Mechanistic Insight into the 2° Alcohol Oxidation Mediated by an Efficient CuI/L-Proline-TEMPO Catalyst—A Density Functional Theory Study


Abstract
:
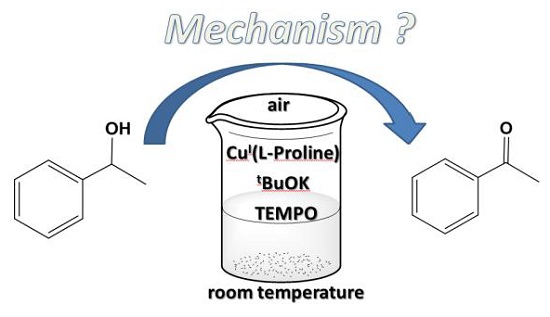
1. Introduction
2. Results and Discussion
2.1. Reaction Models
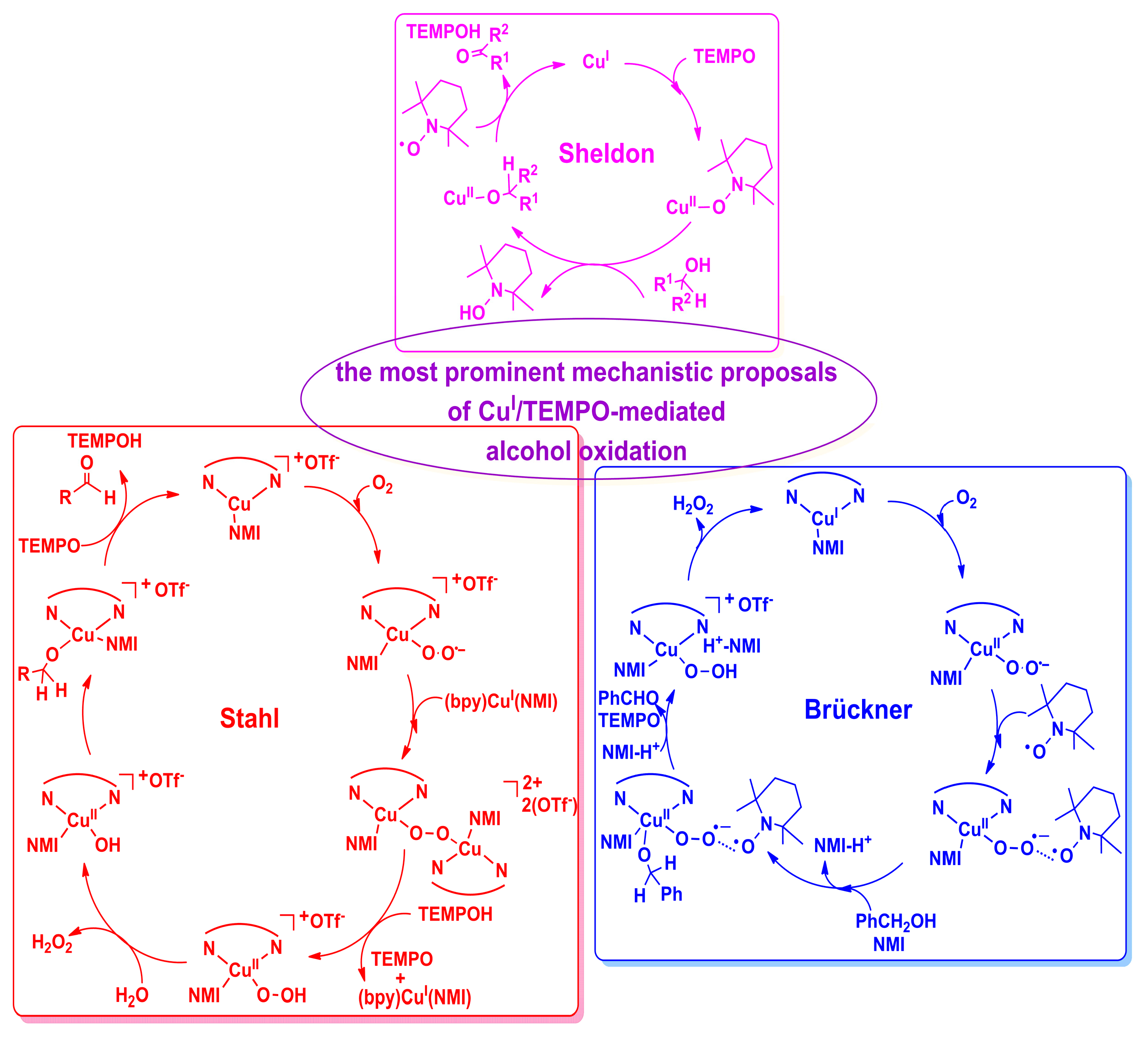
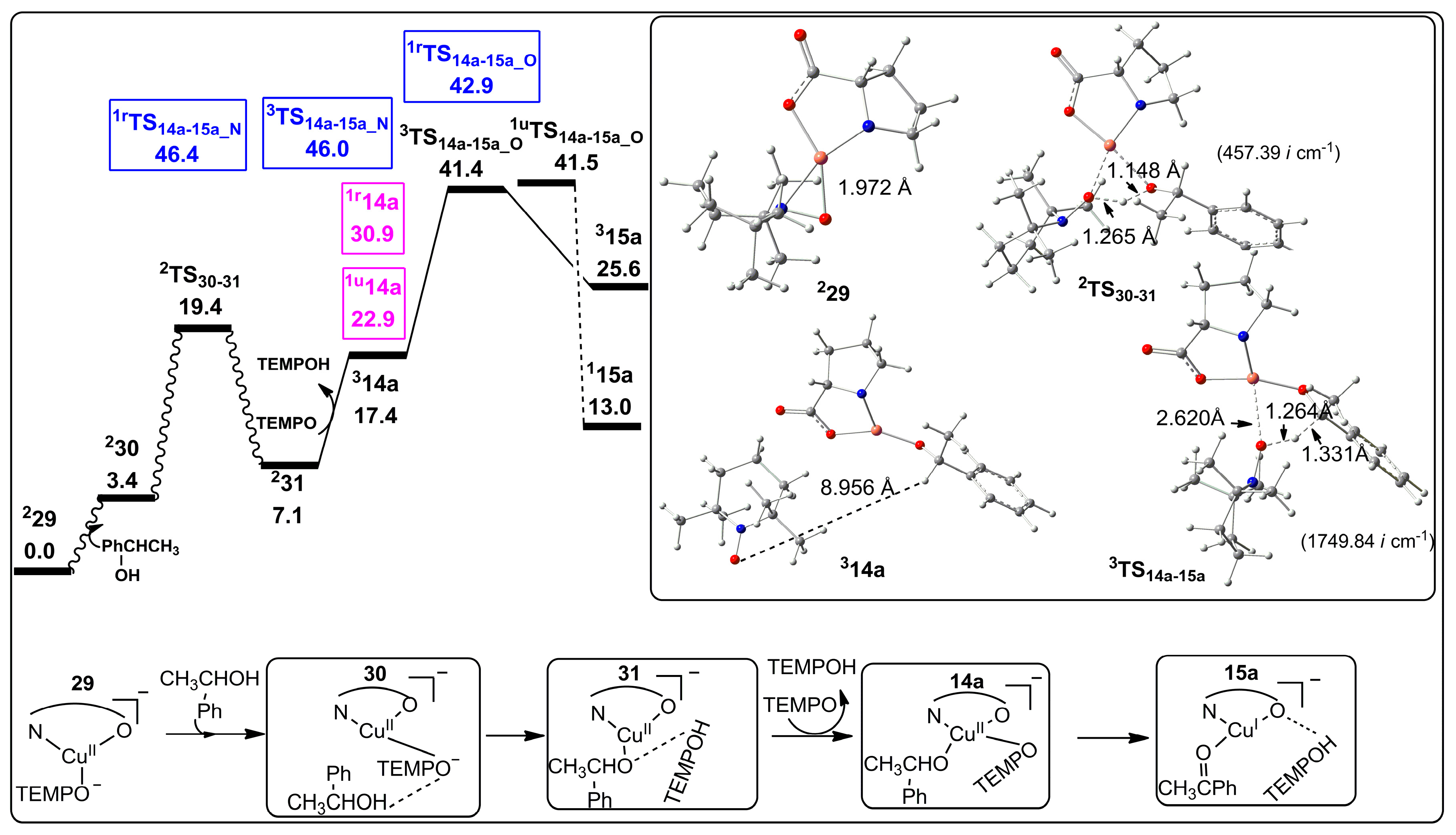
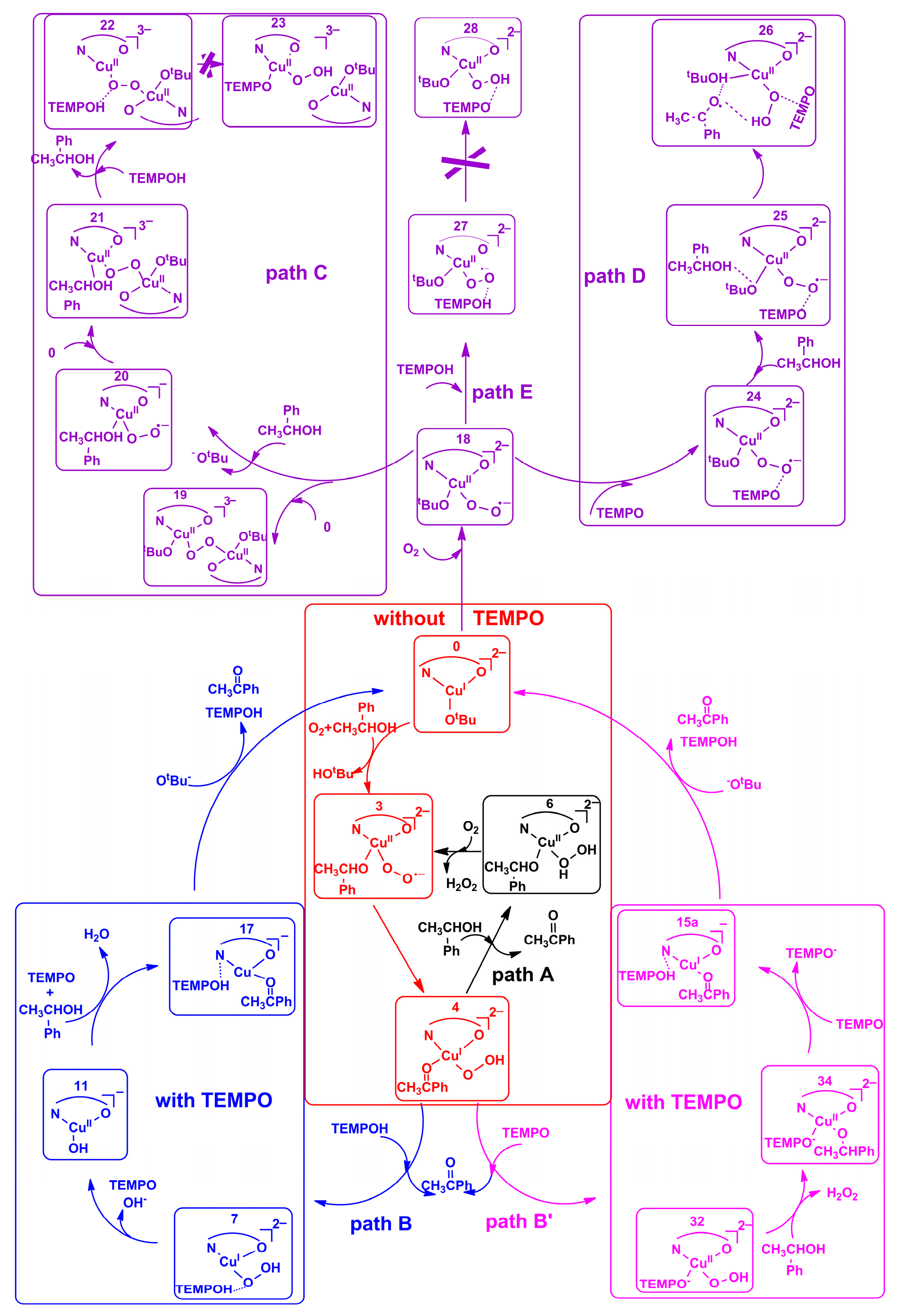
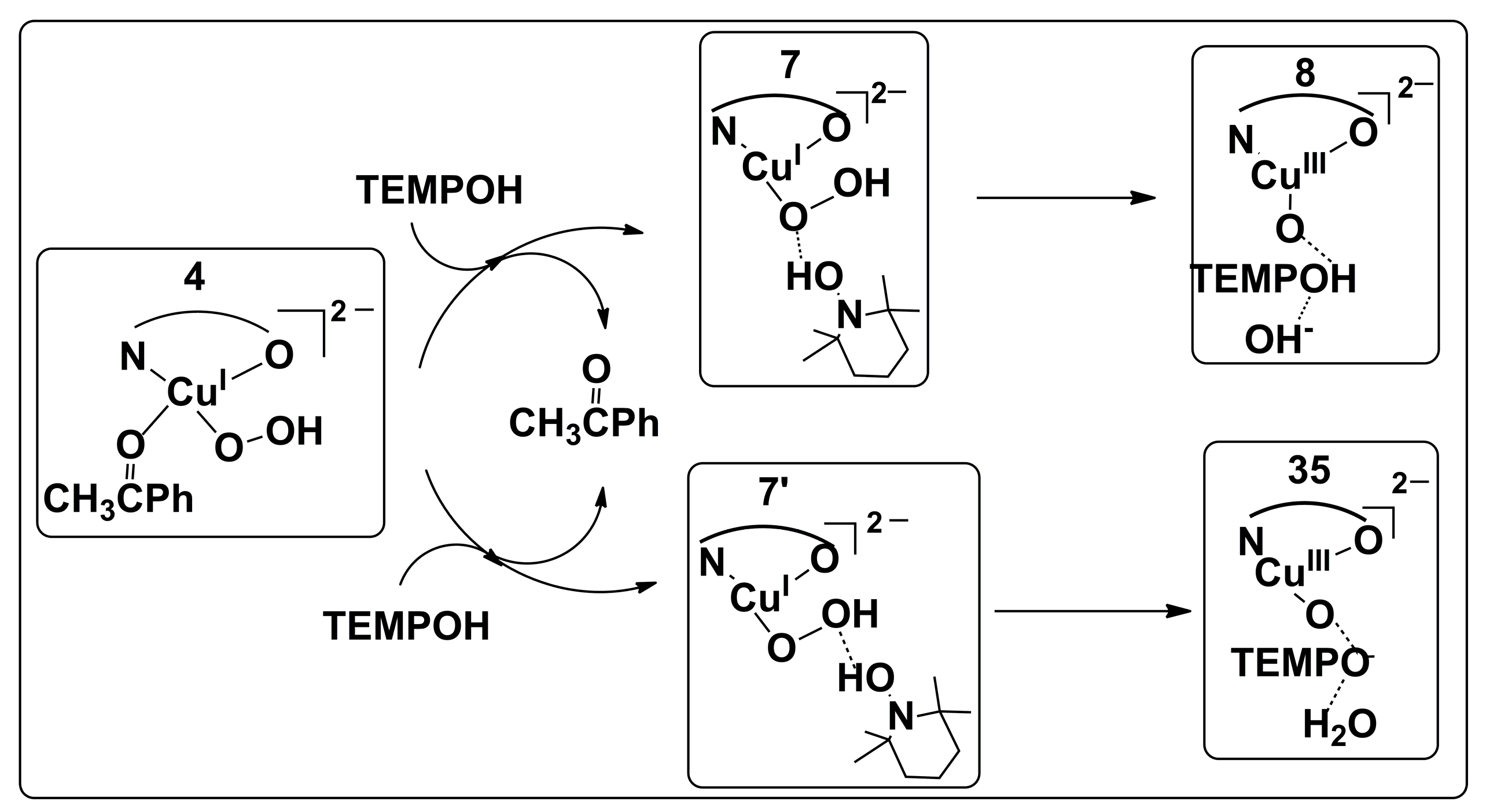
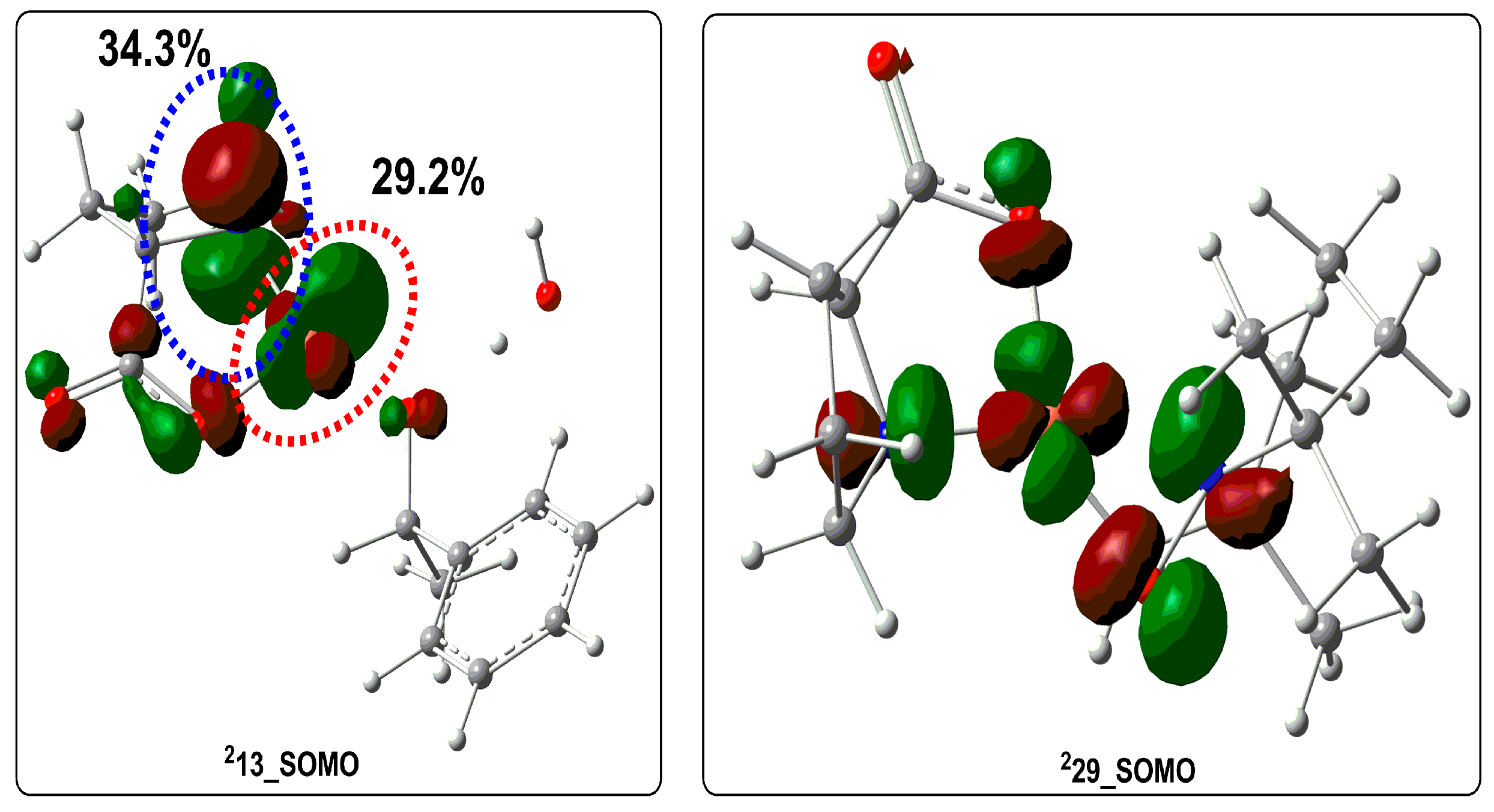
2.2. Possible Reaction Mechanisms
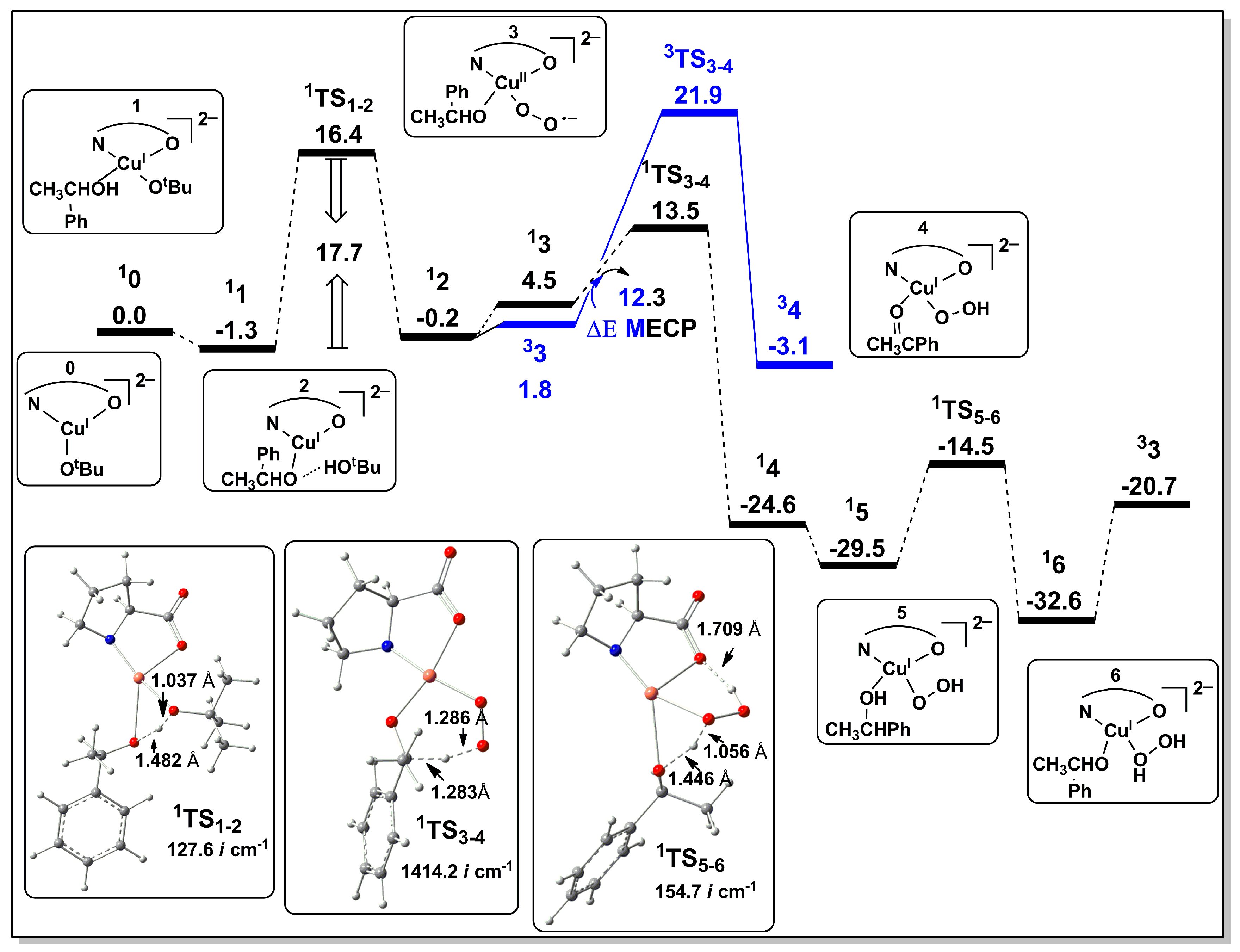
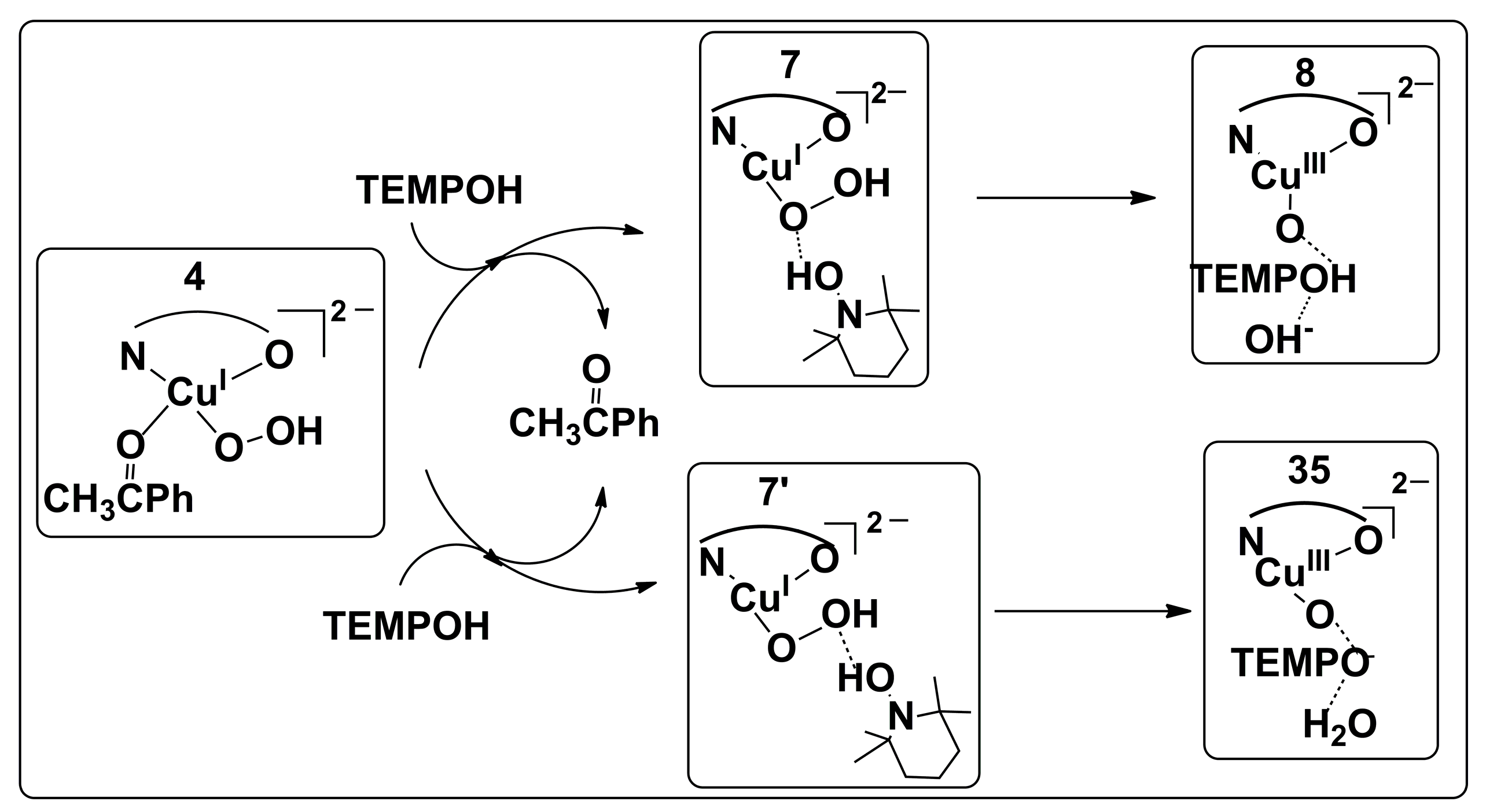
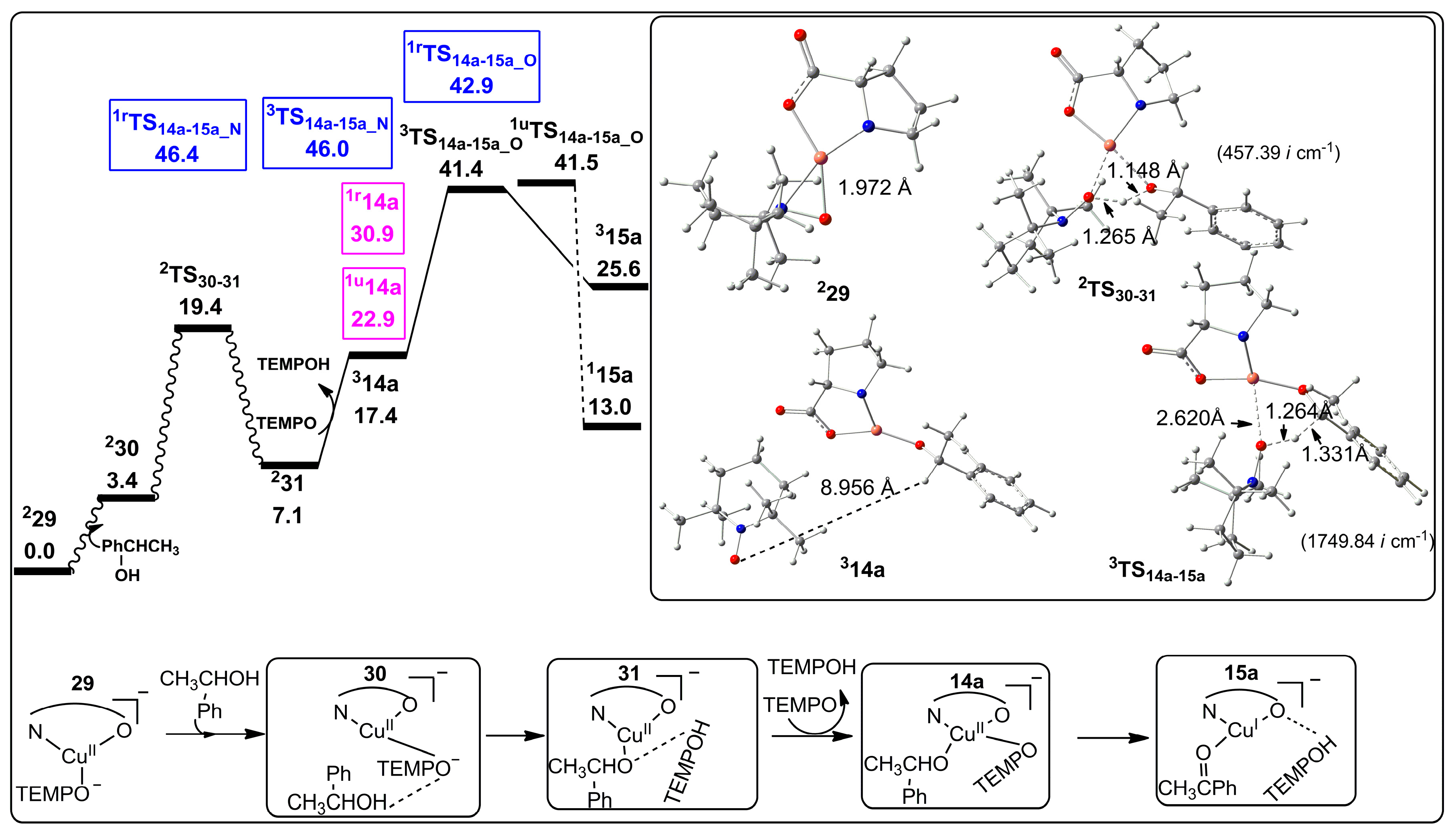
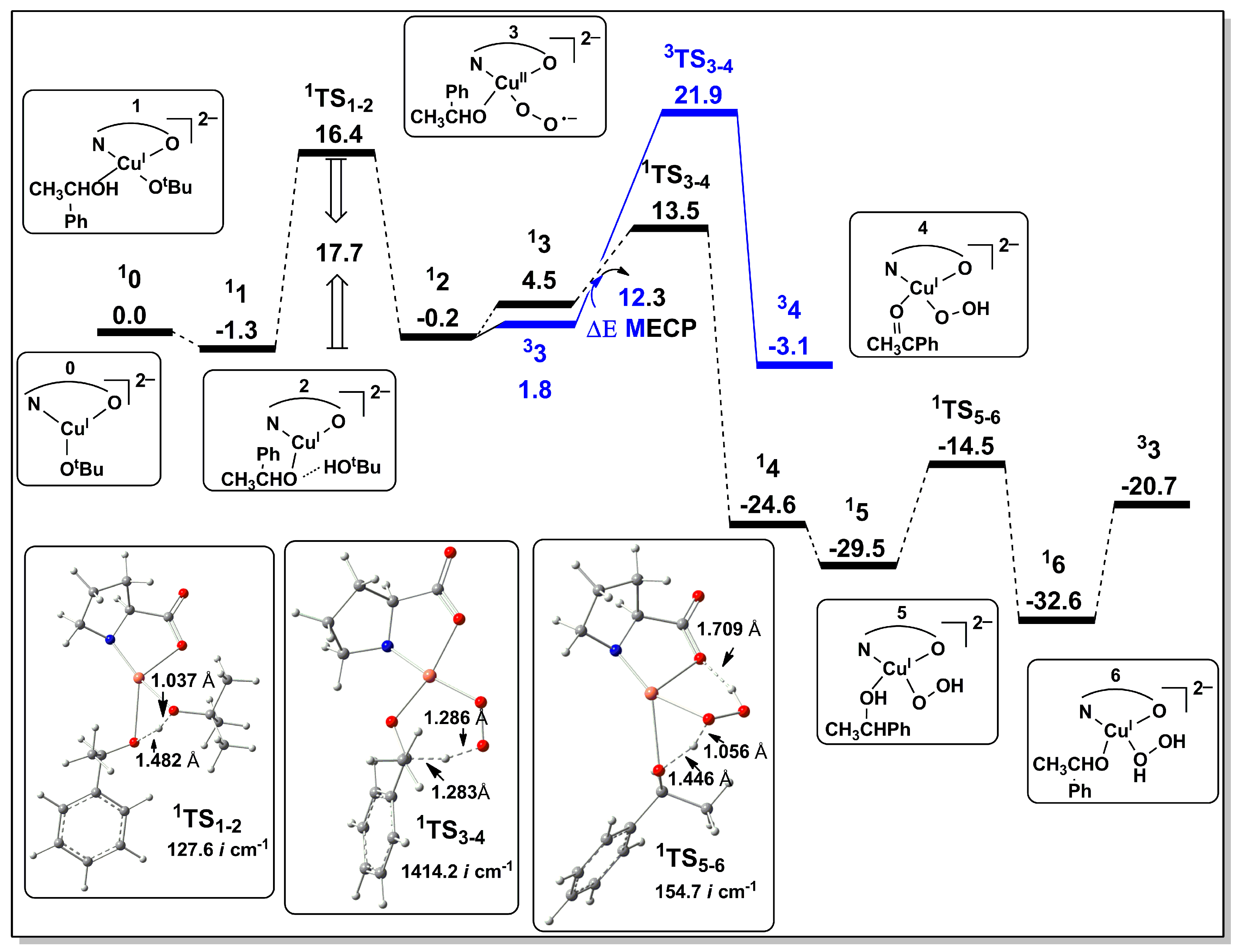
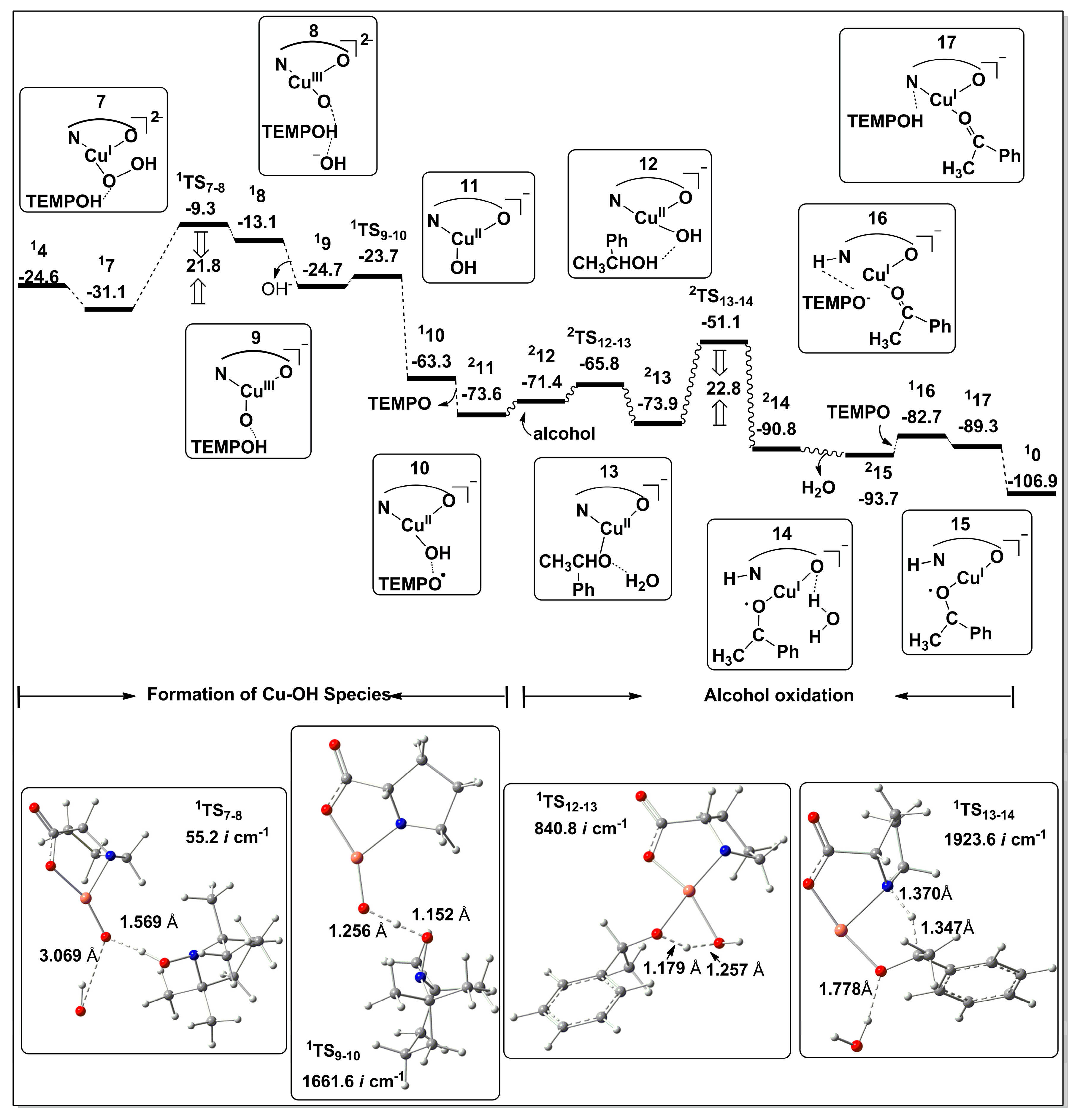
2.2.1. Catalyst Model A
2.2.2. Catalyst Model B
2.2.3. Preliminary Mechanistic Assessment
3. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Parmeggiani, C.; Cardona, F. Transition metal based catalysts in the aerobic oxidation of alcohols. Green Chem. 2012, 14, 547–564. [Google Scholar] [CrossRef]
- Tojo, G.; Fernandez, M. Oxidations Mediated by TEMPO and Related Stable Nitroxide Radicals (Anelli Oxidation). In Oxidation of Alcohols to Aldehydes and Ketones; Tojo, G., Ed.; Springer: New York, NY, USA, 2010; pp. 241–253. [Google Scholar]
- Mallat, T.; Baiker, A. Oxidation of alcohols with molecular oxygen on solid catalysts. Chem. Rev. 2004, 104, 3037–3058. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.J.; Sigman, M.S. Recent advances in homogeneous transition metal-catalyzed aerobic alcohol oxidations. Tetrahedron 2006, 62, 8227–8241. [Google Scholar] [CrossRef]
- Brink, G.J.T.; Arends, I.W.C.E.; Sheldon, R.A. Green, Catalytic oxidation of alcohols in water. Science 2000, 287, 1636–1639. [Google Scholar] [CrossRef] [PubMed]
- Sigman, M.S.; Jensen, D.R. Ligand-modulated palladium-catalyzed aerobic alcohol oxidations. Acc. Chem. Res. 2006, 39, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Tang, S.; Lei, A.W. Oxidant controlled Pd-catalysed selective oxidation of primary alcohols. Chem. Commun. 2013, 49, 1324–1326. [Google Scholar] [CrossRef] [PubMed]
- Guan, B.T.; Xing, D.; Cai, G.X.; Wan, X.B.; Yu, N.; Fang, Z.; Yang, L.P.; Shi, Z.J. Highly selective aerobic oxidation of alcohol catalyzed by a gold(I) complex with an anionic ligand. J. Am. Chem. Soc. 2005, 127, 18004–18005. [Google Scholar] [CrossRef] [PubMed]
