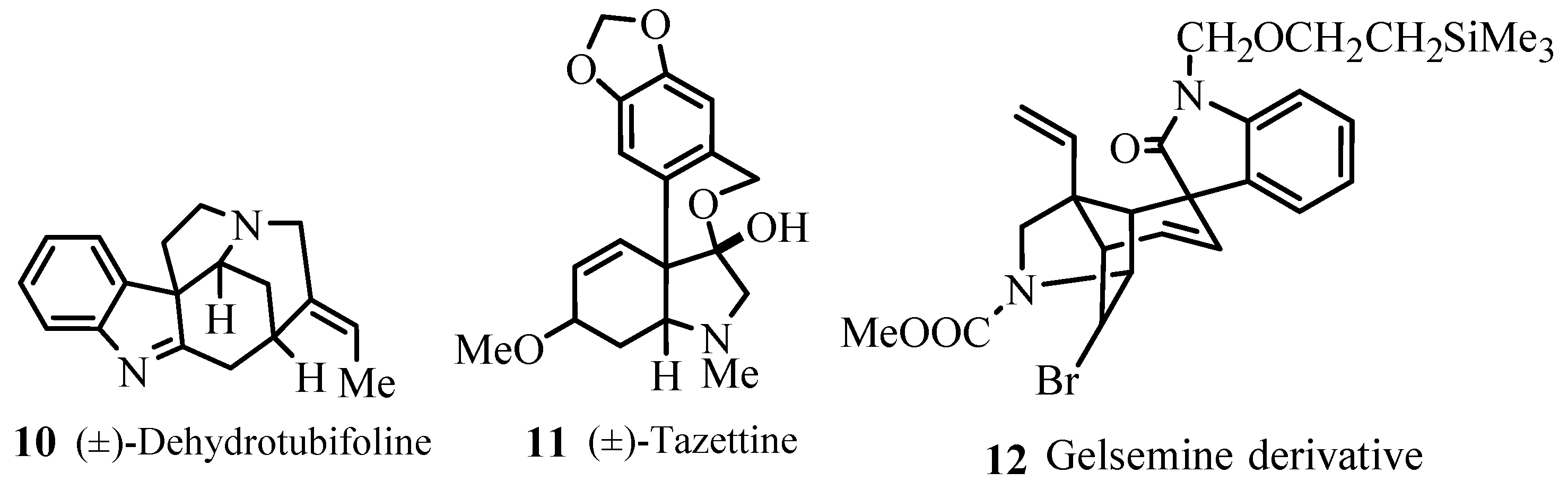
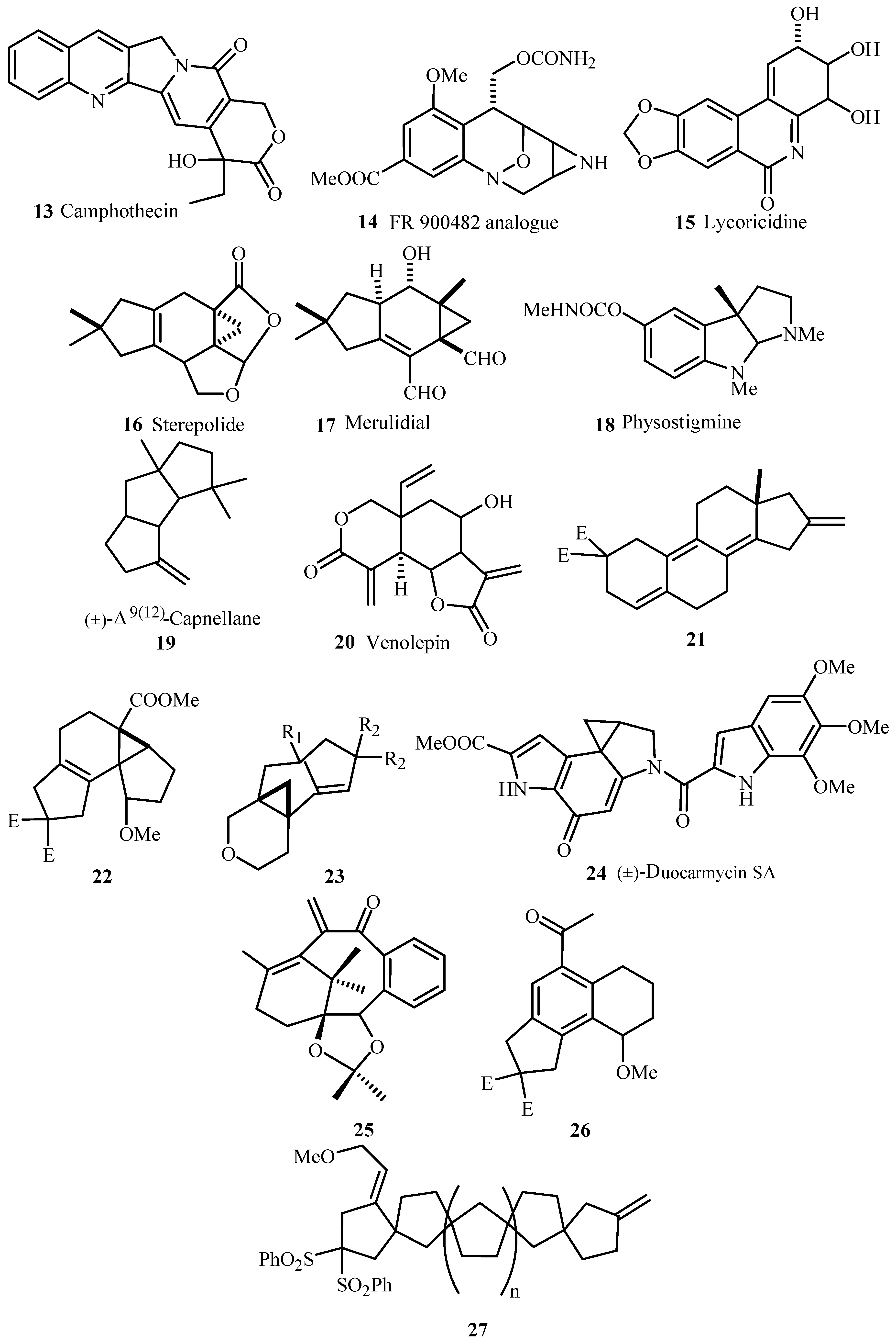
Figure 4.
Ligands and catalysts employed for Heck reaction reported in review by Kobetić and Biliškov [14].
Figure 4.
Ligands and catalysts employed for Heck reaction reported in review by Kobetić and Biliškov [14].
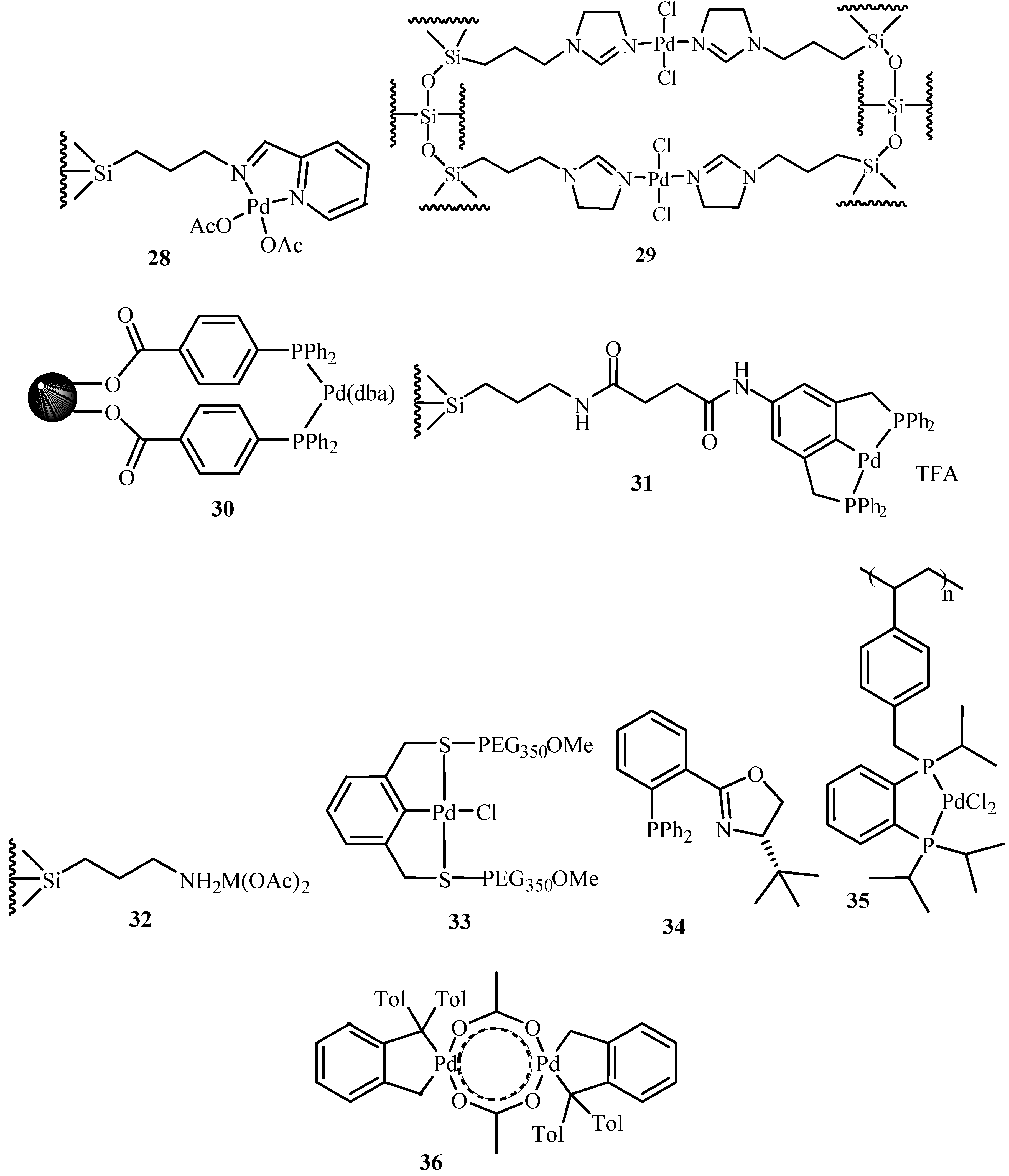
Figure 5.
MOM-Rhazinal (MOM: Methoxymethyl ether).
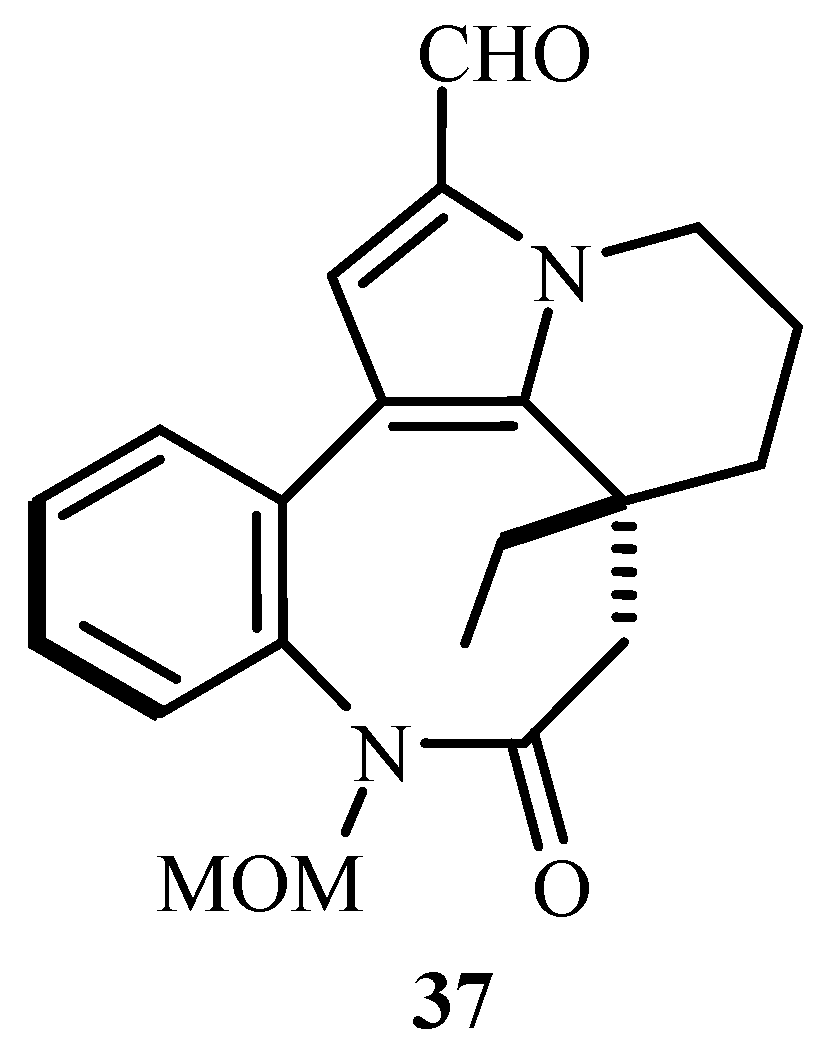
Figure 6.
