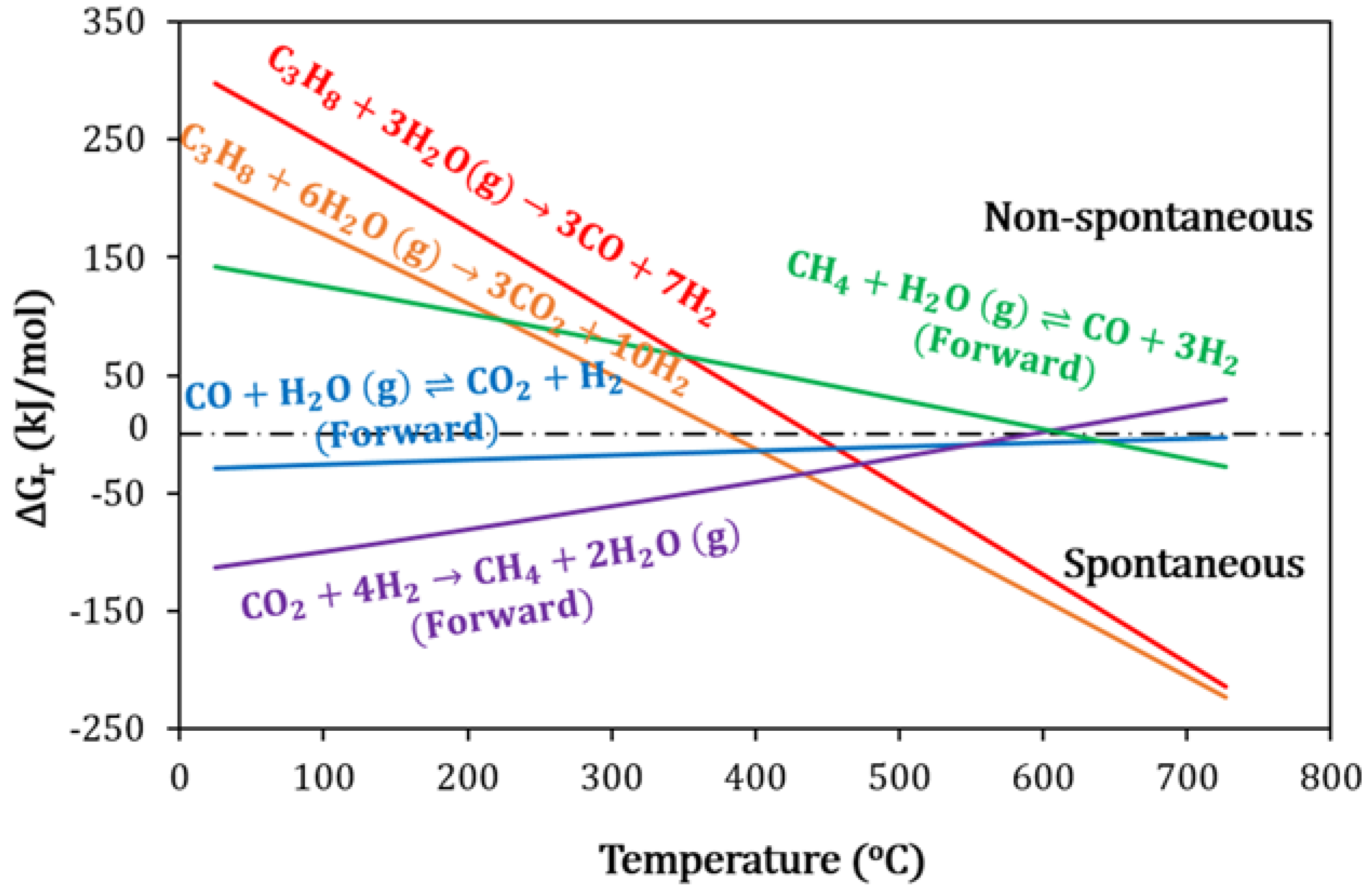
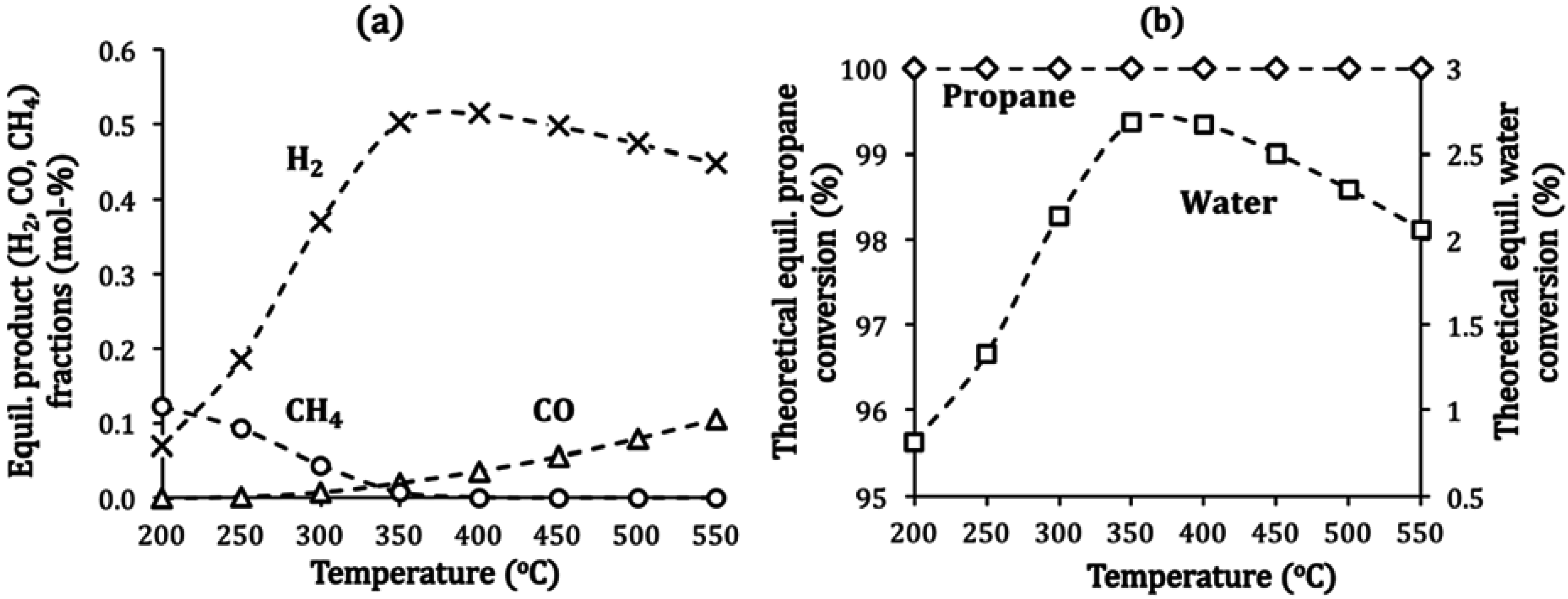
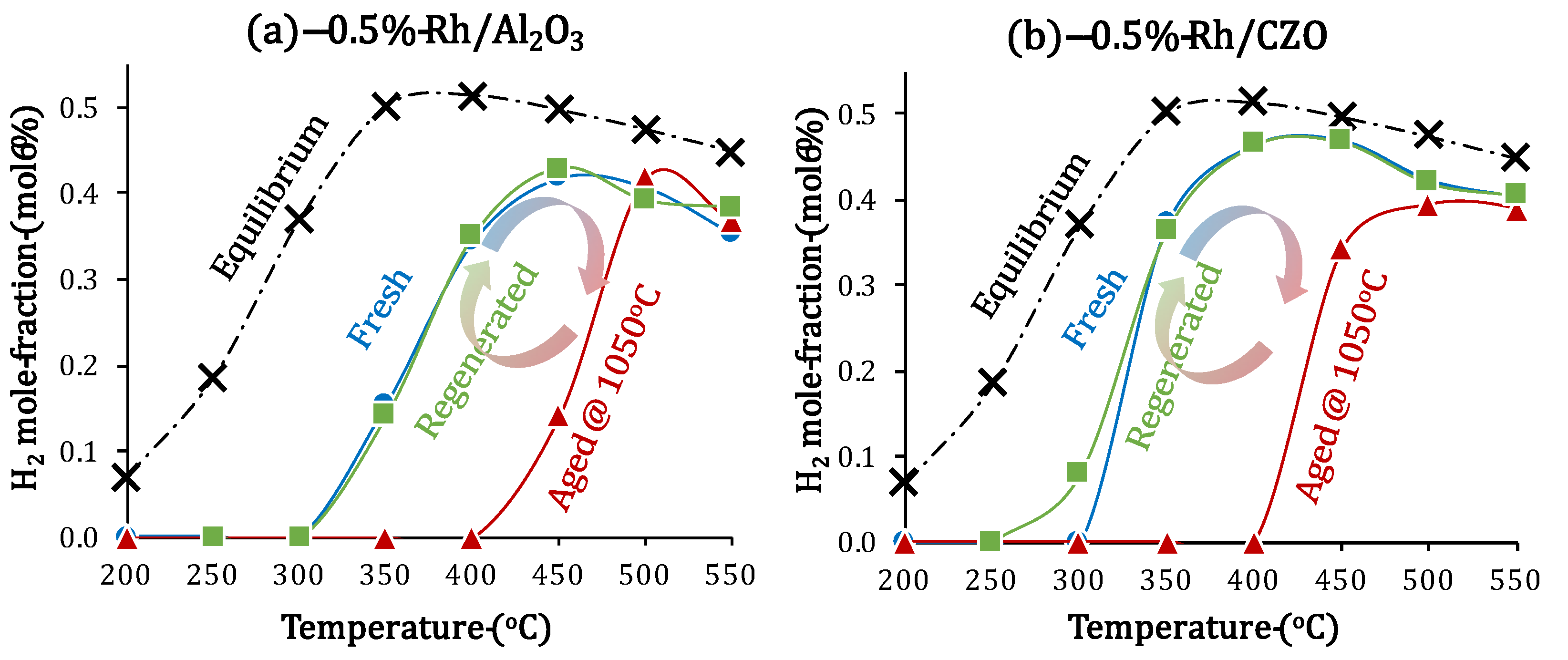
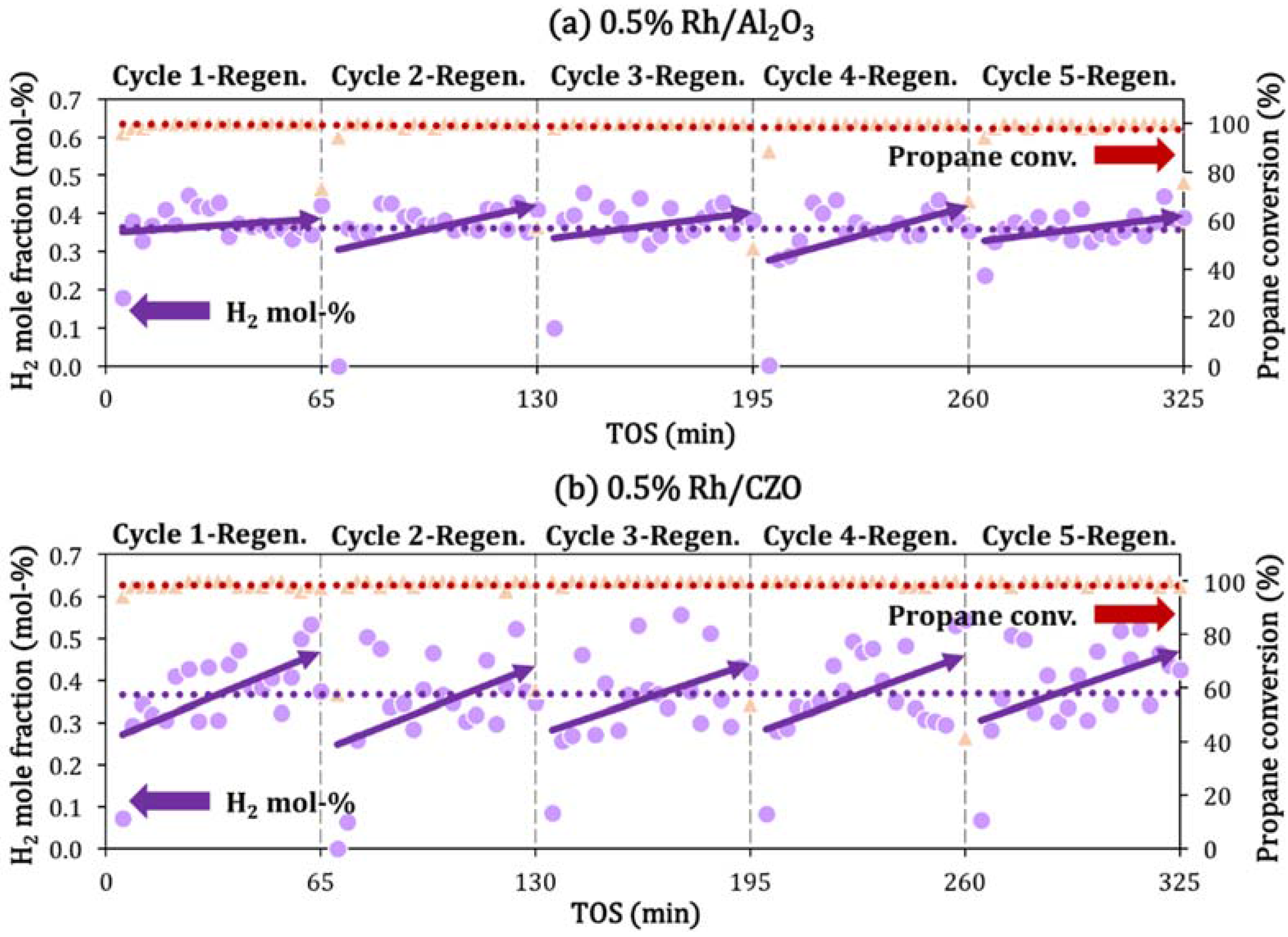
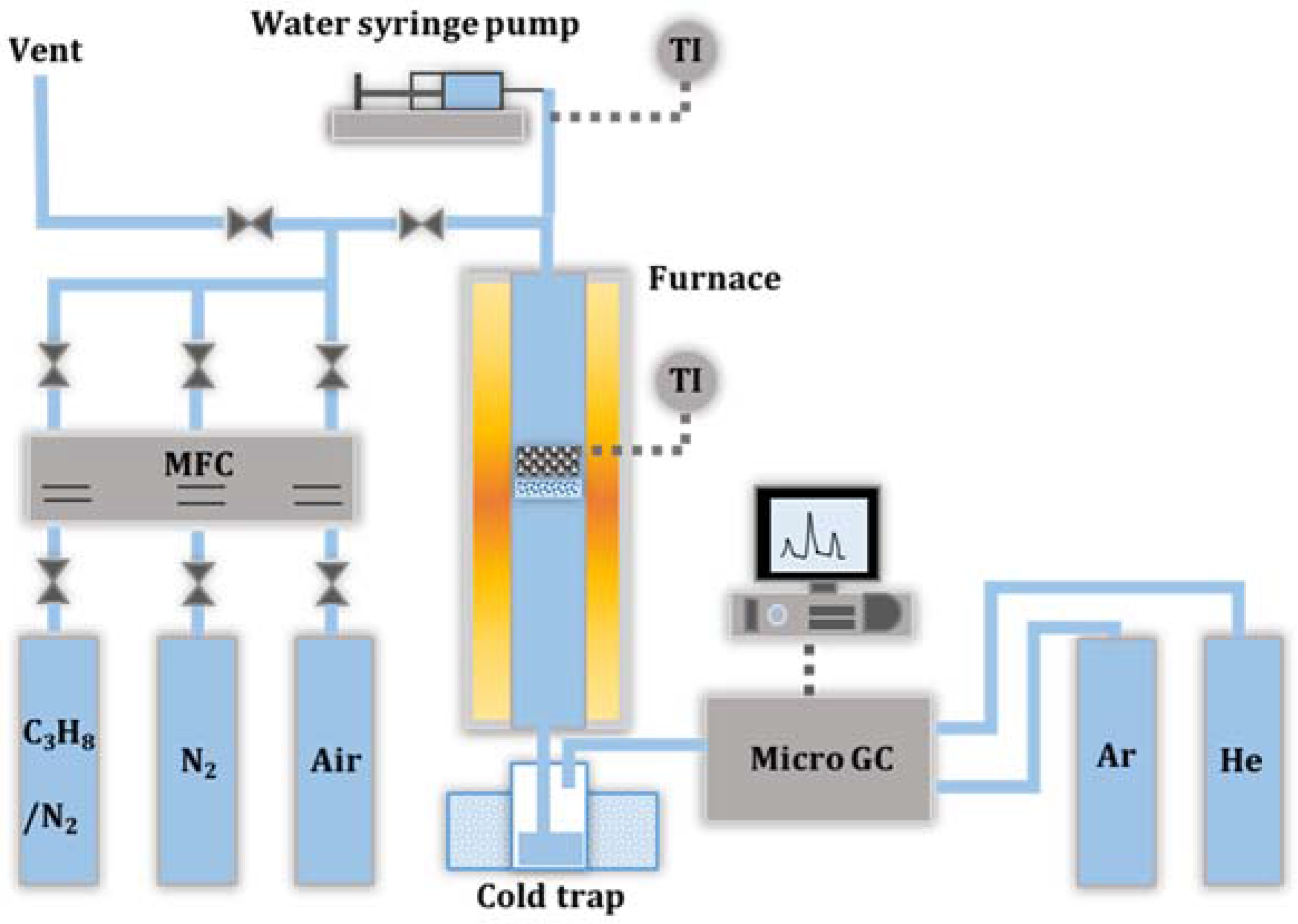
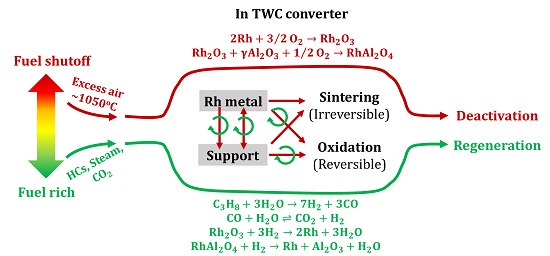
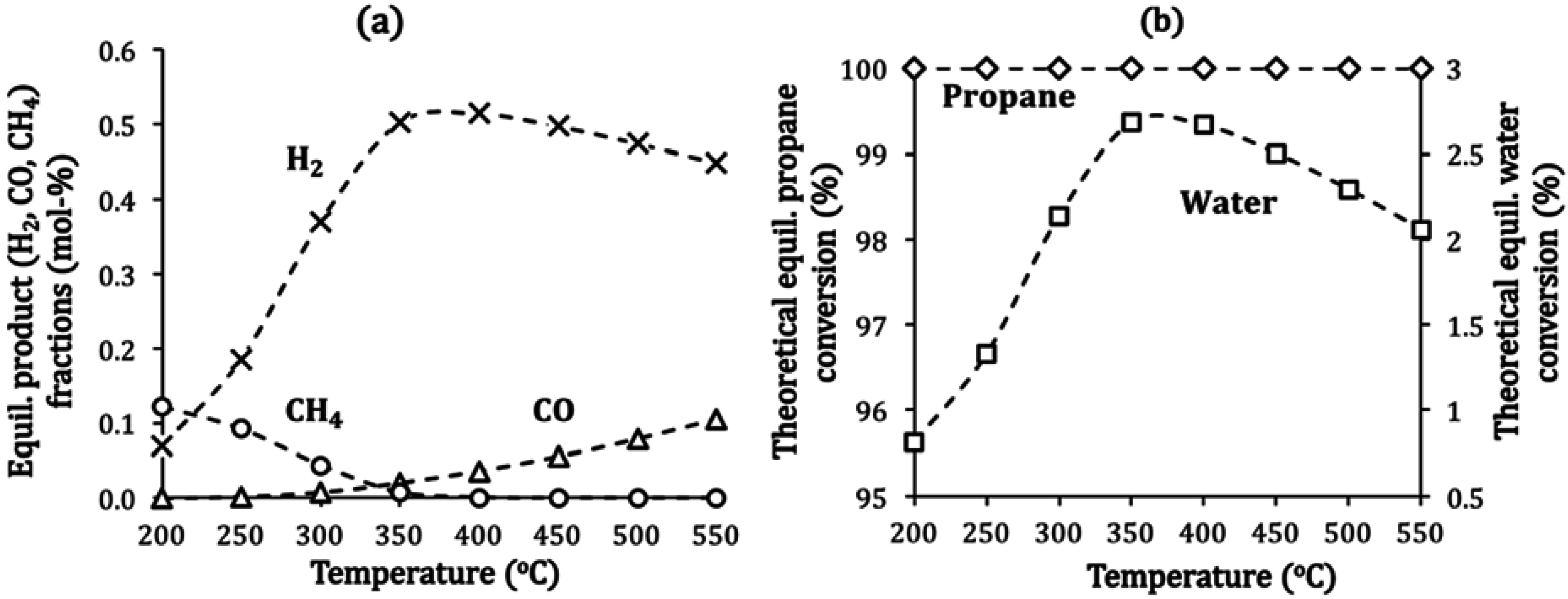