- Miyamura, H.; Matsubara, R.; Miyazaki, Y.; Kobayashi, S. Aerobic oxidation of alcohols at room temperature and atmospheric conditions catalyzed by reusable gold nanoclusters stabilized by the benzene rings of polystyrene derivatives. Angew. Chem. Int. Ed. 2007, 46, 4151–4154. [Google Scholar] [CrossRef] [PubMed]
- Karimi, B.; Esfahani, F.K. Gold nanoparticles supported on the periodic mesoporous organosilicas as efficient and reusable catalyst for room temperature aerobic oxidation of alcohols. Adv. Synth. Catal. 2012, 354, 1319–1326. [Google Scholar] [CrossRef]
- Dijksma, A.; Marino-Gonzalez, A.; Payeras, A.M.; Arends, I.W.C.E.; Sheldon, R.A. Efficient and selective aerobic oxidation of alcohols into aldehydes and ketones using ruthenium/TEMPO as the catalytic system. J. Am. Chem. Soc. 2001, 123, 6826–6833. [Google Scholar] [CrossRef]
- Lenz, R.; Ley, S.V. Tetra-n-propylammonium perruthenate (TPAP)-catalysed oxidations of alcohols using molecular oxygen as a co-oxidant. J. Chem. Soc. Perkin Trans. 1 1997, 1, 3291–3292. [Google Scholar] [CrossRef]
- Hasan, M.; Musawir, M.; Kozhevnikov, I.V. Oxidation of primary alcohols to aldehydes with oxygen catalysed by tetra-n-propylammonium perruthenate. J. Mol. Catal. A 2002, 180, 77–84. [Google Scholar] [CrossRef]
- Mizuno, N.; Yamaguchi, K. Selective aerobic oxidations by supported ruthenium hydroxide catalysts. Catal. Today 2008, 132, 18–26. [Google Scholar] [CrossRef]
- Semmelhack, M.F.; Schmid, C.R.; Cortes, D.A.; Chou, C.S. Oxidation of alcohols to aldehydes with oxygen and cupric ion, mediated by nitrosonium ion. J. Am. Chem. Soc. 1984, 106, 3374–3376. [Google Scholar] [CrossRef]
- Jiang, N.; Ragauskas, A.J. Cu(II)-catalyzed selective aerobic oxidation of alcohols under mild conditions. J. Org. Chem. 2006, 71, 7087–7090. [Google Scholar] [CrossRef] [PubMed]
- Figiel, P.J.; Sibaouih, A.; Ahmad, J.U.; Nieger, M.; Räisänen, M.T.; Leskelä, M.; Repo, T. Aerobic oxidation of benzylic alcohols in water by 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO)/Copper(II) 2-N-arylpyrrolecarbaldimino complexes. Adv. Synth. Catal. 2009, 351, 2625–2632. [Google Scholar] [CrossRef]
- Mannam, S.; Alamsetti, S.K.; Sekar, G. Aerobic, Chemoselective oxidation of alcohols to carbonyl compounds catalyzed by a DABCO-copper complex under mild conditions. Adv. Synth. Catal. 2007, 349, 2253–2258. [Google Scholar] [CrossRef]
