3. Experimental
3.1. Materials and Methods
Commercial K
4[Fe(CN)
6]∙3H
2O was dehydrated under vacuum prior to the use (ca. 30 °C/10 Torr; Note: the reaction proceeds equally well with hydrated K
4[Fe(CN)
6] but the anhydrous salt was utilized to avoid possible variations in the reaction stoichiometry resulting from the use of partly dehydrated materials). Anhydrous tetrahydrofuran, dichloromethane and methanol were dried using a PureSolv MD5 Solvent Purification System (Innovative Technology Inc., Amesbury, MA, USA). Anhydrous
N,
N-dimethylformamide (Sigma-Aldrich, St. Louis, MO, USA) and solvents (Lach-Ner, Neratovice, Czech Republic) used for aqueous workup, column chromatography and crystallizations were used as received. Compound
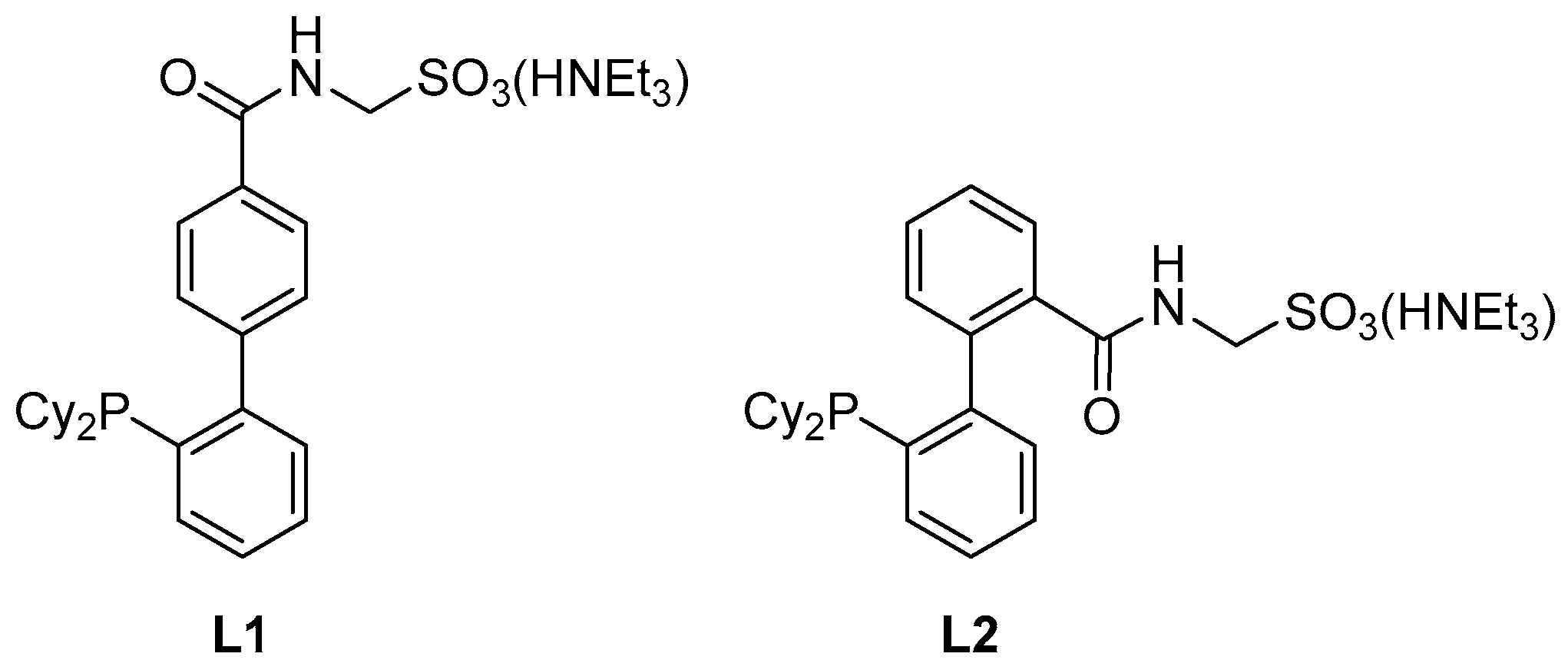
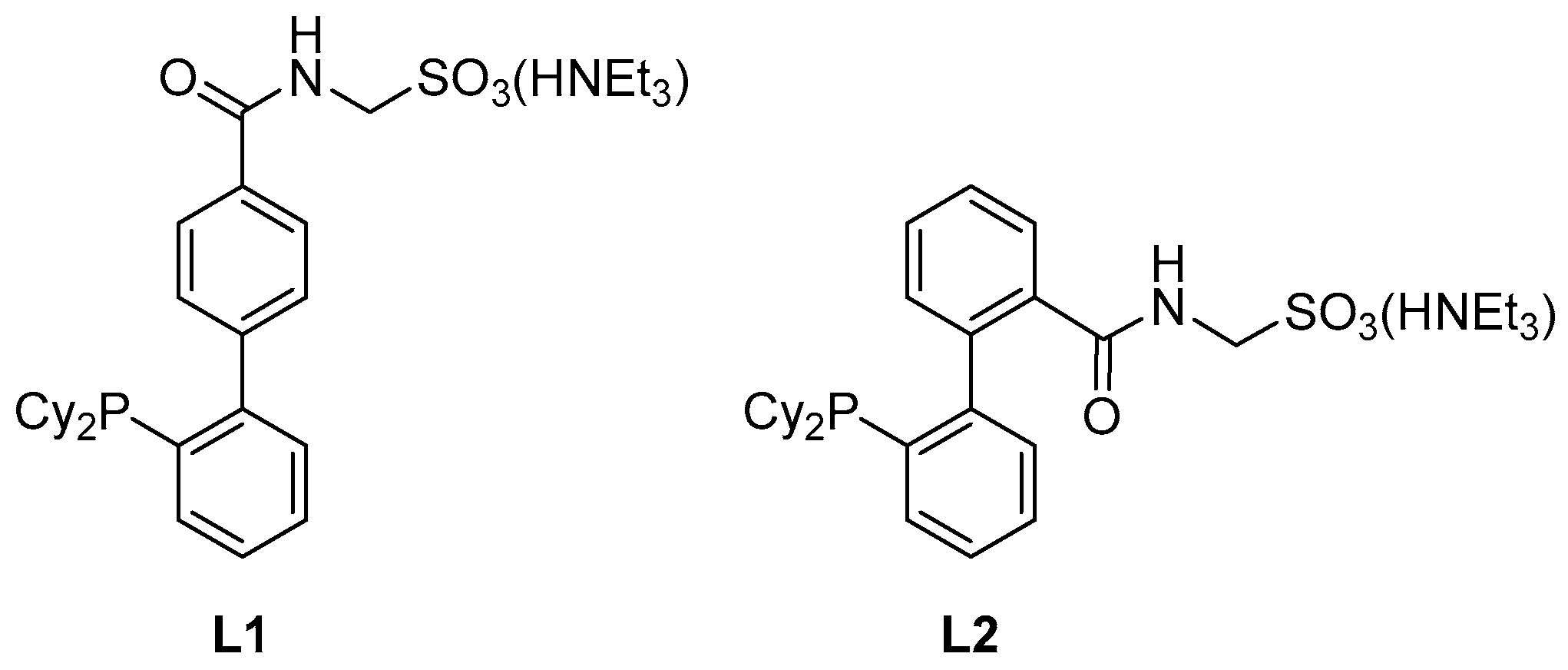
L1 [
8] was prepared according to the literature.
NMR spectra were recorded at 298 K on a Bruker AVANCE III 400 spectrometer (1H, 400.13 MHz; 13C{1H}, 100.62 MHz; 19F, 376.46 MHz; and 31P, 161.97 MHz), Bruker AVANCE III 600 spectrometer (1H, 600.17 MHz; and 13C, 150.93 MHz) (Billerica, MA, USA) or Varian UNITY Inova 400 spectrometer (1H, 399.95 MHz; 13C, 100.58 MHz; and 31P, 161.90 MHz) (Palo Alto, CA, USA). Chemical shifts (δ/ppm) are given relative to internal tetramethylsilane (1H and 13C NMR), to external 85% aqueous H3PO4 (31P NMR) or to external neat CFCl3 (19F NMR). Mass spectra were recorded on an Esquire 3000 (Bruker) or an LTQ Orbitrap XL instrument (Thermo Fisher Scientific, Waltham, MA, USA; high resolution analyses). Infrared spectra were collected in Nujol mulls with an FTIR Thermo Nicolet 760 instrument (Thermo Fisher Scientific, Waltham, MA, USA) in the range 400–4000 cm−1.