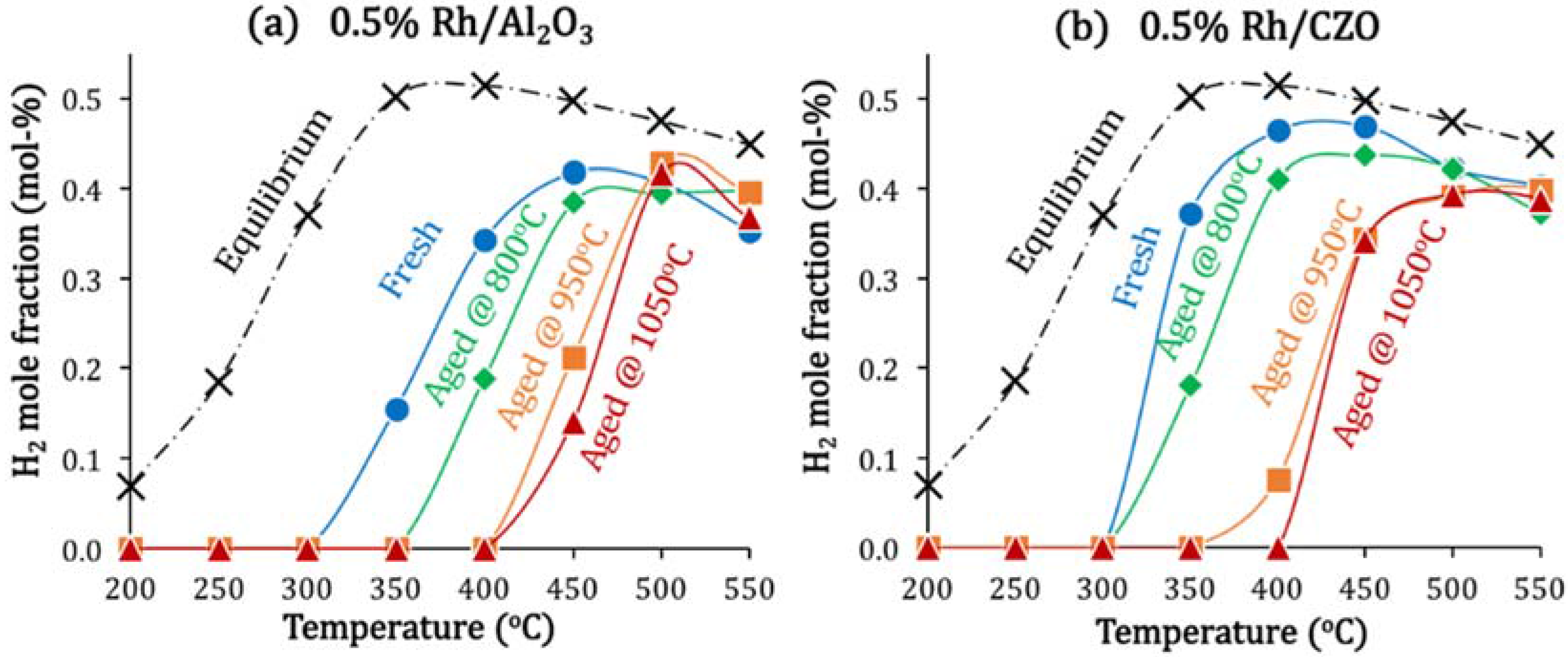
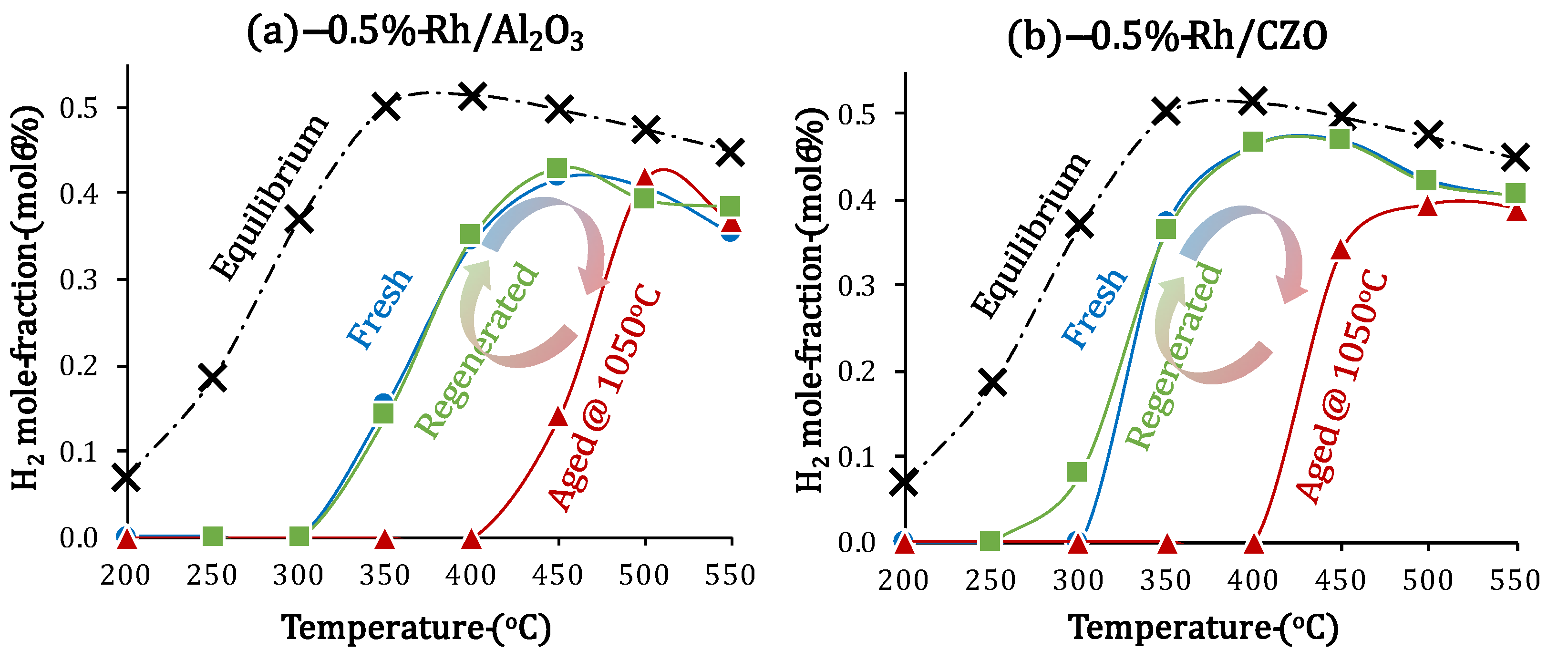
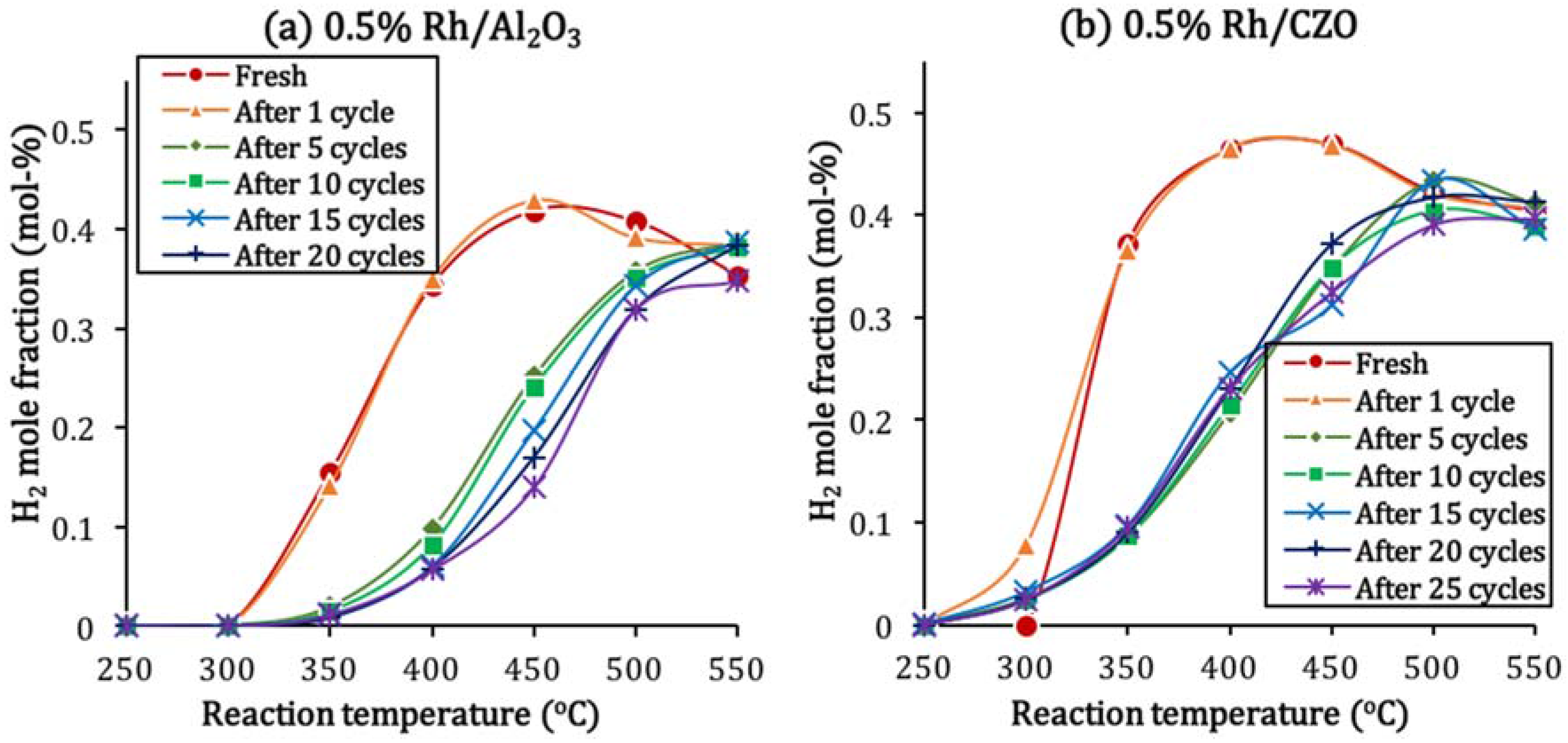
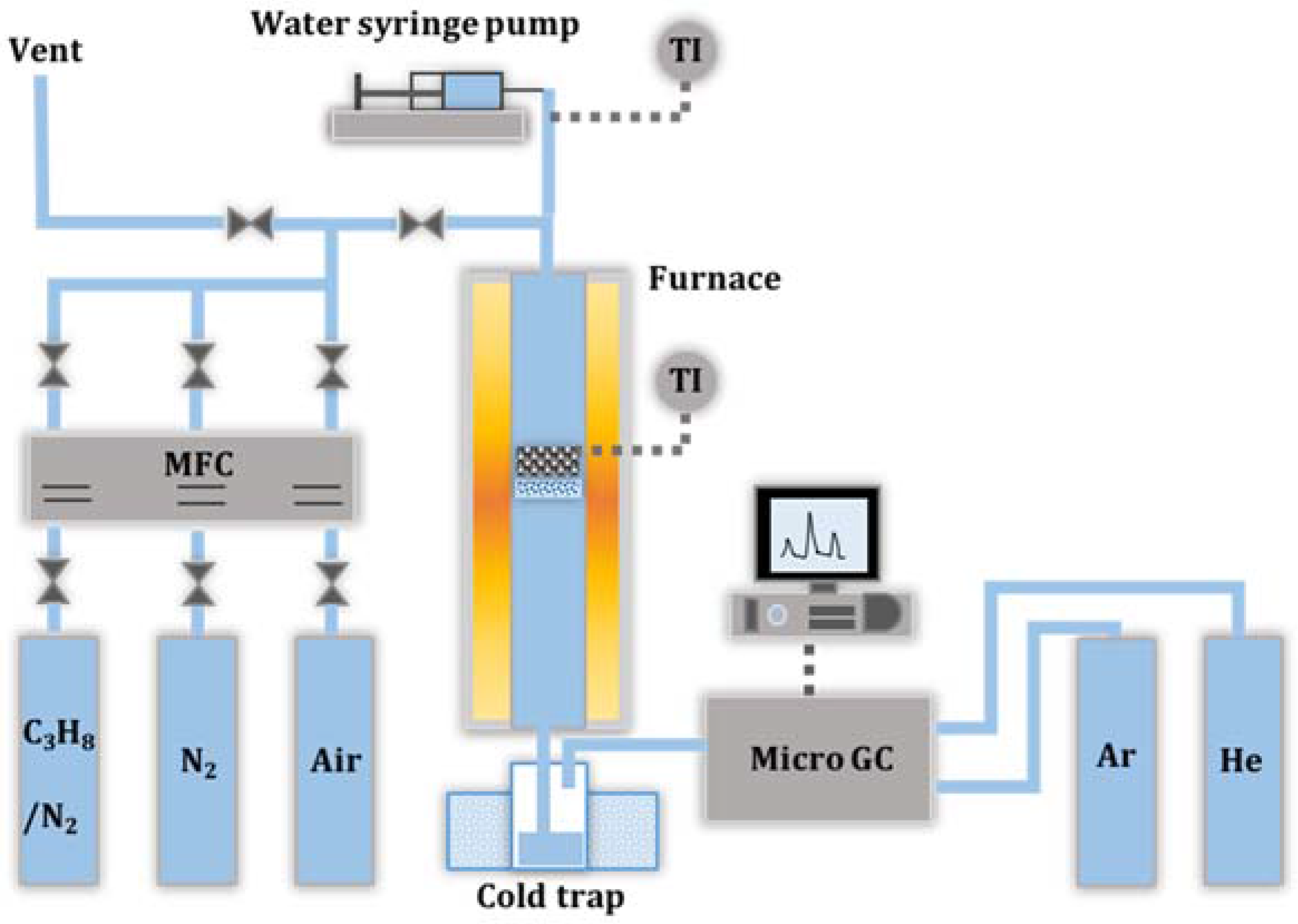
2.4. Catalyst Deactivation and Regeneration Mechanisms
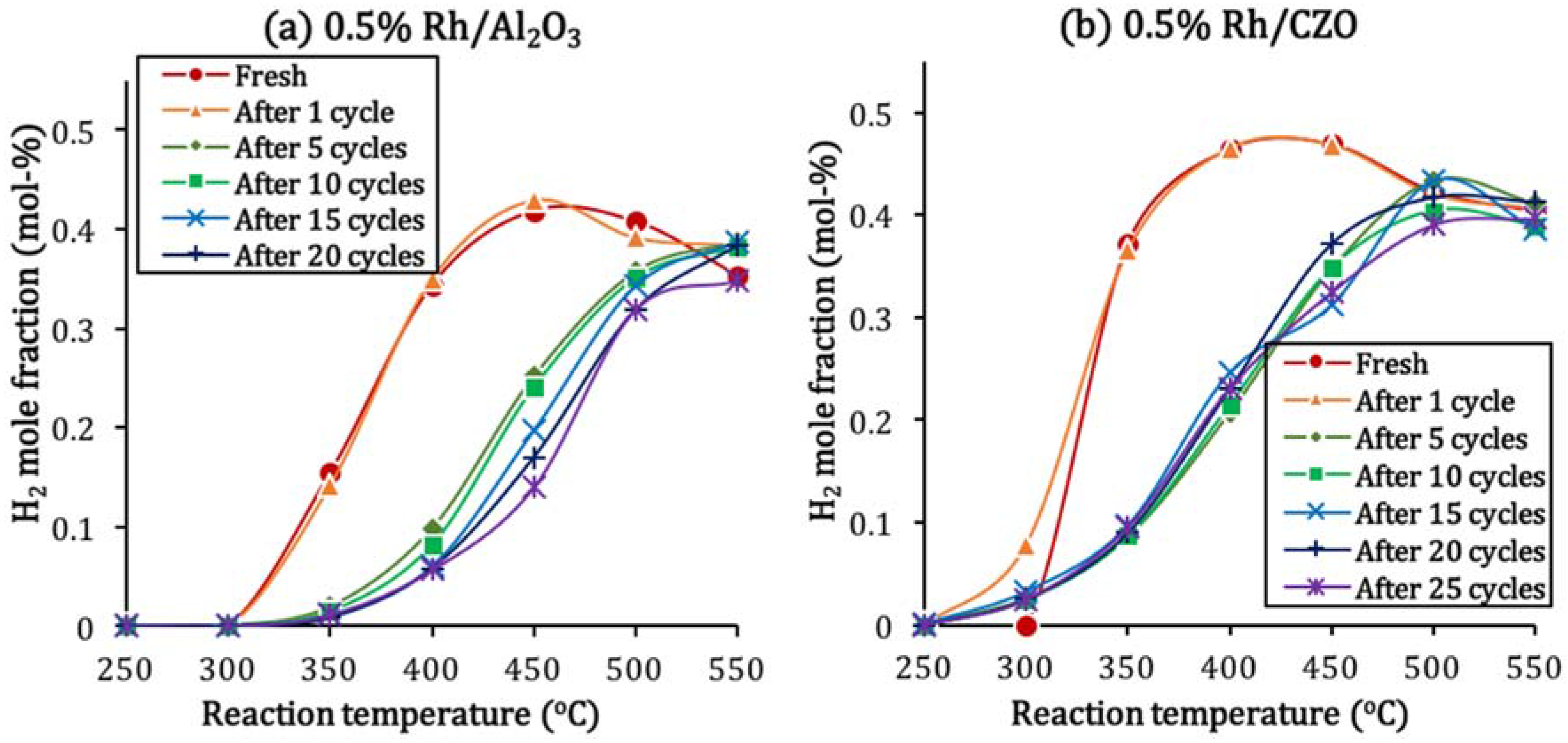
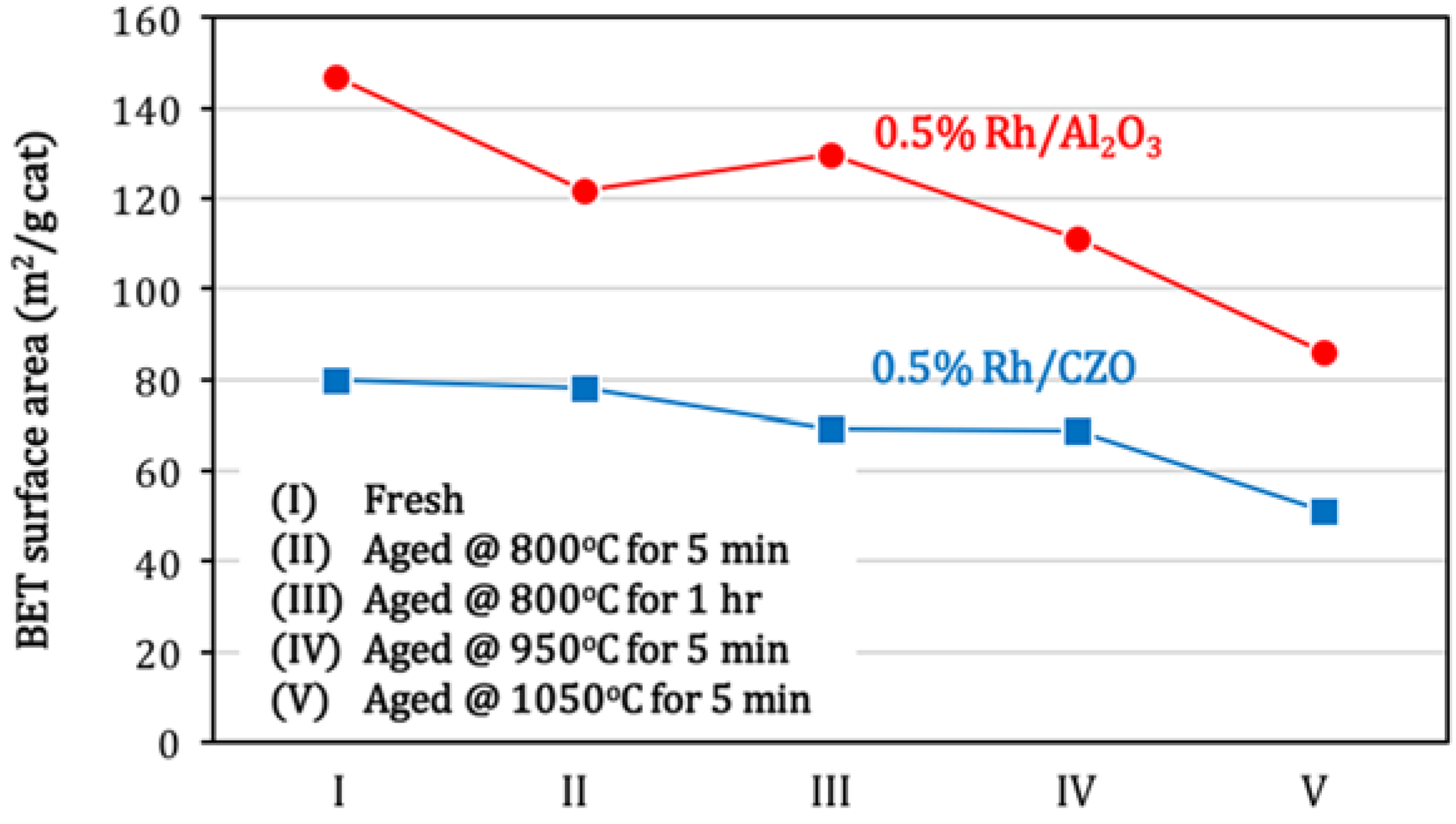