- Marko, I.E.; Gautier, A.; Dumeunier, R.; Philippart, F.; Brown, S.M.; Urch, C.J. Efficient, Copper-catalyzed, aerobic oxidation of primary alcohols. Angew. Chem. Int. Ed. 2004, 43, 1588–1591. [Google Scholar] [CrossRef] [PubMed]
- Hoover, J.M.; Stahl, S.S. Highly Practical Copper(I)/TEMPO Catalyst system for chemoselective aerobic oxidation of primary alcohols. J. Am. Chem. Soc. 2011, 133, 16901–16910. [Google Scholar] [CrossRef] [PubMed]
- Steves, J.E.; Preger, Y.; Martinelli, J.R.; Welch, J.C.; Root, T.W.; Hawkins, J.M.; Stahl, S.S. Process development of CuI/ABNO/NMI-catalyzed aerobic alcohol oxidation. Org. Process Res. Dev. 2015, 19, 1548–1553. [Google Scholar] [CrossRef]
- Wang, L.Y.; Bie, Z.X.; Shang, S.S.; Lv, Y.; Li, G.S.; Niu, J.Y.; Gao, S. Bioinspired aerobic oxidation of alcohols with a bifunctional ligand based on bipyridine and TEMPO. RSC Adv. 2016, 6, 35008. [Google Scholar] [CrossRef]
- Markó, I.E.; Giles, P.R.; Tsukazaki, M.; Brown, S.M.; Urch, C.J.; Galano, A. Copper-catalyzed oxidation of alcohols to aldehydes and ketones: an efficient, aerobic alternative. Science 1996, 274, 2044–2046. [Google Scholar] [CrossRef] [PubMed]
- Markó, I.E.; Gautier, A.; Mutonkole, J.-L.; Dumeunier, R.; Ates, A.; Urch, C.J.; Brown, S.M. Neutral, non-racemising, catalytic aerobic oxidation of alcohols. J. Organomet. Chem. 2001, 624, 344–347. [Google Scholar] [CrossRef]
- Betzemeier, B.; Cavazzini, M.; Quici, S.; Knochel, P. Copper-catalyzed aerobic oxidation of alcohols under fuorous biphasic conditions. Tetrahedron Lett. 2000, 41, 4343–4346. [Google Scholar] [CrossRef]
- Steves, J.E.; Stahl, S.S. Copper(I)/ABNO-catalyzed aerobic alcohol oxidation: Alleviating steric and electronic constraints of Cu/TEMPO catalyst systems. J. Am. Chem. Soc. 2013, 135, 15742–15745. [Google Scholar] [CrossRef] [PubMed]
- Ryland, B.L.; Stahl, S.S. Practical aerobic oxidations of alcohols and amines with homogeneous copper/TEMPO and related catalyst systems. Angew. Chem. Int. Ed. 2014, 53, 8824–8838. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, M.; Miles, K.C.; Stahl, S.S. Electrocatalytic alcohol oxidation with TEMPO and bicyclic nitroxyl derivatives: Driving force trumps steric effects. J. Am. Chem. Soc. 2015, 137, 14751–14757. