1. Introduction
Supported bimetallic catalysts have drawn attention in heterogeneous catalysis research due to, compared with their monometallic counterparts, their superior catalytic properties [
1]. Bimetallic nanoparticles have been studied in several important chemical reactions [
2]. The impregnation method is commonly used for the synthesis of bimetallic catalysts because of its simplicity. However, nanoparticles produced using such an approach usually have inherently non-uniform sizes and/or compositions. The uncertainty about the composition and size of the synthesized bimetallic nanoparticles makes it difficult to establish meaningful structure–reactivity relationships [
3,
4]. It is thus imperative to exploit novel synthesis strategies that produce supported bimetallic nanoparticles with specific compositions and ultrastability. Several new types of catalyst synthesis methods have been developed to ensure appropriate metal-metal interaction to form the compositions of each nanoparticle. For example, electroless deposition [
5,
6,
7] and strong electrostatic adsorption [
8,
9,
10,
11,
12] approaches have been extensively studied.
An inherent problem for developing nanocatalysts, especially bimetallic or multimetallic nanoparticle based catalysts, is their lack of stability under practical catalytic reaction conditions [
13,
14]. Supported metal/alloy nanoparticles experience rapid growth during their use, especially at elevated reaction temperatures [
15]. Such sintering effect decreases the number of surface active sites and consequently leads to deactivation of the nanocatalysts of interest. In order to maintain the sizes of the metal/alloy nanoparticles during catalytic reactions, it is highly desirable to strongly anchor the metal/alloy nanoparticles onto the surfaces of the support materials.
Heteroepitaxy, the oriented growth of one crystal/film on another with certain crystallographic orientation relationship, has been extensively studied in surface science and semiconductor fields [
16]. It is expected that the enhanced interfacial adhesion between an epitaxially grown metal nanoparticle and a well-defined nanoscale facet of the support can inhibit, or at least reduce, sintering of metal/alloy nanoparticles caused by particle migration and coalescence processes. To grow all metal nanoparticles epitaxially in a supported metal catalyst requires the fabrication of support materials that possess the same or similar surface structure. When such high-surface-area crystalline supports are synthesized, it is possible to grow all metal/alloy nanoparticles epitaxially with the same type of interfacial structure and most likely with the same or similar shape, which is important for the fundamental study of the structure—performance relationships of supported metal/alloy nanoparticle catalysts.
Epitaxial growth of metals on clean and flat substrates has been researched for many years. For instance, metals can epitaxially grow on metal oxides through atomic layer deposition [
15], pulsed laser deposition [
17], UHV sputtering deposition [
18,
19,
20,
21], and annealing [
22]. Unfortunately, vapor-phase deposition techniques suffer from several disadvantages, such as the requirement of ultra-high vacuum conditions and the use of special equipment [
23]. Furthermore, most of the vapor-phase deposition techniques only work for planar surfaces and cannot be applied to deposit metal/alloy nanoparticles onto high-surface-area supports. The wet chemistry synthetic route remains the main method for producing supported metal/alloy nanocatalysts due to its relatively low cost, easy-operation, versatility, and scalability [
24].
We have now developed a general strategy to synthesize ultrastable nanostructured metal/alloy catalysts by using the recent advancement in controllable synthesis of nanostructured support materials. In this paper, we illustrate this general synthesis method for fabricating ZnO nanowire supported PtZn nanoalloy catalysts. PtZn alloy nanoparticles have been synthesized through many methods including the reaction of Zn vapor with Pt particles supported on carbon [
25], electrodeposition of Zn onto Pt particles [
26], and heating Pt particles on ZnO supports under a reducing atmosphere [
27,
28,
29,
30]. A recent review article published by Long
et al. [
31] gives an elaborate survey of the development of Pt-based catalysts, the important role of the structure, size, shape, and morphology of Pt nanoparticles, and their applications for energy conversion.
In this work, we selected highly stable ZnO nanowires that primarily consist of low-energy and stable {10−10} nanoscale facets. Successful synthesis of epitaxially anchored PtZn nanoparticles onto ZnO nanowires depends on (1) the cleanness and flatness of the ZnO {10−10} nanoscale facets; (2) the uniform deposition of the Pt precursor species onto the ZnO {10−10} facets; and (3) the control of the reduction processes to promote the formation and epitaxial growth of PtZn nanoparticles on the ZnO {10−10} facets. When these processes are optimized, epitaxially anchored PtZn bimetallic nanoparticles can be reliably and repeatedly produced. We have tested the synthesized PtZn/ZnO nanocatalysts for water-gas-shift (WGS) reaction and attributed their long-term stability to the epitaxial anchoring of the PtZn nanoparticles.
2. Results and Discussion
The preparation processes of supported metal/alloy nanoparticle catalysts generally consists of three steps: (1) contact of a metal precursor with the support material; (2) oxidation/calcination of the metal precursor/support composite to eliminate the undesirable molecular species; and (3) formation of the active nanoparticles via various types of reduction processes. It is usually during the reduction step that the metal/alloy nanoparticles evolve into their thermodynamically stable shapes and size distributions. Importantly, the reduction step often leads to a wide distribution of nanoparticles or nanoalloys with non-uniform composition. Furthermore, during the reduction processes, the surface structure and chemistry of the support materials may be modified which in turn can significantly affect the characteristics of the supported metal/alloy nanoparticles. Precise and careful control of the metal precursor distribution and the movement of the metal atoms during the reduction processes is essential to preparing supported nanoparticle catalysts with size-, shape- and composition-control.
Deposition-precipitation is a common method for preparing supported metal/alloy catalysts where the metal precursor complexes are deposited onto the support materials, which are then suspended in the precipitating solution. Such a catalyst preparation method utilizes the fact that precipitation of the metal complexes onto the support surfaces requires a lower supersaturation than the formation of the new metal-containing phases directly in the solution phase. It is critical to maintain the supersaturation at a constant and moderate level. The control of the release rate of the precipitating agent, for example, by the controlled gradual addition of the precipitating agent is crucial to the homogeneous deposition of the metal precursor complexes. Deposition of metal-containing species from a precursor solution onto the support surfaces can be tuned through the fine control of the solution’s pH value. The specific addition sequence of the various components, the droplet sizes, the appropriate turbulence of the solution, and the solution temperature, etc. affects the structure and chemistry of the final supported metal/alloy catalysts. The deposition-precipitation method works well for non-porous support materials or those materials that contain meso- and macropores. Such a method is excellent for individually separable particles of the support such as individually separable ZnO nanowires or other nanostructured support materials. We used the deposition-precipitation method and post-treatment manipulation to synthesize our ultrastable, epitaxially anchored PtZn nanoparticles supported on ZnO nanowires.
The BET (Brunauer-Emmett-Teller) surface area of the pristine ZnO nanowires was determined to be 17.6 m2/g. After depositing the Pt species on the ZnO nanowires and the calcination treatment, the BET surface area of the Pt/ZnO NW catalyst was measured to be 16.3 m2/g, slightly lower than that of the pristine ZnO nanowires. The nominal Pt loading was 1.0 wt. % with a goal to disperse small Pt nanoparticles uniformly onto the ZnO nanowires. ICP-MS measurement of the actual Pt loading yielded a value of about 0.8 wt. % of Pt in the final catalysts, slightly lower than the nominal loading level. Some Pt species were lost during the precipitation-deposition and probably the subsequent treatment processes. The calcined and reduced catalysts are labeled as Pt/ZnO NW-X and Pt/ZnO NW-HX, respectively, where “X” represents the calcination or reduction temperature. Unless otherwise stated the calcination and reduction time was 4 and 2 h, respectively.