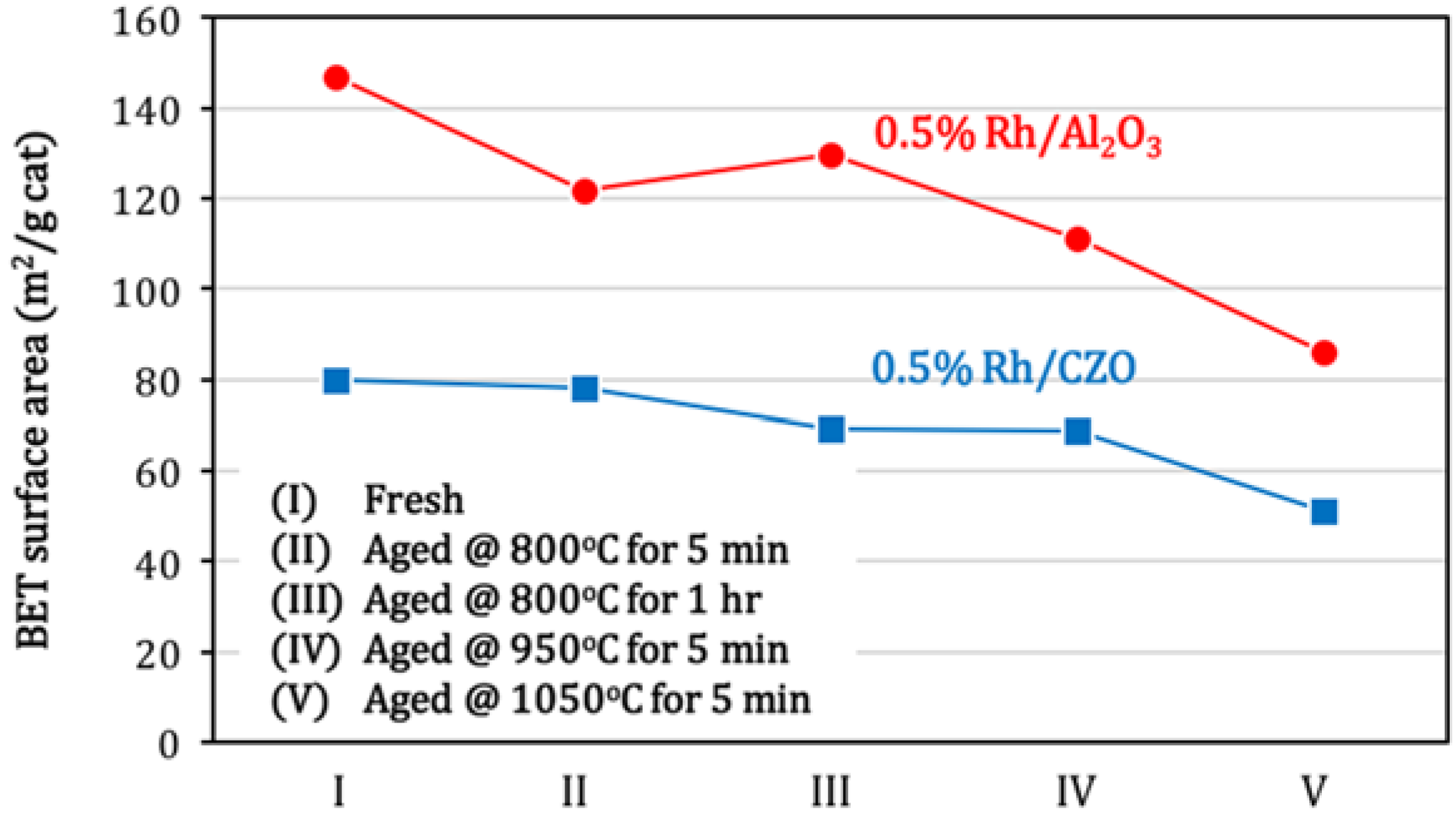
Figure 10 shows the BET surface areas of fresh and aged 0.5% Rh/Al
2O
3 and 0.5% Rh/CZO at different aging treatments. In agreement with the catalyst activity result as shown in
Figure 6, most significant losses in catalyst surface areas occurred at 950 °C and 1050 °C. The Al
2O
3 support exhibited higher intrinsic surface area, but a slightly higher percentage of sintering relative to the CZO support (with 41.4% and 35.9% for Rh/Al
2O
3 and Rh/CZO respectively after 1050 °C aging). ZrO
2 in CZO support is likely the main contributor to the thermostability of Rh/CZO [
50].