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.F.; Han, X.W.; Luan, Y.X.; Wang, Y.; Wen, X.; Ding, C.R. L-Proline: an efficient N,O-bidentate ligand for copper-catalyzed aerobic oxidation of primary and secondary benzylic alcohols at room temperature. Chem. Commun. 2013, 49, 7908–7910. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A. Recent advances in green catalytic oxidations of alcohols in aqueous media. Catal. Today 2015, 247, 4–13. [Google Scholar] [CrossRef]
- Hoover, J.M.; Ryland, B.L.; Stahl, S.S. Mechanism of copper(I)/TEMPO-catalyzed aerobic alcohol oxidation. J. Am. Chem. Soc. 2013, 135, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Ryland, B.L.; McCann, S.D.; Brunold, T.C.; Stahl, S.S. Mechanism of alcohol oxidation mediated by copper(II) and nitroxyl radicals. J. Am. Chem. Soc. 2014, 136, 12166–12173. [Google Scholar] [CrossRef] [PubMed]
- Rabeah, J.; Bentrup, U.; Stöβer, R.; Brückner, A. Selective alcohol oxidation by a copper TEMPO catalyst: mechanistic insights by simultaneously coupled operando EPR/UV-Vis/ATR-IR spectroscopy. Angew. Chem. Int. Ed. 2015, 127, 11791–11794. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.N.; Aschi, M.; Schwarz, H.; Koch, W. The singlet and triplet states of phenyl cation. A hybrid approach for locating minimum energy crossing points between non-interacting potential energy surfaces. Theor. Chem. Acc. 1998, 99, 95–99. [Google Scholar] [CrossRef]
- Harvey, J.N.; Aschi, M. Spin-forbidden dehydrogenation of methoxy cation: A statistical view. Phys. Chem. Chem. Phys. 1999, 1, 5555–5563. [Google Scholar] [CrossRef]
- Poli, R.; Harvey, J.N. Spin forbidden chemical reactions of transition metal compounds. New ideas and new computational challenges. Chem. Soc. Rev. 2003, 32, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Shaik, S. How to conceptualize catalytic cycles? The energetic span model. Acc. Chem. Res. 2011, 44, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S. A refinement of everyday thinking: The energetic span model for kinetic assessment of catalytic cycles. WIREs Comput. Mol. Sci. 2012, 2, 795–815. [Google Scholar] [CrossRef]
- Kozuch, S.; Sason, S. A combined kinetic-quantum mechanical model for assessment of catalytic cycles: application to cross-coupling and heck reactions. J. Am. Chem. Soc. 2006, 128, 3355–3365. [Google Scholar] [CrossRef] [PubMed]
