Experimental Research of an Active Solution for Modeling In Situ Activating Selective Catalytic Reduction Catalyst

Abstract
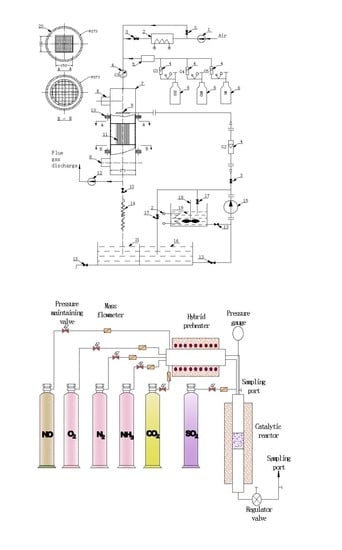
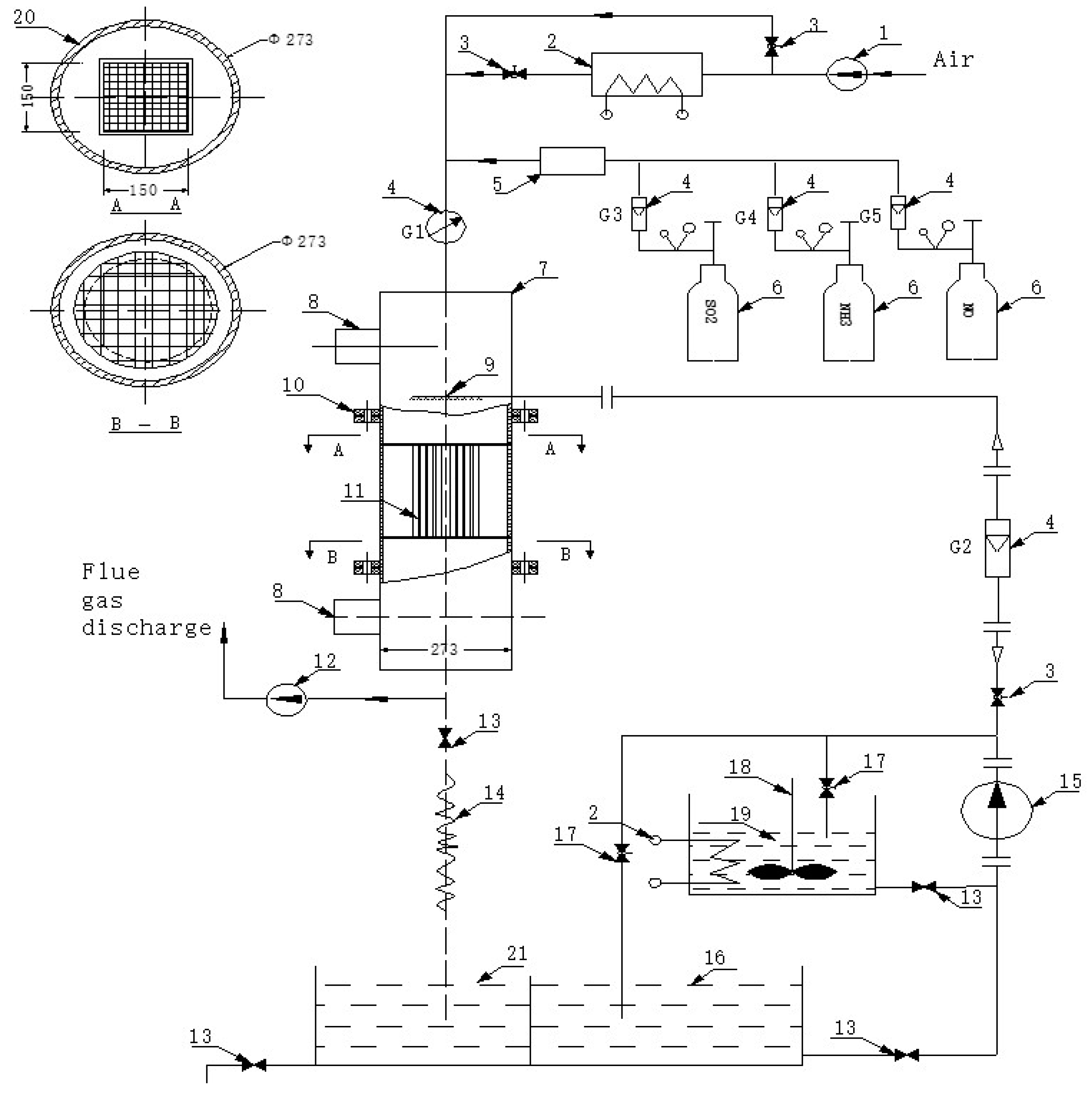
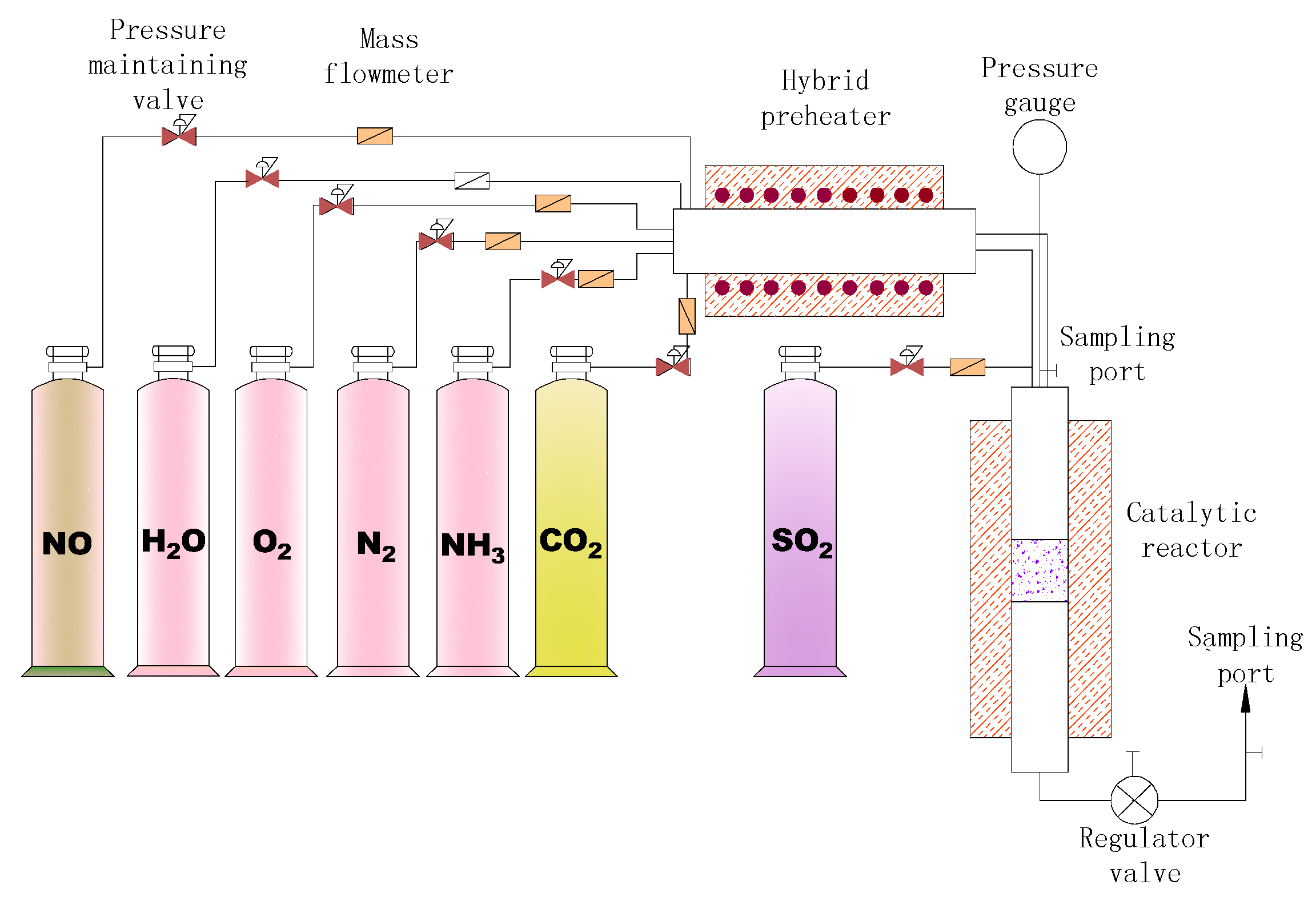
:
1. Introduction
2. Experimental Materials and Methods
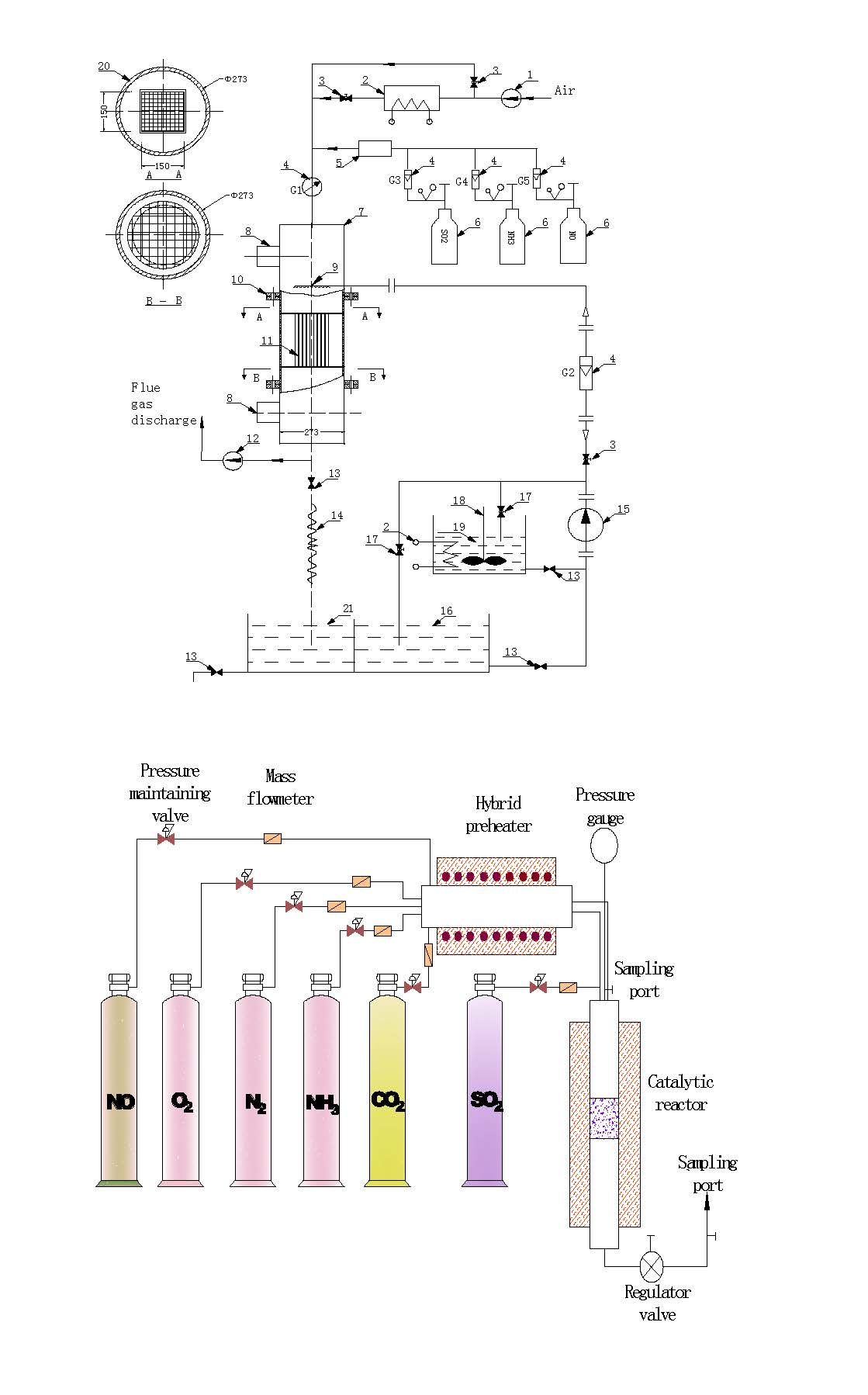
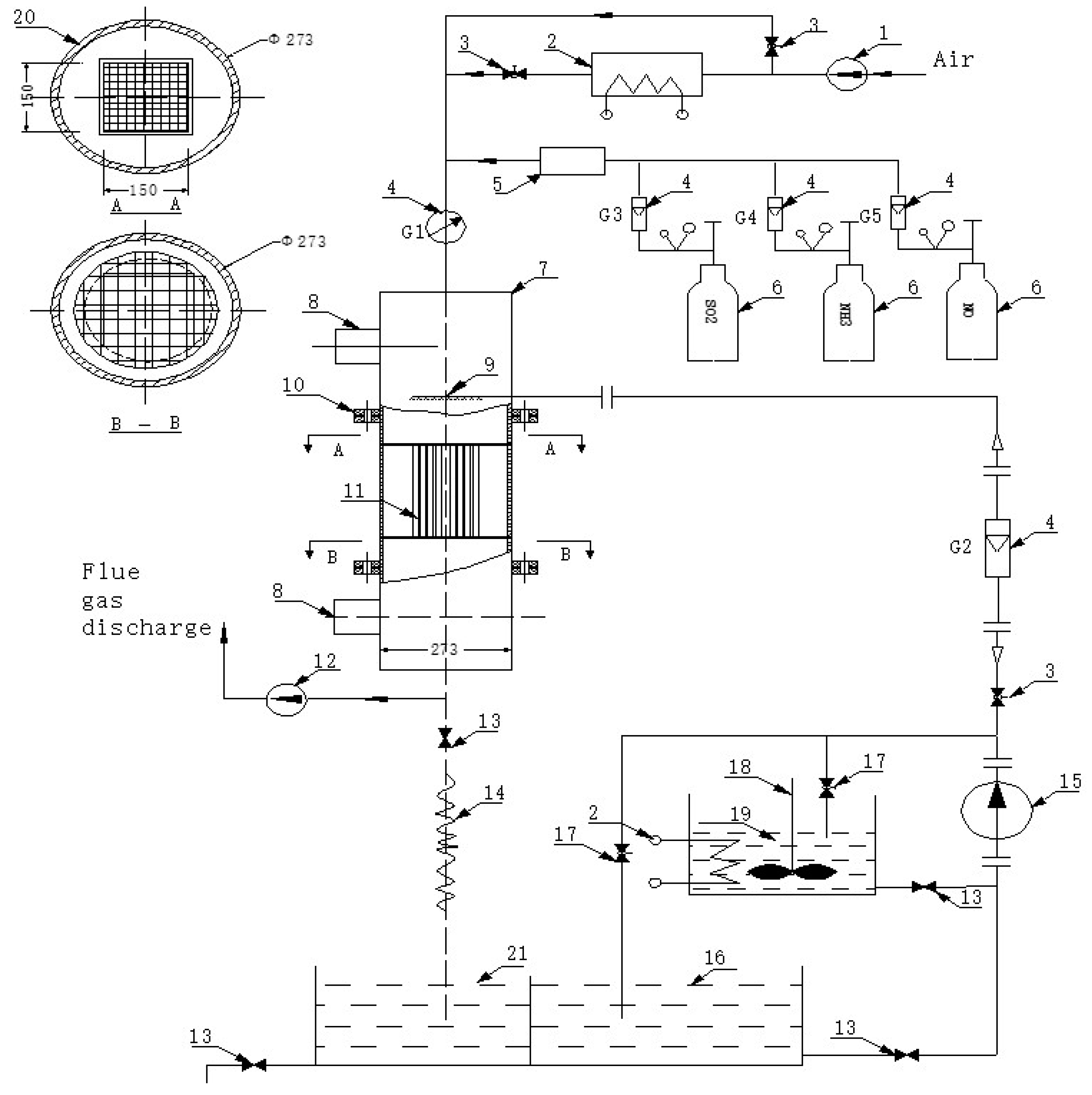
2.1. Equipment
2.1.1. Cleaning
2.1.2. Activation
2.1.3. Drying and Roasting
2.2. Materials
2.2.1. Catalyst Preparation
2.2.2. Activating Solution Preparation
2.2.3. Catalyst Activation
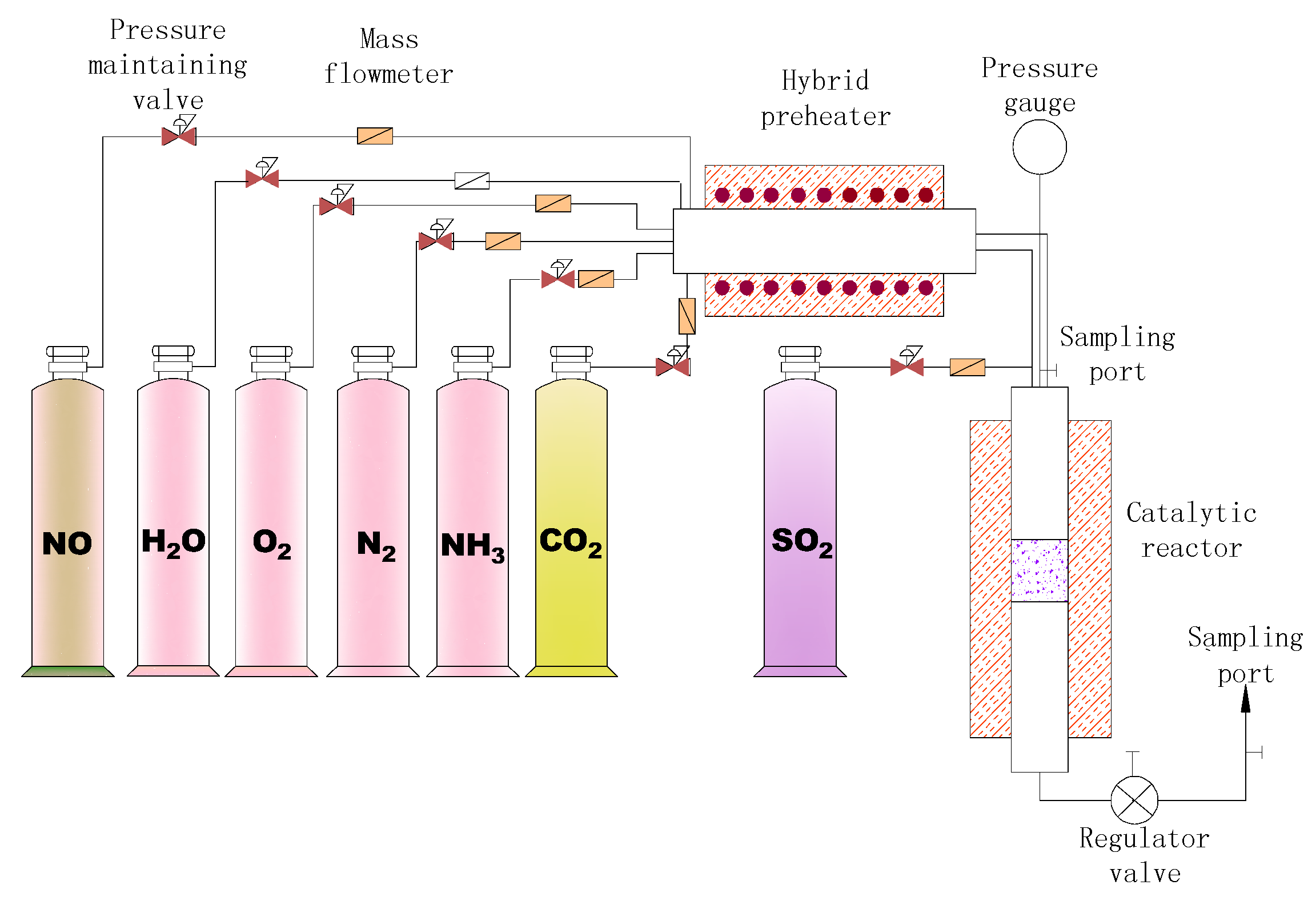
2.3. Methodology
2.3.1. Catalytic Performance Test
2.3.2. Activity Evaluations
2.3.3. Catalyst Characteristics
Pore Structure and Morphology
Carbon Surface Chemistry
3. Results and Discussion
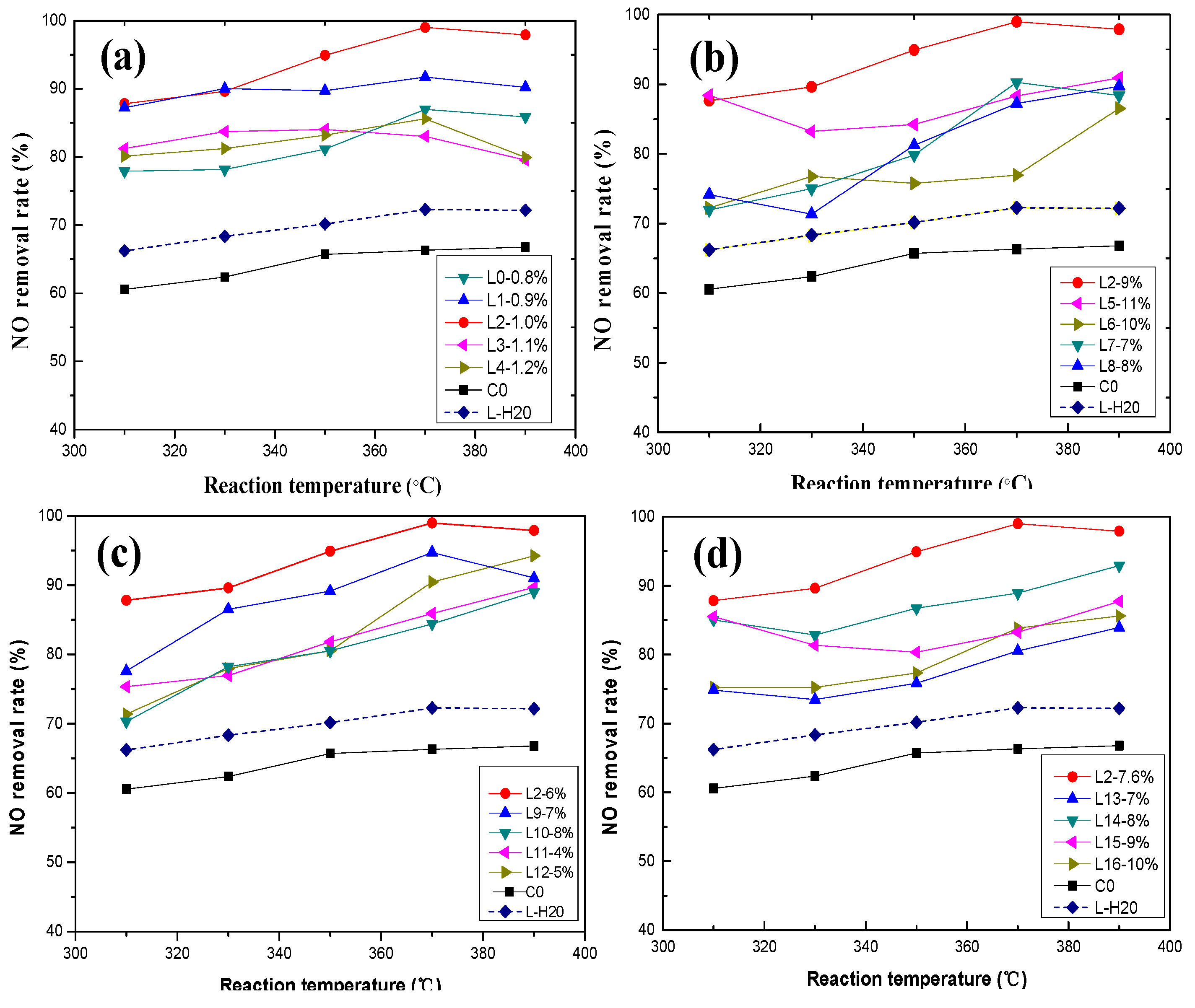
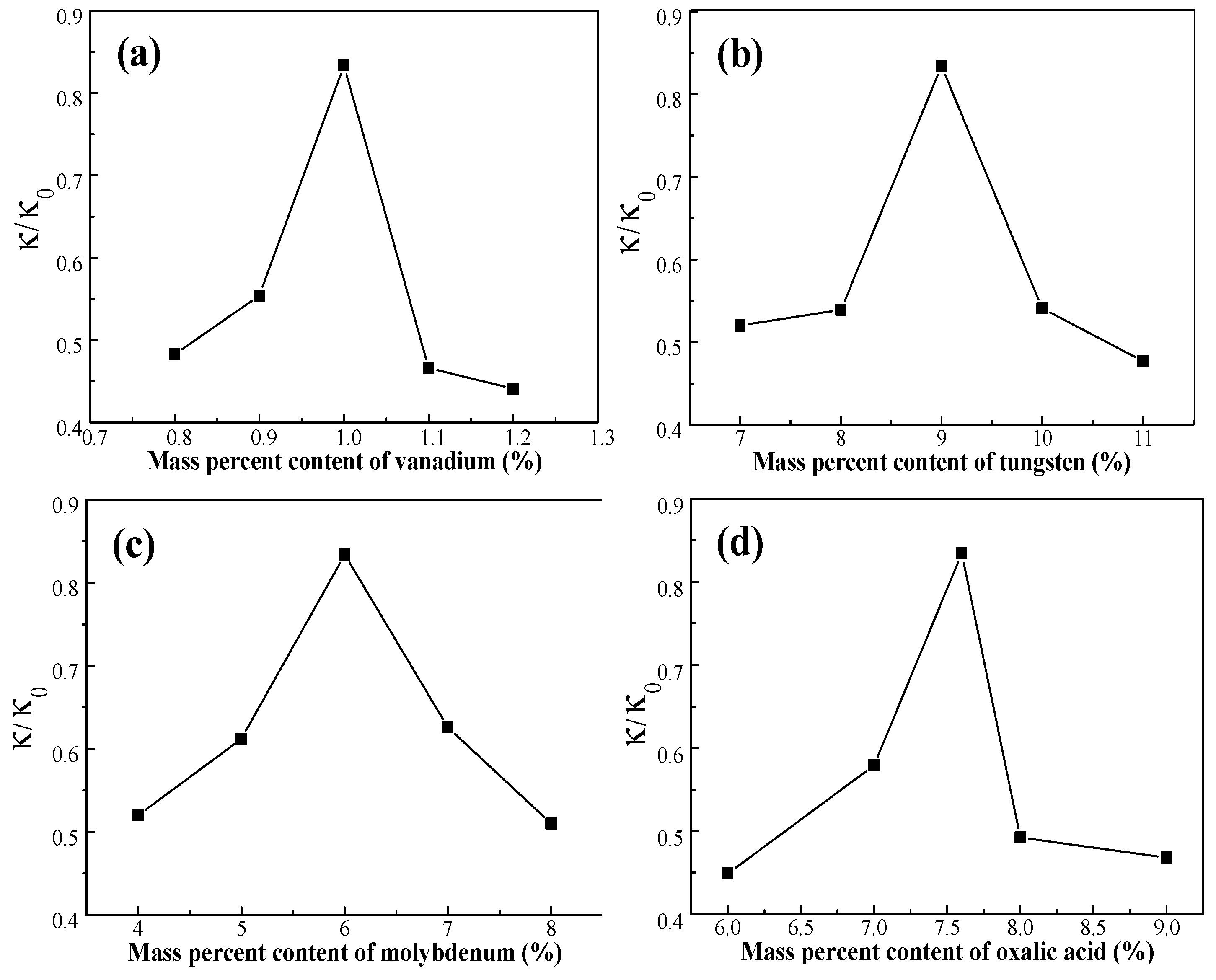
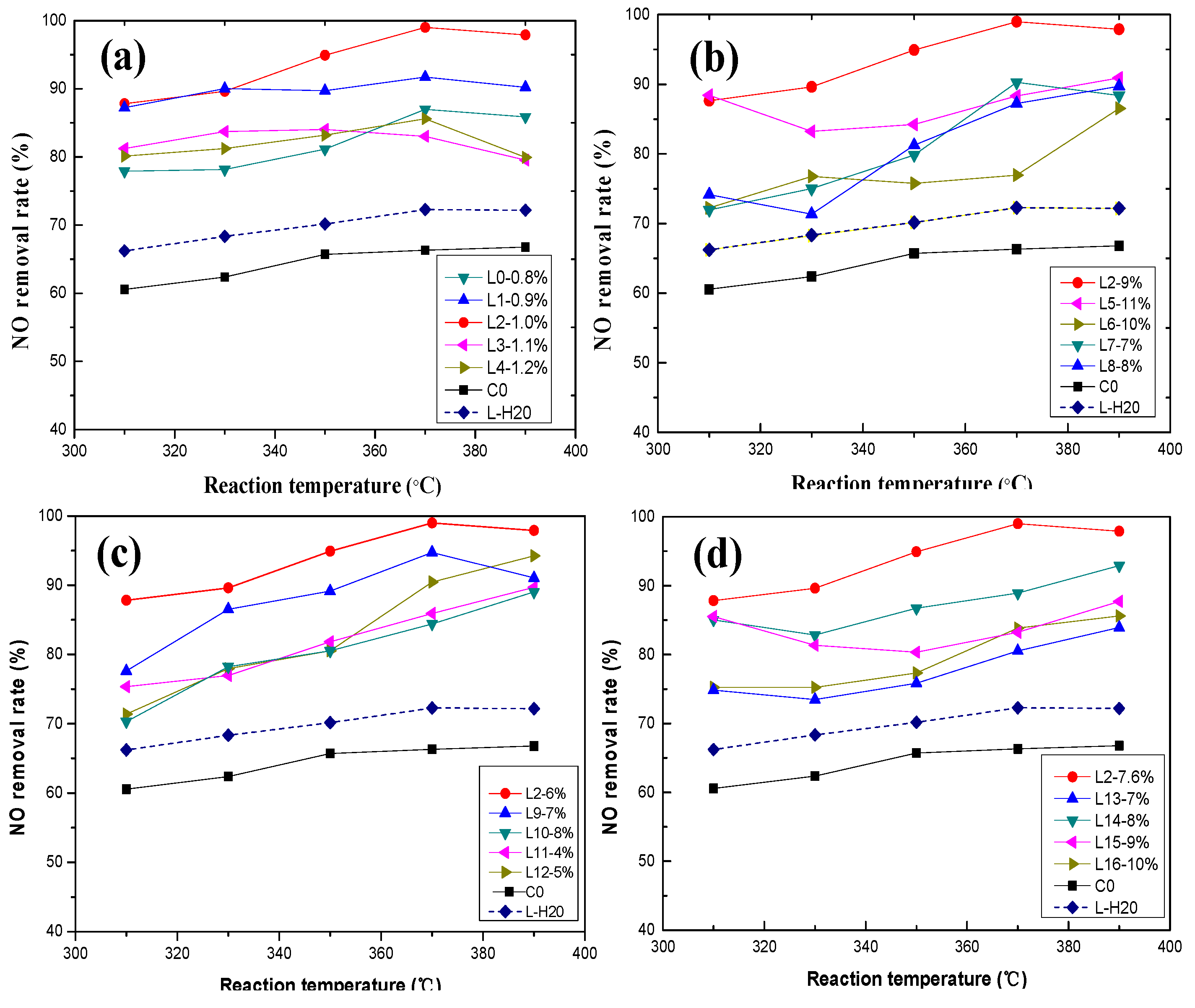
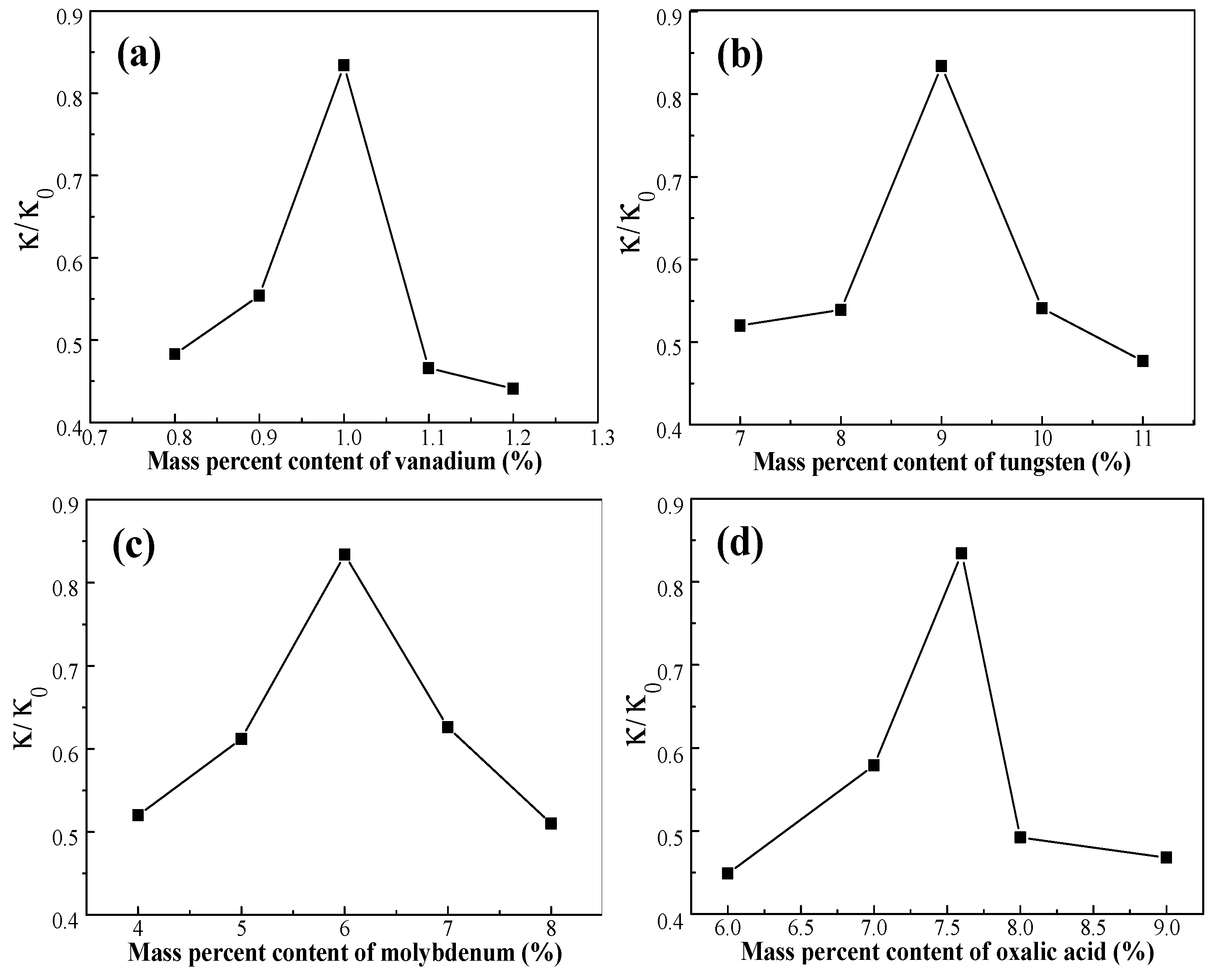
3.1. Effect of Active Ingredients on NO Removal Rate
3.1.1. Effect of Vanadium
3.1.2. Effect of Tungsten
3.1.3. Effect of Molybdenum Composition
3.1.4. Effect of Oxalic Acid
3.2. Activity Analyses of the Activated Catalyst
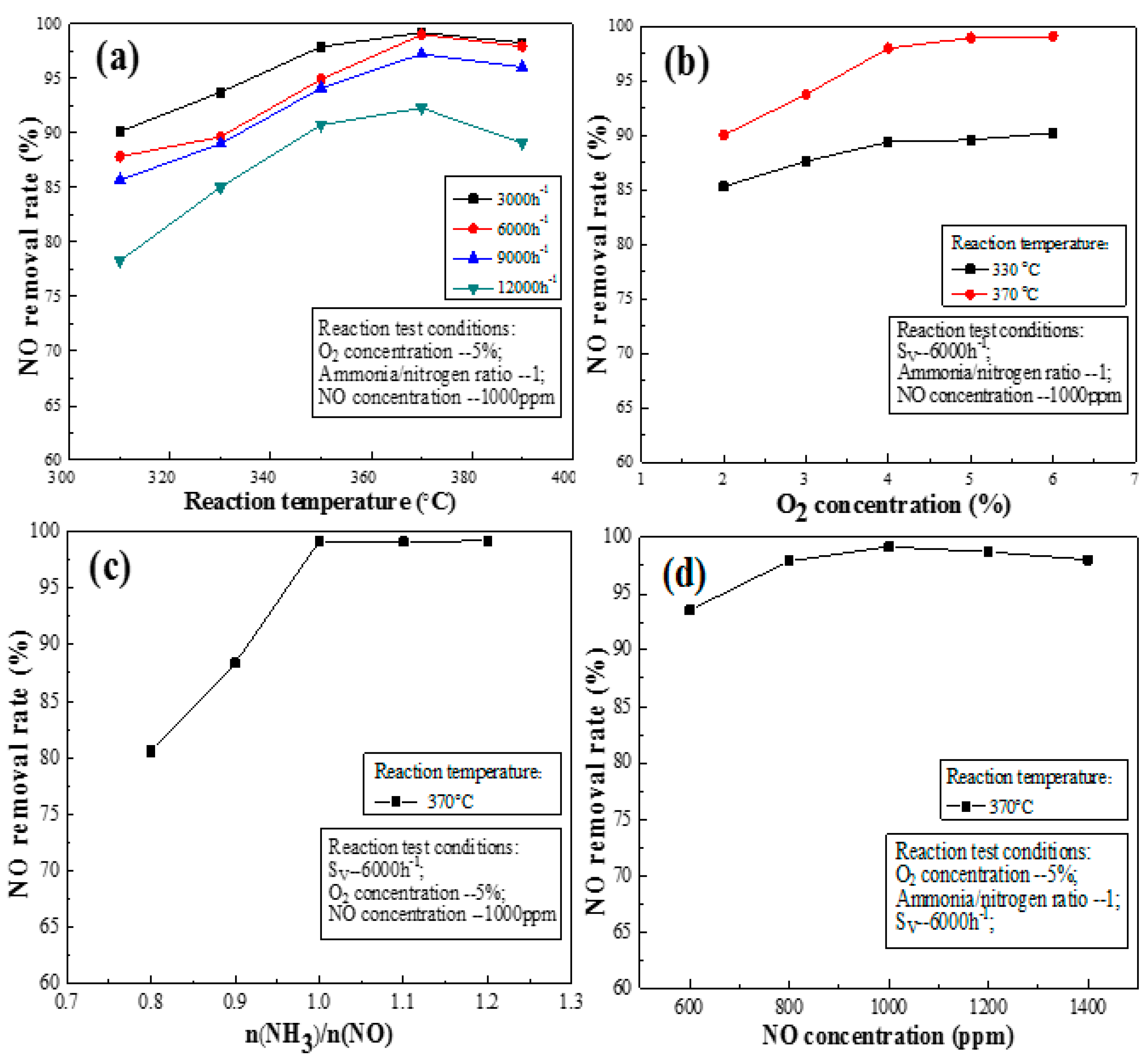
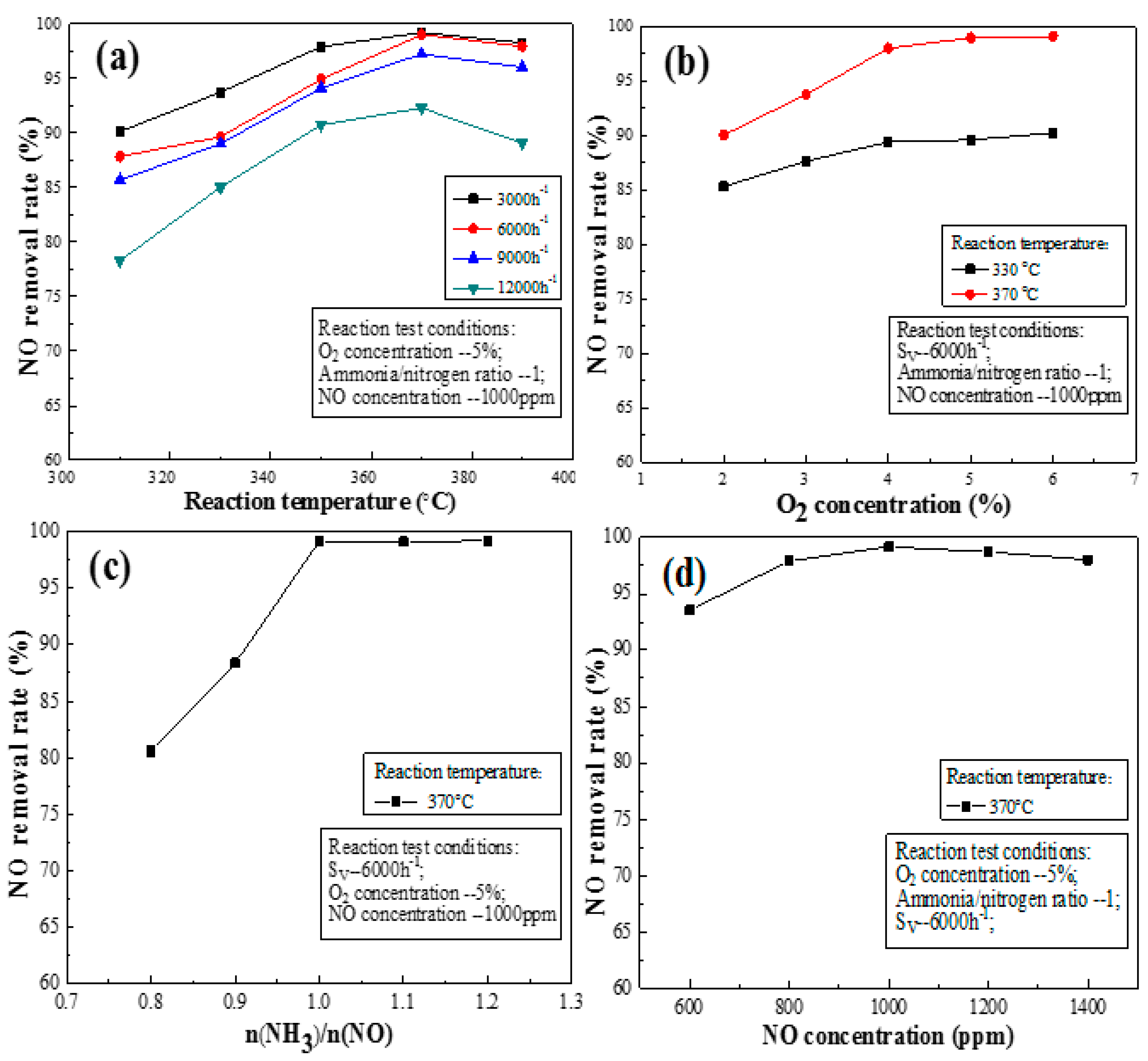
3.3. Effects of Activated Catalysts on the NO Removal Rate under Flue Gas Conditions
3.3.1. Space Velocity
3.3.2. Oxygen Concentration
3.3.3. Ratio of Ammonia to Nitrogen
3.3.4. Initial Concentration of NO
3.4. Characterization of the Catalyst
3.4.1. SEM
3.4.2. BET
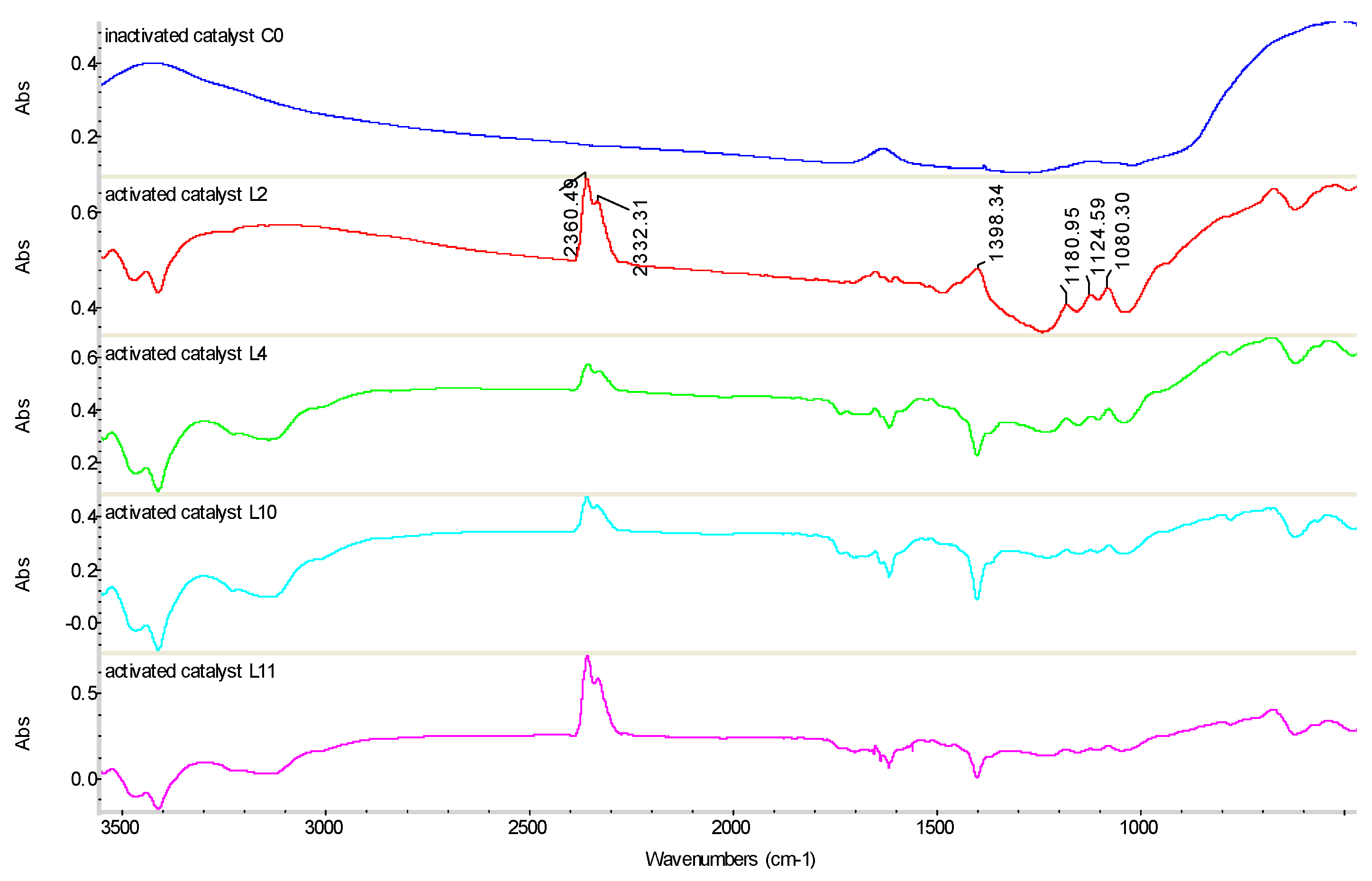
3.4.3. FT-IR
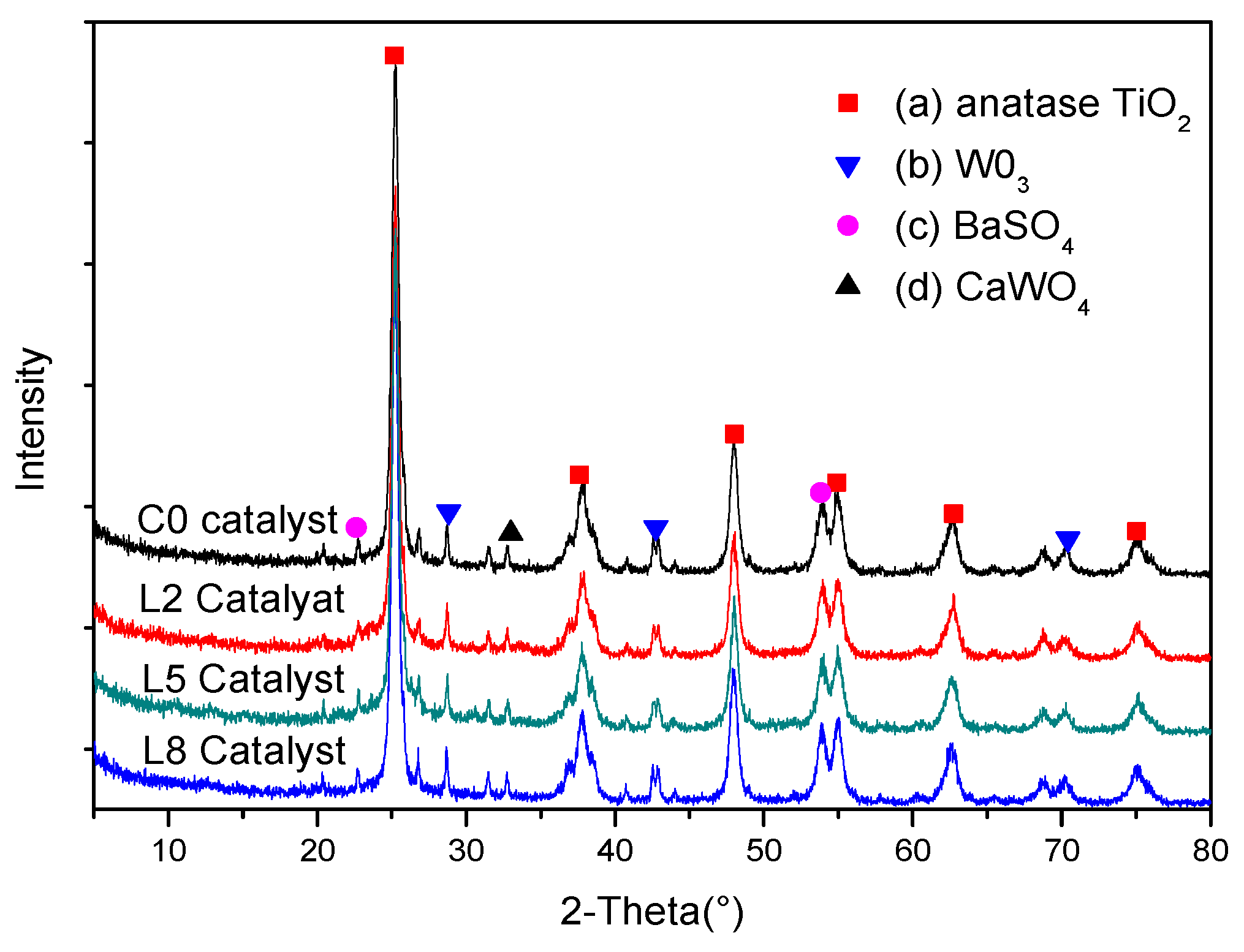
3.4.4. XRD
3.4.5. EDS
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Qi, C.; Bao, W.; Wang, L.; Li, H. Study of the V2O5-WO3/TiO2 Catalyst Synthesized from Waste Catalyst on Selective Catalytic Reduction of NOx by NH3. Catalysts 2017, 7, 110. [Google Scholar] [CrossRef]