N-heterocyclic carbene (NHC) ligands of diazabutadienes reviewed by Hillier et al. [17].
Figure 6.
N-heterocyclic carbene (NHC) ligands of diazabutadienes reviewed by Hillier et al. [17].
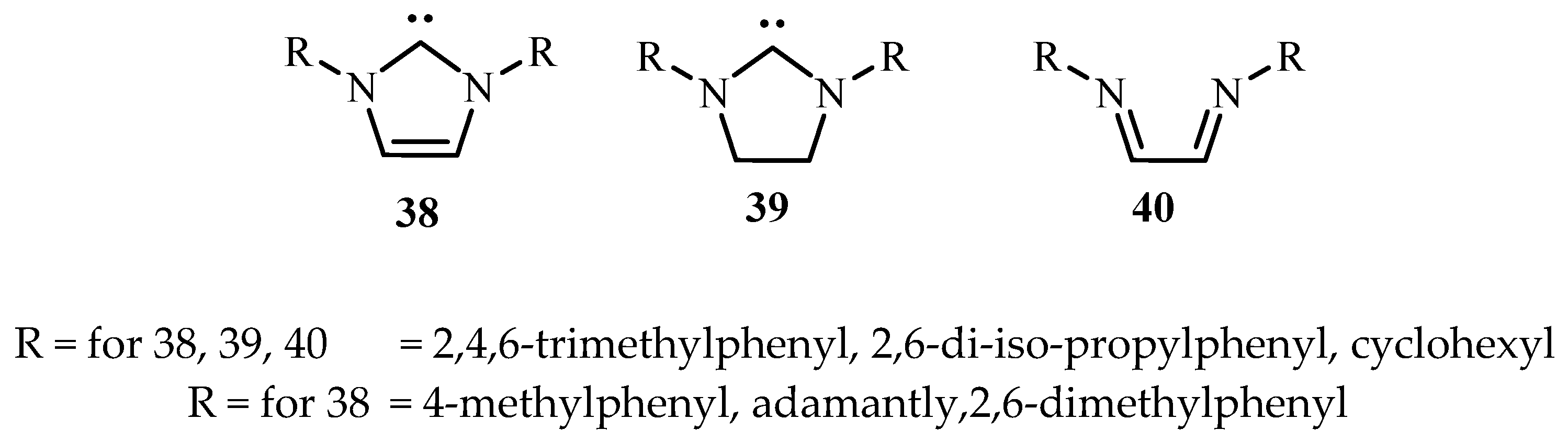
Figure 7.
N-heterocyclic carbene ligands and metal complexes reviewed by Herrmann [18].
Figure 7.
N-heterocyclic carbene ligands and metal complexes reviewed by Herrmann [18].
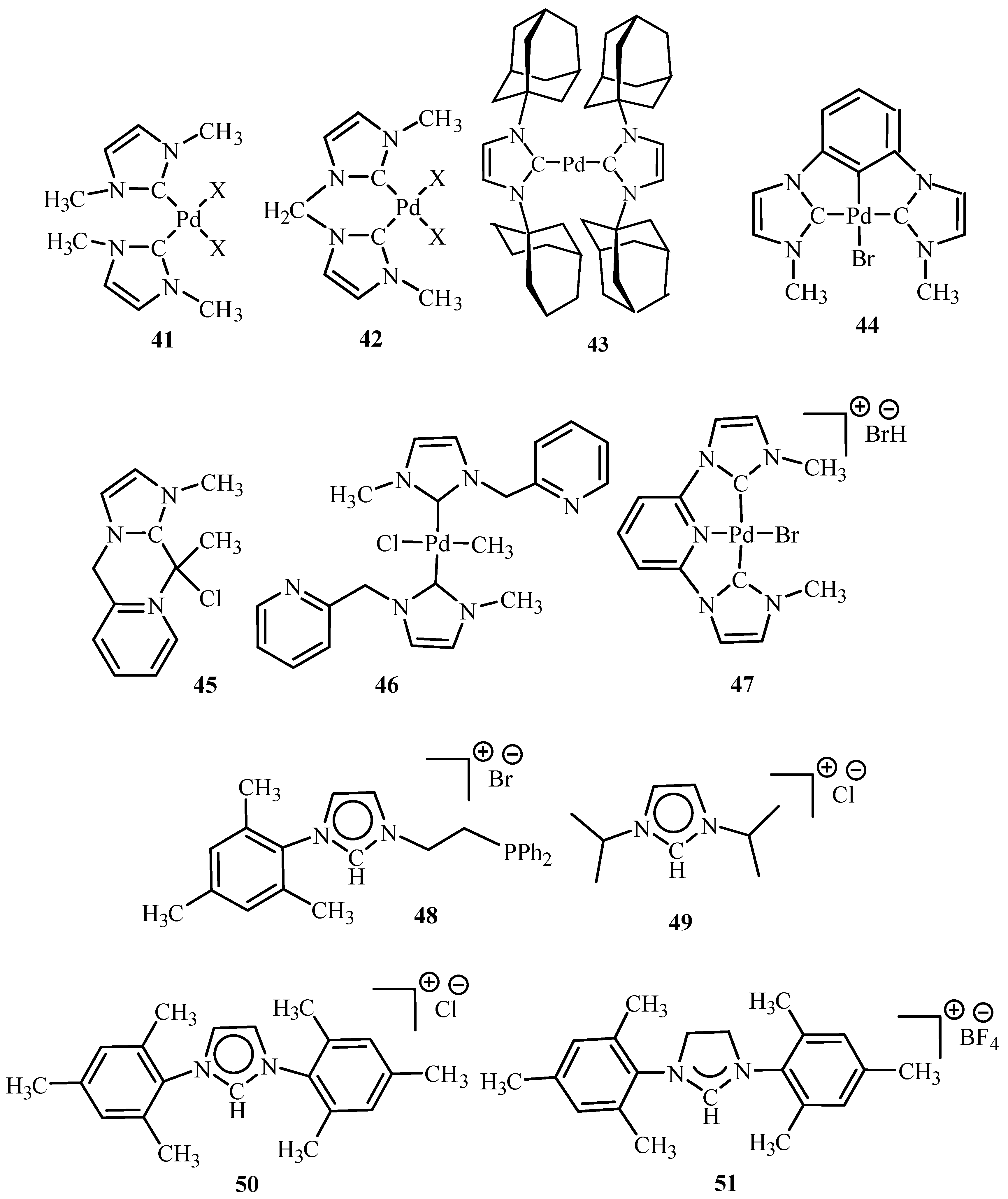
Figure 8.
Palladium complexes with phosphorus and N-heterocyclic carbene ligands reviewed by Herrmann et al. [19].
Figure 8.
Palladium complexes with phosphorus and N-heterocyclic carbene ligands reviewed by Herrmann et al. [19].
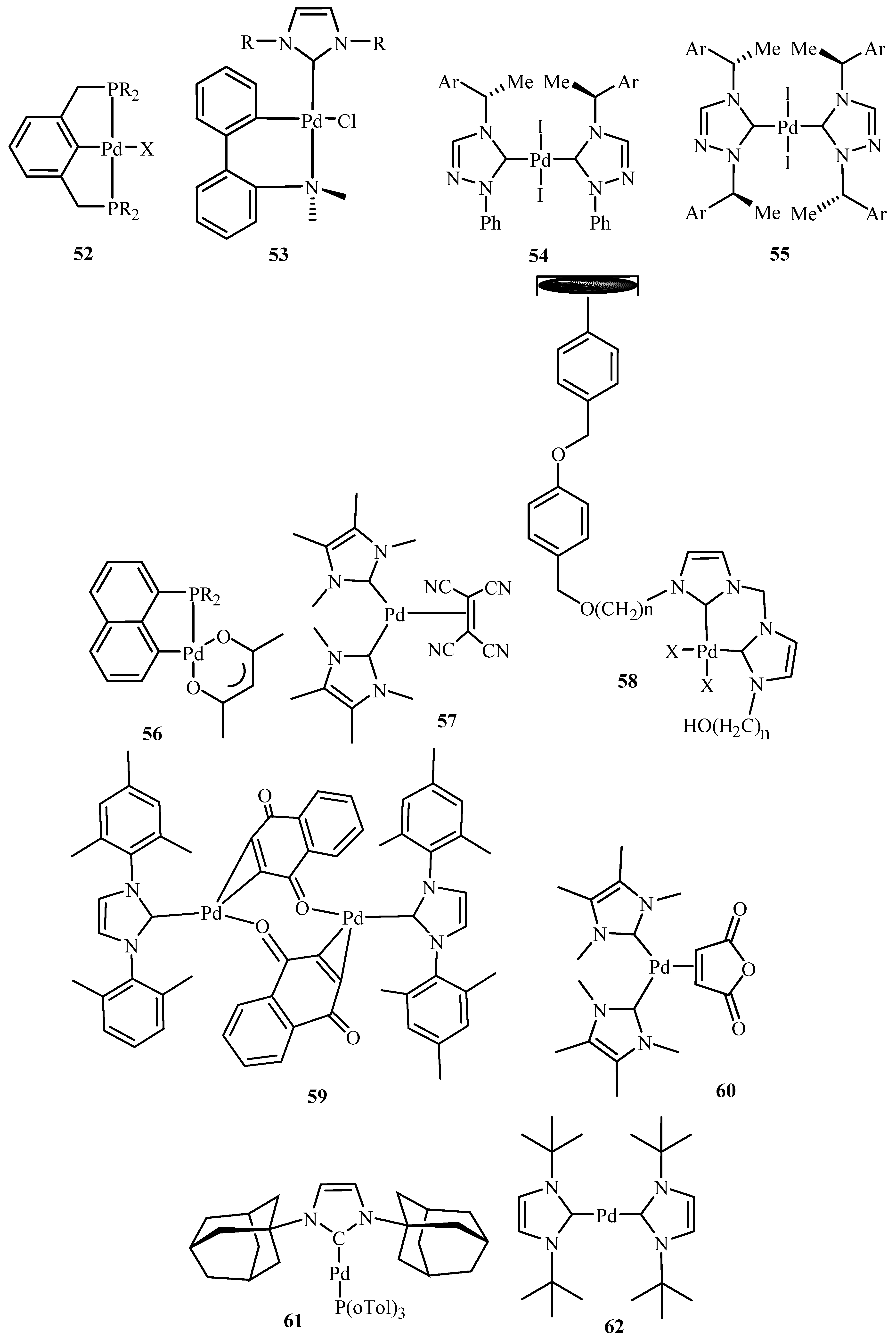
Figure 9.
Ligands and palladacycles reviewed by Beletskaya and Cheprakov [20].
Figure 9.
Ligands and palladacycles reviewed by Beletskaya and Cheprakov [20].
