
Reaction Mechanisms of CO2 Reduction to Formaldehyde Catalyzed by Hourglass Ru, Fe, and Os Complexes: A Density Functional Theory Study


Abstract
:
1. Introduction
2. Results and Discussion
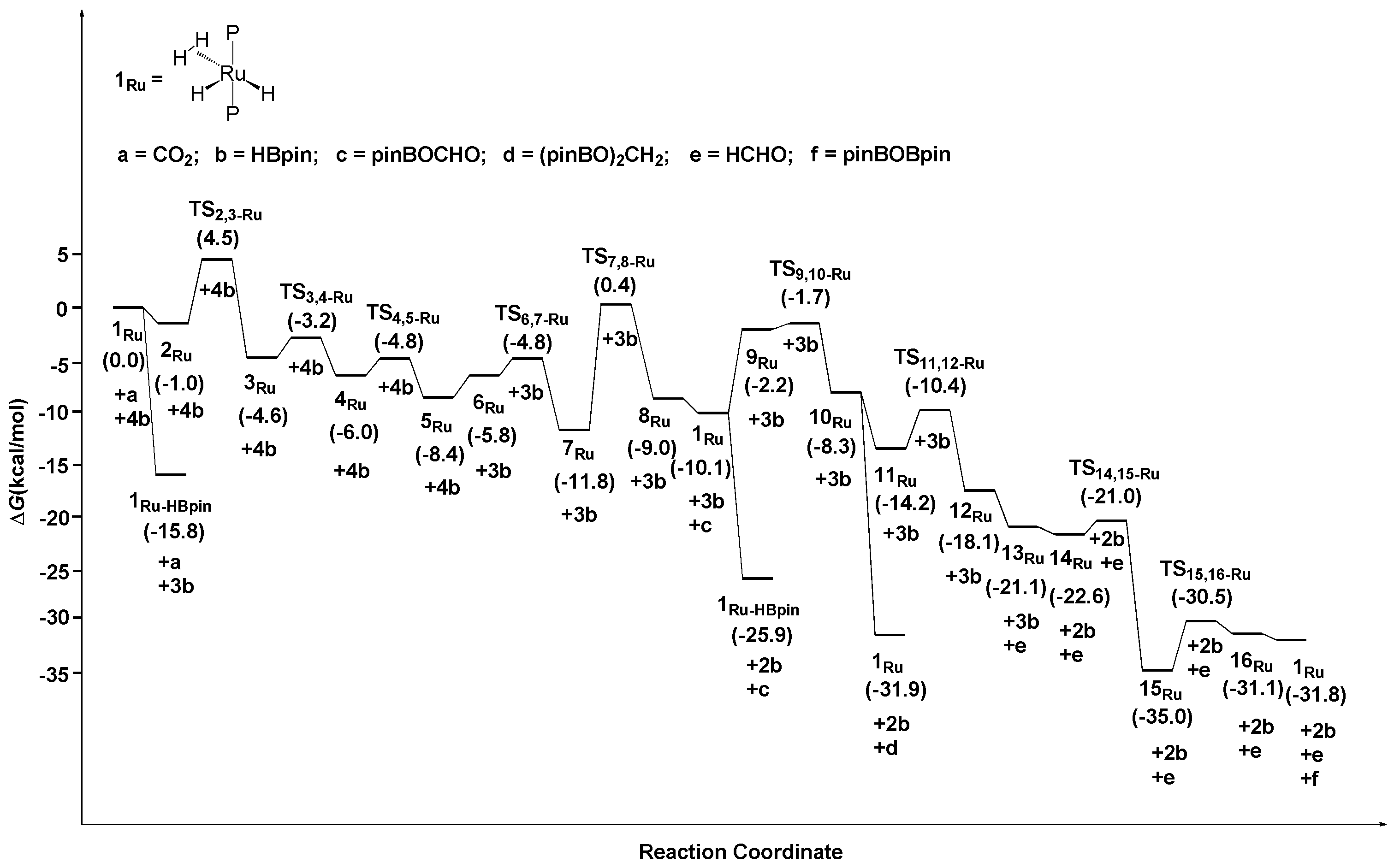
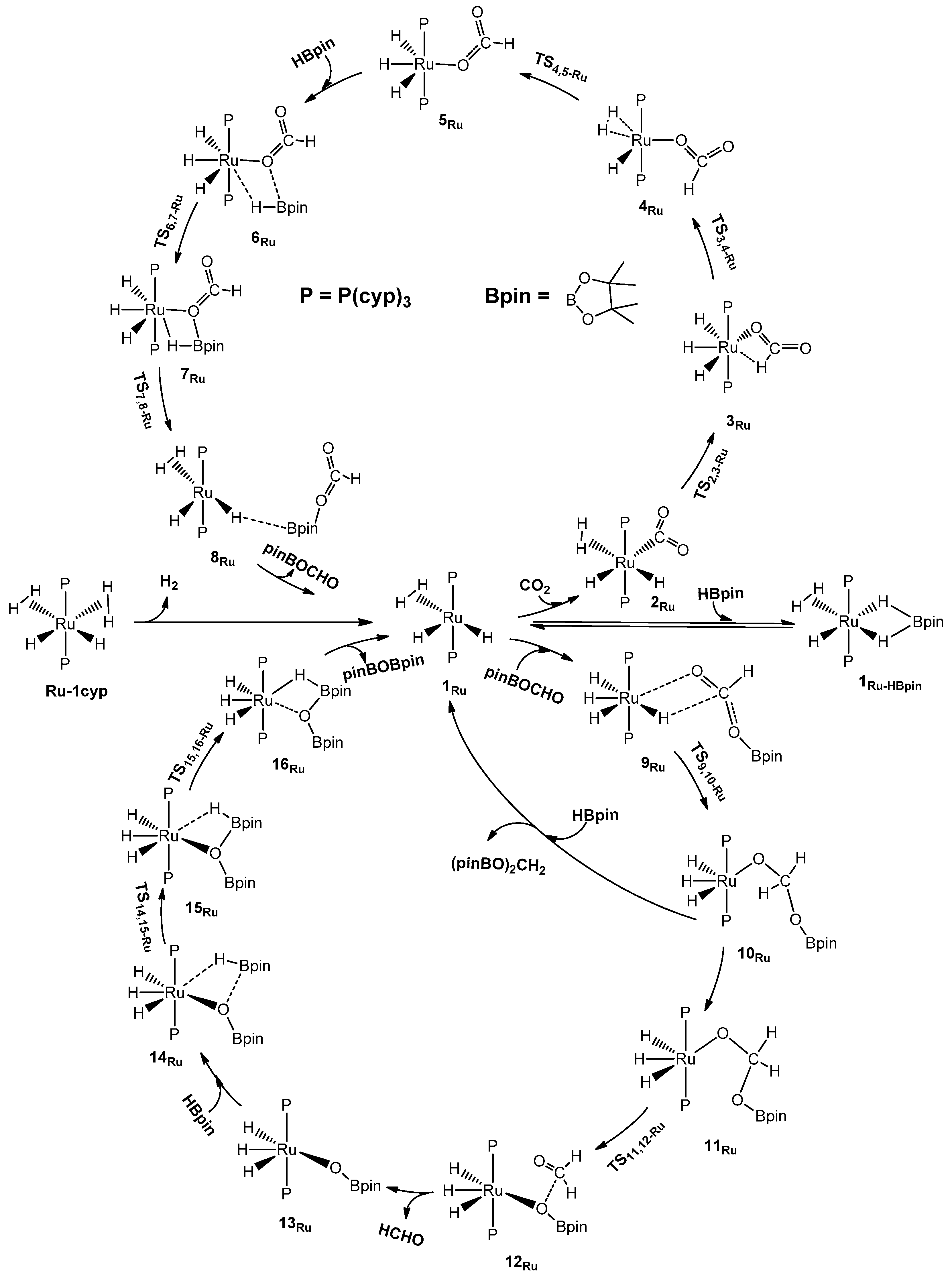
2.1. The Mechanism for the Reduction of Carbon Dioxide to Formaldehyde Catalyzed by the Ru Complex
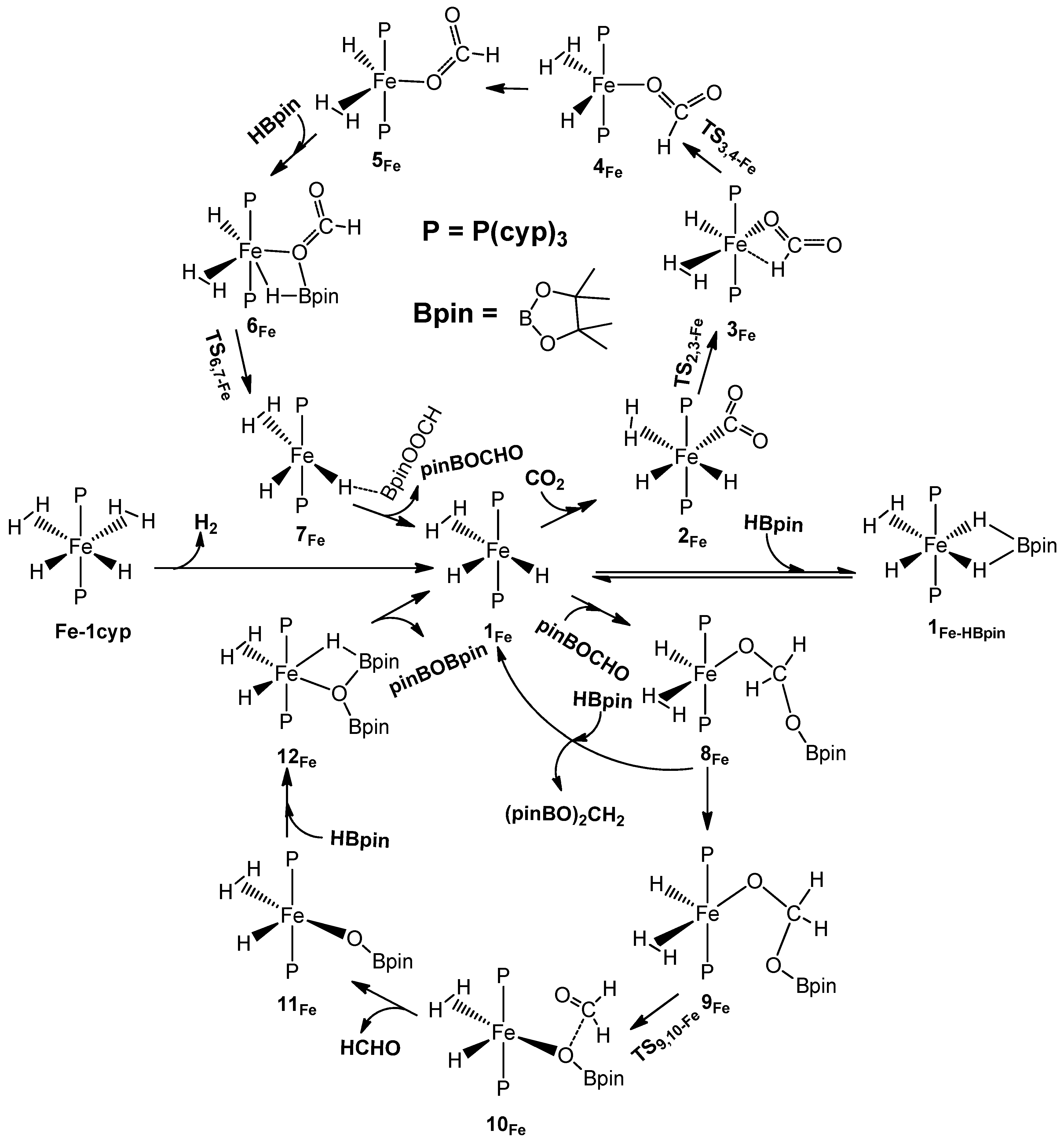
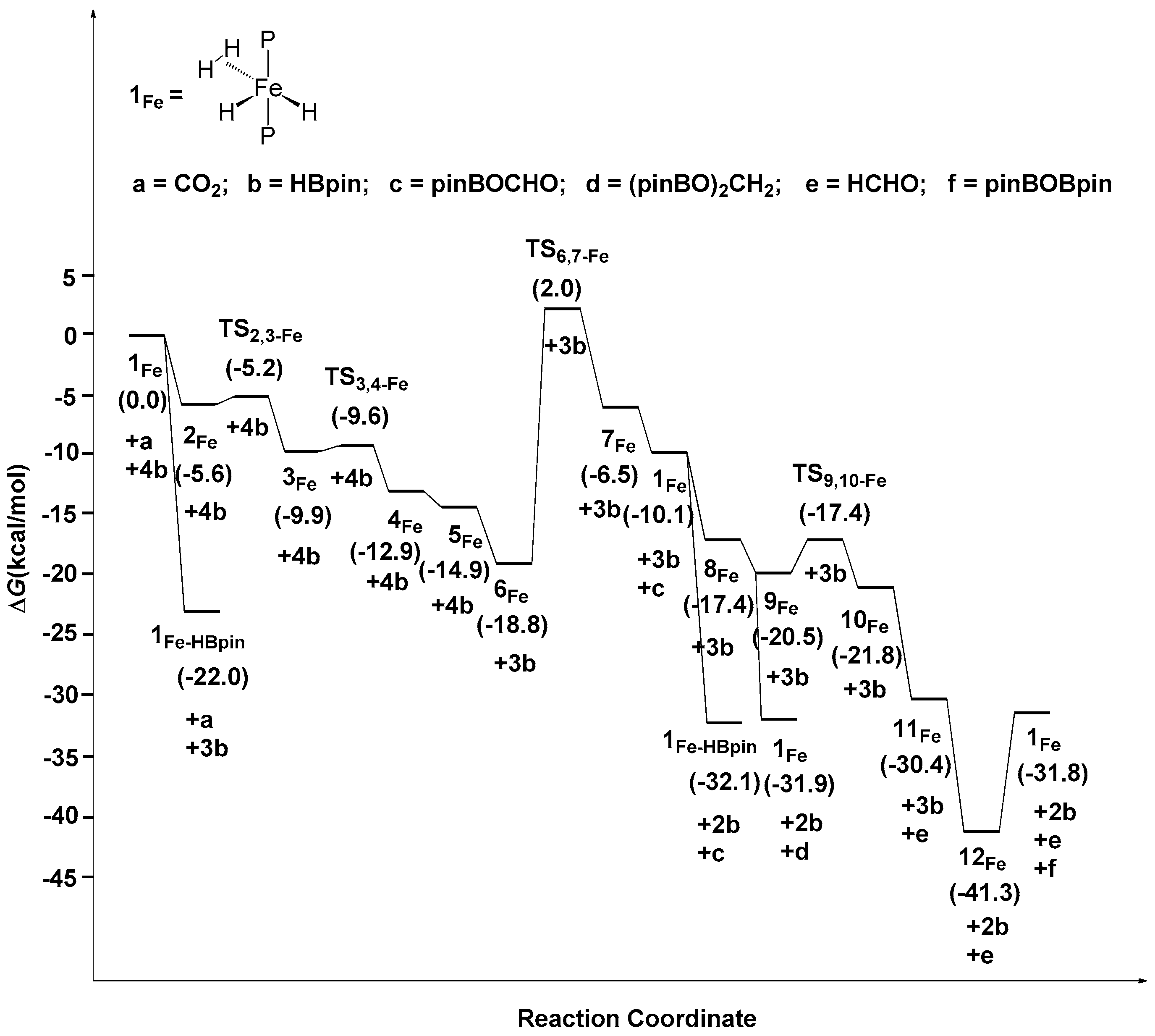
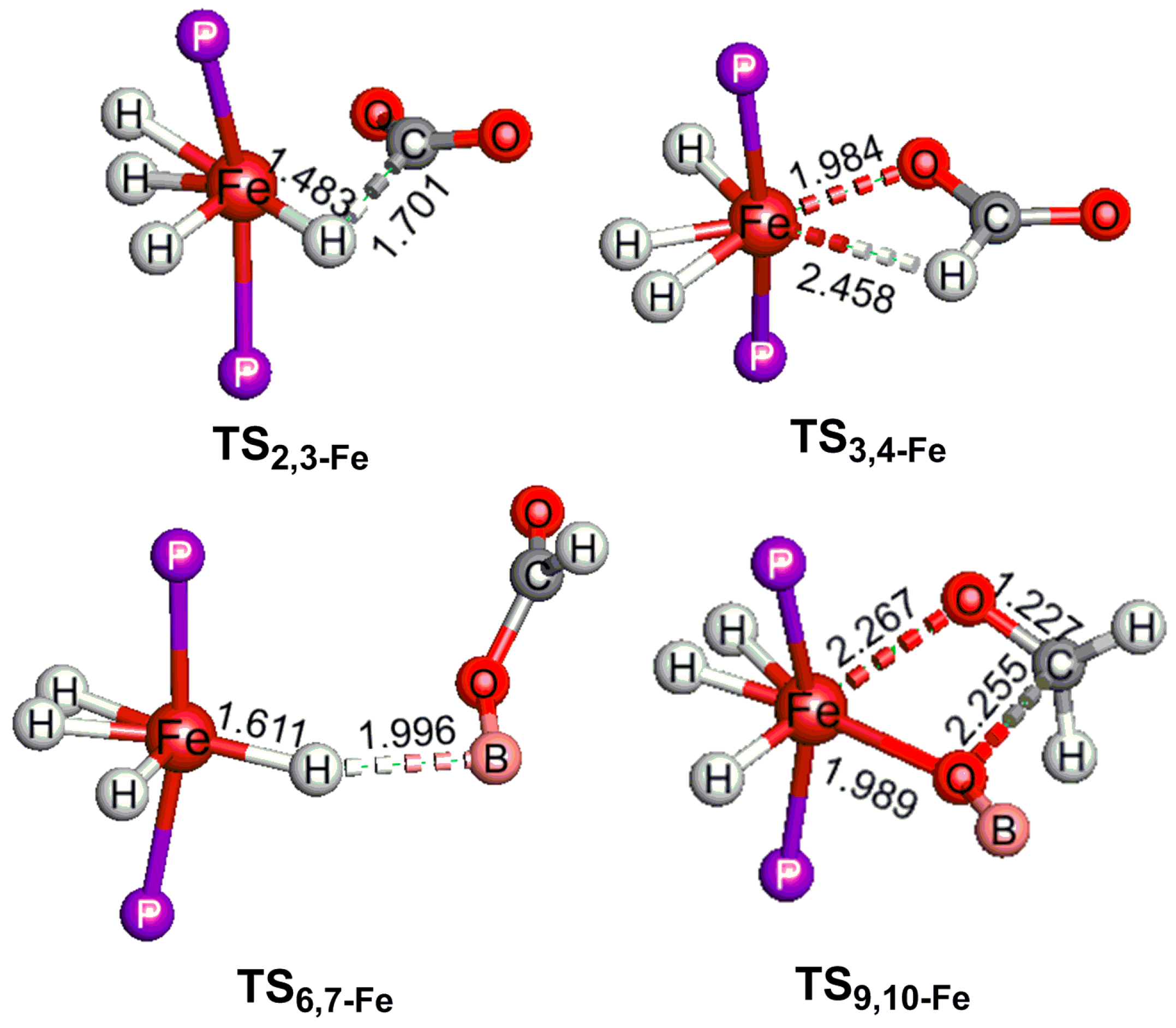
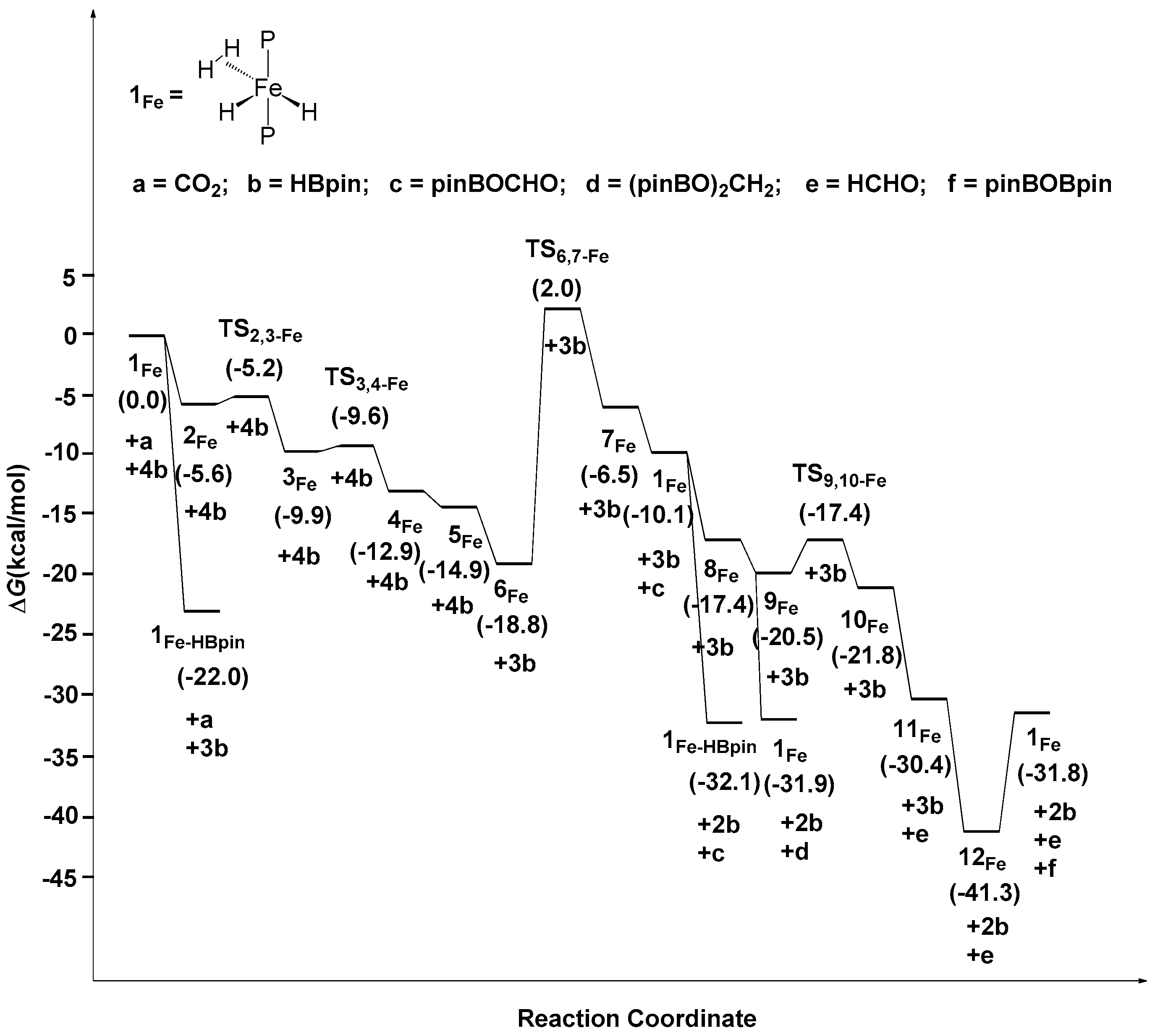
2.2. The Mechanism for the Reduction of Carbon Dioxide to Formaldehyde Catalyzed by the Fe Complex
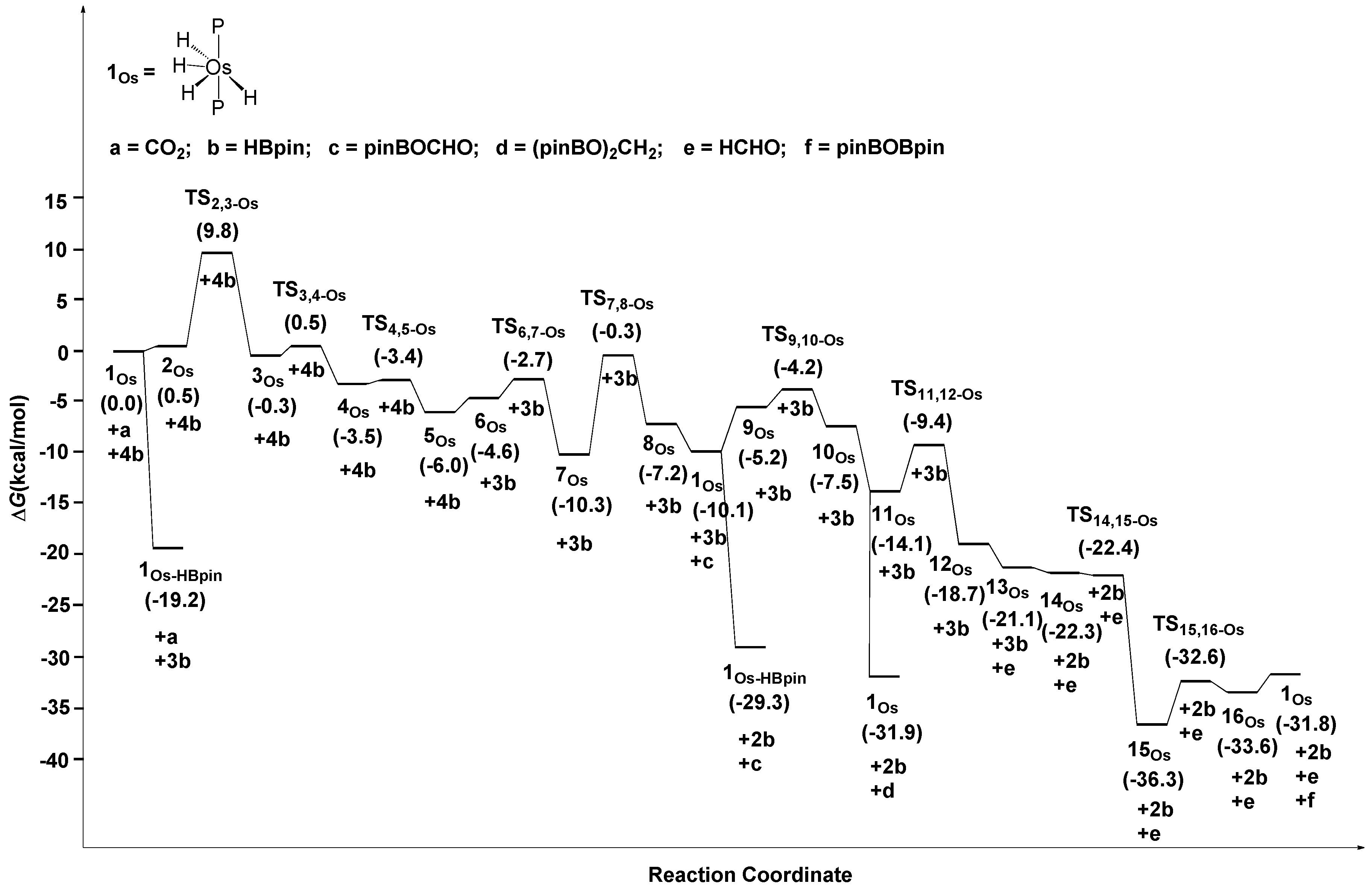
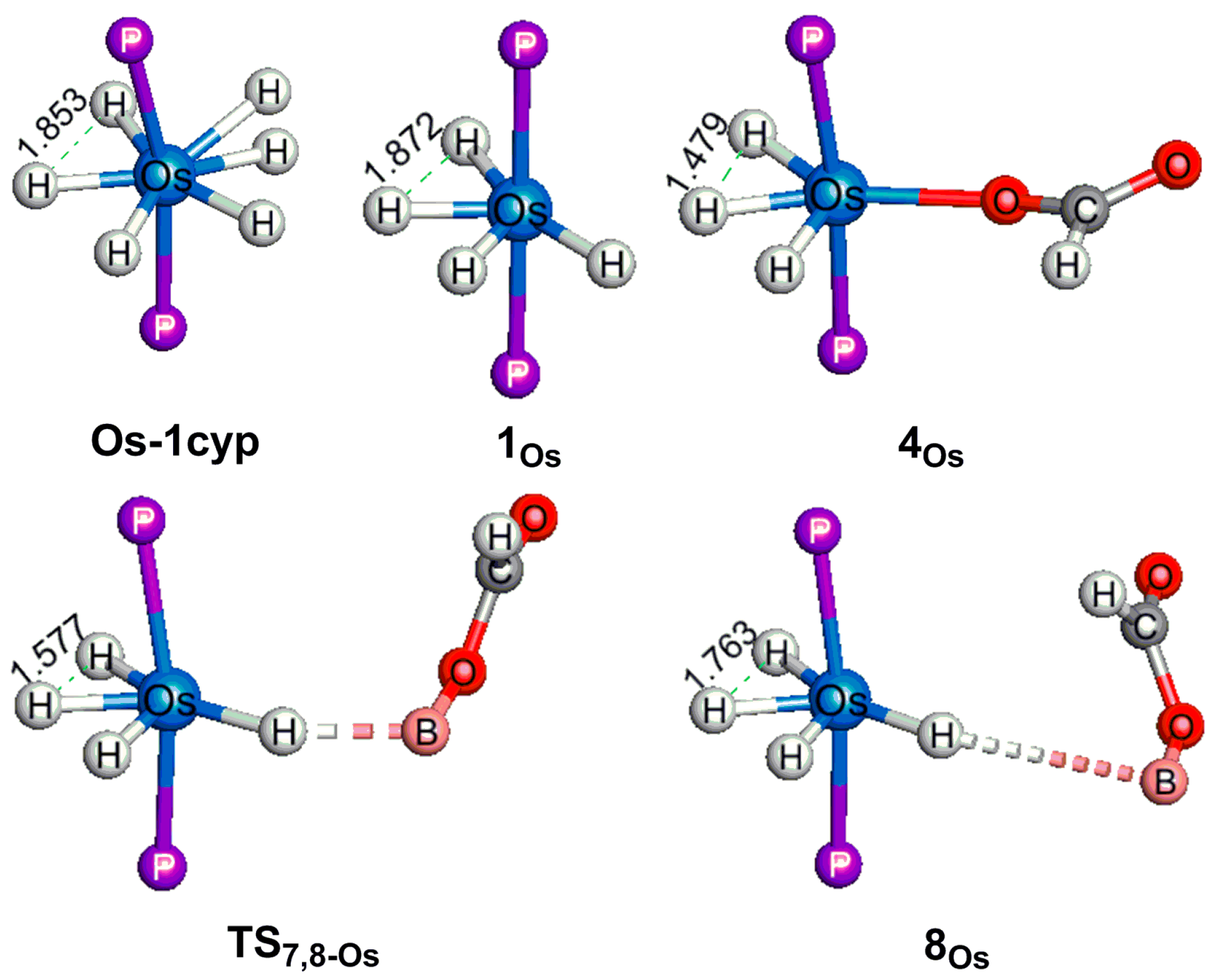
2.3. The Mechanism for the Reduction of Carbon Dioxide to Formaldehyde Catalyzed by the Os Complex
2.4. Structures and Reactivity
3. Computational Details
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kleeberg, C.; Cheung, M.S.; Lin, Z.; Marder, T.B. Copper-mediated reduction of CO2 with pinB-SiMe2Ph via CO2 insertion into a coppersilicon bond. J. Am. Chem. Soc. 2011, 133, 19060–19063. [Google Scholar] [CrossRef] [PubMed]