- Liang, Z.; Ma, X.; Lin, H.; Tang, Y. The energy consumption and environmental impacts of SCR technology in China. Appl. Energy 2011, 88, 1120–1129. [Google Scholar] [CrossRef]
- Bao, J.; Mao, L.; Zhang, Y.; Fang, H.; Shi, Y.; Yang, L.; Yang, H. Effect of Selective Catalytic Reduction System on Fine Particle Emission Characteristics. Energy Fuels 2016, 30, 1325–1334. [Google Scholar] [CrossRef]
- Schwaemmle, T.; Heidel, B.; Brechtel, K.; Scheffknecht, G. Study of the effect of newly developed mercury oxidation catalysts on the DeNOx -activity and SO2-SO3-conversion. Fuel 2012, 101, 179–186. [Google Scholar] [CrossRef]
- Guo, X. Poisoning and Sulfation on Vanadia SCR Catalyst. Ph.D. Thesis, Brigham Young University, Provo, UT, USA, 2006. [Google Scholar]
- Peng, Y.; Li, J.; Si, W.; Luo, J.; Wang, Y.; Fu, J.; Li, X.; Crittenden, J.; Hao, J. Deactivation and regeneration of a commercial SCR catalyst: Comparison with alkali metals and arsenic. Appl. Catal. B Environ. 2015, 168–169, 195–202. [Google Scholar] [CrossRef]
- Zheng, Y.J.; Jensen, A.D.; Johnsson, J.E.; Thøgersen, J.R. Deactivation of V2O5-WO3-TiO2, SCR catalyst at biomass fired power plants: Elucidation of mechanisms by lab- and pilot-scale experiments. Appl. Catal. B Environ. 2008, 83, 186–194. [Google Scholar] [CrossRef]
- Benson, S.A.; Laumb, J.D.; Crocker, C.R.; Pavlish, J.H. SCR catalyst performance in flue gases derived from subbituminous and lignite coals. Fuel Process. Technol. 2005, 86, 577–613. [Google Scholar] [CrossRef]
- Xu, W.; He, H.; Yu, Y. Deactivation of a Ce/TiO2 Catalyst by SO2 in the Selective Catalytic Reduction of NO by NH3. J. Phys. Chem. C 2009, 113, 4426–4432. [Google Scholar] [CrossRef]
- Yu, J.; Guo, F.; Wang, Y.; Xu, G. Sulfur poisoning resistant mesoporous Mn-based catalyst for low-temperature SCR of NO with NH3. Appl. Catal. B Environ. 2010, 95, 160–168. [Google Scholar] [CrossRef]
- Shen, B.X. Deactivation of MnO-CeO/ACF Catalysts for Low-Temperature NH-SCR in the Presence of SO. Acta Phys. Chim. Sin. 2010, 26, 3009–3016. [Google Scholar]