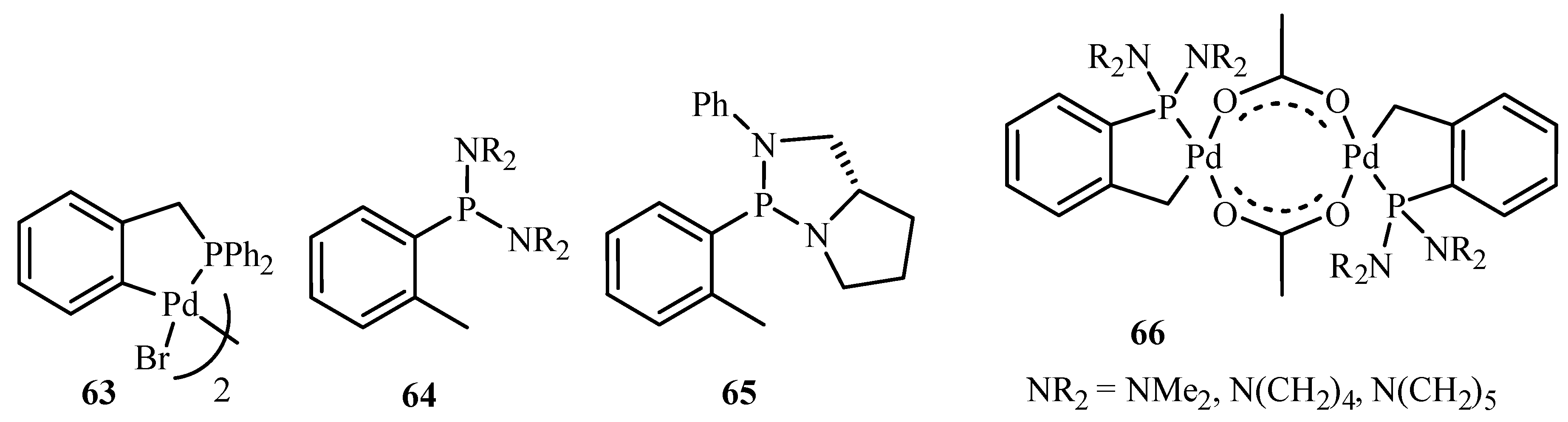
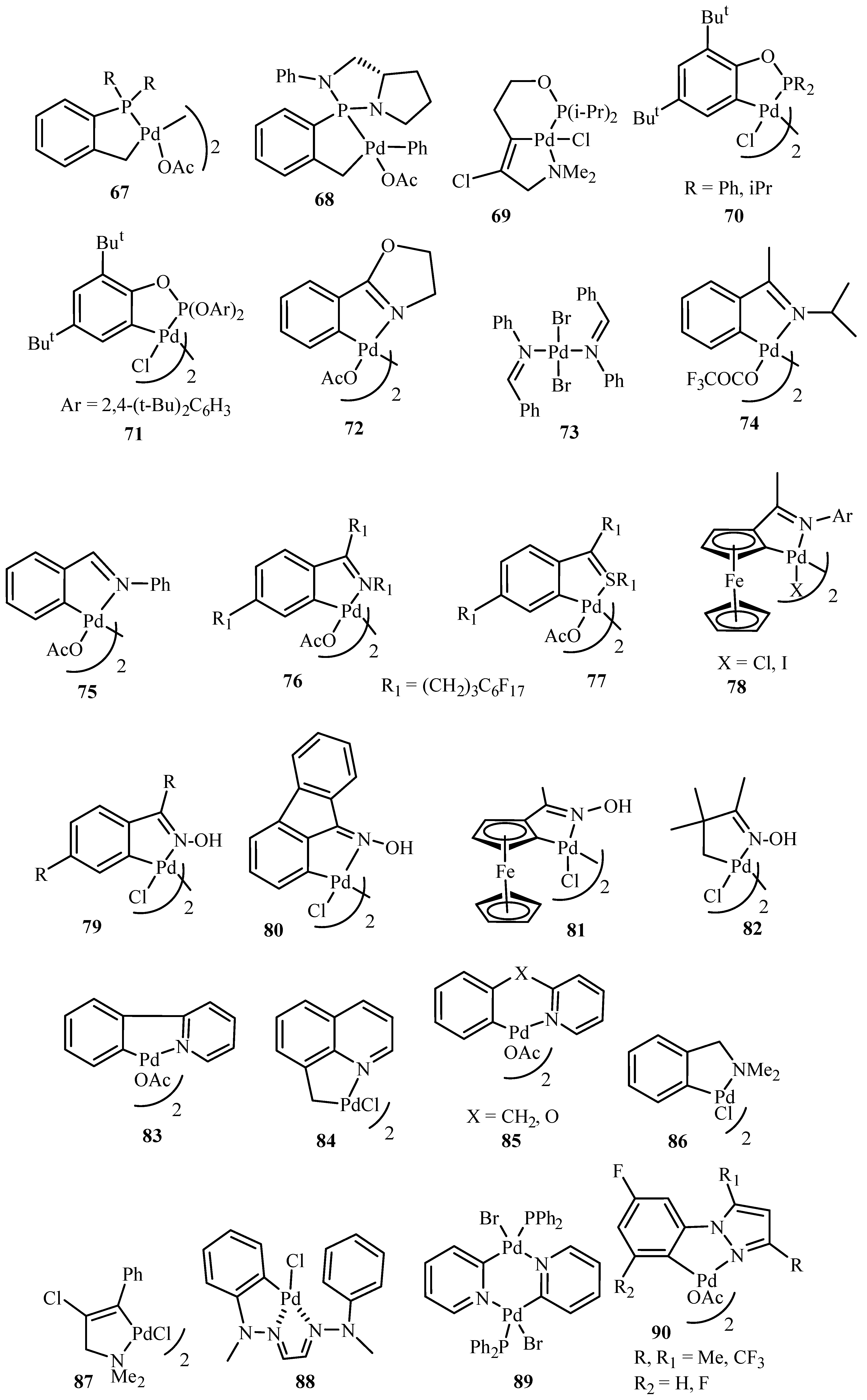
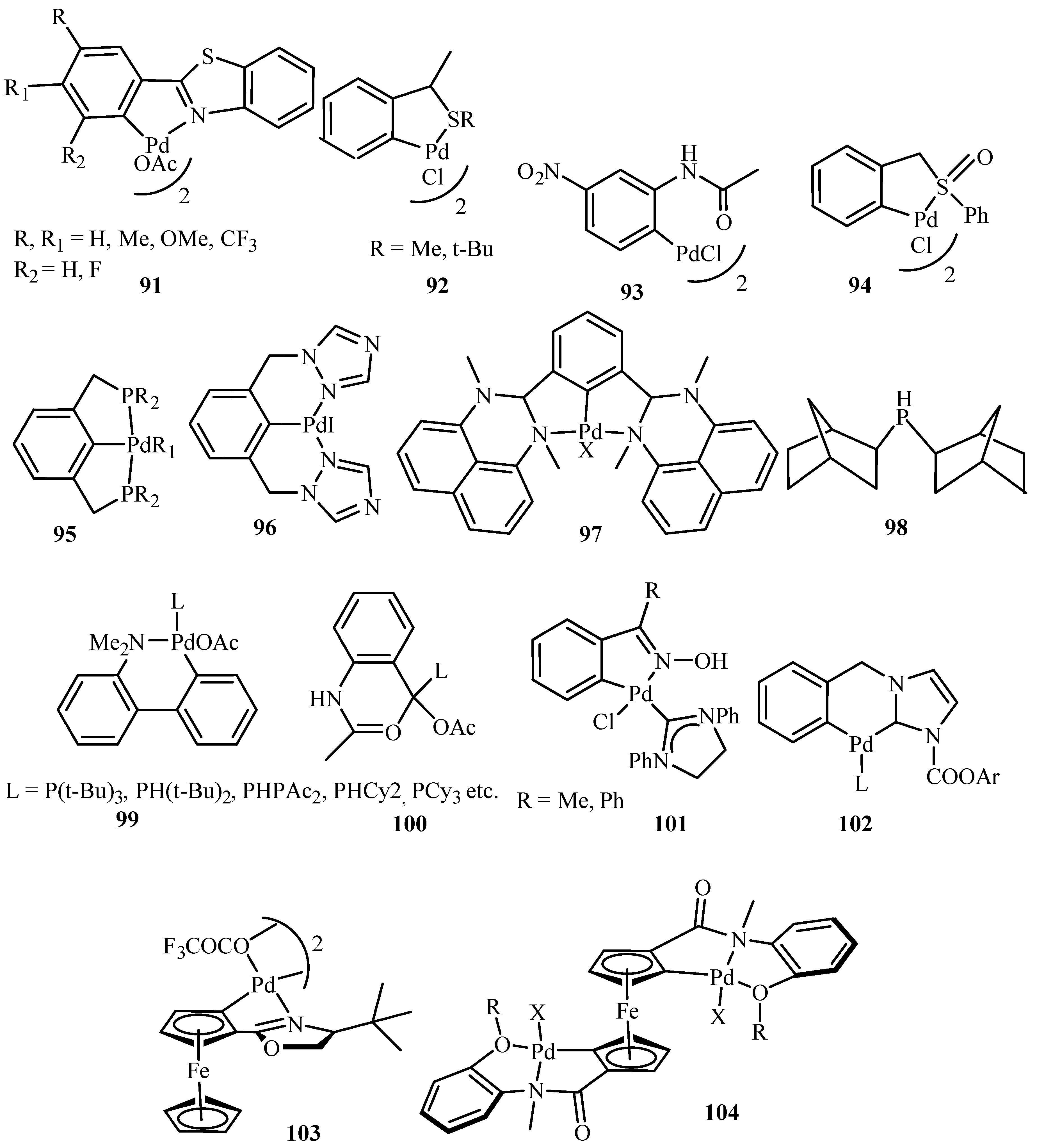
Figure 10.
Metal complexes reviewed by Herrmann et al. [22].
Figure 10.
Metal complexes reviewed by Herrmann et al. [22].
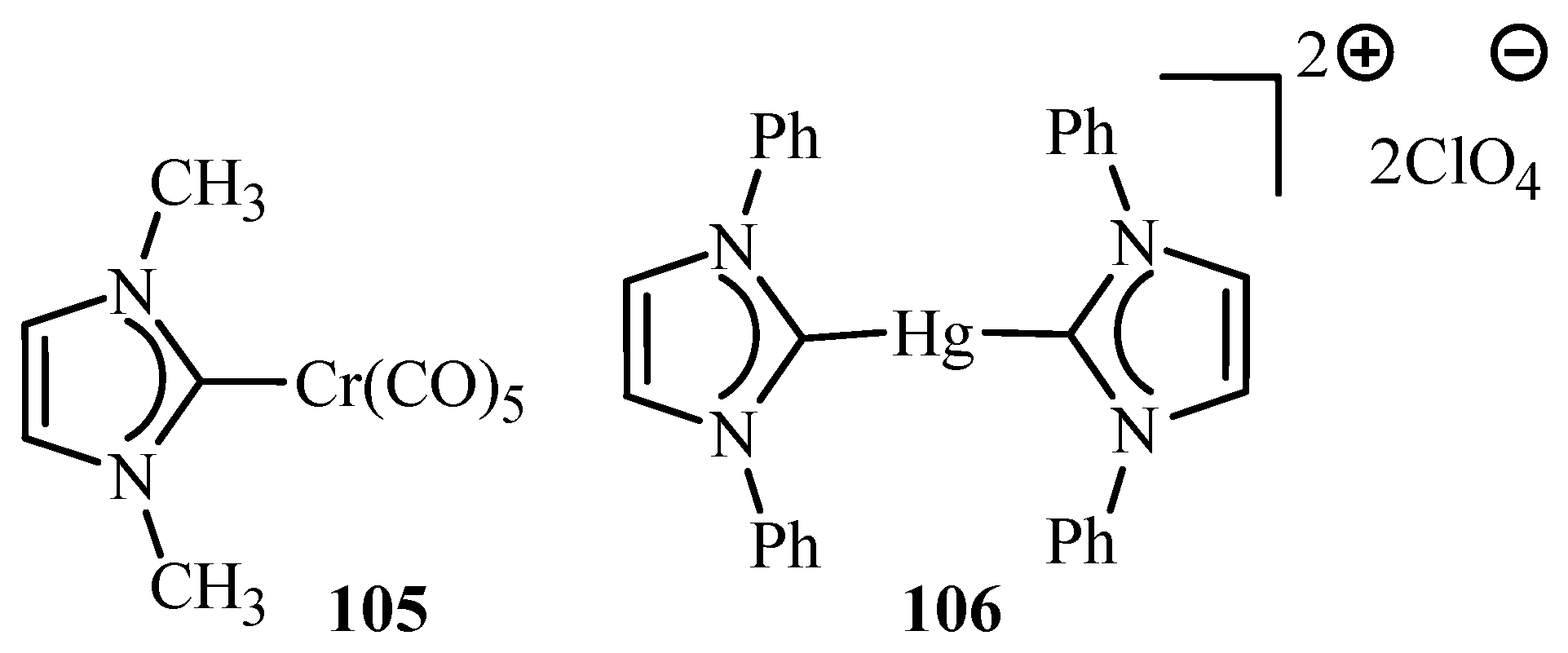
Figure 11.
Palladacycles and coordinatively unsaturated Pd catalysts featuring bulky phosphanes of high denticity in review by Farina [23].
Figure 11.
Palladacycles and coordinatively unsaturated Pd catalysts featuring bulky phosphanes of high denticity in review by Farina [23].