- Rezayee, N.M.; Huff, C.A.; Sanford, M.S. Tandem amine and ruthenium-catalyzed hydrogenation of CO2 to methanol. J. Am. Chem. Soc. 2015, 137, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Bontemps, S.; Vendier, L.; Sabo-Etienne, S. Ruthenium-catalyzed reduction of carbon dioxide to formaldehyde. J. Am. Chem. Soc. 2014, 136, 4419–4425. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, F.A.; Piers, W.E.; Parvez, M. Selective hydrosilation of CO2 to a bis(silylacetal) using an anilido bipyridyl-ligated organoscandium catalyst. Angew. Chem. Int. Ed. 2014, 126, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Moret, S.; Dyson, P.J.; Laurenczy, G. Direct synthesis of formic acid from carbon dioxide by hydrogenation in acidic media. Nat. Commun. 2014, 5, 4017. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J.; Harris, J.E.; Yin, X.; Silverwood, I.; White, A.J.P.; Kazarian, S.G.; Hellgardt, K.; Shaffer, M.S.P.; Williams, C.K. Mononuclear phenolate diamine zinc hydride complexes and their reactions With CO2. Organometallics 2014, 33, 1112–1119. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Yang, X.; Yao, J.; Chen, H. Mechanistic study and ligand design for the formation of zinc formate complexes from zinc hydride complexes and carbon dioxide. Organometallics 2015, 34, 121–126. [Google Scholar] [CrossRef]
- Tlili, A.; Blondiaux, E.; Frogneux, X.; Cantat, T. Reductive functionalization of CO2 with amines: An entry to formamide, formamidine and methylamine derivatives. Green Chem. 2015, 17, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Berkefeld, A.; Piers, W.E.; Parvez, M.; Castro, L.; Maron, L.; Eisenstein, O. Decamethylscandocinium-hydrido-(perfluorophenyl)-borate: Fixation and tandem tris(perfluorophenyl)-borane catalysed deoxygenative hydrosilation of carbon dioxide. Chem. Sci. 2013, 4, 2152–2162. [Google Scholar] [CrossRef]
- Li, Y.; Yan, T.; Junge, K.; Beller, M. Catalytic methylation of C–H bonds using CO2 and H2. Angew. Chem. Int. Ed. 2014, 53, 10476–10480. [Google Scholar] [CrossRef] [PubMed]
- Filonenko, G.A.; Smykowski, D.; Szyja, B.M.; Li, G.N.; Szczygiel, J.; Hensen, E.J.M.; Pidko, E.A. Catalytic hydrogenation of CO2 to formates by a lutidine-derived Ru-CNC pincer complex: Theoretical insight into the unrealized potential. ACS Catal. 2015, 5, 1145–1154. [Google Scholar] [CrossRef]
- Haunschild, R. Theoretical study on the reaction mechanism of carbon dioxide reduction to methanol using a homogeneous ruthenium(II) phosphine catalyst. Polyhedron 2015, 85, 543–548. [Google Scholar] [CrossRef]
- Musashi, Y.; Sakaki, S. Theoretical study of ruthenium-catalyzed hydrogenation of carbon dioxide into formic acid. Reaction mechanism involving a new type of sigma-bond metathesis. J. Am. Chem. Soc. 2000, 122, 3867–3877. [Google Scholar] [CrossRef]