- Aguilar-Romero, M.; Camposeco, R.; Castillo, S.; Marín, J.; Glez, V.R.; García-Serrano, L.A.; Mejía-Centeno, I. Acidity, surface species, and catalytic activity study on V2O5-WO3/TiO2 nanotube catalysts for selective NO reduction by NH3. Fuel 2017, 198, 123–133. [Google Scholar] [CrossRef]
- Wilburn, R.T.; Wright, T.L. SCR Ammonia Slip Distribution in Coal Plant Effluents and Dependence upon SO. Powerpl. Chem. 2004, 6, 295–314. [Google Scholar]
- Tang, F.; Xu, B.; Shi, H.; Qiu, J.; Fan, Y. The poisoning effect of Na+ and Ca2+ ions doped on the V2O5/TiO2 catalysts for selective catalytic reduction of NO by NH3. Appl. Catal. B Environ. 2010, 94, 71–76. [Google Scholar] [CrossRef]
- Chen, L.; Li, J.; Ge, M. The poisoning effect of alkali metals doping over nano V2O5-WO3/TiO2 catalysts on selective catalytic reduction of NOx by NH3. Chem. Eng. J. 2011, 170, 531–537. [Google Scholar] [CrossRef]
- Nicosia, D.; Czekaj, I.; Kröcher, O. Chemical deactivation of V2O5/WO3-TiO2 SCR catalysts by additives and impurities from fuels, lubrication oils and urea solution: Part II. Characterization study of the effect of alkali and alkaline earth metals. Appl. Catal. B Environ. 2008, 77, 228–236. [Google Scholar] [CrossRef]
- Xiong, Z.B.; Hu, Q.; Liu, D.Y.; Wu, C.; Zhou, F.; Wang, Y.Z.; Jin, J.; Lu, C.M. Influence of partial substitution of iron oxide by titanium oxide on the structure and activity of iron–cerium mixed oxide catalyst for selective catalytic reduction of NOx, with NH3. Fuel 2016, 165, 432–439. [Google Scholar] [CrossRef]
- Li, X.; Li, X.; Chen, J.; Hao, J. An efficient novel regeneration method for Ca-poisoning V2O5-WO3/TiO2 catalyst. Catal. Commun. 2016, 87, 45–48. [Google Scholar] [CrossRef]
- Dong, G.; Zhang, Y.; Zhao, Y. Effect of the pH value of precursor solution on the catalytic performance of VO-WO/TiO in the low temperature NH-SCR of NO. J. Fuel Chem. Technol. 2014, 42, 1455–1463. [Google Scholar] [CrossRef]