- Kozuch, S.; Shaik, S. Kinetic-quantum chemical model for catalytic cycles: The Haber-Bosch process and the effect of reagent concentration. J. Phys. Chem. A 2008, 112, 6032–6041. [Google Scholar] [CrossRef] [PubMed]
- Uhe, A.; Kozuch, S.; Shaik, S. Software news and update automatic analysis of computed catalytic cycles. J. Comput. Chem. 2010, 2011, 978–985. [Google Scholar]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.; Belanzoni, P.; Gamez, P.; Reedijk, J.; Baerends, E.J. Activation of the C-H bond by electrophilic attack: Theoretical study of the reaction mechanism of the aerobic oxidation of alcohols to aldehydes by the Cu(bipy)2+/2,2,6,6-Tetramethylpiperidinyl-1-oxy cocatalyst system. Inorg. Chem. 2009, 48, 11909–11920. [Google Scholar] [CrossRef] [PubMed]
- Belanzoni, P.; Michel, C.; Baerends, E.J. Cu(bipy)2+/TEMPO-catalyzed oxidation of alcohols: Radical or nonradical mechanism? Inorg. Chem. 2011, 50, 11896–11904. [Google Scholar] [CrossRef] [PubMed]
- Varela-Álvarez, A.; Liebeskind, L.S.; Musaev, D.G. Mechanistic insights into the aerobic copper(I)-catalyzed cross-coupling of S-Acyl thiosalicylamide thiol esters and boronic acids. Organometallics 2012, 31, 7958–7968. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A Gen. Phys. 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K. The path of chemical reactions-The IRC approach. Acc. Chem. Res. 1981, 14, 363–368. [Google Scholar] [CrossRef]
- Fukui, K. A Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. [Google Scholar] [CrossRef]
- Lu, G.; Fang, C.; Xu, T.; Dong, G.B.; Liu, P. Computational study of Rh-catalyzed carboacylation of olefins: Ligand-promoted rhodacycle isomerization enables regioselective C−C bond functionalization of benzocyclobutenones. J. Am. Chem. Soc. 2015, 137, 8274–8283. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.F.; Qu, S.L.; Tao, Y.; Deng, X.; Wang, Z.X. Mechanistic insight into ketone α-alkylation with unactivated olefins via C−H activation promoted by metal-organic cooperative catalysis (MOCC): Enriching the MOCC chemistry. J. Am. Chem. Soc. 2015, 137, 6279–6291. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewicz, A.; Miera, G.G.; Marcos, R.; Norrby, P.-O.G.; Martın-Matute, B. Mechanistic studies on the alkylation of amines with alcohols catalyzed by a bifunctional iridium complex. ACS Catal. 2015, 5, 3704–3716. [Google Scholar] [CrossRef]