- Chong, C.C.; Kinjo, R. Catalytic hydroboration of carbonyl derivatives, imines, and carbon dioxide. ACS Catal. 2015, 5, 3238–3259. [Google Scholar] [CrossRef]
- Bontemps, S. Boron-mediated activation of carbon dioxide. Coord. Chem. Rev. 2015, 308, 117–130. [Google Scholar] [CrossRef]
- Suh, H.-W.; Guard, L.M.; Hazari, N. A mechanistic study of allene carboxylation with CO2 resulting in the development of a Pd(II) pincer complex for the catalytic hydroboration of CO2. Chem. Sci. 2014, 5, 3859–3872. [Google Scholar] [CrossRef]
- Courtemanche, M.-A.; Légaré, M.-A.; Maron, L.; Fontaine, F.-G. Reducing CO2 to methanol using frustrated lewis pairs: On the mechanism of phosphine–borane-mediated hydroboration of CO2. J. Am. Chem. Soc. 2014, 136, 10708–10717. [Google Scholar] [CrossRef] [PubMed]
- Courtemanche, M.-A.; Légaré, M.-A.; Maron, L.; Fontaine, F.-G. A highly active phosphine–borane organocatalyst for the reduction of CO2 to methanol using hydroboranes. J. Am. Chem. Soc. 2013, 135, 9326–9329. [Google Scholar] [CrossRef] [PubMed]
- Declercq, R.; Bouhadir, G.; Bourissou, D.; Légaré, M.-A.; Courtemanche, M.-A.; Nahi, K.S.; Bouchard, N.; Fontaine, F.-G.; Maron, L. Hydroboration of carbon dioxide using ambiphilic phosphine–borane catalysts: On the role of the formaldehyde adduct. ACS Catal. 2015, 5, 2513–2520. [Google Scholar] [CrossRef]