- Topsøe, N.Y.; Topsøe, H.; Dumesic, J.A. Vanadia-Titania Catalysts for Selective Catalytic Reduction (SCR) of Nitric-Oxide by Ammonia 1. Combined Temperature-Programmed In-Situ FTIR and Online Mass-Spectroscopy Studies. J. Catal. 1995, 151, 226–240. [Google Scholar] [CrossRef]
- Djerad, S.; Tifouti, L.; Crocoll, M.; Weisweiler, W. Effect of vanadia and tungsten loadings on the physical and chemical characteristics of V2O5-WO3/TiO2 catalysts. J. Mol. Catal. A Chem. 2004, 208, 257–265. [Google Scholar] [CrossRef]
- Giakoumelou, I.; Fountzoula, C.; Kordulis, C.; Boghosian, S. Molecular structure and catalytic activity of V2O5/TiO2, catalysts for the SCR of NO by NH3: In situ Raman spectra in the presence of O2, NH3, NO, H2, H2O and SO2. J. Catal. 2006, 239, 1–12. [Google Scholar] [CrossRef]
- Kobayashi, M.; Hagi, M. V2O5-WO3/TiO2-SiO2-SO42− catalysts: Influence of active components and supports on activities in the selective catalytic reduction of NO by NH3 and in the oxidation of SO2. Appl. Catal. B Environ. 2006, 63, 607–612. [Google Scholar] [CrossRef]
- Oliveri, G.; Ramis, G.; Busca, G.; Escribano, V.S. Thermal stability of vanadia–titania catalysts. J. Mater. Chem. 1993, 3, 1239–1249. [Google Scholar] [CrossRef]
- Alemany, L.J.; Lietti, L.; Ferlazzo, N.; Forzatti, P.; Busca, G.; Giamello, E.; Bregani, F. Reactivity and Physicochemical Characterization of V2O5-WO3/TiO2 De-NOx Catalysts. J. Catal. 1995, 155, 117–130. [Google Scholar] [CrossRef]
- Sun, C.; Dong, L.; Yu, W.; Liu, L.; Li, H.; Gao, F.; Dong, L.; Chen, L. Promotion effect of tungsten oxide on SCR of NO with NH3, for the V2O5-WO3/Ti0.5 Sn0.5 O2, catalyst: Experiments combined with DFT calculations. J. Mol. Catal. A Chem. 2011, 346, 29–38. [Google Scholar] [CrossRef]
- Wachs, I.E.; Kim, T.; Ross, E.I. Catalysis science of the solid acidity of model supported tungsten oxide catalysts. Catal. Today 2006, 116, 162–168. [Google Scholar] [CrossRef]