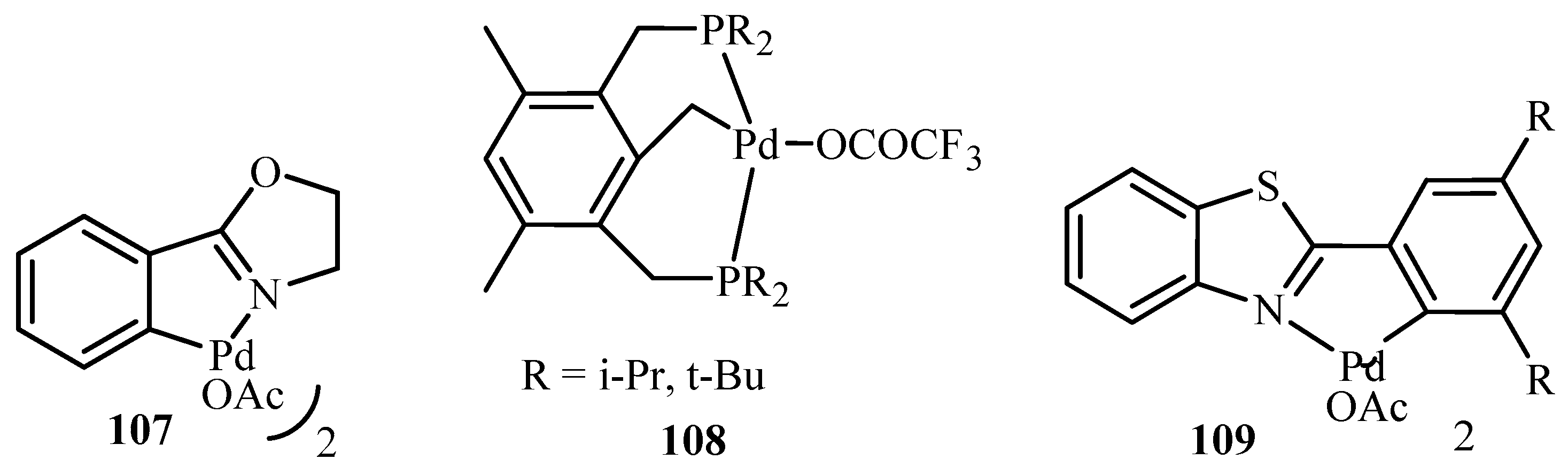
Figure 12.
Ligands reviewed by Zapf and Beller [26].
Figure 12.
Ligands reviewed by Zapf and Beller [26].
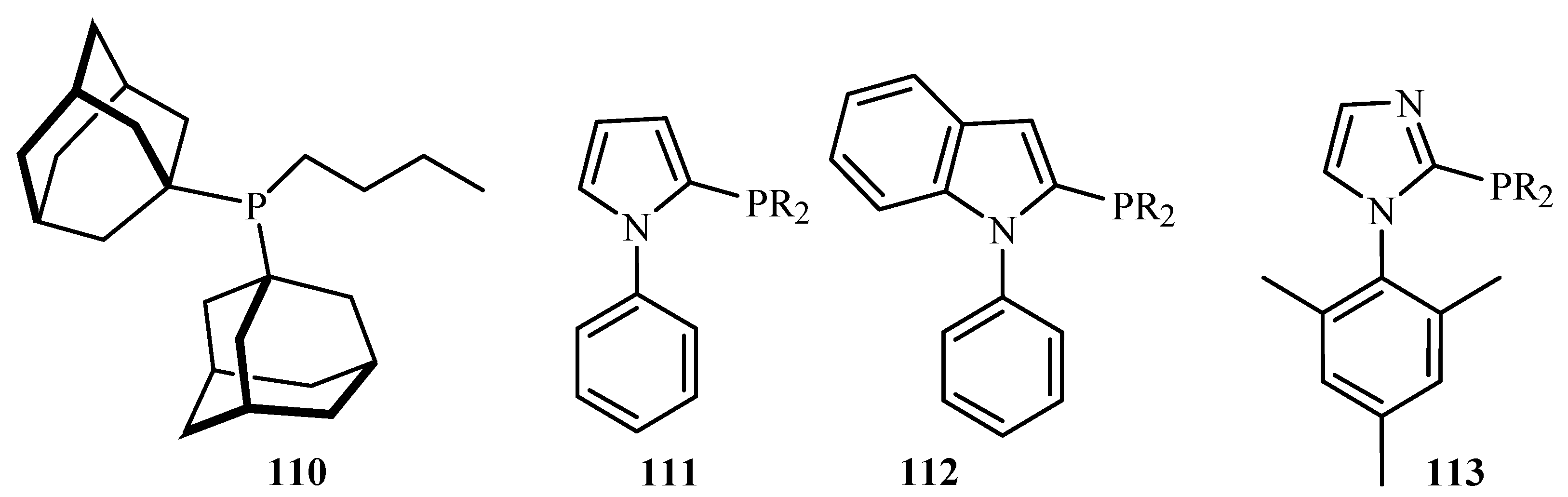
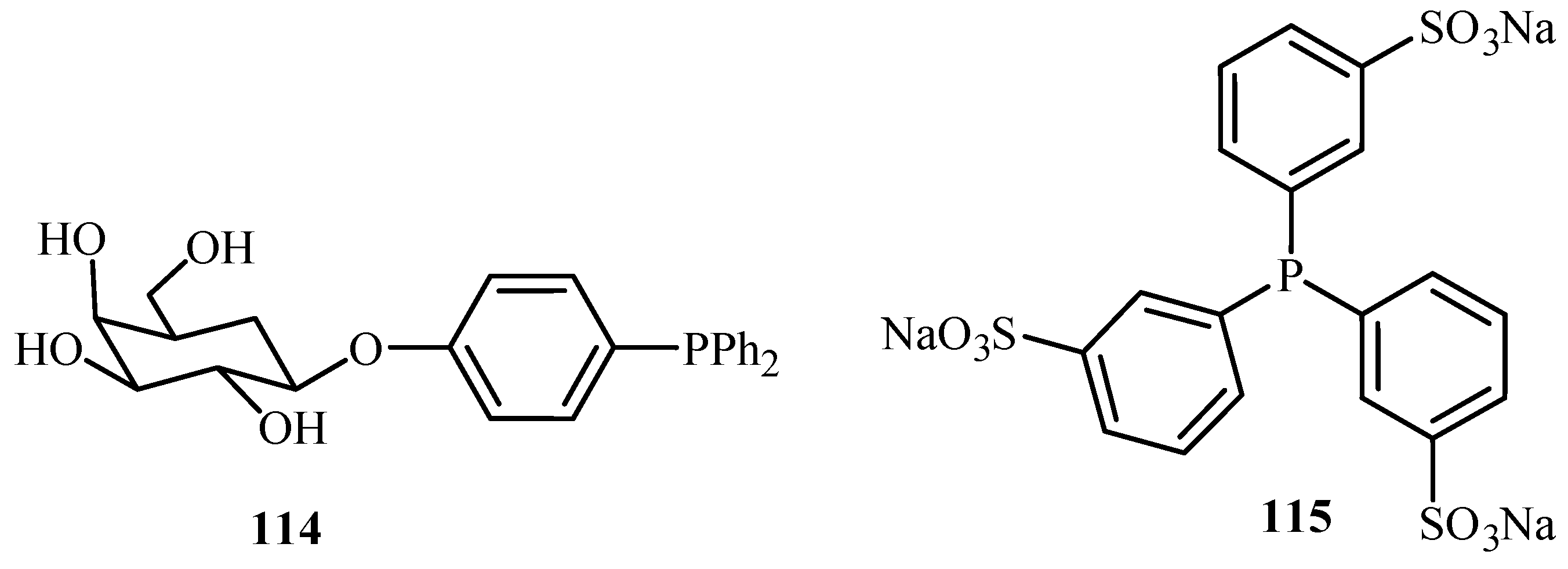
Figure 13.
Molecule made via solid-phase synthesis using Heck reaction reviewed by Franzén [27].
Figure 13.
Molecule made via solid-phase synthesis using Heck reaction reviewed by Franzén [27].
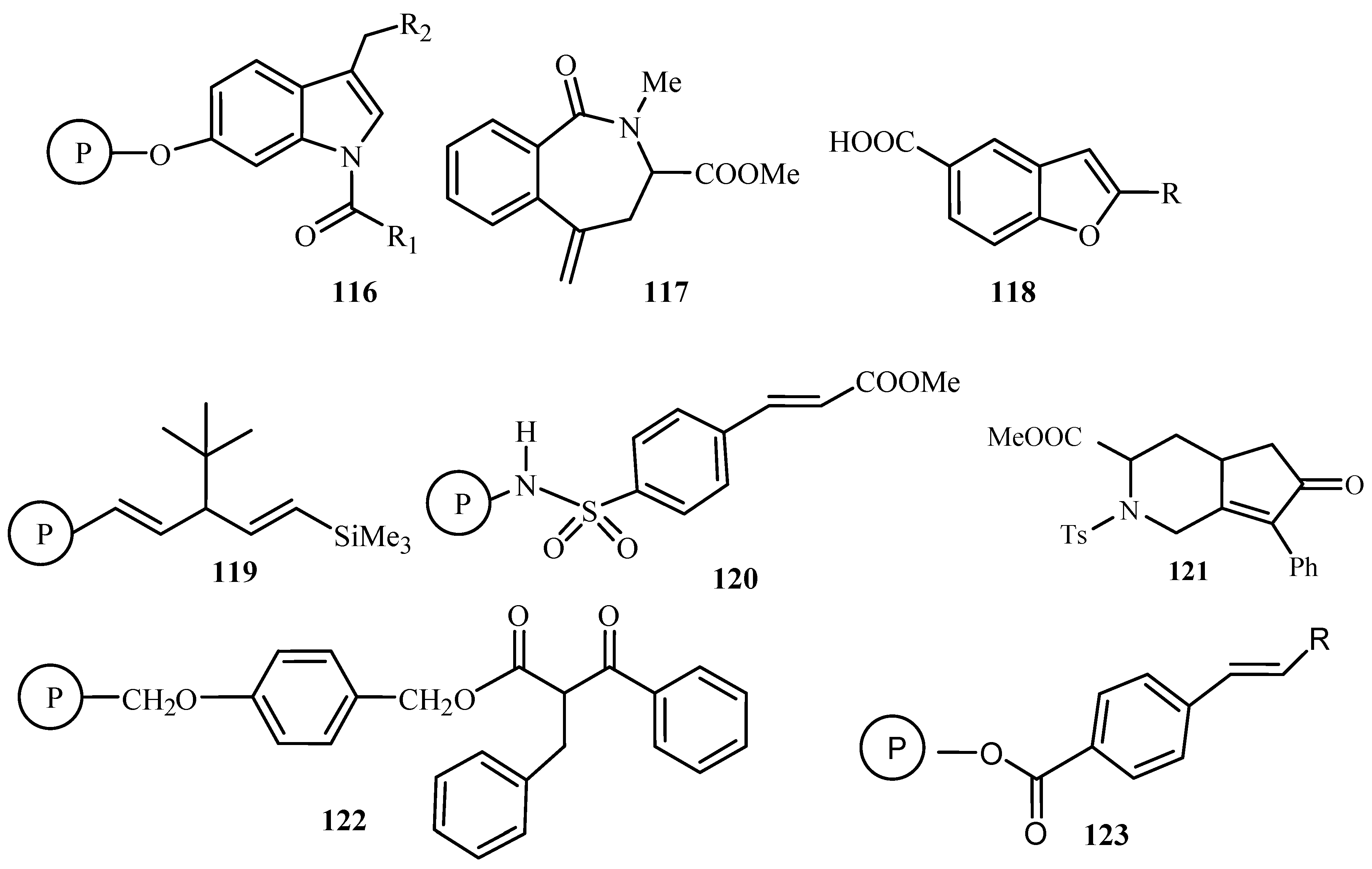
Scheme 2.
Reaction scheme for C-H activation and transmetallation.
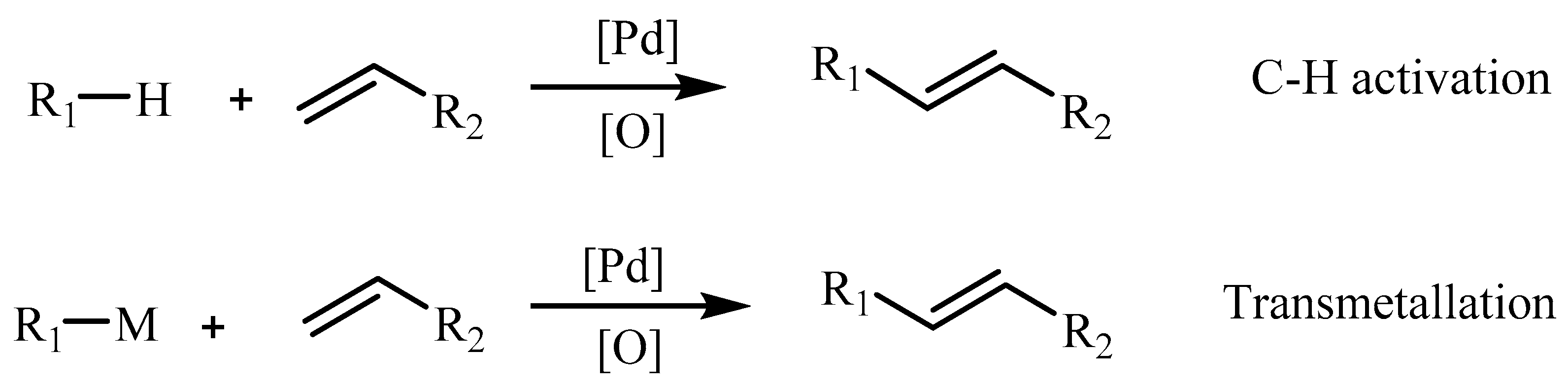
Scheme 3.