- Lu, Z.; Hausmann, H.; Becker, S.; Wegner, H.A. Aromaticity as stabilizing element in the bidentate activation for the catalytic reduction of carbon dioxide. J. Am. Chem. Soc. 2015, 137, 5332–5335. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhang, C.; Jiang, J.; Wang, Z.-X.; Guan, H. How does the nickel pincer complex catalyze the conversion of CO2 to a methanol derivative? A computational mechanistic study. Inorg. Chem. 2011, 50, 3816–3825. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.-W.; Guard, L.M.; Hazari, N. Synthesis and reactivity of a masked PSiP pincer supported nickel hydride. Polyhedron 2014, 84, 37–43. [Google Scholar] [CrossRef]
- Anker, M.D.; Arrowsmith, M.; Bellham, P.; Hill, M.S.; Kociok-Kohn, G.; Liptrot, D.J.; Mahon, M.F.; Weetman, C. Selective reduction of CO2 to a methanol equivalent by B(C6F5)3-activated alkaline earth catalysis. Chem. Sci. 2014, 5, 2826–2830. [Google Scholar] [CrossRef] [Green Version]
- Sypaseuth, F.D.; Matlachowski, C.; Weber, M.; Schwalbe, M.; Tzschucke, C.C. Electrocatalytic carbon dioxide reduction by using cationic pentamethylcyclopentadienyl–iridium complexes with unsymmetrically substituted bipyridine ligands. Chem.–Eur. J. 2015, 21, 6564–6571. [Google Scholar] [CrossRef] [PubMed]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO Version 3.1; TCI, University of Wisconsin: Madison, WI, USA, 1998. [Google Scholar]