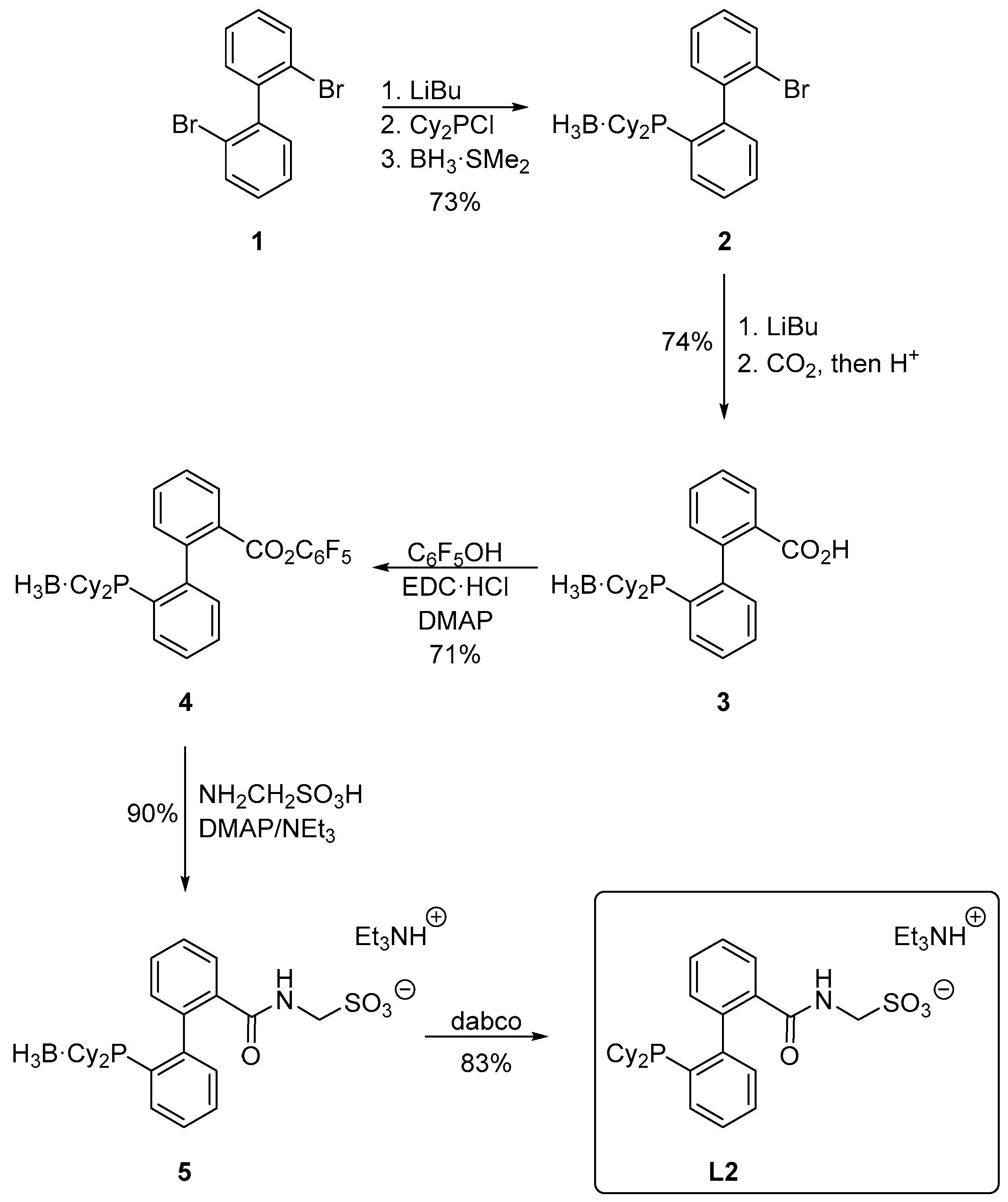
3.2. Preparation of 2′-(Dicyclohexylphosphino)-2-bromo[1,1′-biphenyl]–borane (1:1) (Compound 2)
A two-necked flask was charged with 2,2′-dibromo[1,1′-biphenyl] (1; 4.68 g; 15.0 mmol), purged with argon and sealed with a rubber septum. Anhydrous tetrahydrofuran (60 mL) was introduced and the reaction flask was placed in a dry ice/ethanol bath. A solution of n-butyllithium in hexanes (6.0 mL of 2.5 M, 15.0 mmol) was introduced dropwise with continuous cooling and stirring, yielding a pale yellow solution. After 30 min, chloro-dicyclohexylphosphine (3.6 mL, 16.5 mmol) was added in one portion, causing the reaction mixture to turn purple. The reaction mixture was kept at −78 °C for another 30 min, and then warmed to room temperature whereupon its color changed to colorless. After stirring overnight, neat borane-dimethyl sulfide complex (1.6 mL, 16.5 mmol) was added via a syringe. The reaction mixture was stirred for another 30 min, quenched with methanol (5 mL) and evaporated to dryness. The solid residue was taken up with ethyl acetate (100 mL) and the extract was washed with saturated aqueous NaHCO3 (50 mL), brine (50 mL) and dried over magnesium sulfate. The solvents were removed under vacuum and the solid residue was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:30 v/v). The second band was collected and evaporated to afford compound 2 as a colorless, slowly crystallizing oil. Yield 4.86 g (73%).