- Fierro, J.L.G. (Ed.) Metal Oxides: Chemistry and Applications; CRC Press: Boca Raton, FL, USA, 2006; pp. 1–30. [Google Scholar]
- Kobayashi, M.; Miyoshi, K. WO3-TiO2 monolithic catalysts for high temperature SCR of NO by NH3: Influence of preparation method on structural and physico-chemical properties activity and durability. Appl. Catal. B Environ. 2007, 72, 253–261. [Google Scholar] [CrossRef]
- Peng, Y.; Si, W.; Li, X.; Luo, J.; Li, J.; Crittenden, J.; Hao, J. Comparison of MoO3 and WO3 on arsenic poisoning V2O5/TiO2 catalyst: DRIFTS and DFT study. Appl. Catal. B Environ. 2016, 181, 692–698. [Google Scholar] [CrossRef]
- Yang, S.; Xiong, S.; Liao, Y.; Xiao, X.; Qi, F.; Peng, Y.; Fu, Y.; Shan, W.; Li, J. Mechanism of N2O formation during the low-temperature selective catalytic reduction of NO with NH3 over Mn-Fe Spinel. Environ. Sci. Technol. 2014, 48, 10354–10362. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.; Liu, F.; He, H.; Shi, X.; Zhang, C. ChemInform Abstract: Novel Cerium-Tungsten Mixed Oxide Catalyst for the Selective Catalytic Reduction of NOx with NH3. Chem. Commun. 2011, 47, 8046–8048. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, S.; Li, J.; Ma, L. Promoting effect of MoO3 on the NOx reduction by NH3 over CeO2/TiO2 catalyst studied with in situ DRIFTS. Appl. Catal. B Environ. 2014, 144, 90–95. [Google Scholar] [CrossRef]
- Nova, I.; Lietti, L.; Casagrande, L.; Dall’Acquab, L.; Giamellob, E.; Forzattia, P. Characterization and reactivity of TiO2-supported MoO3 De-Nox SCR catalysts. Appl. Catal. B Environ. 1998, 17, 245–258. [Google Scholar] [CrossRef]
- Zhao, B.; Liu, X.; Zhou, Z.; Shao, H.; Wang, W.; Si, J.; Xu, M. Effect of molybdenum on mercury oxidized by V2O5-MoO3/TiO2 catalysts. Chem. Eng. J. 2014, 253, 508–517. [Google Scholar] [CrossRef]
- Huang, X.; Peng, Y.; Liu, X.; Li, K.; Deng, Y.; Li, J. The promotional effect of MoO3 doped V2O5/TiO2 for chlorobenzene oxidation. Catal. Commun. 2015, 69, 161–164. [Google Scholar] [CrossRef]