- Herbert, M.B.; Suslick, A.; Liu, P.; Zou, L.; Dornan, P.K.; Houk, K.N.; Grubbs, R.H. Cyclometalated c with modified N-chelating groups. Organometallics 2015, 34, 2858–2869. [Google Scholar] [CrossRef]
- Luis, S.; Jonathan, M.G. How reliable are DFT transition structures? Comparison of GGA, hybrid-meta-GGA and meta-GGA functionals. Org. Biomol. Chem. 2011, 9, 689–700. [Google Scholar] [CrossRef]
- Cannon, J.S.; Zou, L.; Liu, P.; Lan, Y.; O’Leary, D.J.; Houk, K.N.; Grubbs, R.H. Carboxylate-assisted C(sp3)−H activation in olefin metathesis-relevant ruthenium complexes. J. Am. Chem. Soc. 2014, 136, 6733–6743. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Liang, Y.; Houk, K.N. Mechanisms and origins of switchable chemoselectivity of Ni-catalyzed C(aryl)−O and C(acyl)−O activation of aryl esters with phosphine ligands. J. Am. Chem. Soc. 2014, 136, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Qu, S.; Wang, Z.-X.; Wang, X. A Computational Mechanistic study of an unprecedented Heck-type relay reaction: insight into the origins of regio- and enantioselectivities. J. Am. Chem. Soc. 2014, 136, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, P. Mechanism and origins of selectivities in the copper-catalyzed dearomatization-induced ortho C−H cyanation of vinylarenes. ACS Catal. 2015, 5, 2944–2951. [Google Scholar] [CrossRef]
- Williams, T.J.; Bray, J.T.W.; Lake, B.R.M.; Willans, C.E.; Rajabi, N.A.; Ariafard, A.; Manzini, C.; Bellina, F.; Whitwood, A.C.; Fairlamb, I.J.S. Mechanistic elucidation of the arylation of non-spectator N-heterocyclic carbenes at copper using a combined experimental and computational approach. Organometallics 2015, 34, 3497–3507. [Google Scholar] [CrossRef]
- Zhao, G.M.; Liu, H.L.; Zhang, D.D.; Huang, X.R.; Yang, X. DFT Study on Mechanism of N-Alkylation of Amino Derivatives with Primary Alcohols Catalyzed by Copper(II) Acetate. ACS Catal. 2014, 4, 2231–2240. [Google Scholar] [CrossRef]
- Ariafard, A.; Zarkoob, F.; Batebi, H.; Stranger, R.; Yates, B. DFT studies on the carboxylation of the C−H bond of heteroarenes by copper(I) complexes. Organometallics 2011, 30, 6218–6224. [Google Scholar] [CrossRef]