1H NMR (CDCl3): δ 0.11–0.77 (br m, 3 H, BH3), 0.88–0.99 (m, 1 H), 1.06–1.27 (m, 5 H), 1.28–1.39 (m, 4 H), 1.48–1.71 (m, 8 H), 1.72–1.82 (m, 3 H), 1.87–1.97 (m, 1 H) (PCy2); 7.18 (dd, 3JHH = 7.5 Hz, 4JHH = 1.7 Hz, 1 H), 7.20–7.23 (m, 1 H), 7.29–7.33 (m, 1 H), 7.38 (td, 3JHH = 7.7 Hz, 4JHH = 1.2 Hz, 1 H), 7.46 (tt, 3JHH = 7.7 Hz, 4JHH = 1.5 Hz, 1 H), 7.50 (tt, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, 1 H), 7.70 (dd, 3JHH = 8.0 Hz, 4JHH = 1.2 Hz, 1 H), 7.99–8.04 (m, 1 H) (aromatics). 13C{1H} NMR (CDCl3): δ 25.66, 25.83, 26.46 (d, JPC = 12 Hz), 26.86 (d, JPC = 12 Hz), 26.98 (d, JPC = 12 Hz), 27.10 (d, JPC = 11 Hz), 27.29, 27.54, 28.08, 28.91, 33.68 (d, JPC = 33 Hz), 34.50 (d, JPC = 32 Hz) (PCy2); 124.73, 125.81 (d, 1JPC = 44 Hz), 126.60, 127.70 (d, JPC = 10 Hz), 129.72, 130.23 (d, JPC = 2 Hz), 130.98, 132.44 (d, JPC = 7 Hz), 133.00, 136.24 (d, JPC = 12 Hz), 141.80 (d, JPC = 2 Hz), 145.15 (d, JPC = 2 Hz) (aromatics). 31P{1H} NMR (CDCl3): δ 33.6 (br d). IR (Nujol): 3378 m, 3058 s, 2365 vs, 2274 s, 1923 w, 1728 w, 1698 w, 1587 w, 1563 w, 1449 s, 1423 s, 1345 w, 1328 w, 1300 w, 1275 m, 1256 w, 1210 w, 1180 m, 1165 m, 1146 s, 1121 s, 1088 s, 1063 s, 1038 w, 1025 m, 1001 s, 949 w, 919 w, 887 w, 870 m, 851 m, 817 w, 745 vs, 730 s, 683 m, 649 w, 618 w, 589 m, 581 m, 547 w, 523 m, 508 w, 484 m, 453 s, 428 m cm−1. ESI+ MS: m/z 467.0 ([M + Na]+), 482.9 ([M + K]+). Anal. Calcd. for C24H33BBrP: C 65.04, H 7.51%. Found: C 64.95, H 7.42%.
3.3. Preparation of 2′-(Dicyclohexylphosphino)[1,1′-biphenyl]-2-carboxylic acid–borane (1:1) (Compound 3)
Under argon, bromide 2 (4.43 g, 10.0 mmol) was dissolved in anhydrous tetrahydrofuran (60 mL) in a three-necked reaction flask equipped with a stirring bar. The solution was cooled in a dry ice/ethanol bath and then treated by n-butyllithium (4.4 mL of 2.5 M solution in hexanes, 11.0 mmol). The resulting purple solution was stirred at −78 °C for another 30 min and then poured onto crushed dry ice, whereupon the mixture turned colorless. After warming to room temperature, the solution was acidified with 1.25 M methanolic HCl (15 mL) and concentrated under vacuum. The solid residue was dissolved in ethyl acetate (50 mL), washed with brine (3 × 50 mL) and dried over magnesium sulfate. The solvents were removed under reduced pressure, leaving a crude product, which was purified by flash chromatography (silica gel, ethyl acetate/hexanes, 1:3 v/v). The first band was collected and evaporated to give 3 as a white solid. Yield 3.00 g (74%).
1H NMR (CDCl3): δ –0.45 to 0.65 (br m, 3 H, BH3), 0.94–1.44 (m, 10 H), 1.46–1.83 (m, 11 H), 1.86–2.03 (m, 1 H, PCy2); 7.08–7.14 (m, 1 H), 7.17 (dd, 3JHH = 7.6 Hz, 4JHH = 1.2 Hz, 1 H), 7.40–7.46 (m, 2 H), 7.51 (td, 3JHH = 7.6 Hz, 4JHH = 1.5 Hz, 1 H), 7.57 (td, 3JHH = 7.4 Hz, 4JHH = 1.6 Hz, 1 H), 7.79–7.87 (m, 1 H), 8.12 (dd, 3JHH = 7.8 Hz, 4JHH = 1.4 Hz, 1 H) (aromatics). 13C{1H} NMR (CDCl3): δ 25.84 (d, JPC = 1 Hz), 25.91 (d, JPC = 1 Hz), 26.72 (d, JPC = 11 Hz), 26.86–27.03 (m, 3 C), 27.08 (d, JPC = 11 Hz), 27.27, 27.65, 27.92, 34.12 (d, JPC = 33 Hz), 34.47 (d, JPC = 33 Hz) (PCy2); 124.47 (d, JPC = 44 Hz), 126.74 (d, JPC = 9 Hz), 128.17, 129.08, 129.76 (d, JPC = 2 Hz), 131.24 (d, JPC = 8 Hz), 131.35, 131.83 (2 C), 134.10 (d, JPC = 7 Hz), 143.48 (d, JPC = 3 Hz), 147.31 (d, JPC = 7 Hz) (aromatics); 171.16 (C=O). 31P{1H} NMR (CDCl3): δ 29.5 (s). The signals due to residual dichloromethane are observed at δH 5.30 and δC 53.42. IR (Nujol): 2413 m, 2360 s, 2340 s, 1733 w, 1688 vs, 1600 w, 1588 w, 1575 s, 1308/1296 s, 1275/1266 s, 1209 w, 1180 w, 1162 w, 1150 m, 1110 m, 1066 s, 1049 m, 1004 m, 953 m, 887 m, 583 m, 821 m, 770/762 s, 733 s, 707 m, 685 m, 669 w, 658 s, 593 m, 521 m. ESI– MS: m/z 406.9 ([M − H]−). HRMS calc. for C25H33O2BP ([M − H]−): 407.23167, found 407.23087. Anal. Calcd. for C25H34BO2P∙0.7CH2Cl2: C 65.99, H 7.63%. Found: C 65.89, H 7.59%.