The representative SEM images of the pristine ZnO nanowires and the Pt/ZnO NW-H500 catalyst (
Figure 1a,b) show their typical morphology with lengths of tens of micrometers and diameters of tens of nanometers. The XRD patterns (
Figure 1c) show that the pristine ZnO nanowires possess a wurtzite structure and that the structure of the ZnO nanowires did not change after being deposited with the Pt species, the calcination, and the reduction treatments. The XRD patterns do not display, however, any diffraction peaks that represent Pt or PtZn phases, presumably due to the high dispersion of the Pt/PtZn species and/or the low Pt loading level.
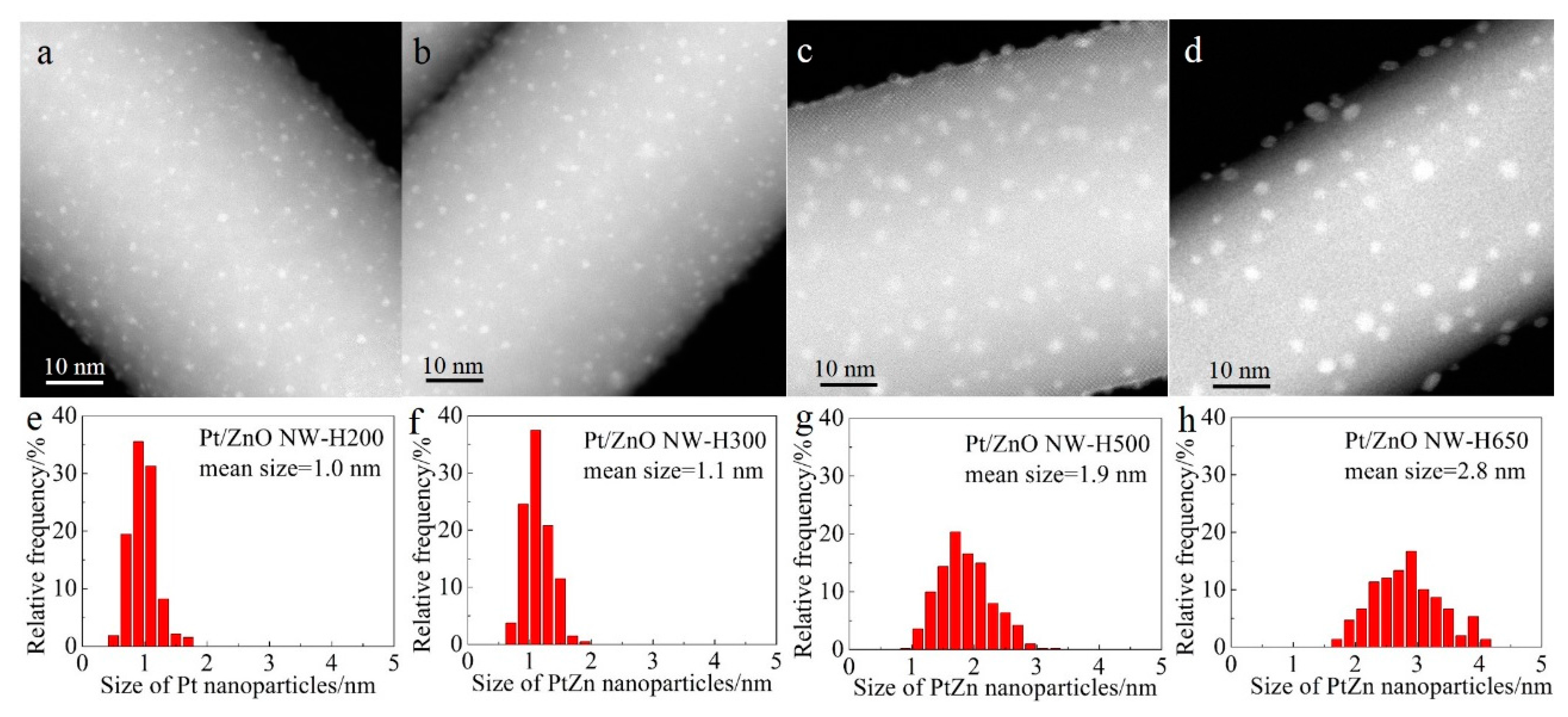
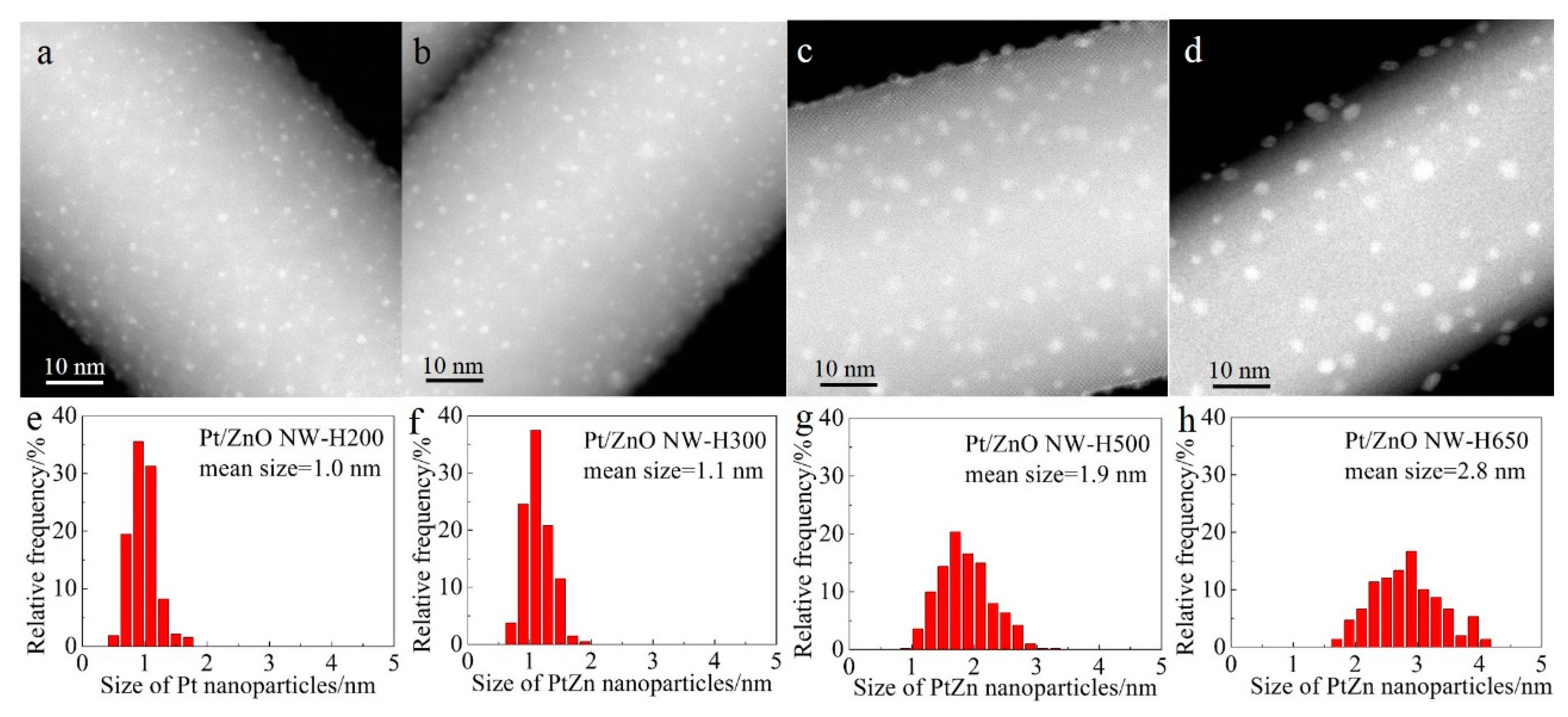
Figure 2 shows the representative high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images and the corresponding size distributions of the Pt/ZnO NW catalysts that had been calcined at 400 °C and reduced at different temperatures. In HAADF-STEM images the signal intensity is determined by the sample thickness and the atomic number Z of the probed elements of interest [
32]. Therefore, columns of Pt atoms are brighter than the columns of Zn atoms provided that the imaged area is of similar thickness along the incident electron beam direction. Small Pt nanoparticles supported on ZnO NWs should give a high image contrast, as shown in
Figure 2. The Pt nanoparticles seem to be uniformly distributed along the surfaces of the ZnO NWs. The good dispersion is attributed to the strong interaction between the platinum precursor complexes and the ZnO NWs that possess a very high isoelectric point. Oxychlorinated species may have formed during the calcination step.