- Jin, G.; Werncke, C.G.; Escudié, Y.; Sabo-Etienne, S.; Bontemps, S. Iron-catalyzed reduction of CO2 into methylene: Formation of C–N, C–O, and C–C bonds. J. Am. Chem. Soc. 2015, 137, 9563–9566. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-adjusted ab initio pseudopotentials for the second and third row transition elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Martin, J.M.L.; Sundermann, A. Correlation consistent valence basis sets for use with the Stuttgart–Dresden–Bonn relativistic effective core potentials: The atoms Ga–Kr and In–Xe. J. Chem. Phys. 2001, 114, 3408–3420. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J. Self-consistent molecular orbital methods. XII. Further extensions of Gaussian-type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Yang, X. Hydrogenation of carbon dioxide catalyzed by PNP pincer iridium, iron, and cobalt complexes: A computational design of base metal catalysts. ACS Catal. 2011, 1, 849–854. [Google Scholar] [CrossRef]
- Yang, X. Metal hydride and ligand proton transfer mechanism for the hydrogenation of dimethyl carbonate to methanol catalyzed by a pincer ruthenium complex. ACS Catal. 2012, 2, 964–970. [Google Scholar] [CrossRef]
- Sun, Y.; Hu, L.; Chen, H. Comparative assessment of DFT performances in Ru- and Rh-promoted σ-bond activations. J. Chem. Theory Comput. 2015, 11, 1428–1438. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Sun, X.; Geng, C.; Zhao, H.; Li, J. Benchmark study on methanol C–H and O–H bond activation by bare [FeIVO]2+. J. Phys. Chem. A 2014, 118, 7146–7158. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.; Webster, C.E.; Hall, M.B. JIMP2, Version 0.091, a Free Program for Visualizing and Manipulating Molecules; Texas A&M University: College Station, TX, USA, 2006. [Google Scholar]
- Reed, A.E.; Weinstock, R.B.; Weinhold, F. Natural population analysis. J. Chem. Phys. 1985, 83, 735–746. [Google Scholar] [CrossRef]
- Wiberg, K.B. Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stages | Complexes | e | Bond | WBI | ΔG‡/ΔG° (kcal/mol) |
|---|---|---|---|---|---|
| Stage 1 | 2Fe | −1.892 | M–H bond | 0.5876 | 0.4 |
| TS2,3-Fe | −1.782 | 0.5420 | |||
| 2Ru | −1.538 | 0.6273 | 5.5 | ||
| TS2,3-Ru | −1.439 | 0.4674 | |||
| 2Os | −1.540 | 0.6887 | 9.3 | ||
| TS2,3-Os | −1.518 | 0.5094 | |||
| Stage 2 | 6Fe | −1.209 | M–O bond | 0.3517 | 20.8 |
| TS6,7-Fe | −0.974 | 0.0287 | |||
| 7Ru | −1.411 | 0.2704 | 12.2 | ||
| TS7,8-Ru | −1.104 | 0.0275 | |||
| 7Os | −1.427 | 0.2995 | 10.0 | ||
| TS7,8-Os | −1.111 | 0.0271 | |||
| Stage 3 | 1Fe | −1.280 | M–H bond | 0.7154 | −7.3 |
| 8Fe | −1.279 | 0.2182 | |||
| 9Ru | −1.867 | 0.6661 | 0.5 | ||
| TS9,10-Ru | −1.850 | 0.5757 | |||