Figure 10.
BET surface areas of fresh and aged (a) 0.5% Rh/Al2O3 and (b) 0.5% Rh/CZO, as a function of aging conditions. Aged samples were obtained by aging fresh catalysts (I) in air at the following conditions: (II) 800 °C for 5 min; (III) 800 °C for 1 h; (IV) 950 °C for 5 min; or (V) 1050 °C for 5 min. The aging processes were followed by cooling in air to room temperature. As a reference, BET surface areas of support materials were measured 142.9 m2/g and 60.3 m2/g respectively for fresh Al2O3 and CZO.
Figure 10.
BET surface areas of fresh and aged (a) 0.5% Rh/Al2O3 and (b) 0.5% Rh/CZO, as a function of aging conditions. Aged samples were obtained by aging fresh catalysts (I) in air at the following conditions: (II) 800 °C for 5 min; (III) 800 °C for 1 h; (IV) 950 °C for 5 min; or (V) 1050 °C for 5 min. The aging processes were followed by cooling in air to room temperature. As a reference, BET surface areas of support materials were measured 142.9 m2/g and 60.3 m2/g respectively for fresh Al2O3 and CZO.
The metal dispersions of fresh and aged catalysts are shown in
Table 3. Fresh Rh catalysts showed higher metal dispersions on CZO than on Al
2O
3. Soria and Duarte
et al. [
51,
52] reported that the enhancement of metal dispersion in Rh/CeO
2 system was achieved by the Rh-Ce interaction (ceria stabilized Rh
+ species formed on the support). The higher metal dispersions likely enhance its activity in catalytic steam reforming of propane. After aging, loss of active metal sites occurs for both Rh catalysts, and is accelerated with increasing aging temperature from 800 °C to 1050 °C. Barbier
et al. [
53], reported that the decrease of Rh surface area in Rh/Al
2O
3 system was mainly linked to the diffusion of Rh
3+ into the alumina matrix, while the presence of Ce
xO
y stabilizes Rh and prevents Rh
3+ from dissolving into the support.
Table 3.
Metal dispersions (%) of fresh and aged 0.5% Rh/Al2O3 and 0.5% Rh/CZO catalysts as measured by room temperature CO chemisorption a. After simulated fuel shutoff, loss of active metal sites occurred for both Rh catalysts, and was accelerated with aging temperature from 800 °C to 1050 °C.
Table 3.
Metal dispersions (%) of fresh and aged 0.5% Rh/Al2O3 and 0.5% Rh/CZO catalysts as measured by room temperature CO chemisorption a. After simulated fuel shutoff, loss of active metal sites occurred for both Rh catalysts, and was accelerated with aging temperature from 800 °C to 1050 °C.
| Catalyst | Metal Dispersion (%) |
|---|
| Fresh | Aged @ 800 °C | Aged @ 1050 °C |
|---|
| 0.5%Rh/Al2O3 | 30.1 | 15.8 | 8.0 |
| 0.5%Rh/CZO | 80.7 | 40.2 | 27.9 |
However, there likely exists an overestimation of the metal particle dispersion by measuring the CO chemisorption of Rh/CZO, due to the formation of carbonate species on CeO
2 surface even at low temperature (323 K) and by the likelihood of multiple CO molecules adsorbing on the Rh itself. Some preliminary TEM result as below (
Table 4). After aging and regeneration, negligible metal crystallite size grow was observed with both Rh/Al
2O
3 and Rh/CZO, which supports our result that metal sintering was not the major deactivation mode in Rh-TWC. The dramatically reduced CO chemisorption capacity of the aged Rh-TWC together with the TEM images suggests that the main deactivation mode during 1050 °C aging was metal-support interaction.
Table 4.
Mean metal particle sizes (nm) of fresh and aged TWCs as measured using TEM images.
Table 4.
Mean metal particle sizes (nm) of fresh and aged TWCs as measured using TEM images.
| Catalyst | Active Particle Mean Size (nm) * |
|---|
| Fresh | After Air Aging @ 1050 °C | After Regeneration |
|---|
| 0.5% Rh/Al2O3 | 4.4 | 5.7 | 5.4 |
| 0.5% Rh/CZO | 8.5 | 9.2 | 9.3 |
During air aging, the oxidation state of Rh increased in both Rh/Al
2O
3 and Rh/CZO,
i.e., Rh
0 → Rh
3+ (Reaction 6). Meanwhile, strong metal-support interactions with the formation of Rhodium Aluminate (Rh(AlO
2)
y) took place in Rh/Al
2O
3 sample (Reaction 3) [
50].
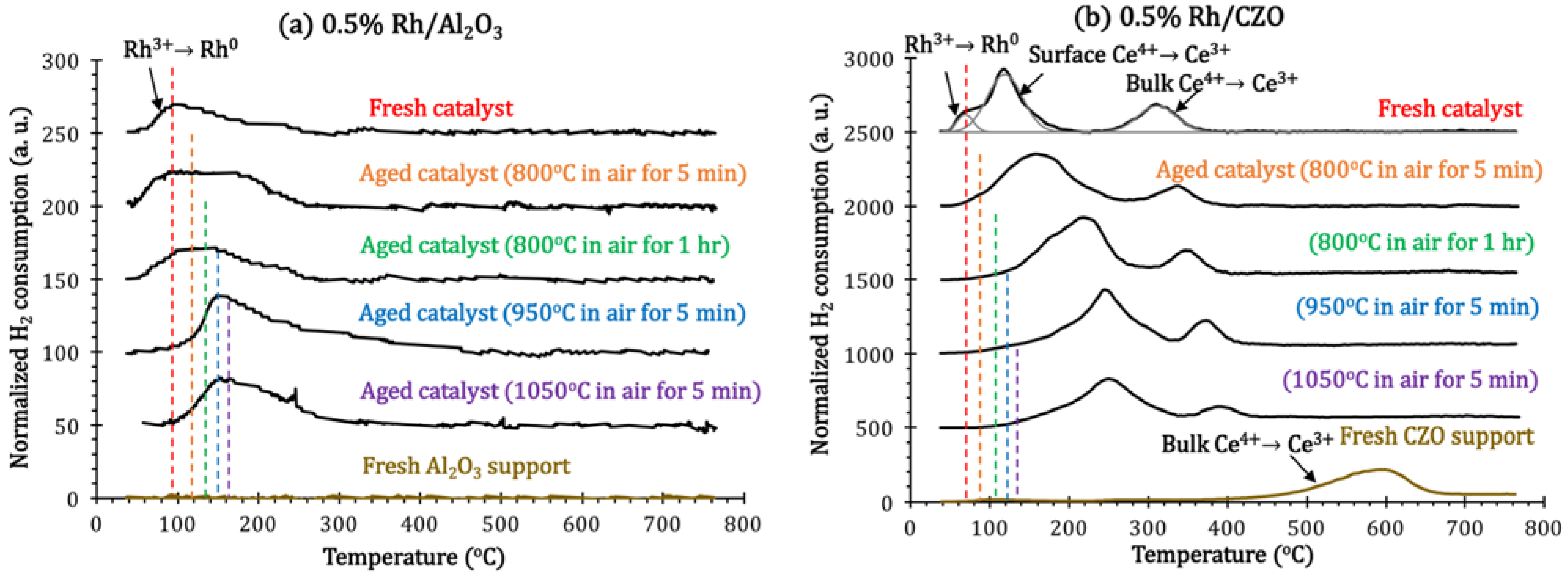
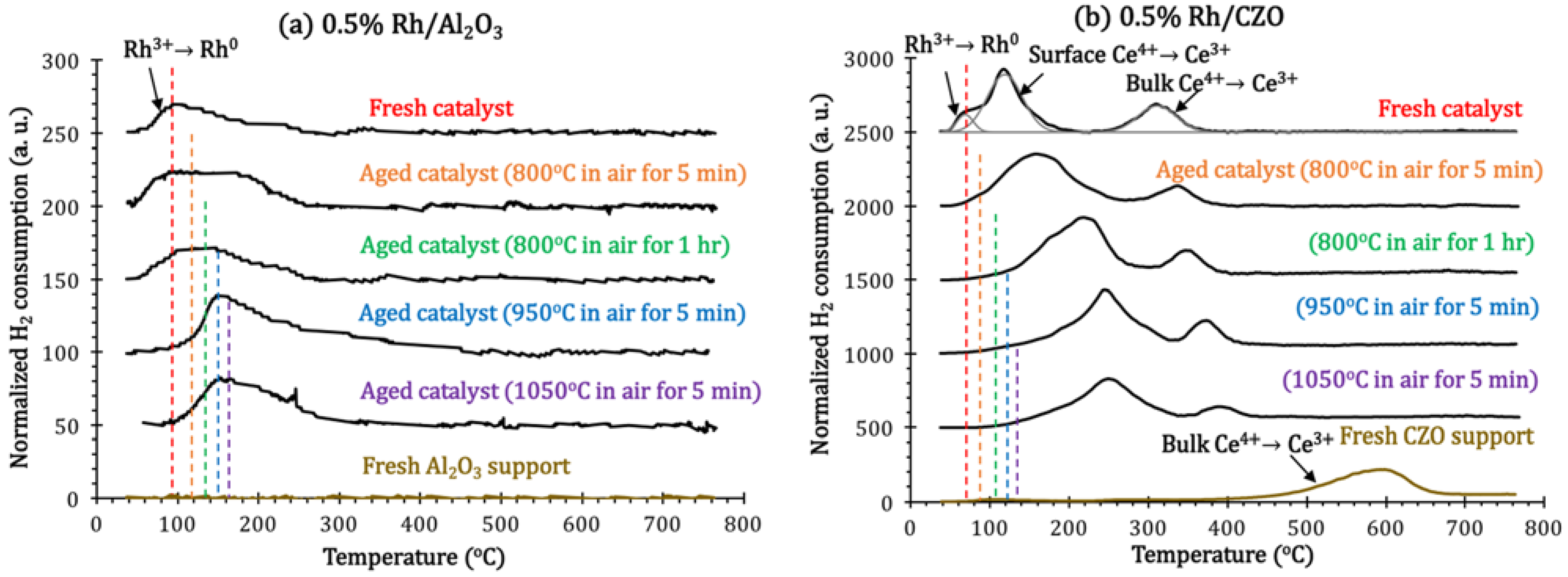
H
2-TPR was used to study the catalyst redox property after aging at different conditions (
Figure 11). The lower the temperature of the H
2 consumption peak, the easier the reduction. The reductions of Rh
3+ to Rh
0 in both catalysts occurred around 100 °C. H
2 reaction pathways on Rh sites include H
2 spill over and dissociation on Rh
0 sites (Reaction 7), and subsequent reduction of Rh
3+ → Rh
0 (Reaction 8) [
54,
55,
56].