Figure 2 also shows that, after calcination at 400 °C and reduction at 200 °C, the Pt species were highly dispersed on the ZnO NWs as nanoclusters (~1.0 nm) with a narrow size distribution (
Figure 2a,e). Raising the reduction temperature from 200 °C to 300 °C slightly increased the size of the Pt nanoclusters. By increasing the reduction temperature to 500 °C and 650 °C, the average sizes of the Pt nanoparticles increased to 1.9 nm and 2.8 nm, respectively (
Figure 2c,d,g,h) and the particle size distributions broadened as well, suggesting that high-temperature reduction promoted the sintering of Pt clusters.
Higher magnification HAADF-STEM images (
Figure 3a,b) revealed that, after reduction at 200 °C and 300 °C, the Pt/PtZn species randomly dispersed on the surfaces of the ZnO NWs, and some PtZn nanoparticles may have been formed with highly disordered or amorphous structure However, after being reduced at 500 °C (
Figure 3c), the small clusters sintered to form nanoparticles that were oriented and aligned with respect to the ZnO {10−10} surfaces, resulting in a fixed crystallographic epitaxial relationship between the nanoparticles and the ZnO nanowires. Moreover, from
Figure 3c, we can clearly see that the small nanoparticles consist of dots with alternate bright and grey contrasts: the bright dots represent columns of Pt atoms, and the grey dots represent columns of Zn atoms along the incident electron beam direction. It has been reported that, during the H
2 reduction at temperatures >250 °C, Pt nanoparticles interact with the ZnO support and that PtZn nanoalloys form [
27,
29,
30,
33]. Therefore, it is reasonable to conjecture that, after high temperature reduction, PtZn alloy nanoparticles were formed in our catalysts.
Detailed analysis of
Figure 3c and other similar images can provide the specific alloy structure of the observed nanoparticles. The digital diffractogram (
Figure 3d) obtained from
Figure 3c displays spots of both the ZnO nanowire support (triangle) and the PtZn nanoparticles (ellipse). Analysis of the spot symmetry and the spacings (by using the spots of the ZnO as an internal reference) confirmed that the observed alloy particles are face-centered tetragonal PtZn (a = b = 4.03 Å, c = 3.51 Å). Furthermore, from the digital diffractogram one can conclude that the PtZn nanoparticles grew with a fixed crystallographic relationship with respect to the ZnO nanowires. The PtZn {110} planes were aligned along the ZnO {11−20} planes, while the PtZn {1−1−1} planes grew parallel with the ZnO {1−101} planes. There is an angle of ~5° between the PtZn {00−1} planes and the ZnO {1−10−1} planes. The epitaxial relationship between the PtZn nanoparticles and the ZnO NWs is designated as ZnO [11−20] (1−101)//PtZn [110] (1−1−1).
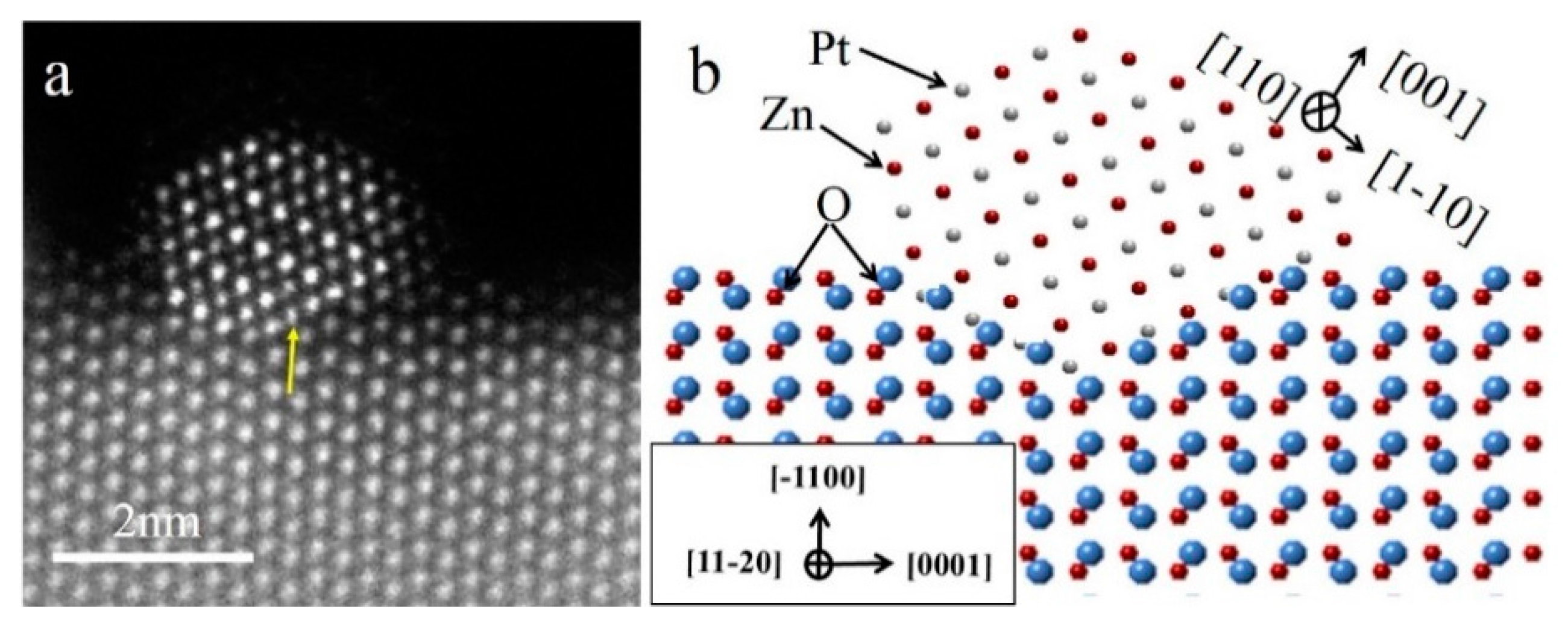
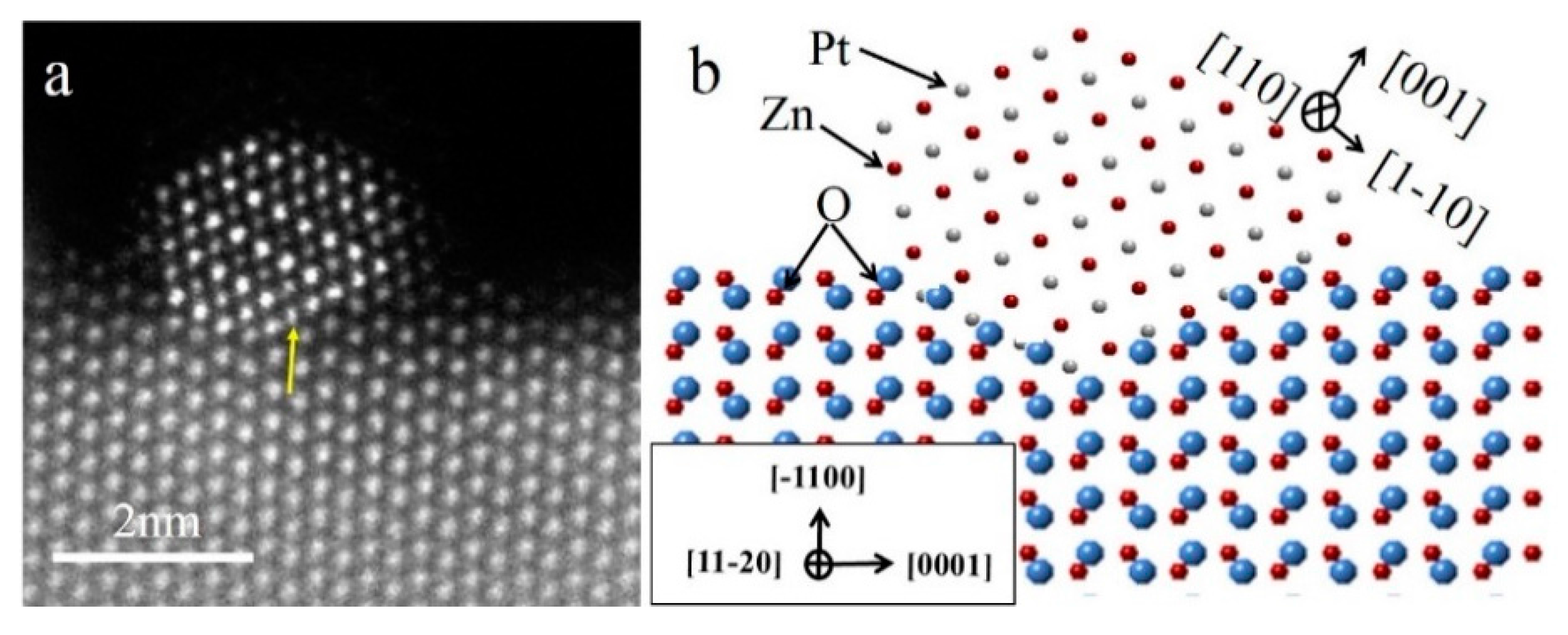
In order to study the atomic arrangement of the PtZn alloy nanoparticles and their relationship with respect to the ZnO NWs, the Pt/ZnO NW-H500 catalyst was further examined by HAADF-STEM with atomic scale resolution (