- Khodayari, R.; Odenbrand, C.U.I. Regenaration of commercial TiO2-V2O5-WO3 SCR catalysts used in biofuel plants. Appl. Catal. B Environ. 2001, 30, 87–99. [Google Scholar] [CrossRef]
- Dunn, J.P.; Koppula, P.R.; Stenger, H.G.; Wachs, I.E. Oxidation of sulfur dioxide to sulfur trioxide over supported vanadia catalysts. Appl. Catal. B Environ. 1998, 19, 103–117. [Google Scholar] [CrossRef]
- Busca, G.; Lietti, L.; Ramis, G.; Berti, F. Chemical and mechanistic aspects of the selelctive catalytic reduction of NOx by ammonia over oxide catalysts: A Review. Appl. Catal. B Environ. 1998, 18, 1–36. [Google Scholar] [CrossRef]
- Djerad, S.; Crocoll, M.; Kureti, S.; Weisweiler, W. Effect of oxygen concentration on the NOx reduction with ammonia over V2O5-WO3/TiO2 catalyst. Catal. Today 2006, 113, 208–214. [Google Scholar] [CrossRef]
- Lee, C. Modeling urea-selective catalyst reduction with vanadium catalyst based on NH3 temperature programming desorption experiment. Fuel 2016, 173, 155–163. [Google Scholar] [CrossRef]
- Lei, Z.; Liu, X.; Jia, M. Modeling of Selective Catalytic Reduction (SCR) for NO Removal Using Monolithic Honeycomb Catalyst. Energy Fuels 2009, 23, 6146–6151. [Google Scholar] [CrossRef]
- Kling, Å.; Andersson, C.; Myringer, Å.; Eskilsson, D.; Järås, S.G. Alkali deactivation of high-dust SCR catalysts used for NOx, reduction exposed to flue gas from 100 MW-scale biofuel and peat fired boilers: Influence of flue gas composition. Appl. Catal. B Environ. 2007, 69, 240–251. [Google Scholar] [CrossRef]
- Reddy, B.M.; Ganesh, I.; Chowdhury, B. Design of stable and reactive vanadium oxide catalyst supported on binary oxides. Catal. Today 1999, 49, 115–121. [Google Scholar] [CrossRef]
- Cai, Y.P. Investigation of the Reaction Network and Catalytic Sites in Selective Catalytic Reduction Ognitric Oxide with Ammonia over Vanadia Catalysts. Ph.D. Dissertation, The Ohio State University, Columbus, OH, USA, 1993. [Google Scholar]