3.4. Synthesis of 2′-(Dicyclohexylphosphino)[1,1′-biphenyl]-2-carboxylic acid–borane (1:1), Pentafluorophenyl Ester (Compound 4)
Under an argon atmosphere, acid 3 (2.043 g, 5.0 mmol), pentafluorophenol (1.104 g, 6.0 mmol), N-[3-(dimethylamino)propyl]-N′-ethylcarbodiimide hydrochloride (1.054 g, 5.5 mmol) and 4-dimethylaminopyridine (0.122 g, 1.0 mmol) were dissolved in dry dichloromethane (40 mL) and the reaction mixture was stirred overnight. On the following day, the reaction was quenched with brine (25 mL), the organic layer was separated, washed with brine (2 × 25 mL), dried over magnesium sulfate and vacuum-evaporated. The solid residue was purified by column chromatography (silica gel, ethyl acetate/hexanes, 1:30 v/v) to give ester 4 as a white solid. Yield 2.04 g (71%).
1H NMR (CDCl3): δ –0.45 to 0.55 (br m, 3 H, BH3), 1.01–1.40 (m, 10 H), 1.43–1.85 (m, 11 H), 2.01–2.14 (m, 1 H, PCy2); 7.17–7.21 (m, 1 H), 7.26 (dd, 3JHH = 7.5 Hz, 4JHH = 1.4 Hz, 1 H), 7.41–7.50 (m, 2 H), 7.59 (td, 3JHH = 7.6 Hz, 4JHH = 1.5 Hz, 1 H), 7.66 (td, 3JHH = 7.5 Hz, 4JHH = 1.5 Hz, 1 H), 7.72–7.78 (m, 1 H), 8.33 (dd, 3JHH = 7.8 Hz, 4JHH = 1.4 Hz, 1 H) (aromatics). 13C{1H} NMR (CDCl3): δ 25.90 (m, 2 C), 26.73–25.97 (m, 4 C), 27.01 (d, JPC = 7 Hz), 27.14 (d, JPC = 11 Hz), 27.50, 27.56, 34.21 (d, JPC = 33 Hz), 34.70 (d, JPC = 33 Hz) (PCy2); 124.56 (d, JPC = 43 Hz), 126.86, 127.11 (d, JPC = 8 Hz), 128.50, 130.01 (d, JPC = 2 Hz), 131.23 (d, JPC = 8 Hz), 131.61, 132.47, 132.83, 133.29 (d, JPC = 5 Hz), 137.81 (dm, 1JFC = 253 Hz, Cmeta of C6F5), 139.33 (dm, 1JFC = 253 Hz, Cpara of C6F5), 141.28 (dm, 1JFC = 252 Hz, Cortho of C6F5), 144.47 (d, JPC = 3 Hz), 147.29 (d, JPC = 9 Hz) (aromatics); 161.85 (C=O) ppm. 19F{1H} NMR (CDCl3): δ–162.87 (m, Fmeta), −158.47 (t, 3JFF = 22 Hz, Fpara), −151.85 (m, Fortho) (C6F5). 31P{1H} NMR (CDCl3): δ 27.9 (s). IR (Nujol): 2358/2342 s, 1770 s, 1733 w, 1716 w, 1698 m, 1558 w, 1540 w, 1521 vs, 1267 m, 1227 s, 1180 w, 1145 m, 1128 m, 1065 m, 1030 s, 1004 m, 966 m, 974 w, 853 w, 758 m, 696 w, 669 m, 656 w, 626 w, 592 w, 524 w cm−1. ESI+ MS: m/z 597.0 ([M + Na]+), 612.9 ([M + K]+). Anal. Calcd. for C31H33BF5O2P: C 64.82, 5.79%. Found: C 64.92, 5.82%.