Since Al
2O
3 is non-reducible, H
2 consumption peaks in fresh and aged Rh/Al
2O
3 are only assigned to Rh reductions. The results indicate that Rh in fresh Rh/Al
2O
3 was already partially oxidized with a H
2 consumption peak at 90 °C. After aging in air with (800–1050 °C), the TPR profiles of Rh/Al
2O
3 samples shifted to higher reducing temperatures, while the H
2 consumption peak area continued to increase. Rh was “released” from metal-support interaction by H
2 (Reaction 5), but this became increasingly difficult at higher aging temperature.
Figure 11.
Normalized H2 consumption in H2-Temperature Programmed Reduction (H2-TPR) measurements of fresh and aged (a) 0.5% Rh/Al2O3 and (b) 0.5% Rh/CZO, as a function of reducing temperature. Aged samples for measurements were respectively achieved by aging the fresh ones in air at the following conditions: 800 °C for 5 min, 800 °C for 1 h, 950 °C for 5 min, or 1050 °C for 5 min.
Figure 11.
Normalized H2 consumption in H2-Temperature Programmed Reduction (H2-TPR) measurements of fresh and aged (a) 0.5% Rh/Al2O3 and (b) 0.5% Rh/CZO, as a function of reducing temperature. Aged samples for measurements were respectively achieved by aging the fresh ones in air at the following conditions: 800 °C for 5 min, 800 °C for 1 h, 950 °C for 5 min, or 1050 °C for 5 min.
The precise stoichiometry of Rh:O is dependent on many factors including metal loading, metal dispersion, aging temperature, and aging oxygen partial pressure. Hwang
et al. [
50], reported the phase diagram for the variation of rhodium oxide species on the dispersion of rhodium samples and the oxidation temperature.
The quantitative H
2 consumption for the fresh and aged 0.5% Rh/Al
2O
3 samples during H
2-TPR is shown in
Table 5. The H
2 consumptions correspond well to our statement that the fresh 0.5% Rh/Al
2O
3 sample was already partially oxidized.
N(H
2)/
N(Rh),
i.e., the ratio between consumed H
2 molecules and reduced Rh atoms, increases with increasing aging temperature, and approaches 1.5 after high temperature (950 °C) aging, suggesting almost complete oxidation of Rh
0 to Rh
3+ in severely aged 0.5% Rh/Al
2O
3. The H
2 consumption by rhodium oxides in 0.5% Rh/CZO is very difficult to quantify because the reduction of Ce
4+ to Ce
3+ also consumes H
2. For 0.5% Rh/CZO samples, qualitative TPR analysis and semi-quantitative XPS analysis (later text) are sufficient for the Rh oxidation state study.
Table 5.
Reduction temperature (TR) and H2 consumption during H2-TPR for fresh and aged 0.5% Rh/Al2O3 samples.
Table 5.
Reduction temperature (TR) and H2 consumption during H2-TPR for fresh and aged 0.5% Rh/Al2O3 samples.
| Sample | TR (°C) | H2 Consumption (μmol H2/gcat) | N (H2)/N (Rh) |
|---|
| Fresh | 90 | 31.78 | 0.65 |
| Aged @ 800 °C for 5 min | 118 | 59.78 | 1.23 |
| Aged @ 800 °C for 1 h | 135 | 72.85 | 1.50 |
| Aged @ 950 °C for 5 min | 150 | 70.49 | 1.45 |
| Aged @ 1050 °C for 5 min | 162 | 71.57 | 1.47 |
For fresh CZO support, one broad reduction peak is shown at 400–660 °C, which is assigned to bulk surface Ce
4+ to Ce
3+, with the following global reaction (Reaction 9) [
57,
58,
59].
The reduction of Rh
3+ to Rh
0 on CZO (71 °C) is easier than for fresh Rh/Al
2O
3 (91 °C). The presence of Rh in fresh Rh/CZO allows the reduction of Ce
4+ to Ce
3+ to occur at a lower temperature (100–420 °C), with the Ce
4+ reduction peak split into two side peaks (at 110 °C and 308 °C respectively). The lower reduction temperature of Ce
4+ (at 110 °C) occurs following the surface reduction of Rh
3+, suggesting that the Ce
4+ sites being reduced were most likely the ones in close contact with the Rh sites. It has been reported that the redox properties of both Rh and Ce are enhanced when Rh is deposited on Ce
xO
y [
60]. The Rh–O–Ce bond is likely formed, creating Rh
δ+/Rh
0 (0 < δ < 1) and Ce
4+/Ce
3+ redox couple. Electrons transfer more efficiently during H
2 reduction [
61,
62,
63]. The introduction of Zr into the Ce
xO
y crystal lattice, now widely practiced for OSC, stabilizes the Rh–Ce interaction via improving mobility of oxygen in Ce
xO
y or maintaining Ce
xO
y dispersion in nanometer scale [
64,
65,
66,
67,
68,
69,
70,
71]. Furthermore, the Rh–O–Ce bond can be very easily dissociated [
60], which makes the interaction between Rh and Ce
xO
y much weaker than that between Rh and Al
2O
3. After complete reduction of Rh
δ+ to Rh
0, electrons transfer from dissociated H
2 on Rh
0, allowing easier reduction of the bulk Ce
4+ to Ce
3+. The schematic mechanism of the redox reaction pathways and the promotional metal-support interaction within 0.5% Rh/CZO during H
2-TPR are sketched in
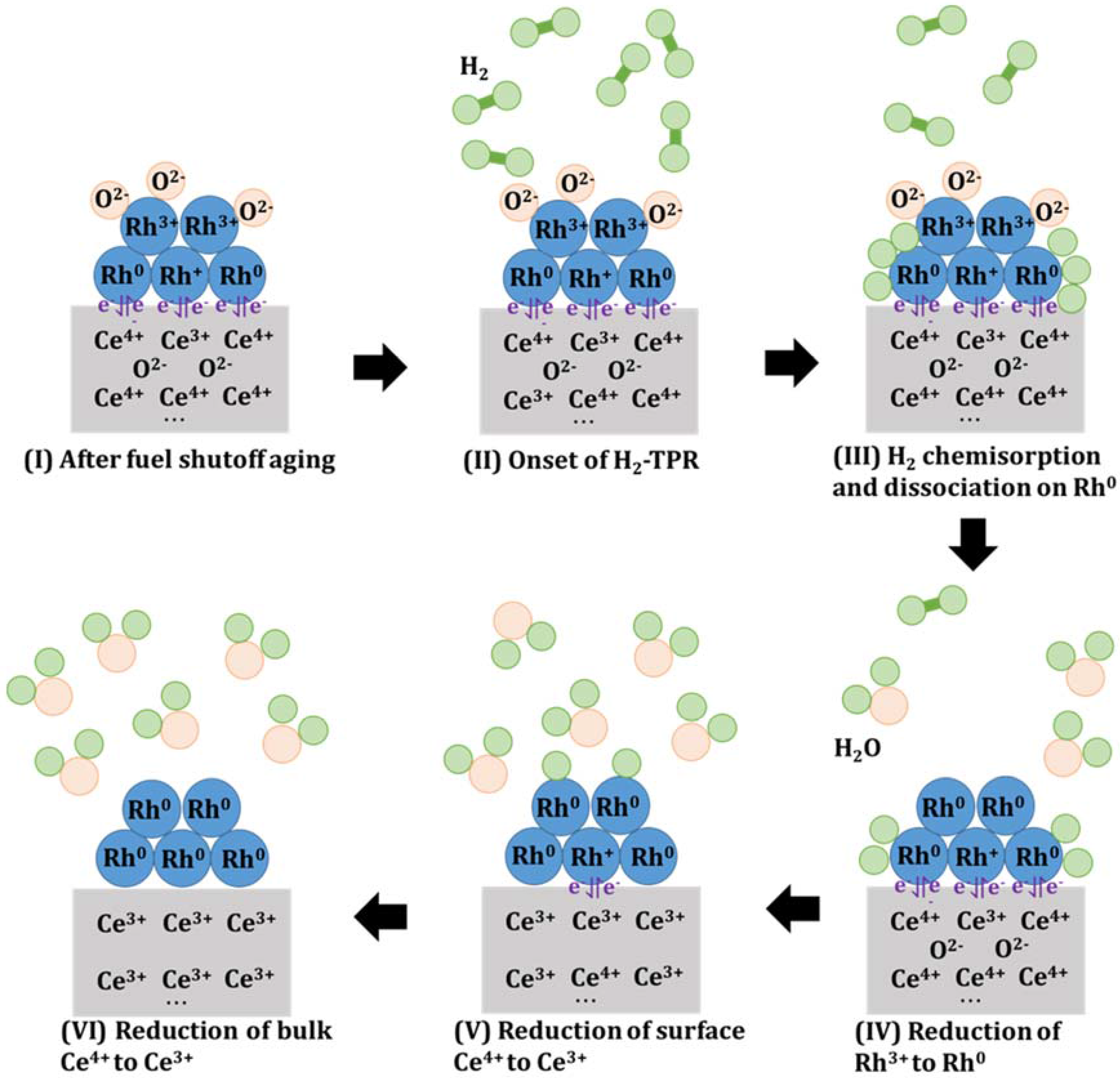
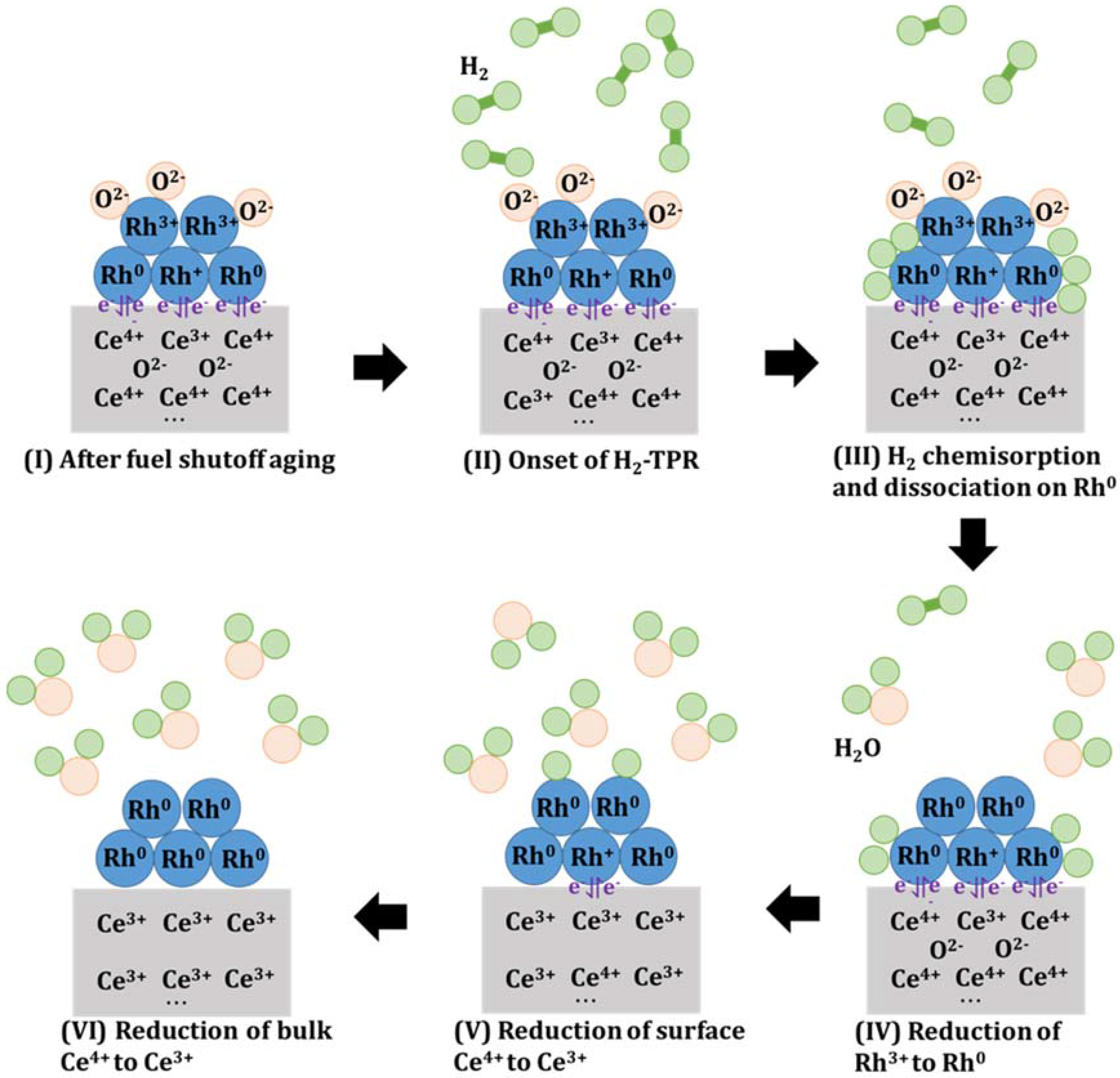
Figure 12.