Collapsing of intermediate to form conjugate addition product or substitution product via β-hydride elimination.
Scheme 3.
Collapsing of intermediate to form conjugate addition product or substitution product via β-hydride elimination.
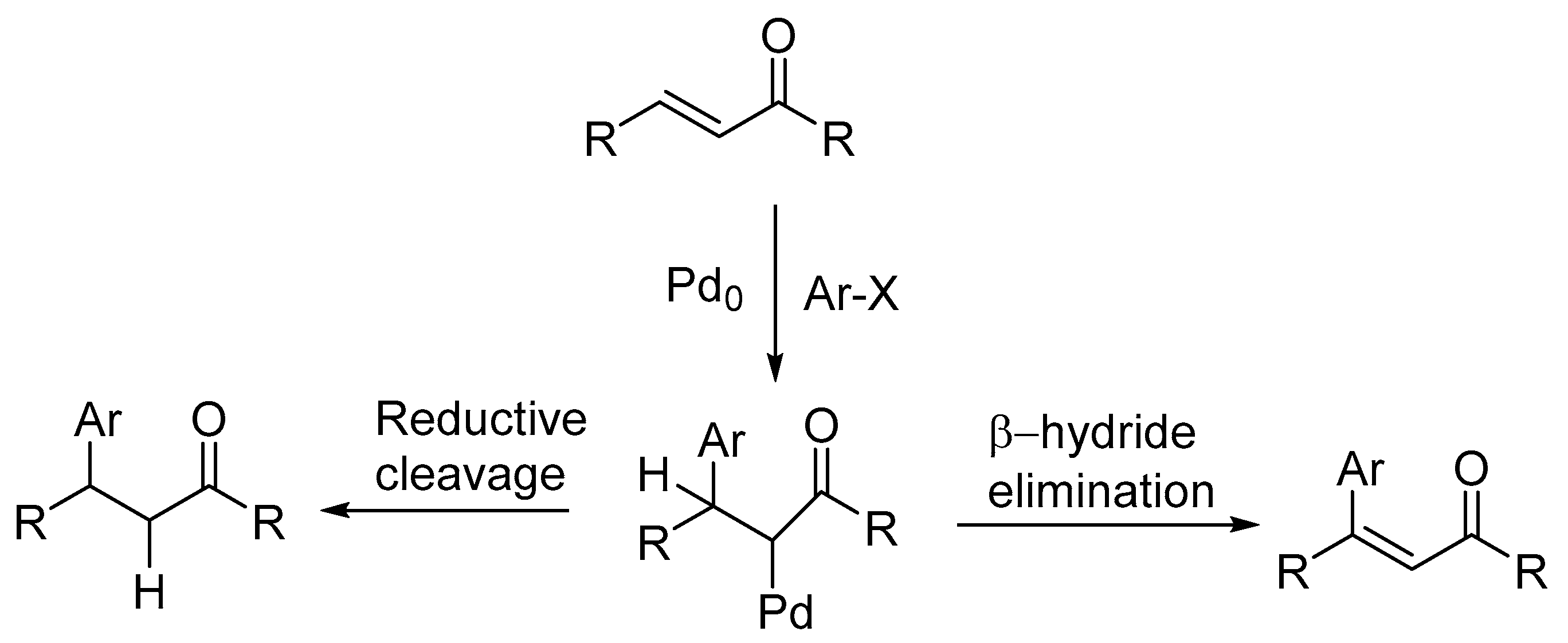
Figure 14.
Scopadulcic acid.

Figure 15.
Carbohydrate based ligands reviewed by Diéguez [51].
Figure 15.
Carbohydrate based ligands reviewed by Diéguez [51].
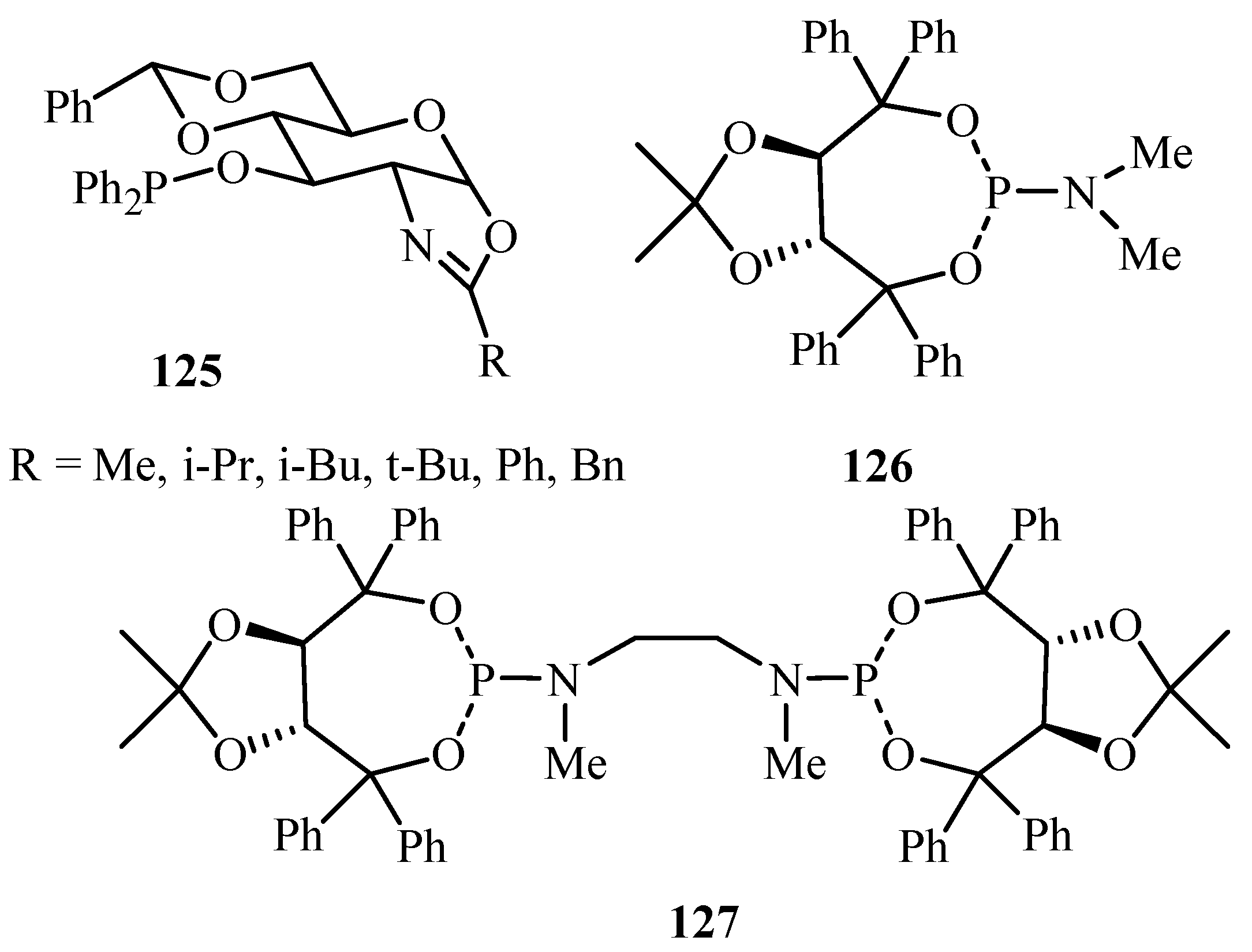
Scheme 4.
Asymmetric inter- and intramolecular Heck reaction.
Figure 16.
Chelation controlled inter- and intramolecular Heck reaction.
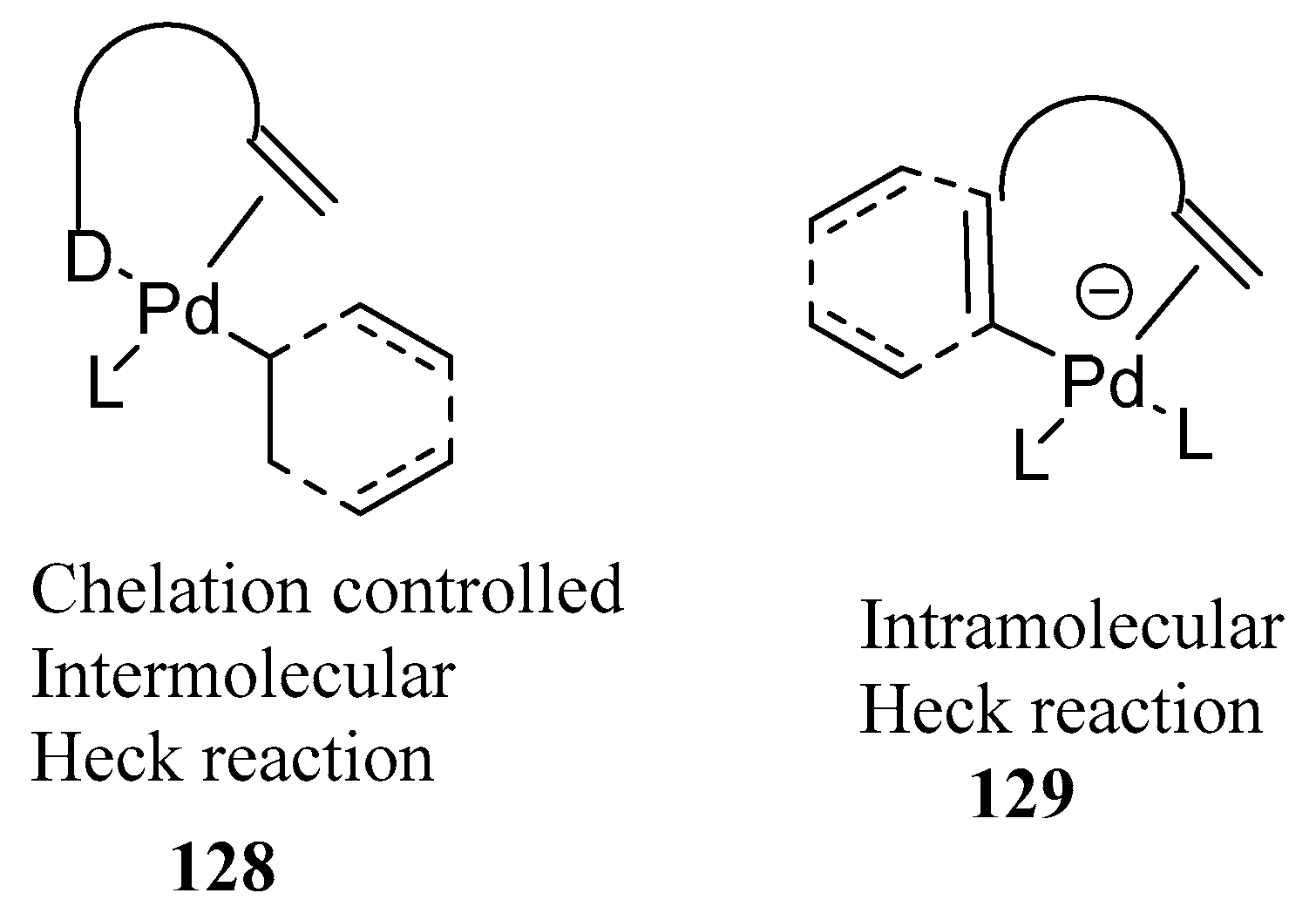
Figure 17.
Water-soluble phosphines reviewed by Genet and Savignac [62].
Figure 17.
Water-soluble phosphines reviewed by Genet and Savignac [62].