Figure 4). Since the PtZn alloy nanoparticles grew epitaxially onto the ZnO NWs with a fixed crystallographic relationship when the ZnO NWs were oriented to the [
11,,
12,
13,
14,
15,
16,
17,
18,
19,
20] zone axis, the associated PtZn alloy nanoparticles were automatically oriented to the {110} zone axis. Such an epitaxial relationship makes it possible to thoroughly investigate the atomic structures of the interfacial regions between the PtZn alloy nanoparticles and the ZnO NWs. The lattice mismatches between the PtZn {1−1−1} and the ZnO {1−101} planes, and the PtZn {00−1} and the ZnO {1−10−1} planes, were about 10% and 30%, respectively. Such large mismatches across heterointerfaces usually do not promote epitaxial growth. However, by introducing interfacial dislocations, epitaxial growth between heterostructures can proceed even with large mismatches, and the growth processes can be explained by invoking the domain matching epitaxy (DME) mechanism [
17,
34,
35]. The presence of an interfacial dislocation at the interface between the small PtZn alloy nanoparticles and the ZnO NWs is clearly imaged in
Figure 4a (indicated by the yellow arrow).
Figure 4b schematically illustrates the faceted PtZn nanoparticle and its crystallographic orientational relationship with respect to the ZnO NW. The ingrowth of the PtZn nanoparticles into the ZnO NWs can be understood in terms of Pt-induced reduction of the ZnO at the contact interface and the subsequent inter-diffusion between Zn and Pt atoms to form PtZn alloy nanoparticles. The driving force is the lowering of the total system’s energy by formation of PtZn alloy nanoparticles with an epitaxial relationship with the ZnO NW. The dominant {111} faceting of the PtZn nanoparticles manifests the fact that the close-packed PtZn {111} surfaces possessed the smallest surface energy. The specific shape of the PtZn nanoparticles may have also been affected by the interfacial strain and dislocations. Epitaxially anchored metal/alloy nanoparticles usually exhibit high structural stability during catalytic reactions [
36].
Figure 5 shows the atomic resolution HAADF-STEM images of a Pt/ZnO NW-H650 catalyst. The HAADF-STEM images show the oriented growth of the PtZn alloy nanoparticles (
Figure 5a) and the interfacial structure between the PtZn and ZnO NW (
Figure 5b). The bright-field phase contrast STEM image (
Figure 5c) clearly shows that the PtZn nanoparticles were covered by a thin layer of material, most likely ZnO
x. The presence of the thin surface layer on the PtZn alloy nanoparticles suggests the SMSI (strong metal-support interaction) effect reported for the titania (TiO
2) supported group VIII system when reduced at temperatures higher than 500 °C under a H
2 atmosphere [
37,
38]. It is interesting to note that, even after calcination at temperatures as high as 650 °C, the PtZn nanoparticles still maintained their epitaxial relationship with the ZnO NWs and did not grow significantly.