In agreement with previous literature [
72], increasing the aging temperature, the reduction of both Rh and Ce in Rh/CZO shifted to higher temperature values, suggesting decreases in hydrogen dissociation capability after aging.
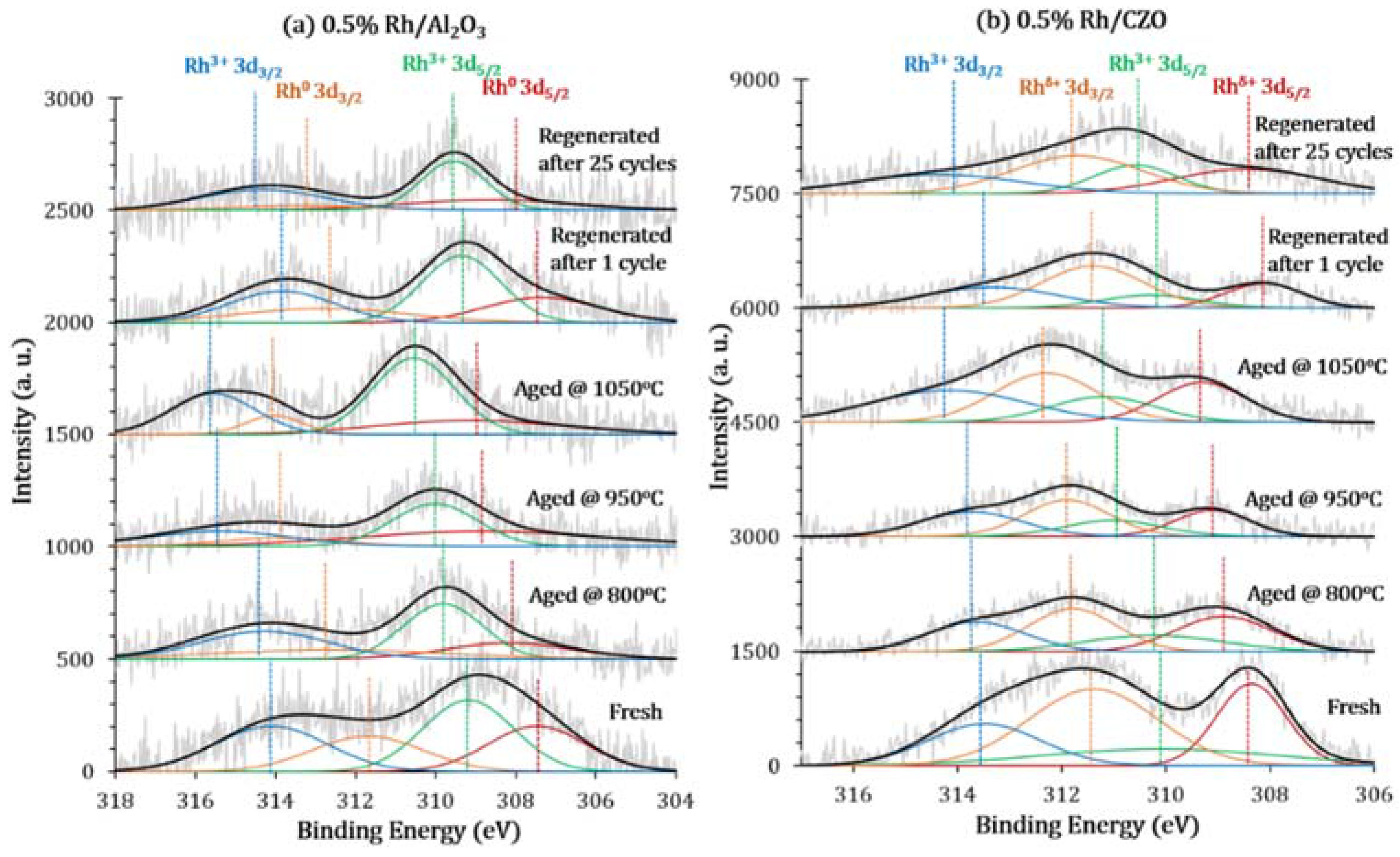
The XPS Rh 3d electron orbitals were used to identify and semi-quantify the Rh oxidation states, by comparing the binding energy values and relative ratio of the corresponding states.
Figure 13 profiles the XPS Rh 3d spectra of both catalysts, and
Table 6 summarizes the peak information details. Rh 3d
3/2 and Rh 3d
5/2 peaks, resulting from spin-orbital splitting, with different binding energies (BE) corresponding to the Rh valence states were assigned [
73,
74,
75,
76,
77,
78,
79,
80]. For a fresh sample, Rh 3d
5/2 peak with BE of at 307.5 eV–308.4 eV is attributed to Rh
0 valence state, while Rh 3d
5/2 peak at 309.2 eV–310.1 eV is attributed to Rh
3+ valence state.
Figure 12.
Speculative schematic of proposed redox reaction mechanism and interaction between Rhδ+/Rh0 and Ce4+/Ce3+ redox couple during H2-TPR of 0.5% Rh/CZO. The redox reactions followed the order described below. (I) After simulated fuel shutoff aging in air at 800 °C, 950 °C, or 1050 °C, surface Rh sites are oxidized to Rh2O3, while the Rh sites in close contacts with CexOy remained in reduced states (Rhδ+, 0 < δ < 1), with Rhδ+/Rh0 and Ce4+/Ce3+ redox couple formed for enhancing electron transfer efficiency; (II) H2 flow through the sample; (III) At low temperature regime around 100 °C to 120 °C, H2 was chemisorbed and dissociated on the Rh0 sites, followed by (IV) Reduction of Rh3+ to Rh0; (V) Reduction of surface Ce4+ sites to Ce3+ promoted by the Rhδ+/Rh0 and Ce4+/Ce3+ redox couple; (VI) Reduction of bulk Ce4+ sites to Ce3+ when more H2 molecules were chemisorbed and dissociated on Rh0.
Figure 12.
Speculative schematic of proposed redox reaction mechanism and interaction between Rhδ+/Rh0 and Ce4+/Ce3+ redox couple during H2-TPR of 0.5% Rh/CZO. The redox reactions followed the order described below. (I) After simulated fuel shutoff aging in air at 800 °C, 950 °C, or 1050 °C, surface Rh sites are oxidized to Rh2O3, while the Rh sites in close contacts with CexOy remained in reduced states (Rhδ+, 0 < δ < 1), with Rhδ+/Rh0 and Ce4+/Ce3+ redox couple formed for enhancing electron transfer efficiency; (II) H2 flow through the sample; (III) At low temperature regime around 100 °C to 120 °C, H2 was chemisorbed and dissociated on the Rh0 sites, followed by (IV) Reduction of Rh3+ to Rh0; (V) Reduction of surface Ce4+ sites to Ce3+ promoted by the Rhδ+/Rh0 and Ce4+/Ce3+ redox couple; (VI) Reduction of bulk Ce4+ sites to Ce3+ when more H2 molecules were chemisorbed and dissociated on Rh0.
Figure 13.
X-ray Photoelectron Spectroscopy (XPS) multiplex spectra in Rh 3d region (with BE of 318 eV–304 eV) of fresh, aged, and regenerated (
a) 0.5% Rh/Al
2O
3 and (
b) 0.5% Rh/CZO powder catalysts, and with aging temperature varied. Aged samples were achieved by aging the fresh catalysts in air at 800 °C, 950 °C or 1050 °C for 5 min. Regenerated samples were achieved by regenerating the aged ones (1050 °C for 5 min) using the method as described in
Section 3.3.
Figure 13.
X-ray Photoelectron Spectroscopy (XPS) multiplex spectra in Rh 3d region (with BE of 318 eV–304 eV) of fresh, aged, and regenerated (
a) 0.5% Rh/Al
2O
3 and (
b) 0.5% Rh/CZO powder catalysts, and with aging temperature varied. Aged samples were achieved by aging the fresh catalysts in air at 800 °C, 950 °C or 1050 °C for 5 min. Regenerated samples were achieved by regenerating the aged ones (1050 °C for 5 min) using the method as described in
Section 3.3.
Table 6.