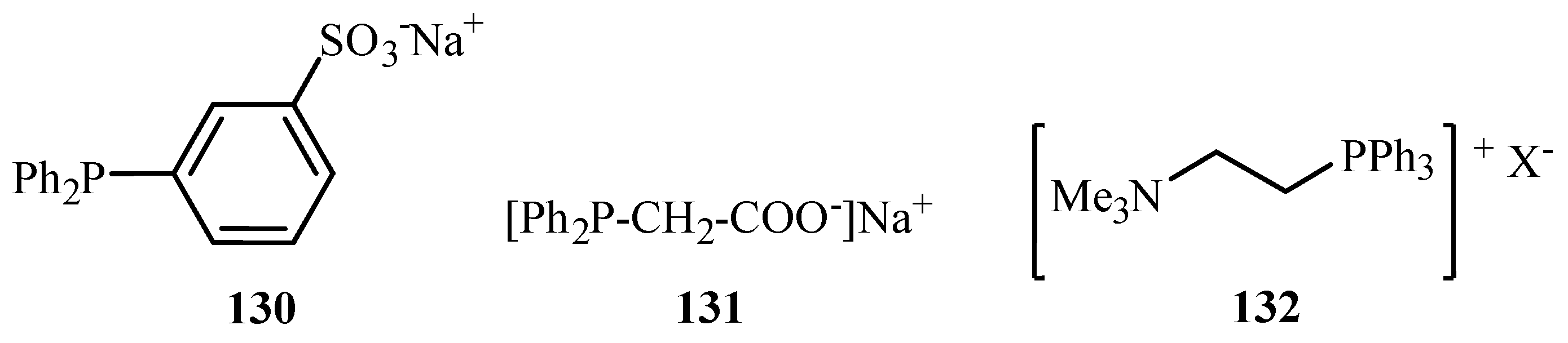
Figure 18.
N-heterocyclic carbene ligand and transition metal complex for catalysis in water reviewed by Velazquez and Verpoort [64].
Figure 18.
N-heterocyclic carbene ligand and transition metal complex for catalysis in water reviewed by Velazquez and Verpoort [64].
Scheme 5.
Mechanism based on Pd(0)/Pd(II) cycle by Cabri and Candiani [66].
Scheme 5.
Mechanism based on Pd(0)/Pd(II) cycle by Cabri and Candiani [66].

Scheme 6.
Olefin substituents via coordination-insertion process.
Scheme 7.
Intermediates generated in Heck reaction mechanism.
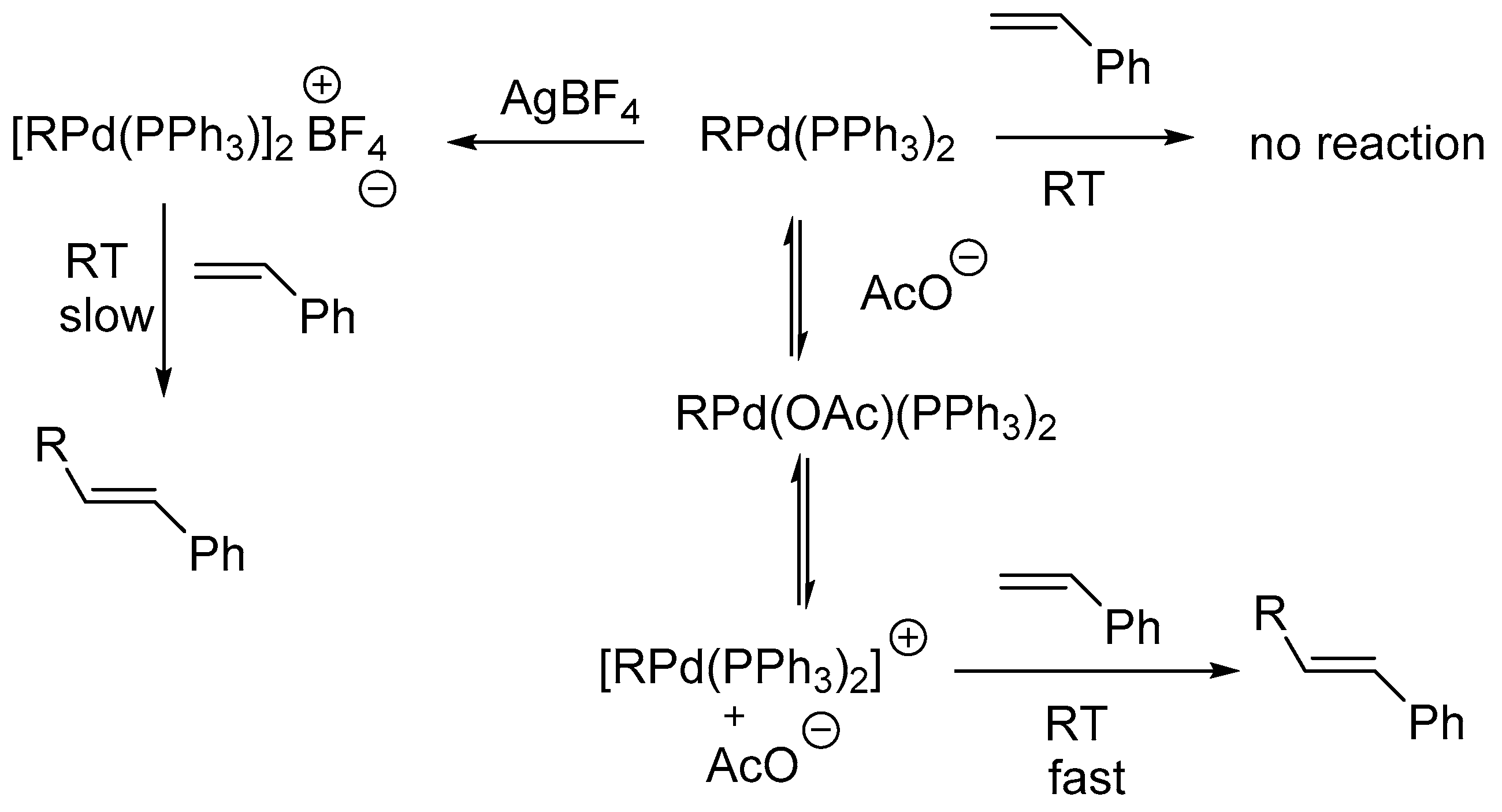
Scheme 8.
A new catalytic cycle reviewed by Amatore et al. [69].
Scheme 8.
A new catalytic cycle reviewed by Amatore et al. [69].
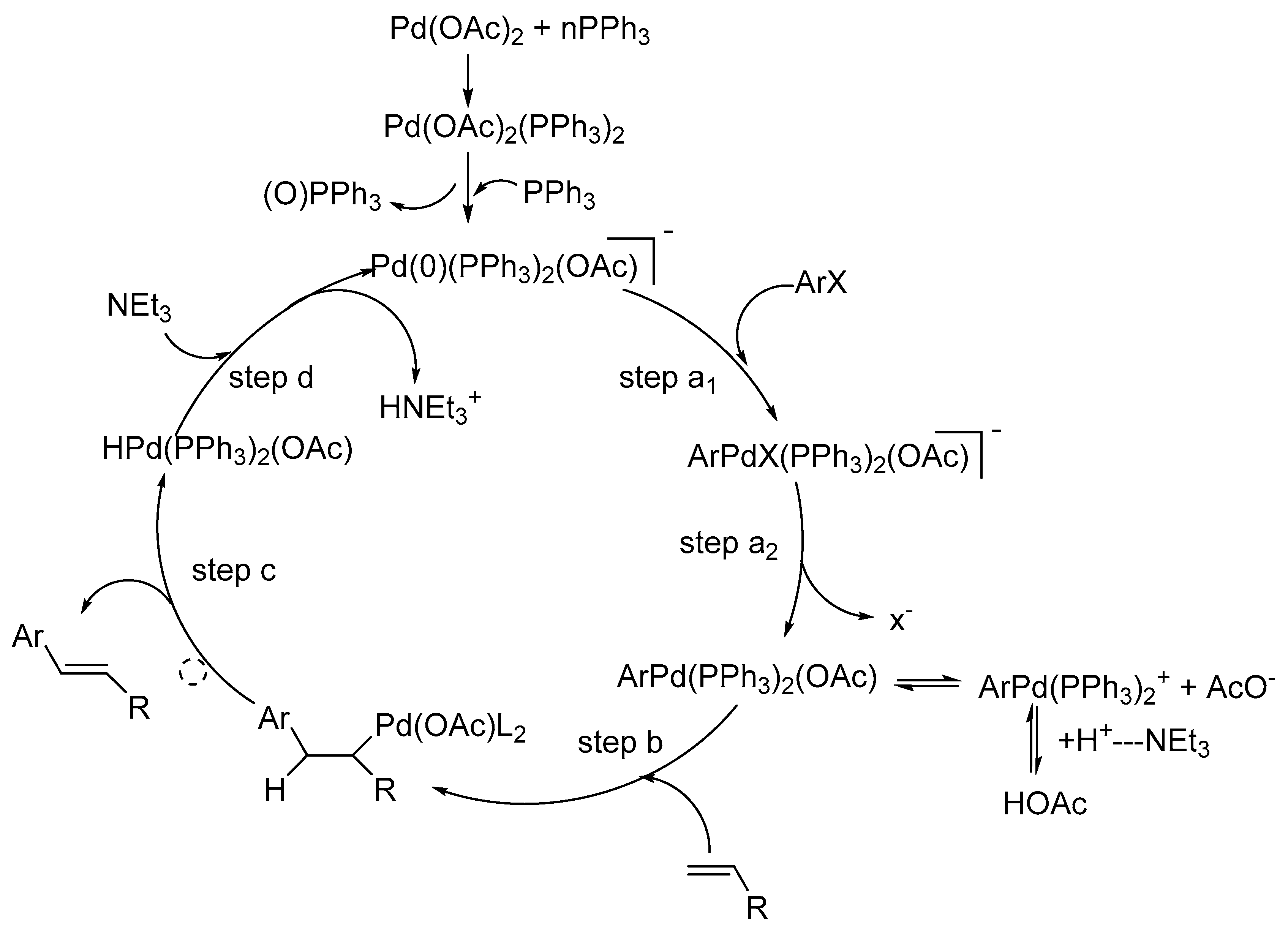
Figure 19.
Intermediate anionic pentacoordinated arylpalladium(II) complexes formed in Heck reaction.
Figure 19.
Intermediate anionic pentacoordinated arylpalladium(II) complexes formed in Heck reaction.

Scheme 9.
An alternative mechanism for Heck reaction via Pd(II)/Pd(IV) cycle.

Scheme 10.
Synthetic route for Octyl-4-methoxy cinnamate by Dead Sea Company.
Scheme 11.
Synthetic route for Naproxen by Albemarle.
Scheme 12.
Synthetic route for Prosulfuron intermediate by Ciba-Geigy/Novartis.

Figure 20.
Fine chemical products produced on a scale in excess of 1 ton/year using Heck or Heck type reaction reviewed by deVries [78].
Figure 20.
Fine chemical products produced on a scale in excess of 1 ton/year using Heck or Heck type reaction reviewed by deVries [78].