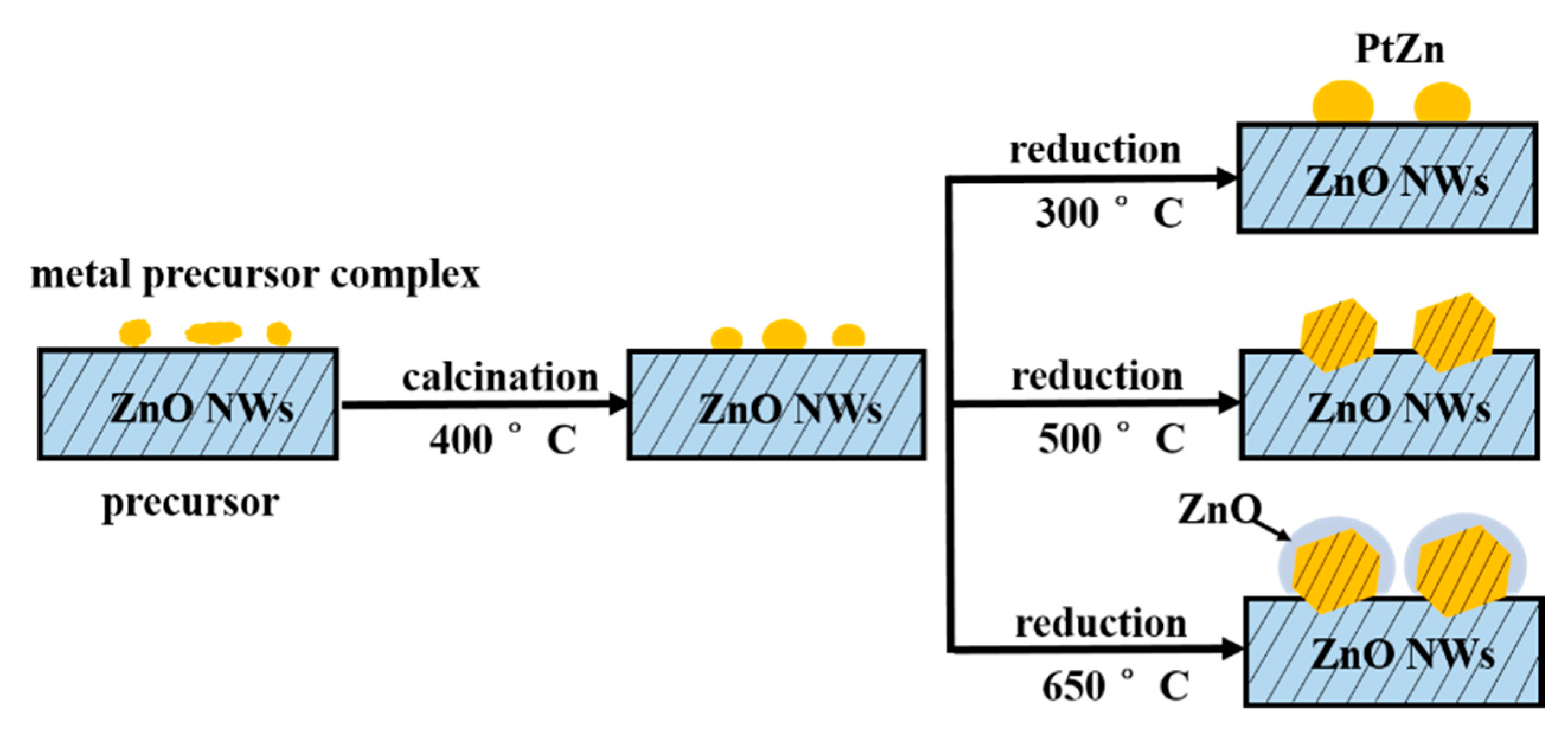
The formation processes of the epitaxial growth of the PtZn alloy nanoparticles onto ZnO nanowires are illustrated in
Scheme 1. Metal complexes are initially coated onto the ZnO nanowires. Then, calcination in air oxidatively removes the undesirable ligands that originate from the deposition of the metal precursor complexes onto the ZnO nanowires. When the Pt/ZnO system is reduced in H
2, the Pt-facilitated H
2 dissociation reduces the ZnO to ZnO
x or Zn; subsequently, the Pt and Zn interact to form PtZn bimetallic nanoparticles. During the PtZn nucleation and growth processes, the newly formed PtZn nanoparticles can grow with an epitaxial relationship with respect to the {10−10} facets of the ZnO nanowires. The formation processes of the PtZn alloy nanoparticles depend on the reduction temperature and time. At reduction temperatures at 400 °C or above, PtZn alloy nanoparticles can be easily formed and grow epitaxially. At very high reduction temperatures, ZnO may further interact with the PtZn alloy nanoparticles resulting in a thin layer of ZnO
x species covering the PtZn surfaces. Our goal is to produce the smallest PtZn bimetallic alloy nanoparticles that are epitaxially anchored into the {10−10} nanoscale facets of the ZnO nanowires.
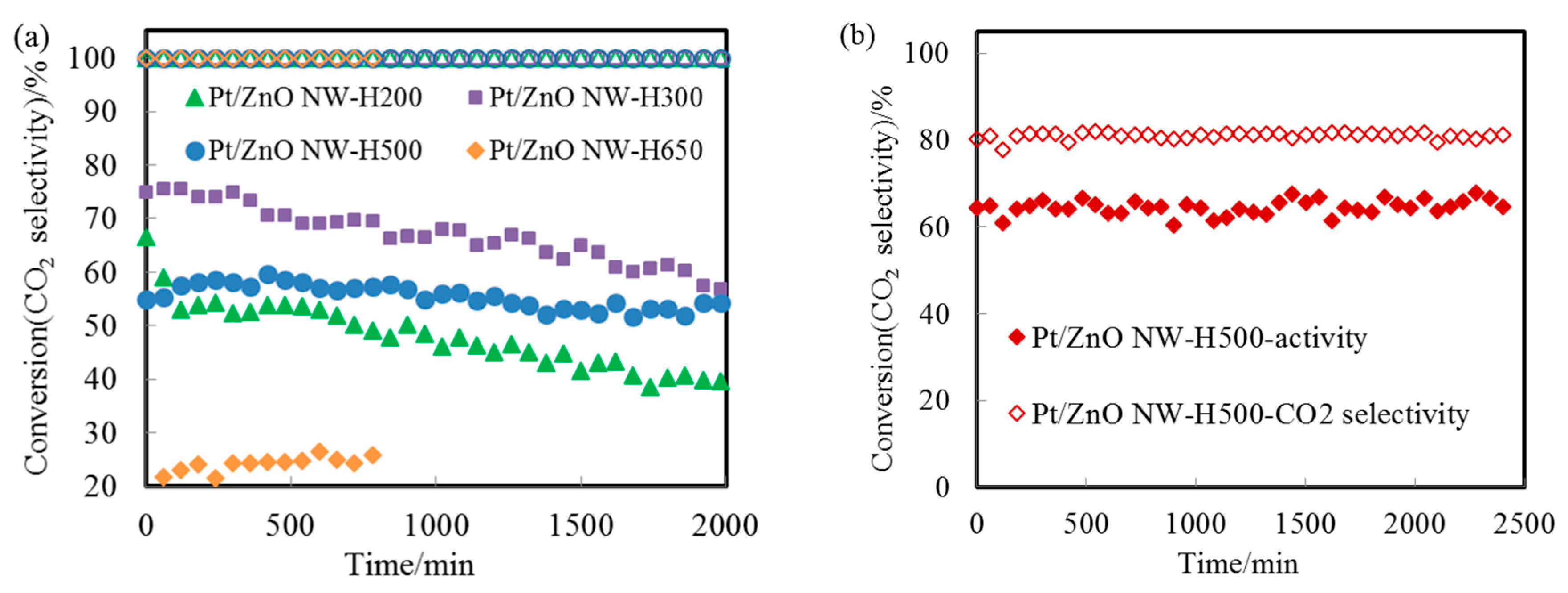
We used the WGS reaction as a probe to evaluate the catalytic performance and the stability of the epitaxial PtZn alloy nanoparticles. We choose this reaction because of its importance for energy applications and because many catalyst systems are not stable during the WGS reaction.