- Liu, F.D.; He, H.; Zhang, C.B.; Shan, W.; Shi, X. Selective catalytic reduction of NO with NH3 over iron titanate catalyst: Catalytic performance and characterization. Appl. Catal. B Environ. 2010, 96, 408–420. [Google Scholar] [CrossRef]
- Peng, Y.; Li, J.; Si, W.; Luo, J.; Dai, Q.; Luo, X.; Liu, X.; Hao, J. Insight into deactivation of commercial SCR catalyst by arsenic: An experiment and DFT study. Environ. Sci. Technol. 2014, 48, 13895. [Google Scholar] [CrossRef] [PubMed]
- Choo, S.T.; Yim, S.D.; Nam, I.S.; Ham, S.W.; Lee, J.B. Effect of promoters including WO and BaO on the activity and durability of VO/sulfated TiO catalyst for NO reduction by NH. Appl. Catal. B Environ. 2003, 44, 237–252. [Google Scholar] [CrossRef]
- Lisi, L.; Lasorella, G.; Malloggi, S.; Russo, G. Single and combined deactivating effect of alkali metals and HCl on commercial SCR catalysts. Appl. Catal. B Environ. 2004, 50, 251–258. [Google Scholar] [CrossRef]
- Zheng, Y.; Jensen, A.D.; Johnsson, J.E. Deactivation of V2O5-WO3-TiO2 SCR catalyst at a biomass-fired combined heat and power plant. Appl. Catal. B Environ. 2005, 60, 253–264. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Channel number | 18 × 18 |
| Square width | 150 mm |
| Channel width | 7.09 mm |
| Specific geometry area | 408 m2/m3 |
| Hardened front length | 25 mm |
| Number | Ammonium Vanadate (wt %) | Ammonium Molybdate (wt %) | Ammonium Tungstate (wt %) | Oxalic Acid (wt %) | Notes | ||||
|---|---|---|---|---|---|---|---|---|---|
| L0–L4 | L0 | 0.8 | 9 | 6 | 7.6 | ||||
| L1 | 0.9 | ||||||||
| L2 | 1.0 | ||||||||
| L3 | 1.1 | ||||||||
| L4 | 1.2 | ||||||||
| L5–L8, LiF | Content of vanadate as in solution LiF | L5 | 7 | 6 | 7.6 | LiF is the number of the optimal solution in L0–L4 | |||
| L6 | 8 | ||||||||
| L7 | 10 | ||||||||
| L8 | 11 | ||||||||
| LiF | 9 | ||||||||
| L9–L12, LiW | Content of vanadate and molybdate as in solution LiW | L9 | 7 | 7.6 | LiW is the number of the optimal solution in L5–L8, LiF | ||||