- Deng, X.; Dang, Y.F.; Wang, Z.X.; Wang, X.T. How does an earth-abundant copper-based catalyst achieve anti-Markovnikov hydrobromination of alkynes? A DFT Mechanistic Study. Organometallics 2016, 35, 1923–1930. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Account. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Kruse, H.; Grimme, S. A geometrical correction for the inter- and intra-molecular basis set superposition error in Hartree-Fock and density functional theory calculations for large systems. J. Chem. Phys. 2012, 136, 154101–154116. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, J.G.; Alessio, M.; Civalleri, B.; Galano, A.; Peintinger, M.F.; Bredow, T.; Grimme, S. Geometrical correction for the inter- and intramolecular basis set superposition error in periodic density functional theory calculations. J. Phys. Chem. A 2013, 117, 9282–9292. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104–154123. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-Type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Accurate Description of van der Waals complexes by density functional theory including empirical corrections. J. Comput. Chem. 2004, 25, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Grunzke, R.; Herres-Pawlis, S. Insights into the influence of dispersion correction in the theoretical treatment of guanidine-quinoline copper(I) Complexes. J. Comput. Chem. 2014, 35, 1943–1950. [Google Scholar] [CrossRef] [PubMed]
- Sousa, S.F.; Pinto, G.R.; Ribeiro, A.J.M.; Coimbra, J.T.S.; Fernandes, P.A.; Ramos, M. Comparative analysis of the performance of commonly available density functionals in the determination of geometrical parameters for copper complexes. J. Comput. Chem. 2013, 34, 2079–2090. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Yury, M.; Åsmund, S.; Giovanni, O. The accuracy of DFT-optimized geometries of functional transition metal compounds: A validation study of catalysts for olefin metathesis and other reactions in the homogeneous phase. Dalton Trans. 2012, 41, 5526–5541. [Google Scholar] [CrossRef]
- Mouesca, J.M.; Mouesca, J.-P.; Noodleman, L.; Bashford, D.; Case, D.A. Density functional Poisson-Boltzmann calculations of redox potentials for iron-sulfur clusters. J. Am. Chem. Soc. 1994, 116, 11898–11914. [Google Scholar] [CrossRef]
- Noodleman, L. Valence bond description of antiferromagnetic coupling in transition metal dimers. J. Chem. Phys. 1981, 74, 5737–5743. [Google Scholar] [CrossRef]