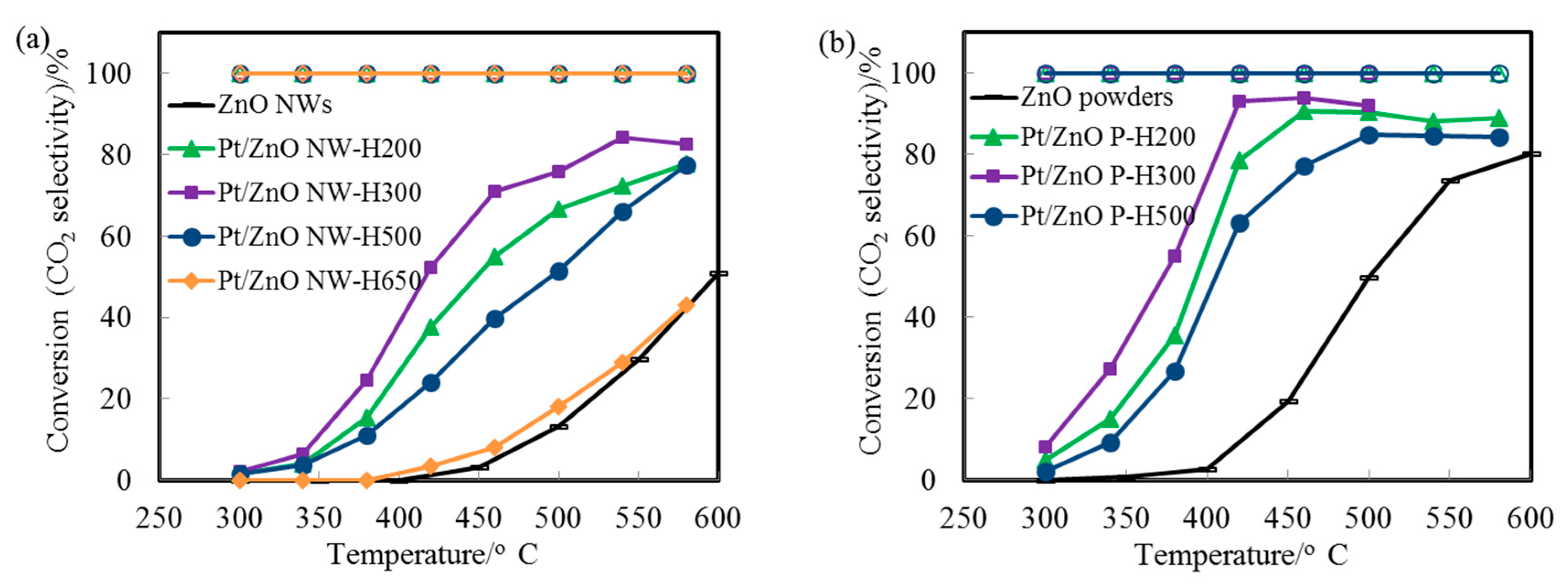
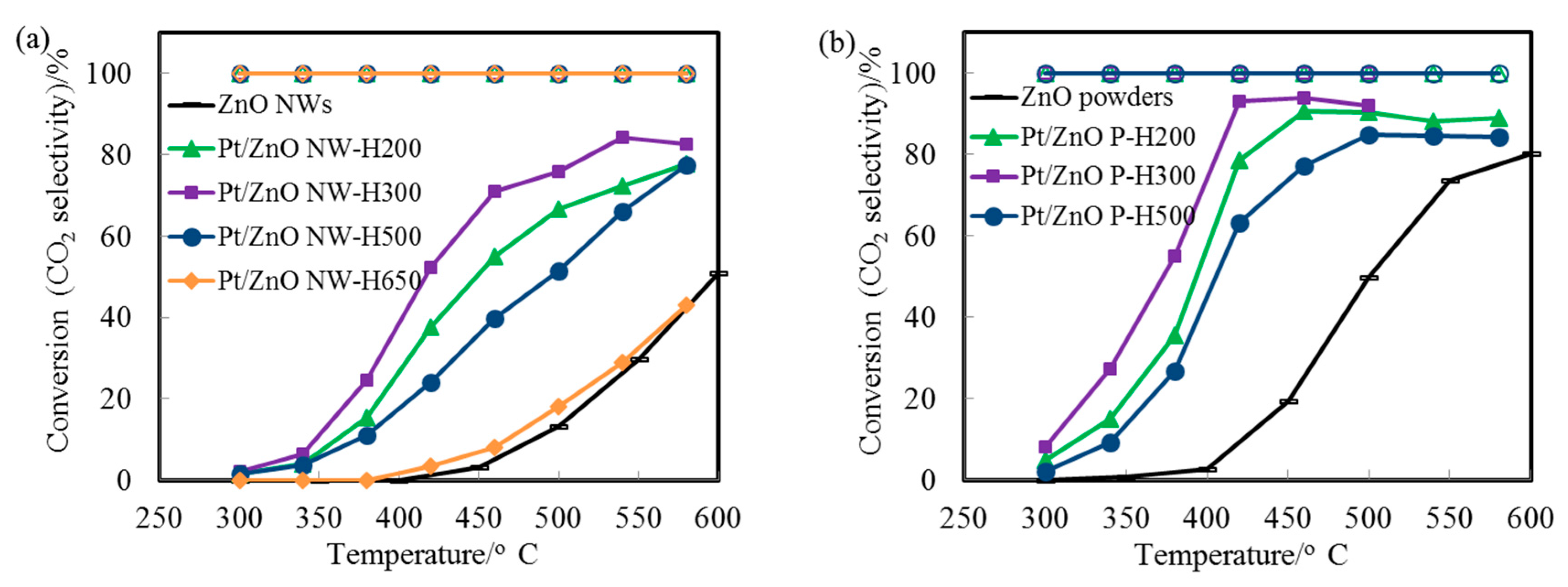
Figure 6a displays the CO conversion profiles as a function of reaction temperature over the Pt/ZnO-NW-H200, Pt/ZnO-NW-H300, and Pt/ZnO-NW-H500 catalysts, respectively. The observed higher activity of the Pt/ZnO NW-H300 (compared to that of the Pt/ZnO NW-H200) is interesting since the sizes of the nanoclusters in the Pt/ZnO NW-H300 catalyst are similar to, or even slightly larger than, the nanoclusters in the Pt/ZnO NW-H200 catalyst. The activity differences must have originated from the effects of the H
2 reduction treatment at different temperatures. Considering that PtZn alloy can form during H
2 reduction at temperatures higher than 250 °C [
27,
29,
30,
33], we propose that the presence of the PtZn alloy nanoparticles in the Pt/ZnO NW-H300 catalyst may have improved the WGS activity. Detailed examinations revealed that there were no detectable PtZn alloy nanoparticles in the Pt/ZnO NW-H200 catalyst.
The initial low activity of the PtZn/ZnO NW-H500 (compared to that of the Pt/ZnO NW-H200 and Pt/ZnO NW-H300 catalysts) can be attributed to the larger sizes of the PtZn alloy nanoparticles. The activity of the Pt/ZnO NW-H650 was much lower than that of the other catalysts, presumably caused by (1) the encapsulation of the PtZn alloy nanoparticles by the ZnO
x/Zn surface coating layers and (2) the significant growth of the PtZn alloy nanoparticles during the high temperature reduction treatment. We have also investigated the WGS reaction on Pt nanoparticles supported on ZnO powders (
Figure 6b). The activity of the ZnO powder-supported PtZn catalyst was higher than that of the Pt/ZnO NW catalysts. PtZn alloy nanoparticles were formed as well and some, but not all, alloy nanoparticles grew epitaxially onto the ZnO powders. The observed higher activity may be related to the presence of ZnO polar surfaces (e.g., ZnO {0001} facets) that may interact with the Pt differently. However, the ZnO powders are not stable at high calcination or reduction temperatures [
36].