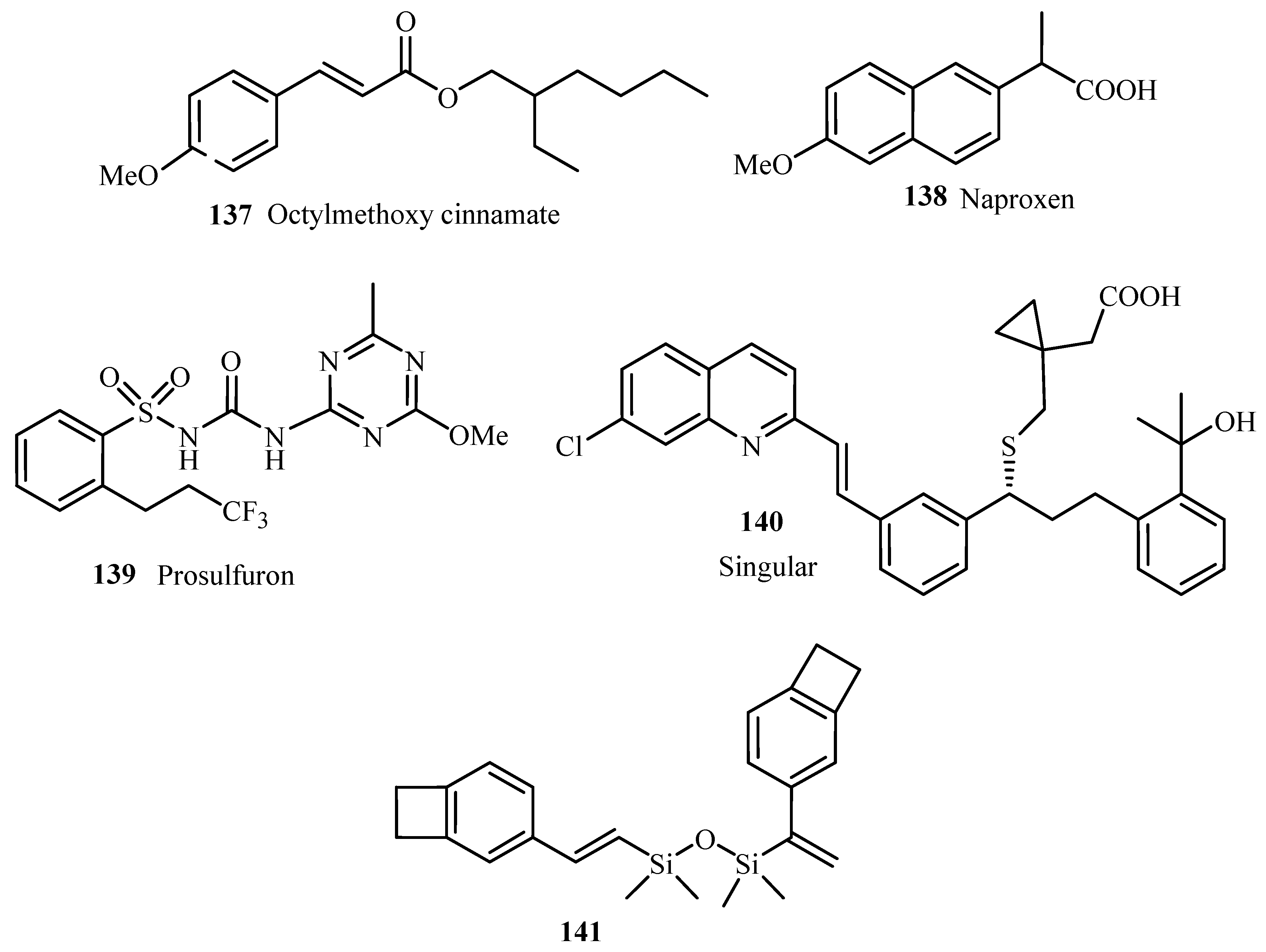
Figure 21.
Additional fine chemical products produced on industrial scale using Heck reaction reviewed by Zapf and Beller [79].
Figure 21.
Additional fine chemical products produced on industrial scale using Heck reaction reviewed by Zapf and Beller [79].
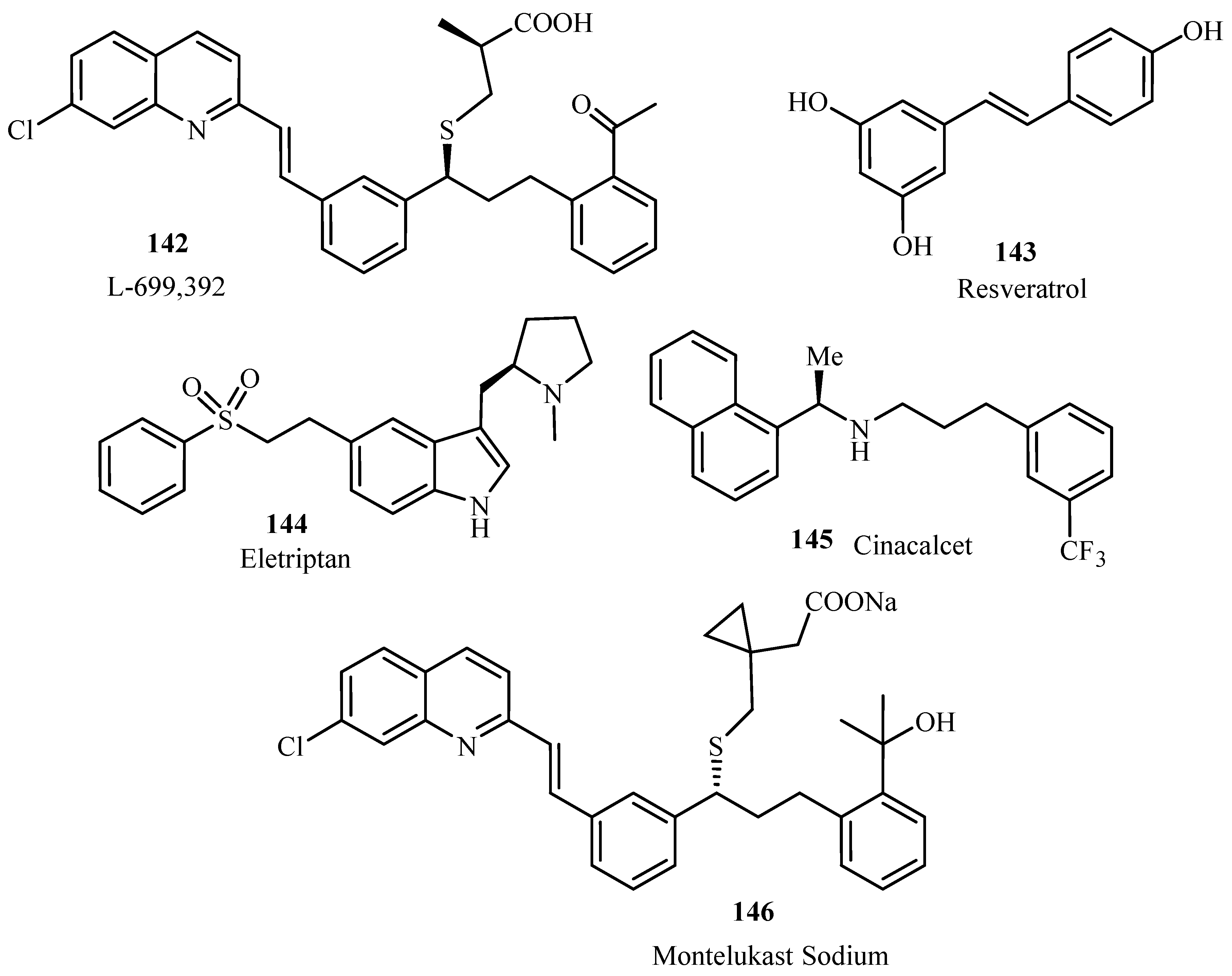
Figure 22.
Cinnamic acid derivatives (CADs) reviewed by Sharma [83].
Figure 22.
Cinnamic acid derivatives (CADs) reviewed by Sharma [83].

Figure 23.
Drug components or drug candidates exploited for inter and intramolecular Heck coupling by Biajoli et al. [85].
Figure 23.
Drug components or drug candidates exploited for inter and intramolecular Heck coupling by Biajoli et al. [85].
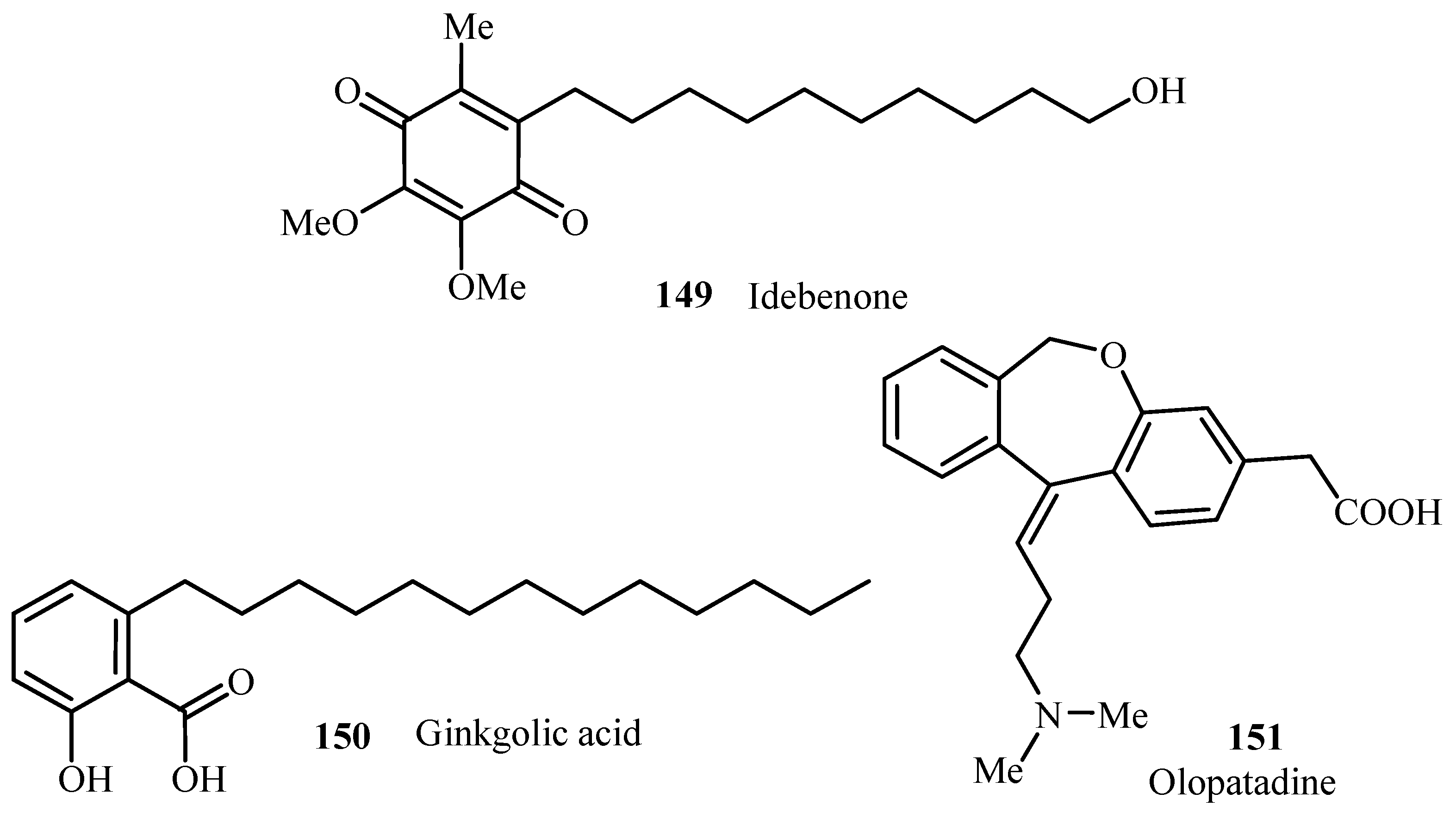

Figure 24.
Lab scale preparation drugs reviewed by Biajoli et al. [85].
Figure 24.
Lab scale preparation drugs reviewed by Biajoli et al. [85].

Figure 25.
Heck reaction in Total synthesis reviewed by Nicolaou et al. [88].
Figure 25.
Heck reaction in Total synthesis reviewed by Nicolaou et al. [88].