3.5. Preparation of 2′-(Dicyclohexylphosphino)-2-{[(sulfonatomethyl)amino]carbonyl}[1,1′-biphenyl]–borane (1:1), Triethylammonium Salt (Compound 5)
Ester 4 (1.148 g, 2.0 mmol), aminomethanesulfonic acid (244 mg, 2.4 mmol) and 4-(dimethylamino)pyridine (2.4 mg, 0.02 mmol) were dissolved in a mixture of dry N,N-dimethylformamide (10 mL) and triethylamine (2.5 mL) under argon. The reaction mixture was stirred in the dark overnight. The solvents were removed under vacuum, leaving a crude product, which was purified by flash column chromatography (silica gel, dichloromethane/methanol/triethylamine, 19:1:0.4 v/v). The second band was collected and evaporated. Subsequent crystallization from hot ethyl acetate afforded analytically pure amide 5 as a white microcrystalline compound. Yield 1.08 g (90%).
1H NMR (CD2Cl2): δ –0.45 to 0.65 (br m, 3 H, BH3), 1.01–1.39 (m, 9 H, PCy2), 1.27 (t, 3JHH = 7.3 Hz, 9 H, CH3 of Et3NH+), 1.42–1.83 (m, 12 H) and 1.97–2.10 (m, 1 H, PCy2); 3.05 (q, 3JHH = 7.3 Hz, 6 H, CH2 of Et3NH+), 3.95 (dd, 2JHH = 13.4 Hz, 3JHH = 5.2 Hz, 1 H, NCHAHB), 4.31 (dd, 2JHH = 13.4 Hz, 3JHH = 7.6 Hz, 1 H, NCHAHB), 6.63 (dd, 3JHH = 7.5 Hz, 3JHH = 5.2 Hz, 1 H, NH), 7.12–7.18 (m, 1 H), 7.24–7.28 (m, 1 H), 7.41–7.50 (m, 4 H), 7.64–7.70 (m, 1 H), 7.78–7.85 (m, 1 H) (aromatics). 13C{1H} NMR (CD2Cl2): δ 8.84 (3 C, CH3 of Et3NH+), 26.33 (d, JPC = 2 Hz), 26.38 (d, JPC = 1 Hz), 27.11, 27.21 (d, JPC = 2 Hz), 27.32, 27.43 (d, JPC = 2 Hz), 27.64 (d, JPC = 2 Hz), 27.73, 28.11, 28.24, 34.38 (d, JPC = 33 Hz) and 34.76 (d, JPC = 33 Hz) (PCy2); 46.36 (3 C, CH2 of Et3NH+), 56.16 (NCH2), 125.62 (d, JPC = 43 Hz), 127.54 (d, JPC = 8 Hz), 128.25, 128.41, 129.62, 130.44 (d, JPC = 2 Hz), 131.84, 132.77 (d, JPC = 7 Hz), 134.82 (d, JPC = 7 Hz), 136.31, 140.11 (d, JPC = 3 Hz), 146.76 (d, JPC = 7 Hz) (aromatics); 168.23 (C=O). 31P{1H} NMR (CDCl3): δ 28.9 (s). IR (Nujol): 3323 s, 2364 s, 2342 m, 1674 s, 1522, 1316 m, 1296 w, 1271 w, 1261 w, 1250 m, 1211 s, 1178 s, 1120 w, 1085 w, 1059 m, 1043 s, 1003 w, 957 w, 917 w, 889 w, 851 w, 804 w, 774 m, 762 s, 857 w, 611 m, 582 w, 568 w, 559 w, 533 w, 515 m, 481 w, 461 w, 443 w, 425 w cm−1. ESI− MS: m/z 500.0 ([M − HNEt3]−), 486.0 ([M − BH3 − HNEt3]−). Anal. Calcd. for C32H52BN2O4PS: C 63.78, H 8.70, N 4.65%. Found: C 63.56, H 8.67, N 4.64%.
3.6. Preparation of 2′-(Dicyclohexylphosphino)-2-{[(sulfonatomethyl)amino]carbonyl}[1,1′-biphenyl], Triethylammonium Salt (Ligand L2)
A reaction flask equipped with a stirring bar was charged with 5 (603 mg, 1.0 mmol) and 1,4-diazabicylco[2.2.2]octane (449 mg, 4.0 mmol), flushed with argon and sealed with a septum. The solids were dissolved in anhydrous methanol (10 mL) and the reaction mixture was heated at 60 °C overnight. The resulting solution was evaporated to dryness and the product was isolated by column chromatography (silica gel, dichloromethane/methanol/triethylamine, 19:1:0.4 v/v). Subsequent crystallization from hot ethyl acetate-heptane (2:1, v/v) afforded solvated L2 as a white crystalline solid. Yield 0.49 g (83%).