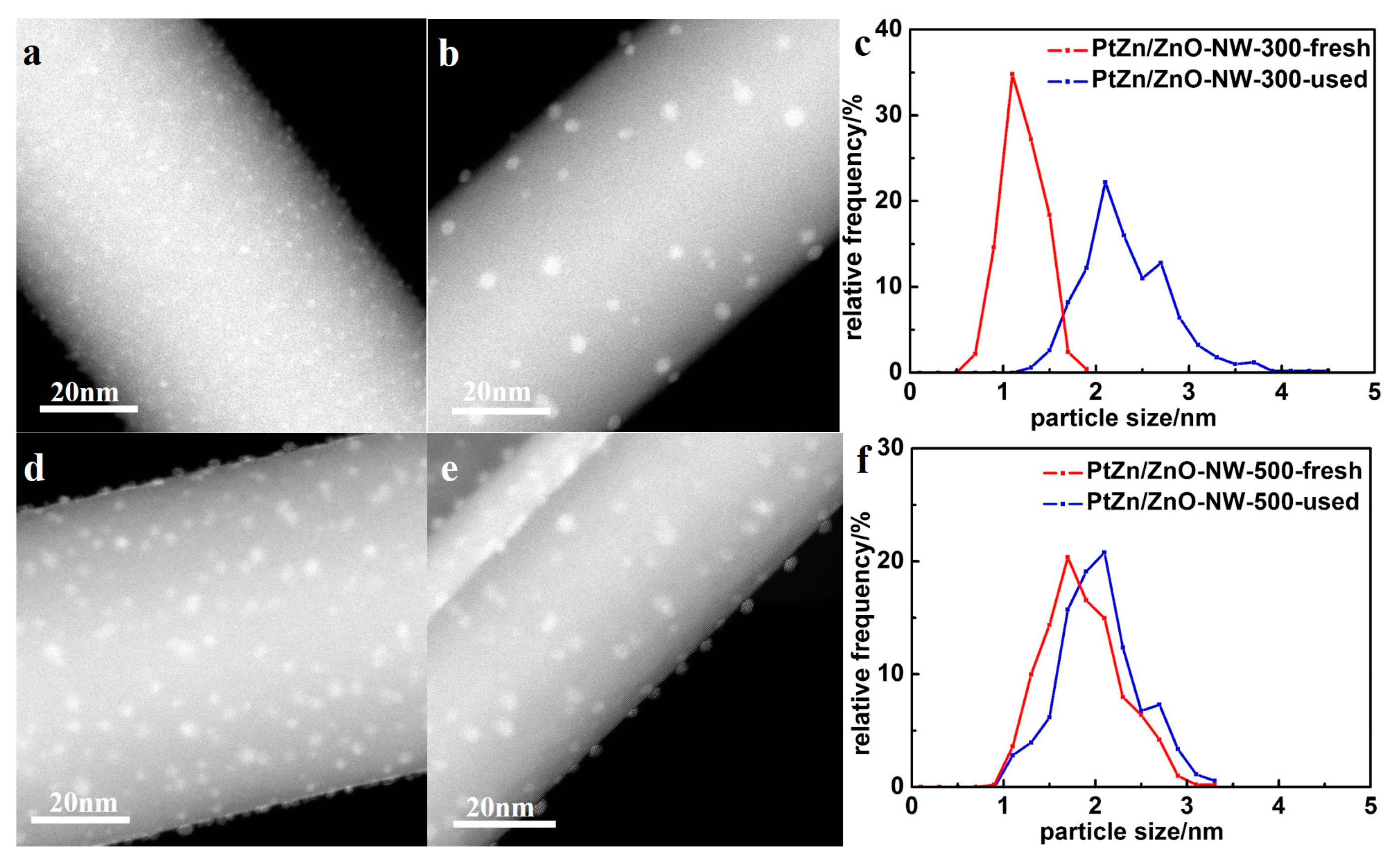
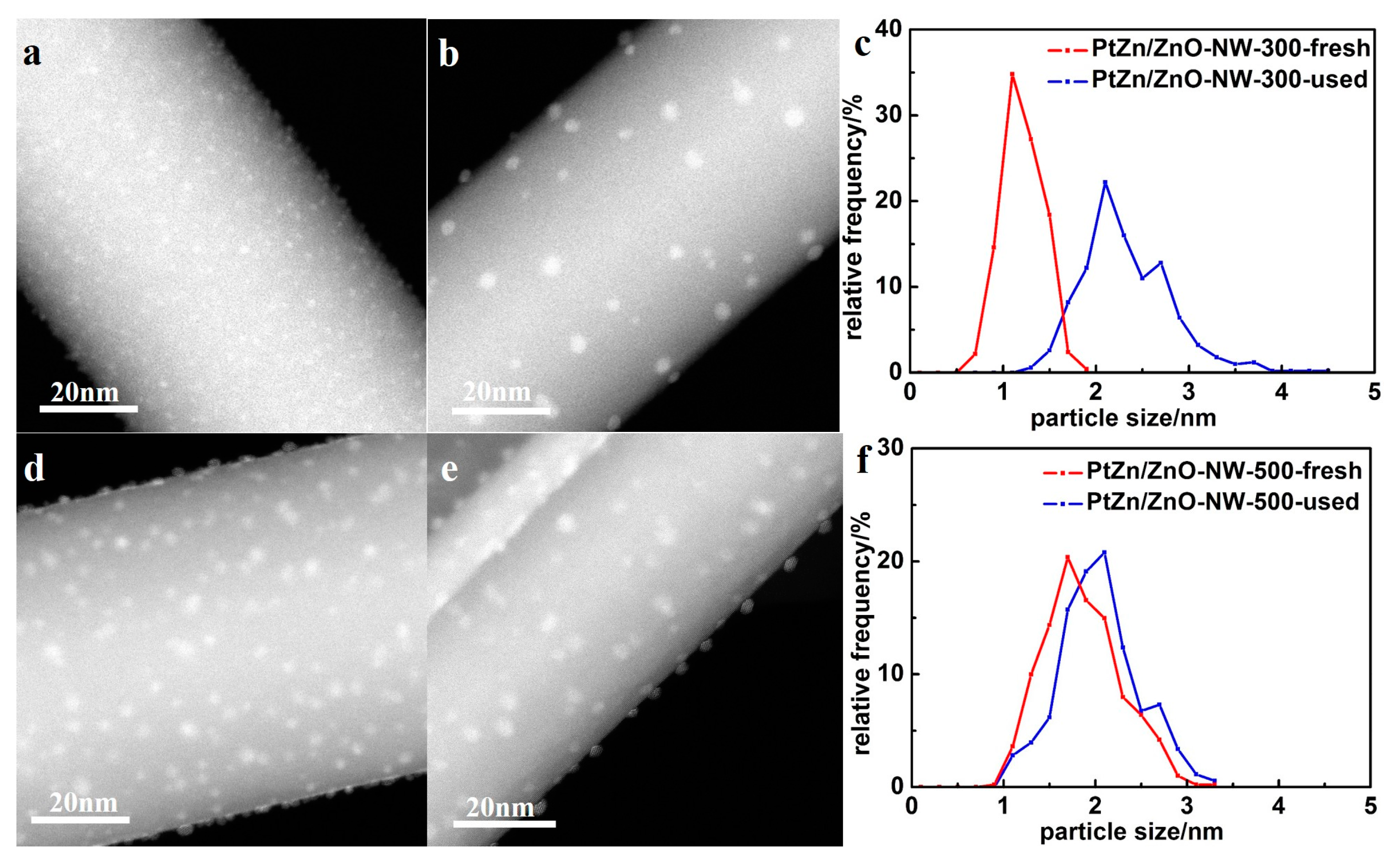
Figure 7a shows the CO conversion at 500 °C with time on stream for Pt/ZnO NW-H200, Pt/ZnO NW-H300, Pt/ZnO NW-H500, and Pt/ZnO NW-H650 catalysts, respectively. The CO conversion of the Pt/ZnO NW-H200 and Pt/ZnO NW-H300 catalysts decreased about 20% during the 30-h test. Such deactivation is most likely caused by the sintering of the small Pt or PtZn nanoclusters during the high temperature WGS reaction. To determine the degree of particle sintering, we measured the Pt/ZnO NW-H300 sample after the 30-h test.