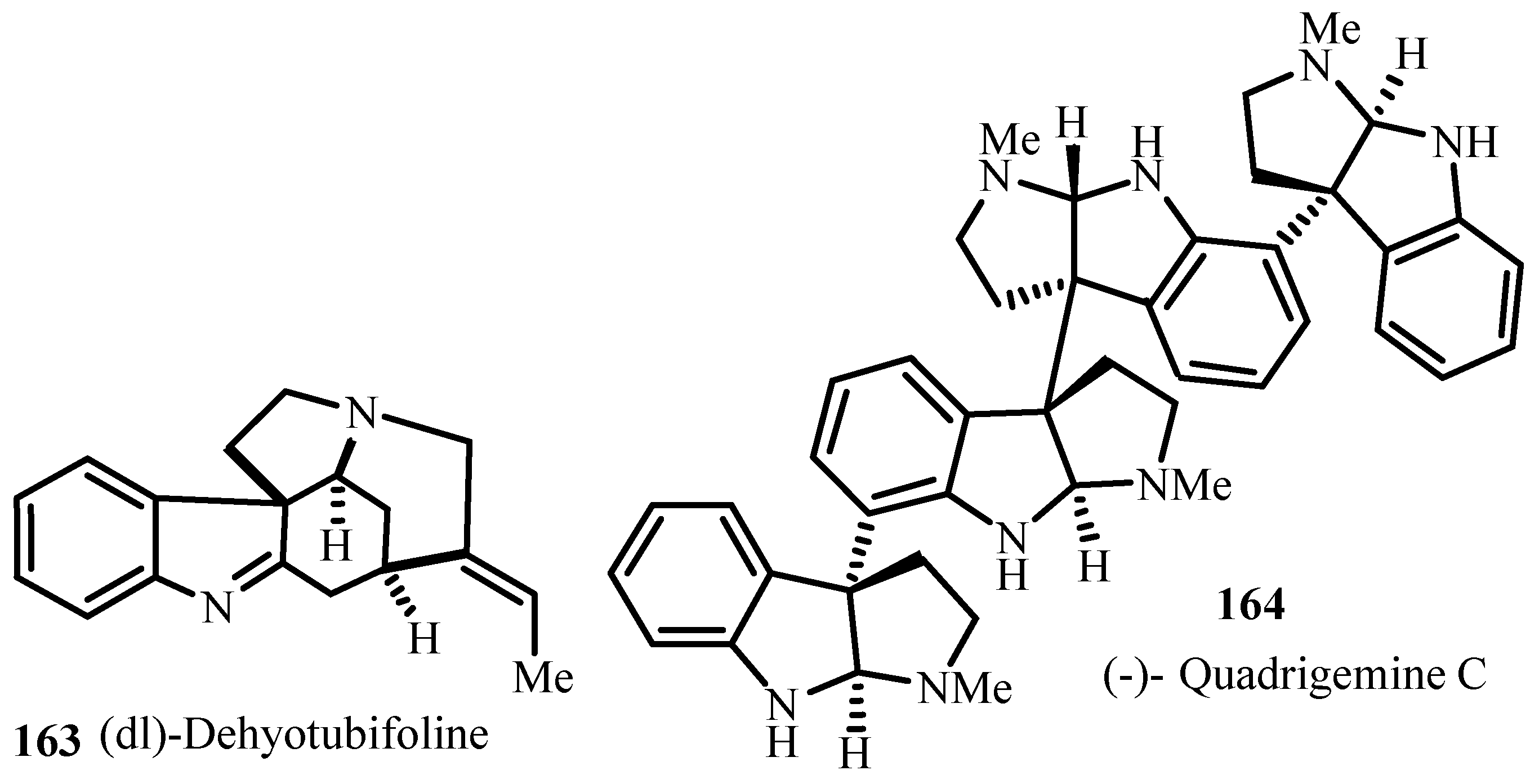
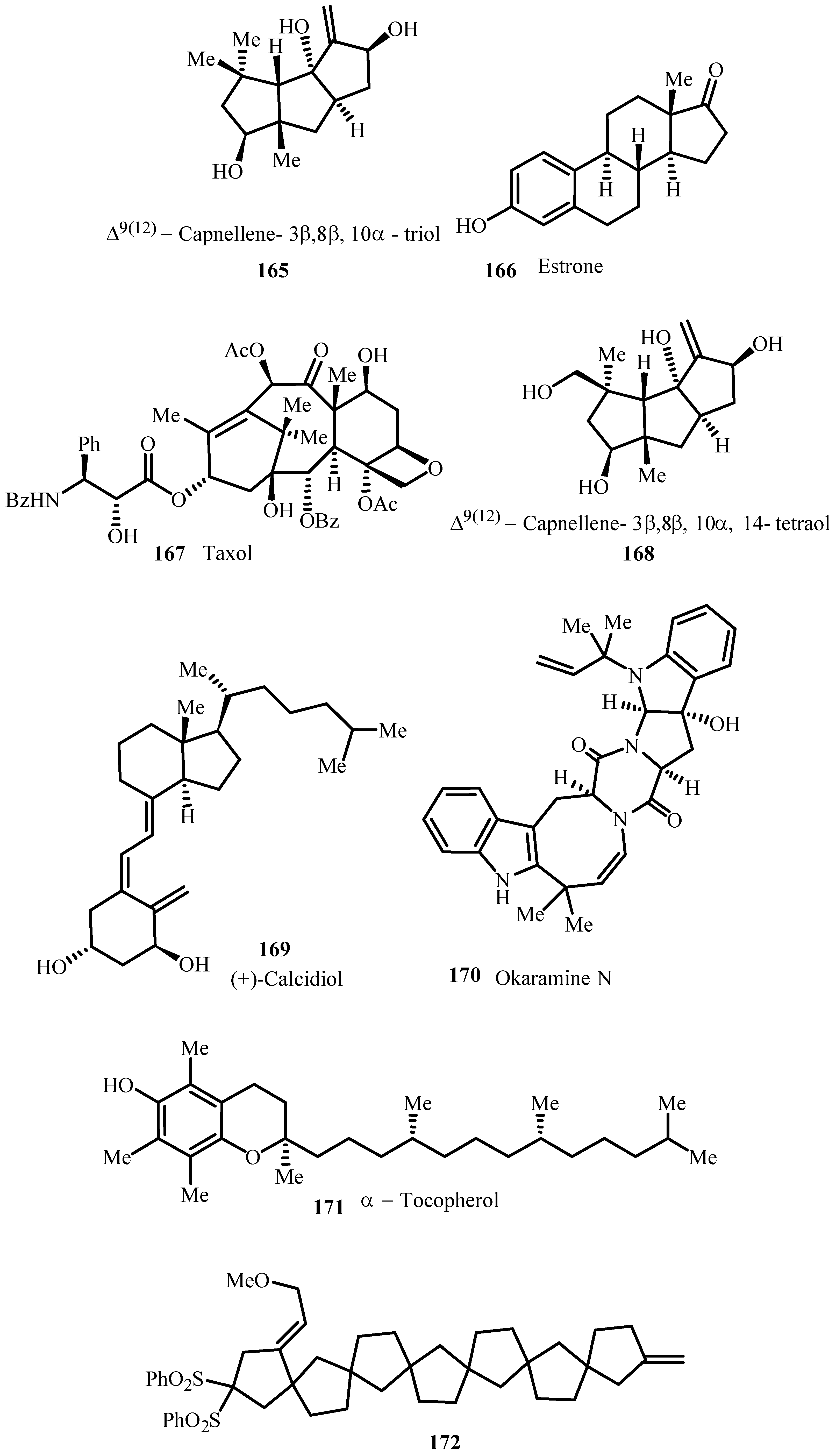
Figure 26.
Glycosides synthesized via Heck reaction reviewed by Wellington and Benner [92].
Figure 26.
Glycosides synthesized via Heck reaction reviewed by Wellington and Benner [92].

Figure 27.
Dendrimer supported catalysts.
Figure 28.
Structure of ionic liquids used for Heck reaction reviewed by Sheldon [101].
Figure 28.
Structure of ionic liquids used for Heck reaction reviewed by Sheldon [101].
Figure 29.
Charge-tagged ligands (CTLs) reviewed by Limberger et al. [106].
Figure 29.
Charge-tagged ligands (CTLs) reviewed by Limberger et al. [106].
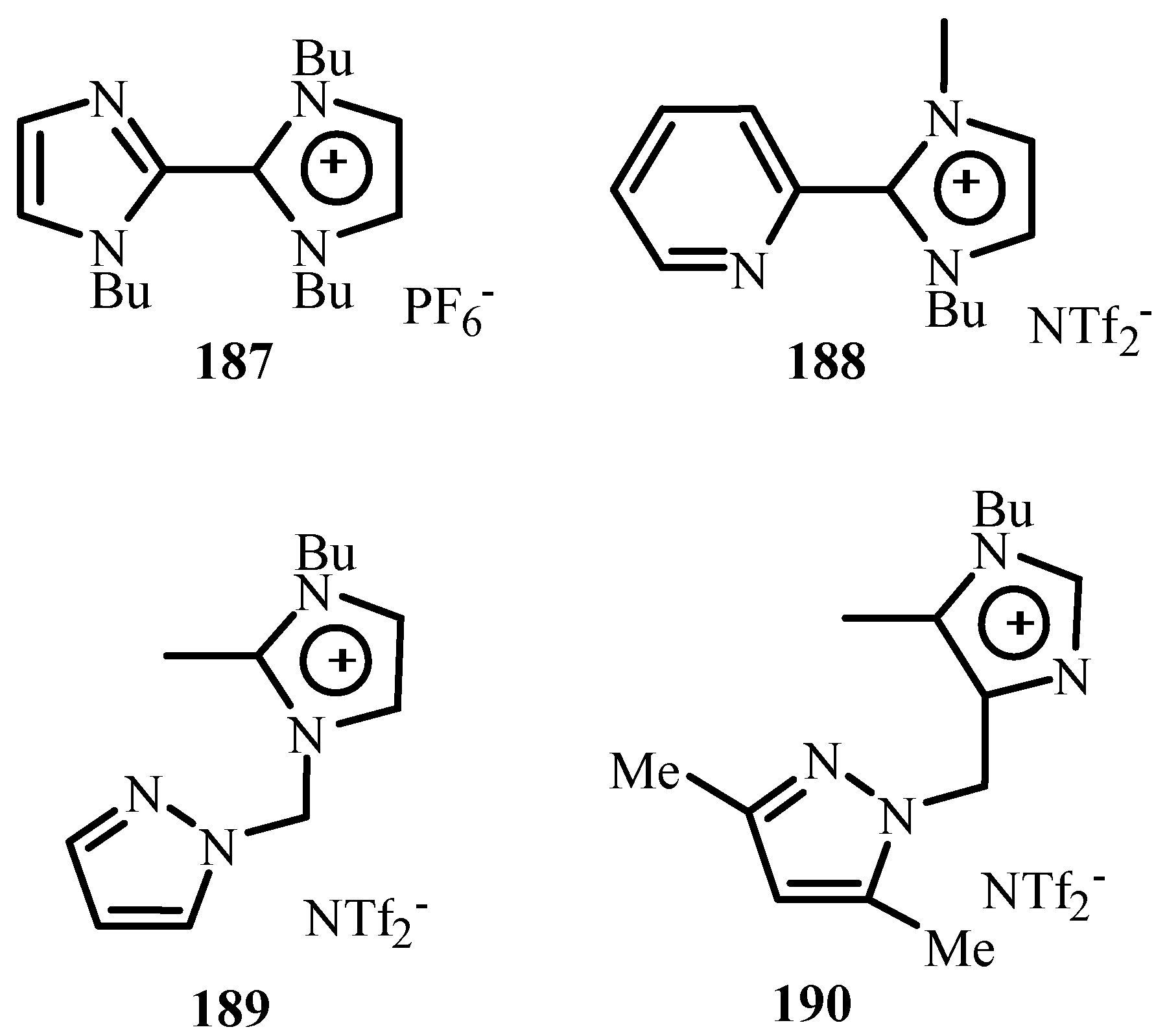
Figure 30.
(tris(m-carboxyphenyl) phosphine trilithium salt (m-TPPTC)) and (p-TPPTC) (TPPTC: tris(m-carboxyphenyl) phosphine trilithium salt).
Figure 30.
(tris(m-carboxyphenyl) phosphine trilithium salt (m-TPPTC)) and (p-TPPTC) (TPPTC: tris(m-carboxyphenyl) phosphine trilithium salt).
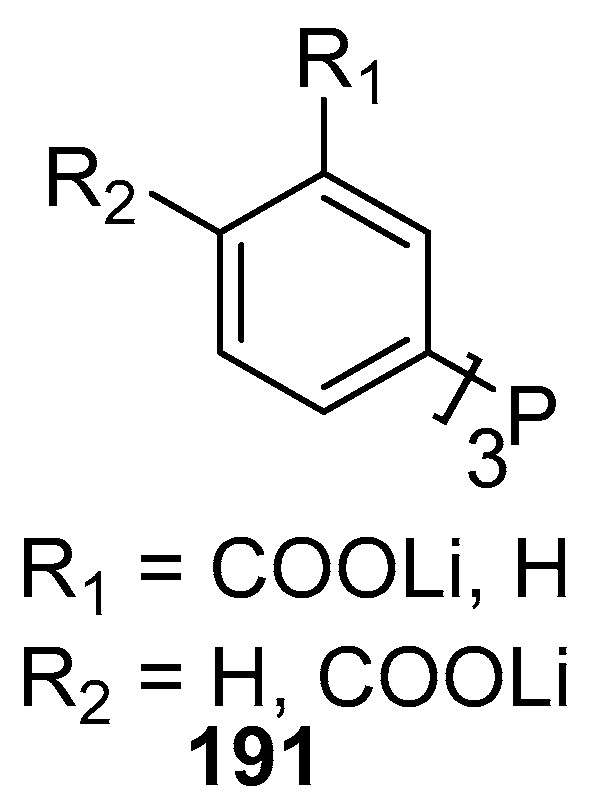
© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jagtap, S. Heck Reaction—State of the Art. Catalysts 2017, 7, 267. https://doi.org/10.3390/catal7090267
AMA Style
Jagtap S. Heck Reaction—State of the Art. Catalysts. 2017; 7(9):267. https://doi.org/10.3390/catal7090267
Chicago/Turabian StyleJagtap, Sangeeta. 2017. "Heck Reaction—State of the Art" Catalysts 7, no. 9: 267. https://doi.org/10.3390/catal7090267
APA StyleJagtap, S. (2017). Heck Reaction—State of the Art. Catalysts, 7(9), 267. https://doi.org/10.3390/catal7090267
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.