1H NMR (CD2Cl2): δ 0.80–1.36 (m, 10 H, PCy2), 1.27 (t, 3JHH = 7.3 Hz, 9 H, CH3 of Et3NH+), 1.37–1.82 (m, 11 H) and 1.92–2.03 (m, 1 H, PCy2); 3.06 (q, 3JHH = 7.3 Hz, 6 H, CH2 of Et3NH+), 4.00 (dd, 2JHH = 13.5 Hz, 3JHH = 6.4 Hz, 1 H, NCHAHB), 4.20 (dd, 2JHH = 13.5 Hz, 3JHH = 6.7 Hz, 1 H, NCHAHB), 6.21 (t, 3JHH = 6.5 Hz, 1 H, NH), 7.13–7.19 (m, 1 H), 7.27–7.32 (m, 1 H), 7.34–7.48 (m, 4 H), 7.54–7.58 (m, 1 H), 7.70–7.75 (m, 1 H) (aromatics). 13C{1H} NMR (CD2Cl2): δ 8.82 (3 C, CH3 of Et3NH+), 26.79, 26.85, 27.41 (d, JPC = 7 Hz), 27.52 (d, JPC = 5 Hz), 27.81 (d, JPC = 2 Hz), 27.90 (d, JPC = 8 Hz), 29.34 (d, JPC = 5 Hz), 30.19 (d, JPC = 13 Hz), 30.46 (d, JPC = 18 Hz), 31.22 (d, JPC = 15 Hz), 33.39 (d, JPC = 12 Hz) and 36.15 (d, JPC = 16 Hz) (PCy2); 46.48 (3 C, CH2 of Et3NH+), 56.30 (NCH2), 127.47, 127.57, 128.27, 128.85, 129.82, 130.32 (d, JPC = 6 Hz), 132.74 (d, JPC = 2 Hz), 133.38 (d, JPC = 3 Hz), 135.24 (d, JPC = 21 Hz), 136.03 (d, JPC = 3 Hz), 141.42 (d, JPC = 7 Hz), 148.93 (d, JPC = 31 Hz) (aromatics); 168.18 (C=O). 31P{1H} NMR (CD2Cl2): δ –12.6 (s). The signals due to solvating ethyl acetate are as follows: δH 1.23 (t, 3JHH = 7.1 Hz, CH3CH2), 2.00 (s, CH3CO), and 4.08 (q, 3JHH = 7.1 Hz, CH3CH2); δC 14.37 (CH3CH2), 21.15 (CH3CO), 60.63 (CH3CH2), and 171.24 (C=O). IR (Nujol): 3404 s, 3309 s, 1735 s, 1663 s, 1595 w, 1585 w, 1575 w, 1572 w, 1533 s, 1306 s, 1243 s, 1205 m, 1177 s, 1158 m, 1032 s, 1004 w, 954 m, 918 w, 888 w, 848 m, 810 w, 796 w, 777 w, 760 m, 750 m, 739 w, 679 w, 658 w, 608 m, 568 w, 557 w, 518 m, 493 w, 437 w cm−1. ESI− MS: m/z 485.9 ([M − HNEt3]−). Anal. Calcd. for C32H49N2O4PS∙0.2AcOEt: C 65.28, H 8.39, N 4.76%. Found: C 64.96, H 8.41, N 4.62%.
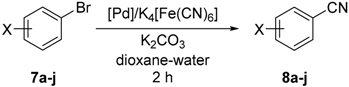
3.7. Pd-Catalyzed Cyanation of Aryl Bromides. General Procedure for Screening Experiments
A solution of ligand L1 or L2 (1 or 2 mol % with respect to aryl bromide) in dry dichloromethane (5 mL) was added to solid palladium precursors (1 mol % with respect to aryl bromide). The resulting mixture was stirred for 10 min and then evaporated under vacuum. Anhydrous potassium hexacyanoferrate(II) (184 mg, 0.5 mmol), potassium carbonate (138 mg, 1.0 mmol) and 4-bromoanisole (187 mg, 1.0 mmol) were added to the reaction vessel, which was then equipped with a magnetic stirring bar, flushed with argon and sealed with a septum. The solvent (1,4-dioxane/water 1:1; 4 mL) was introduced, the septum was replaced by a glass stopper, and the flask was transferred to an oil bath maintained at 100 °C. After stirring for 1 h, the reaction mixture was cooled and diluted with water (2 mL) and chloroform (5 mL). The organic layer was separated and washed with brine (10 mL). The aqueous layer was back-extracted with chloroform (3 × 5 mL). The organic extracts were combined, dried over anhydrous magnesium sulfate, and evaporated under reduced pressure. The conversion was determined by integration of 1H NMR spectrum.