- Ciofini, L.; Daul, C.A. DFT calculations of molecular magnetic properties of coordination compounds. Coord. Chem. Rev. 2003, 238, 187–209. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M.; Pelmenschikov, V. Nickel superoxide dismutase reaction Mechanism Studied by Hybrid Density Functional Methods. J. Am. Chem. Soc. 2006, 128, 7466–7475. [Google Scholar] [CrossRef]
- Bassan, A.; Blomberg, M.R.A.; Siegbalm, P.E.M.; Que, L. A density functional study on a biomimetic non-heme iron catalyst: insights into alkane hydroxylation by a formally HO-FeV=O oxidant. Chem. Eur. J. 2005, 11, 692–705. [Google Scholar] [CrossRef] [PubMed]
- Bachler, V.; Chaudhuri, P.; Wieghardt, K. The symmetry-broken formalism applied to the electronic structure of an iminosemiquinone copper(II) catalyst: A key to the qualitative understanding of its reactivity. Chem. Eur. J. 2001, 7, 404–415. [Google Scholar] [CrossRef]
- Cheng, L.; Li, J.; Zhang, Q.C.; Ma, L.S.; Yang, J.C. DFT studies on the mechanism of alcohol oxidation by the (bpy)CuI-TEMPO/NMI catalytic system. Dalton Trans. 2015, 44, 7395–7403. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
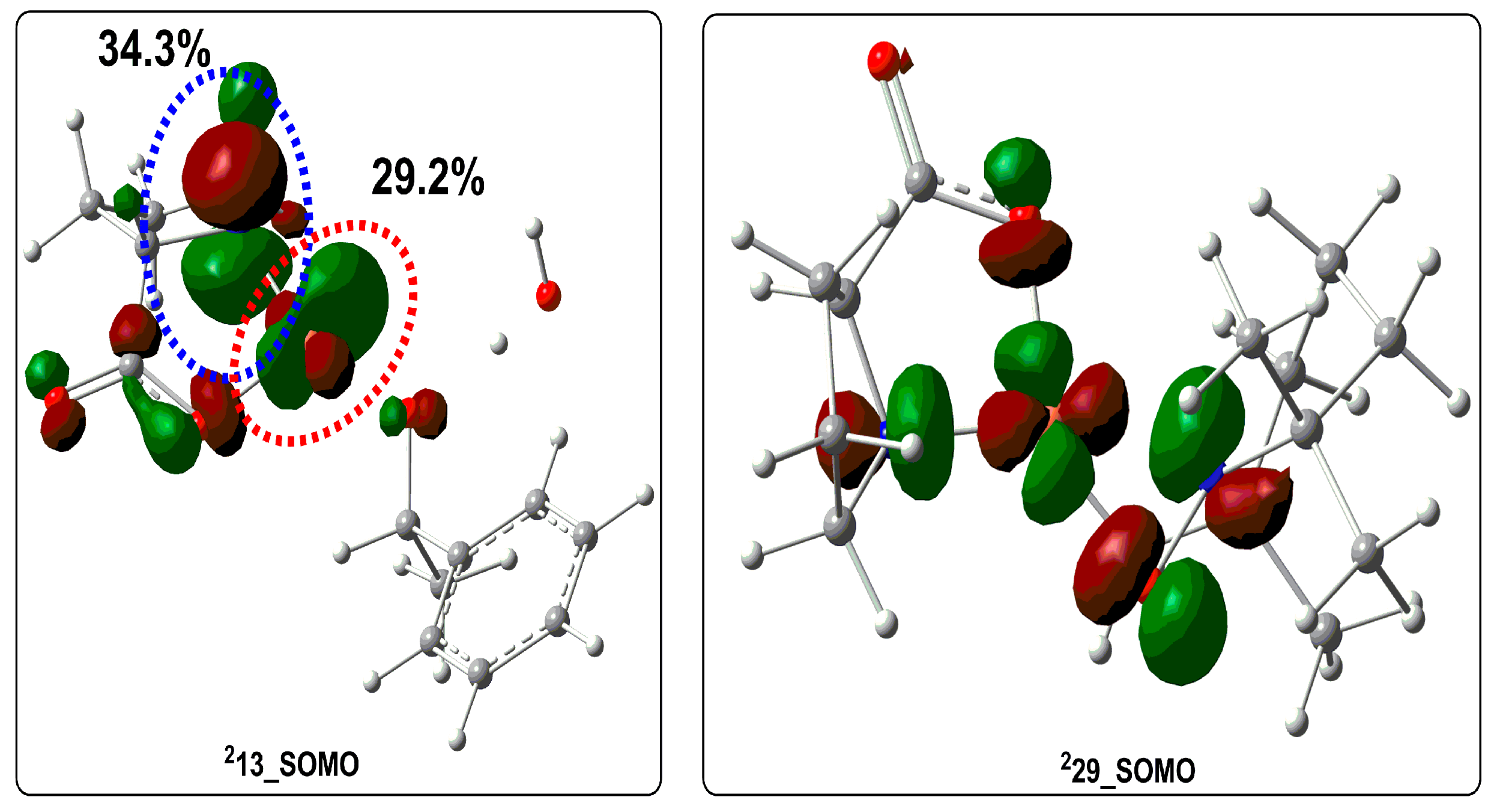
| Structures | Cu–NL2– | Cα–H | Cu–Osub | Cu–NL2– (WBI) | Cα–H (WBI) | Cu–Osub (WBI) | Cu–NL2– Formed % | Cα–H Formed % | Cu–Osub Formed % |
|---|---|---|---|---|---|---|---|---|---|
| 213 | 1.871 Å | 1.104 Å | 1.888 Å | 1.037 | 0.823 | 0.828 | 100 | 100 | 100 |
| 2TS13–14 | 2.189 Å | 1.347 Å | 1.892 Å | 0.466 | 0.451 | 0.789 | 44.9 | 54.8 | 95.3 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Cheng, L.; Wu, Q.; Zhang, Q.; Yang, J.; Liu, J. Mechanistic Insight into the 2° Alcohol Oxidation Mediated by an Efficient CuI/L-Proline-TEMPO Catalyst—A Density Functional Theory Study. Catalysts 2017, 7, 264. https://doi.org/10.3390/catal7090264
Li S, Cheng L, Wu Q, Zhang Q, Yang J, Liu J. Mechanistic Insight into the 2° Alcohol Oxidation Mediated by an Efficient CuI/L-Proline-TEMPO Catalyst—A Density Functional Theory Study. Catalysts. 2017; 7(9):264. https://doi.org/10.3390/catal7090264
Chicago/Turabian StyleLi, Siyu, Lin Cheng, Qi Wu, Qiancheng Zhang, Jucai Yang, and Juming Liu. 2017. "Mechanistic Insight into the 2° Alcohol Oxidation Mediated by an Efficient CuI/L-Proline-TEMPO Catalyst—A Density Functional Theory Study" Catalysts 7, no. 9: 264. https://doi.org/10.3390/catal7090264