Summary of detailed information of XPS spectra as shown in
Figure 12,
i.e., values of binding energy Rh 3d
3/2 for Rh
3+ and Rh
0 oxidation states, and Rh
3+/Rh
0 (or Rh
3+/Rh
δ+) ratios
a for fresh, aged, and regenerated 0.5% Rh/Al
2O
3 and 0.5% Rh/CZO samples.
Table 6.
Summary of detailed information of XPS spectra as shown in Figure 12, i.e., values of binding energy Rh 3d3/2 for Rh3+ and Rh0 oxidation states, and Rh3+/Rh0 (or Rh3+/Rhδ+) ratios a for fresh, aged, and regenerated 0.5% Rh/Al2O3 and 0.5% Rh/CZO samples.
| Catalyst | E (Rh 3d3/2), eV | E (Rh 3d5/2), eV | Rh3+/Rh0 (Rh3+/Rhδ+) b |
|---|
| Rh3+ | Rh0 | Rh3+ | Rh0 |
|---|
| 0.5% Rh/Al2O3 | Fresh | 314.10 | 311.69 | 309.18 | 307.46 | 1.29 |
| 800 °C aged for 5 min | 314.27 | 312.59 | 309.81 | 308.16 | 1.51 |
| 950 °C aged for 5 min | 315.39 | 313.87 | 310.02 | 308.85 | 2.14 |
| 1050 °C aged for 5 min | 315.55 | 314.03 | 310.55 | 309.13 | 3.21 |
| Regenerated (I) c | 313.83 | 312.78 | 309.35 | 307.34 | 1.41 |
| Regenerated (II) d | 314.87 | 313.12 | 309.55 | 308.89 | 2.18 |
| 0.5% Rh/CZO | Fresh | 313.49 | 311.39 | 310.10 | 308.36 | 0.48 |
| 800 °C aged for 5 min | 313.64 | 311.79 | 310.23 | 308.89 | 0.75 |
| 950 °C aged for 5 min | 313.74 | 311.90 | 311.02 | 309.10 | 0.81 |
| 1050 °C aged for 5 min | 314.17 | 312.30 | 311.21 | 309.31 | 0.99 |
| Regenerated (I) c | 313.35 | 311.40 | 310.29 | 308.11 | 0.64 |
| Regenerated (II) d | 314.01 | 311.70 | 310.70 | 308.44 | 0.70 |
Fresh 0.5% Rh/Al
2O
3 displayed (1) an intense Rh
3+ 3d
5/2 peak at 309.2 eV; (2) a small Rh
0 3d
5/2 side peak at 307.5 eV; and (3) Rh
3+/Rh
0 ratio of 1.29. Consistent with the TPR result, the XPS data suggests the Rh sites in the fresh samples were partially oxidized. With increasing aging temperature, Rh 3d peaks shift to higher binding energy, together with increases in Rh
3+/Rh
0 ratio (1.29 → 1.51 → 2.14 → 3.21), suggesting a transition to a higher Rh oxidation state,
i.e., Rh
0 → Rh
3+. It is known that the oxidation process increases Rh oxidation states while the reduction process has an opposite effect [
48]. The non-reducible Rh phase was reported resulting from a diffusion of Rh
3+ ions in subsurface regions of the alumina matrix. The binding energy of the new Rh phase is greater than that in Rh
2O
3, indicating a different state from that of Rh
3+ in Rh
2O
3, which is ascribed to metal-support interaction [
81].
Different from 0.5% Rh/Al
2O
3, low Rh valence state (Rh
δ+, 0 < δ < 1) dominates in fresh 0.5% Rh/CZO (Rh
3+/Rh
δ+ ratio of 0.48). For 0.5% Rh/CZO, Rh
0 3d
5/2 peaks display higher BE values. The small but definite electropositive shifts detected for Rh
0 peaks are ascribed to the existence of both Rh
0 and Rh
δ+ species, giving evidence to the existence of Rh
δ+/Rh
0 and Ce
4+/Ce
3+ redox couple [
63]. This assignment is in agreement with previous FT-IR result [
82,
83], which shows the existence of surface electron deficient Rh
δ+ species present on CZO support. Like Rh/Al
2O
3, the Rh
3+/Rh
δ+ ratio in Rh/CZO increased with aging temperature. It is also important to note that the way Rh3d peak is interpreted largely affects the result. The XPS Rh 3d spectra for Rh/CeO
2 system studied by Force
et al. [
84] was deconvoluted into three peaks, respectively assigned to Rh
0 (306.8 eV), Rh
+ (307.8 eV), Rh
3+ (309.2 eV) states. While other systems have different interpretations [
85]. In our Rh/CZO system, assigning XPS peaks to Rh
0 and Rh
δ+ species is easier for comparison.
Furthermore, the reduced areas under Rh 3d peaks for aged samples suggests Rh sintering and/or Rh dissolution into sintered support during simulated fuel shutoff aging. The characterization result is in agreement with the findings by Kang
et al. [
20]. In their study, the effect of aging atmosphere on the sintering behavior of commercial Pd- or Rh-TWC as well as the TWC performance were investigated under straight oxidizing, reducing, and periodic cycling aging conditions. For Rh-TWC, the diffusion of Rh
2O
3 into the support along with the agglomeration of the Rh metal were found the main causes of catalyst deactivation during high temperature oxidative aging.
XPS Rh 3d spectra of regenerated 0.5% Rh/Al2O3 and 0.5% Rh/CZO in both show that after the first in situ regeneration, the oxidation state of Rh was significantly lowered, exposing more active Rh0 species to the reactant atmosphere. This explains the enhanced reforming activity resulting from H2 reduction (regeneration).
In summary, different types of interactions between Rh and support materials exist in Rh/Al
2O
3 and Rh/CZO during fuel shutoff aging. It is well known that strong interaction between Rh and Al
2O
3 with the formation of Rhodium Aluminate occurs in oxidative aging of Rh/Al
2O
3 [
2]. Compared to aged Rh/Al
2O
3, the metal-support interaction in aged Rh/CZO occurs to a much lesser extent. Haneda
et al. [
86] reported that high-temperature aging can alter the surface properties of Ce
xO
y-ZrO
2 to inhibit the formation of formate species poisoning the catalytic active Rh sites.
The superior regenerability of 0.5% Rh/CZO was believed mainly contributed by the co-existence of Ce
4+/Ce
3+ and Rh
0/Rh
δ+ redox couple [
87,
88,
89,
90]. Wang
et al. [
62], investigated the interaction between Rh and Ce
xO
y in Rh-Ce
xO
y/Al
2O
3 catalyst system, with enhanced electron transfer efficiency during catalytic CO
2 dry reforming of CH
4. Similar promotional effect likely occurred with 0.5% Rh/CZO catalyst during regeneration, as confirmed by TPR and XPS results. The electron transfer pathways during catalyst regeneration are proposed in
Figure 14.
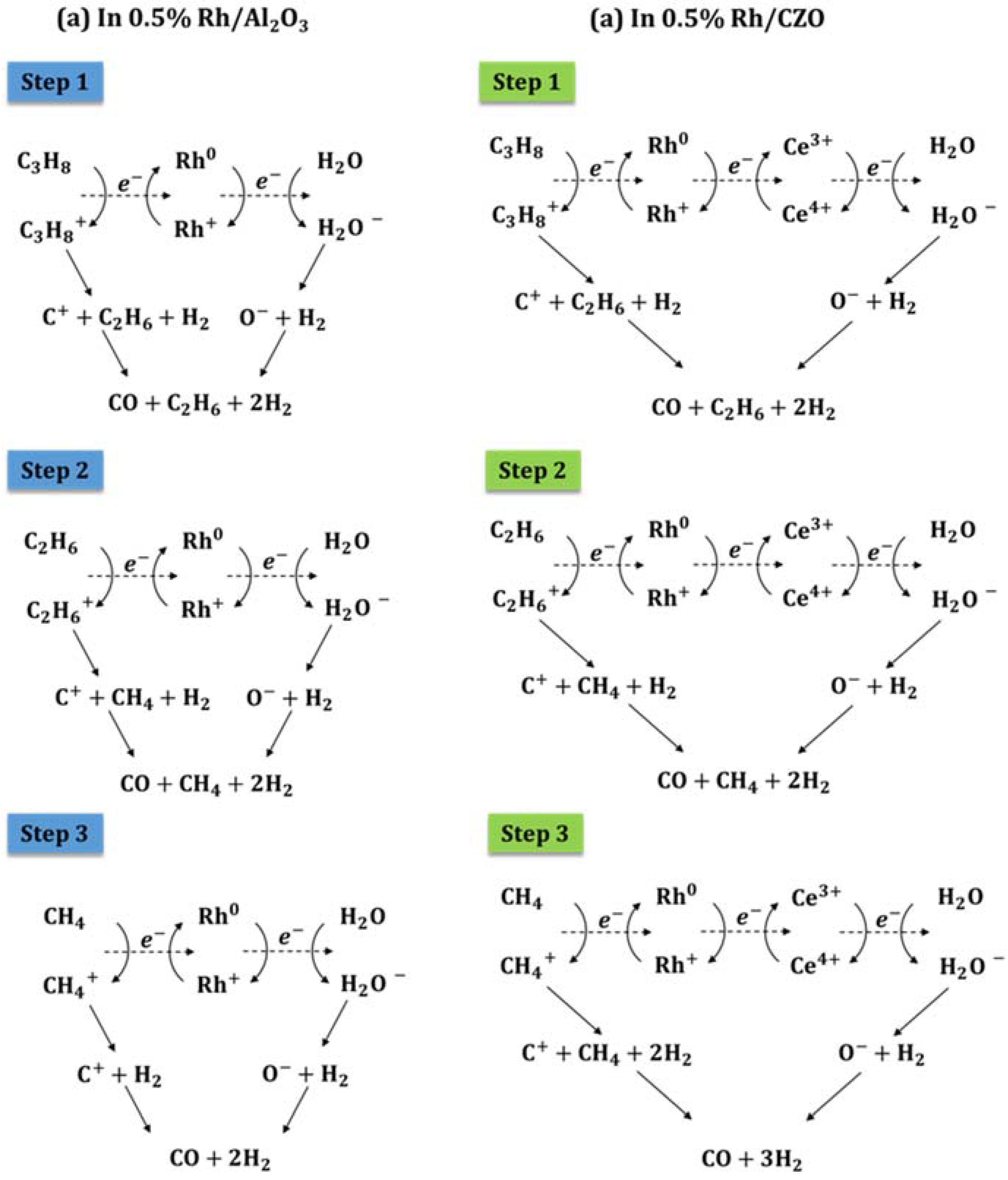
Figure 14.
Proposed reaction mechanism and electron transfer pathways for steam reforming of propane on (a) Rh/Al2O3 and (b) Rh/CZO catalysts.
Figure 14.
Proposed reaction mechanism and electron transfer pathways for steam reforming of propane on (a) Rh/Al2O3 and (b) Rh/CZO catalysts.
For Rh/Al2O3, electrons are first donated by hydrocarbons (reactant C3H8, and product C2H6 and CH4), and then transferred through the redox circle of Rh0 ⇌ Rh+, and finally accepted by H2O. Electron transfer is accompanied by redox reactions and the formation of H2, CO, and intermediate products. For Rh/CZO, the coexistence of the Ce4+/Ce3+ and the Rh0/Rhδ+ redox couple allows availability of Rhδ+ species, to accept the electrons donated by HC more easily. The efficient electron transfer pathway results in the significant catalytic steam reforming performance of Rh/CZO.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}