Figure 8 shows the HAADF-STEM images and the corresponding particle size distributions of the used catalyst. After the stability test, the mean particle size of the Pt/ZnO NW-H300 catalyst increased from approximately 1.1 nm to about 2.3 nm in diameter. On the other hand, the particle sizes in the Pt/ZnO NW-H500 catalyst did not change much at all and showed much better stability. The epitaxial growth stabilized the PtZn alloy nanoparticles both during the catalyst treatments and catalytic reactions.
The catalytic performance of alloy nanoparticles depends strongly on the bonding strength between the alloying elements [
31]. It has been reported that bonding between Pt and Zn produces a large depletion in the density of Pt(5d) states around the Fermi level, with a shift in the centroid of the Pt 5d band and 4f core levels toward higher binding energy [
39]. The formation of the PtZn clusters suggests that bimetallic bonding destabilizes the Pt(5d) orbitals as well the Zn(3d) orbitals. The formation of strong Pt-Zn bonds modifies the adsorption property of Pt toward CO molecules. It has been proposed that the bonding between Pt and Zn weakens the bonding between the Pt atoms and the CO molecules [
39]. Such alloying induced modification of the chemisorption of CO molecules may strongly depend on the faceting and surface composition of the PtZn alloy nanoparticles. The stability of the PtZn alloy nanoparticles should therefore depend on the specific catalytic reactions and the reaction temperature.
The atomic resolution HAADF-STEM images (
Figure 9) clearly show that the epitaxial relationship between the PtZn nanoparticles and the ZnO nanowires did not change at all after the 30-h stability test, further suggesting the excellent stability of the epitaxially anchored PtZn nanoparticles. Furthermore, the nanoscale facets of the PtZn alloy nanoparticles, the interfacial structure between the PtZn alloy nanoparticles and the ZnO nanowires, and the shapes of the PtZn nanoalloys did not change at all during the 30-h test.
We further tested the Pt/ZnO NW-H500 catalyst for methanol steam reforming (MSR) at 400 °C. As shown in
Figure 7b, the epitaxially anchored PtZn nanoparticles on ZnO NWs demonstrated persistent resistance to sintering and maintained excellent durability for the MSR reaction as well.
3. Materials and Methods
3.1. Catalyst Preparation
ZnO nanowires (NWs) were fabricated by a modified vapor transport and condensation method[
40]. Firstly, a uniform mixture of ZnO and C was obtained by mixing ZnO powders (99%, Sigma-Aldrich, Tempe, AZ, USA) and carbon black (Cabot Corp, Alpharetta, GA, USA) in the weight ratio of 1:1 with the addition of deionized water. Then, the mixture was dried in the oven at 200 °C for 10 h to remove water. For the synthesis of ZnO nanowires, about 3 g of the mixture was ground into powders and placed into a high-temperature furnace. Under 10 vol. % O
2/N
2 flowing gas, the mixture was heated to >900 °C, and the ultrapure ZnO NWs were collected downstream.
The ZnO NW supported Pt nanocatalysts were prepared by a modified deposition-precipitation method. The H2PtCl6·6H2O solution was added dropwise into the suspension containing ZnO NWs under constant stirring. Sodium carbonate (Na2CO3) solution was added simultaneously to adjust the pH value at ~7.0. After being stirred and aged for 2 h respectively, the suspension was filtered and washed with deionized water for several times. The resultant solid was dried at 60 °C for 12 h and calcined at 400 °C for 4 h in air. The catalyst was reduced in situ at different temperatures for 2 h in 10 vol. % H2/He prior to catalytic reaction. The supported Pt catalysts reduced at different temperature were denoted as Pt/ZnO NW-HX, where X represents the reduction temperature.
3.2. Characterization Techniques
The actual metal loadings of the catalysts were measured by inductively coupled plasma-mass spectroscopy (ICP-MS) on an Agilent 7500ce instrument (Agilent, Santa Clara, CA, USA). N2 adsorption measurements were performed on a Micromeritics ASAP 2020 instrument (Micromeritics, Shanghai, China) at −196 °C, and the surface area of the support was obtained via the Brunauer–Emmett–Teller (BET) method.
The powder X-ray Diffraction (XRD) measurements were carried out on a PANalytical X’pert PRO X-ray (PANalytical, Shanghai, China) diffractometer using Cu Kα radiation (1.5405 Å) operating at 40 kV and 40 mA. Data recorded at a step size of 0.013° under continuous scan mode.
The morphology of the ZnO NWs was characterized by a field emission scanning electron microscope (Hitachi, S-4800, Hitachi, Tokyo, Japan). HAADF images of the ZnO nanowires and the catalysts were obtained on a JEOL ARM-200F (JEOL, Huntington Beach, CA, USA) aberration-corrected STEM (AC-STEM) operated at 200 kV with a nominal spatial resolution of 0.08 nm in the STEM mode. Samples were prepared through a typical procedure by depositing dispersed catalysts in ethanol and were then transferred onto copper TEM grids.
3.3. Measurement of Catalytic Activity
The activity tests were conducted in a fixed-bed reactor. The catalyst samples were placed in a quartz tube (ID = 4 mm) with the two ends of the catalyst bed filled with quartz wool. For WGS reaction, the stability test were performed at 500 °C. Typically, a 0.05-g catalyst was used, and the reaction feed, composed of 2 vol. % CO/10 vol. % H2O and balance He, was passed through the catalyst bed at desired temperatures with a total flow rate of about 40 mL/min. The gas hourly space velocity (GHSV) was 48,000 mL/h gcat. The effluent gas compositions were on line analyzed by a gas chromatograph (HP 7890, HP, Palo Alto, CA, USA) equipped with a HayeSep DB column. The CO conversion was defined as CCO = [(CO)in − (CO)out]/(CO)in × 100%, where (CO)in and (CO)out were the inlet and outlet molar weight of CO. The selectivity to CO2 was defined as , where and were the molar weight of CO2 and CH4 in the product. No other carbon-containing products were detected.
For the MSR reaction, the stability test were performed at 400 °C. A 0.05-g catalyst was used, and the reaction feed, composed of methanol/water (mole ratio 2/3, 0.6 mL/h in liquid), and He (37 mL/min) was passed through the catalyst bed at desired temperatures. The gas hourly space velocity (GHSV) was 54,000 mL/h gcat. The reaction feed was preheated to 165 °C to vaporize methanol/water mixture. The effluent gas compositions were online analyzed by a gas chromatograph (HP 7890) equipped with a HayeSep DB column (Agilent, Santa Clara, CA, USA). Quantitative analysis of CO, CO2, and methanol was realized by calibrating with standard samples. The methanol conversion was calculated based on the difference between inlet and outlet concentrations. The selectivity of CO2 formation was determined as moles of CO2 per mole of CO2 + CO in the products.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}