| 9Os | −1.832 | 0.7206 | 1.0 | ||
| TS9,10-Os | −1.808 | 0.6002 | |||
| Stage 4 | 9Fe | −0.890 | M–O bond | 0.4354 | 3.1 |
| TS9,10-Fe | −0.840 | 0.2609 | |||
| 11Ru | −1.190 | 0.2583 | 3.8 | ||
| TS11,12-Ru | −1.184 | 0.2616 | |||
| 11Os | −1.177 | 0.4049 | 4.7 | ||
| TS11,12-Os | −1.193 | 0.3061 | |||
| Stage 5 | 11Fe | −0.778 | / | / | −10.9 |
| 12Fe | −1.290 | H–B bond | 0.5236 | ||
| 15Ru | −1.478 | 0.5325 | 4.5 | ||
| TS15,16-Ru | −1.795 | 0.0863 | |||
| 15Os | −1.483 | 0.4898 | 3.7 | ||
| TS15,16-Os | −1.742 | 0.0958 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, C.; Ji, M.; Yang, X.; Yao, J.; Chen, H. Reaction Mechanisms of CO2 Reduction to Formaldehyde Catalyzed by Hourglass Ru, Fe, and Os Complexes: A Density Functional Theory Study. Catalysts 2017, 7, 5. https://doi.org/10.3390/catal7010005
Dong C, Ji M, Yang X, Yao J, Chen H. Reaction Mechanisms of CO2 Reduction to Formaldehyde Catalyzed by Hourglass Ru, Fe, and Os Complexes: A Density Functional Theory Study. Catalysts. 2017; 7(1):5. https://doi.org/10.3390/catal7010005
Chicago/Turabian StyleDong, Chunhua, Mingsong Ji, Xinzheng Yang, Jiannian Yao, and Hui Chen. 2017. "Reaction Mechanisms of CO2 Reduction to Formaldehyde Catalyzed by Hourglass Ru, Fe, and Os Complexes: A Density Functional Theory Study" Catalysts 7, no. 1: 5. https://doi.org/10.3390/catal7010005