| L10 | 8 | ||||||||
| L11 | 4 | ||||||||
| L12 | 5 | ||||||||
| LiW | 6 | ||||||||
| L13–L16, LiM | Content of vanadate, molybdate and tungstate as in solution LiM | L13 | 6 | LiM is the number of the optimal solution in L9–L12, LiW | |||||
| L14 | 7 | ||||||||
| L15 | 8 | ||||||||
| L16 | 9 | ||||||||
| LiM | 7.6 | ||||||||
| Catalyst Sample | Specific Surface Area (m2/g) | Total Pore Volume (cc/g) | Aperture (nm) |
|---|---|---|---|
| C | 52.31 | 0.8022 | 18.6 |
| C0 | 41.85 | 0.3434 | 20.0 |
| L2 | 47.55 | 0.6884 | 17.0 |
| L5 | 50.61 | 0.7502 | 18.1 |
| L8 | 46.80 | 0.6710 | 16.7 |
| L11 | 47.03 | 0.6725 | 16.8 |
| Element | C | C0 | L2 | |||
|---|---|---|---|---|---|---|
| wt % | at % | wt % | at % | wt % | at % | |
| O | 4.3 | 13.38 | 9.25 | 22.94 | 6.87 | 21.33 |
| Al | 0 | 0 | 1.66 | 2.44 | 0 | 0 |
| Si | 0.7 | 1.25 | 6.74 | 9.53 | 2.5 | 4.43 |
| W | 13.72 | 3.72 | 8.96 | 1.93 | 18.64 | 5.04 |
| Mo | 5.52 | 2.87 | 2.48 | 1.03 | 10.59 | 5.49 |
| S | 0 | 0 | 6.37 | 7.89 | 0.10 | 0.15 |
| Ca | 0 | 0 | 4.93 | 4.88 | 0 | 0 |
| Ti | 74.08 | 77.84 | 58.97 | 48.86 | 60.10 | 62.38 |
| V | 0.96 | 0.94 | 0.65 | 0.5 | 1.2 | 1.17 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, T.; Chen, D.; Yin, Y.; Liu, J.; Zeng, X. Experimental Research of an Active Solution for Modeling In Situ Activating Selective Catalytic Reduction Catalyst. Catalysts 2017, 7, 258. https://doi.org/10.3390/catal7090258
Ye T, Chen D, Yin Y, Liu J, Zeng X. Experimental Research of an Active Solution for Modeling In Situ Activating Selective Catalytic Reduction Catalyst. Catalysts. 2017; 7(9):258. https://doi.org/10.3390/catal7090258
Chicago/Turabian StyleYe, Tuo, Donglin Chen, Yanshan Yin, Jing Liu, and Xi Zeng. 2017. "Experimental Research of an Active Solution for Modeling In Situ Activating Selective Catalytic Reduction Catalyst" Catalysts 7, no. 9: 258. https://doi.org/10.3390/catal7090258