3.8. Pd-Catalyzed Cyanation of Aryl Bromides. General Procedure for Preparative Experiments
A reaction vessel was charged (in this order) with palladium acetate (2.2 mg, 1 mol % with respect to the aryl bromide), ligand
L1 (11.8 mg, 2 mol % with respect to the aryl bromide), potassium hexacyanoferrate(II) (184 mg, 0.5 mmol), potassium carbonate (138 mg, 1.0 mmol) and the respective aryl bromide (1.0 mmol). The flask was equipped with a magnetic stirring bar, flushed with argon and sealed with septum. The solvent (1,4-dioxane/water, 1:1; 4 mL) was added, the septum was changed for glass stopper and the flask was transferred into an oil bath kept at 100 °C. After stirring for 2 h, the reaction mixture was cooled and diluted with water (2 mL) and ethyl acetate (5 mL). The organic layer was separated and washed with brine (10 mL). The aqueous layer was extracted with ethyl acetate (3 × 5 mL). The organic extracts were combined, dried over anhydrous magnesium sulfate, and evaporated under reduced pressure to afford pure nitrile, which was analyzed by
1H and
13C NMR spectroscopy. Analytical data of the coupling products matched those reported in the literature [
5,
6].



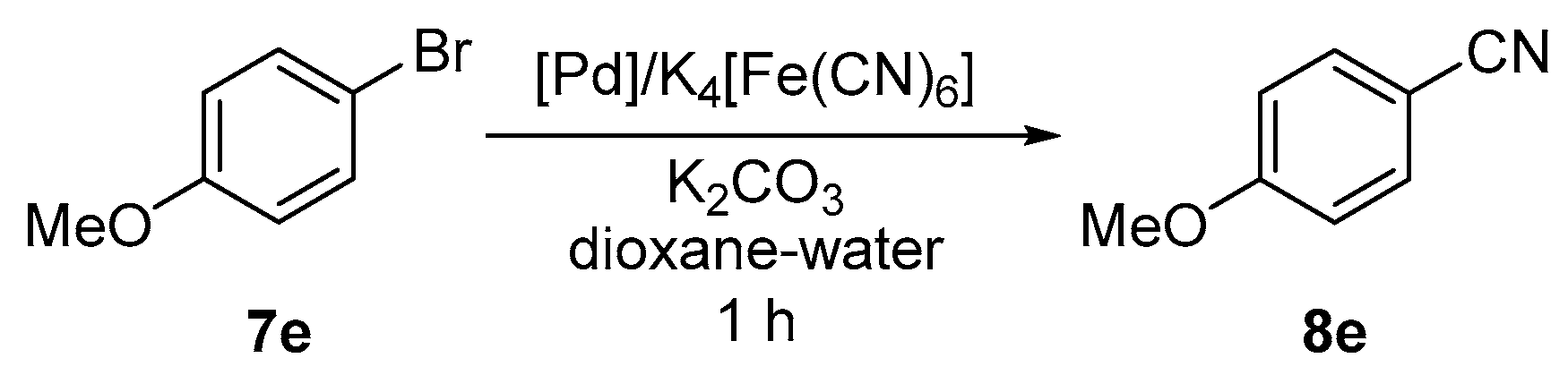

{kind=link}
{kind=link}
{kind=link}
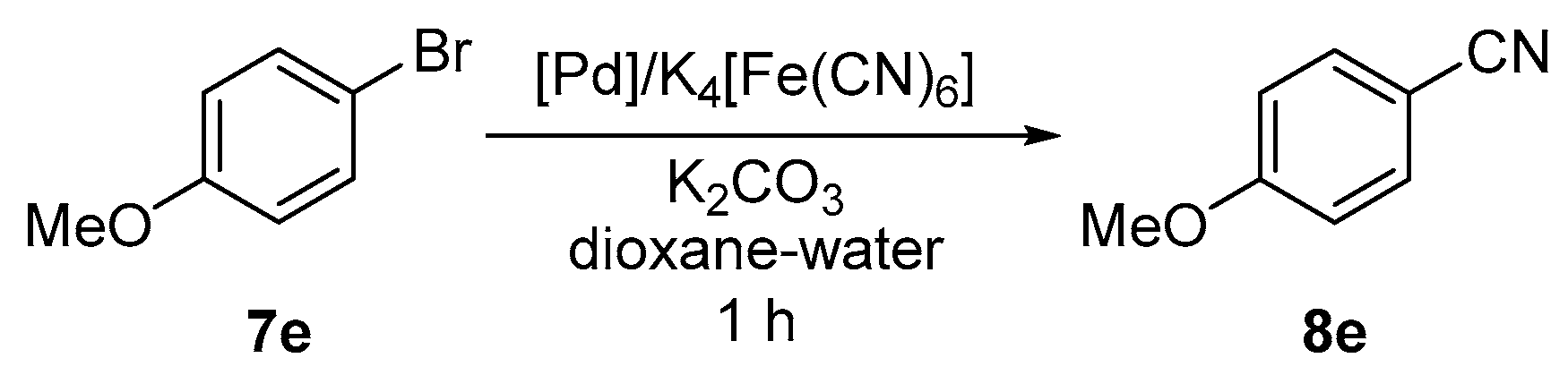{kind=link}