PtRu Nanoparticles Deposited by the Sulfite Complex Method on Highly Porous Carbon Xerogels: Effect of the Thermal Treatment


 ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Textural Properties of Carbon Xerogels and PtRu-Catalysts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m2 g−1) | Vpore p/p0 ≈ 1 (cm3 g−1) | Vmeso BJH (cm3 g−1) | Vmicro (cm3 g−1) | Mean pore size (nm) |
|---|---|---|---|---|---|
| CXG | 528 | 1.79 | 1.66 | 0.14 | 23 |
| PtRu/CXG-COL | 271 | 0.51 | 0.59 | 0.08 | 19 |
| PtRu/CXG-COL-TT200 | 278 | 0.57 | 0.46 | 0.09 | 18 |
| PtRu/CXG-COL-TT400 | 332 | 0.66 | 0.55 | 0.11 | 18 |
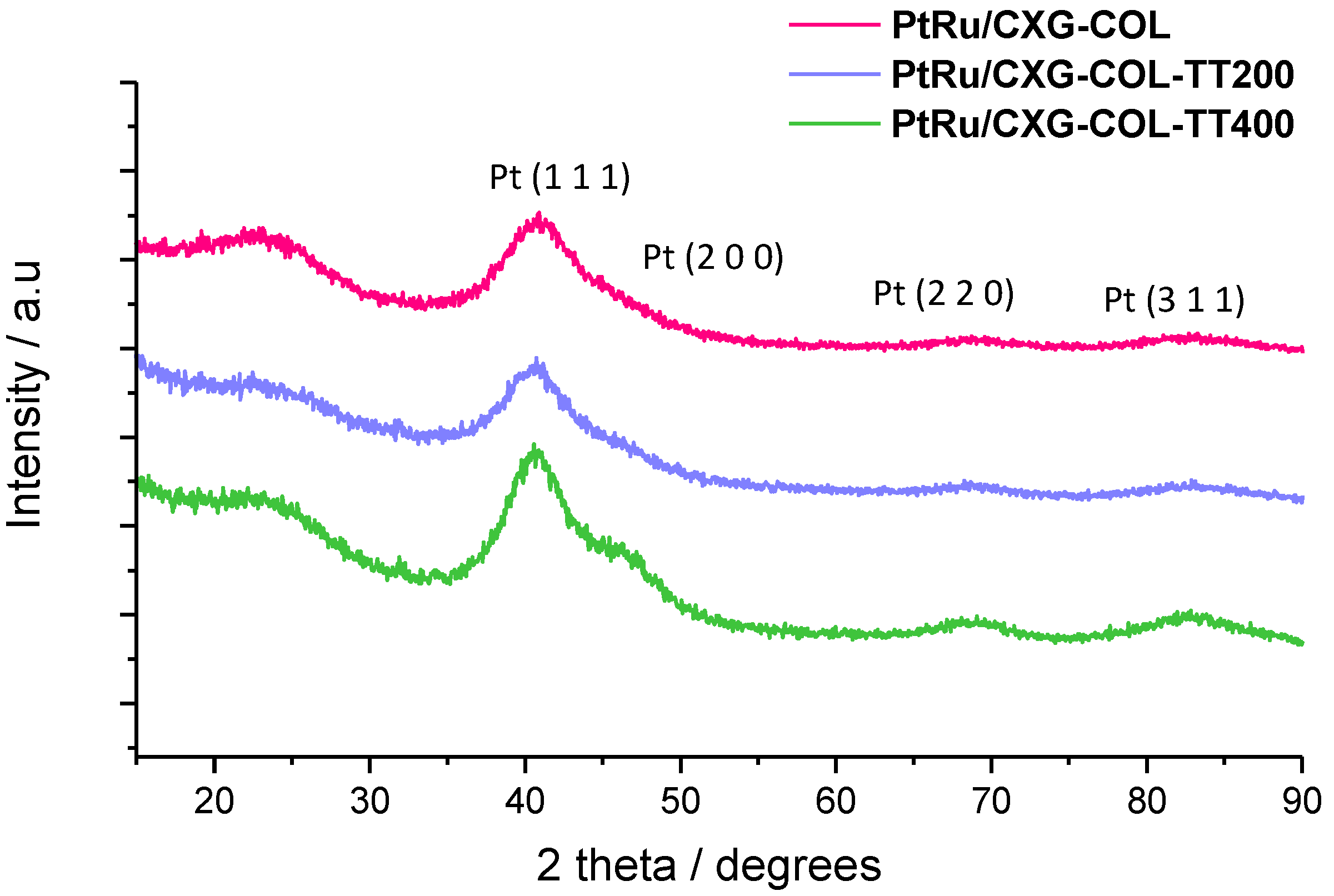
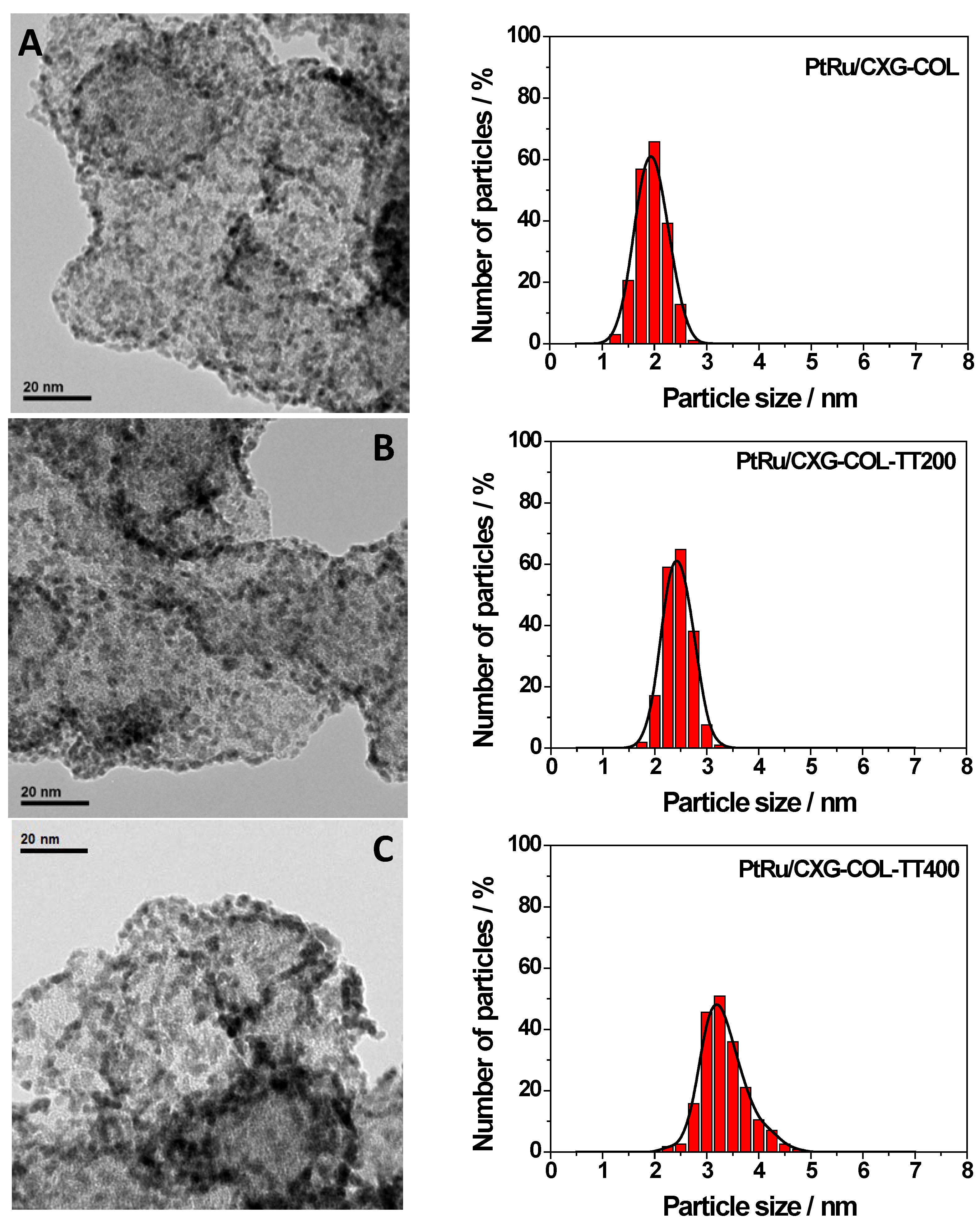
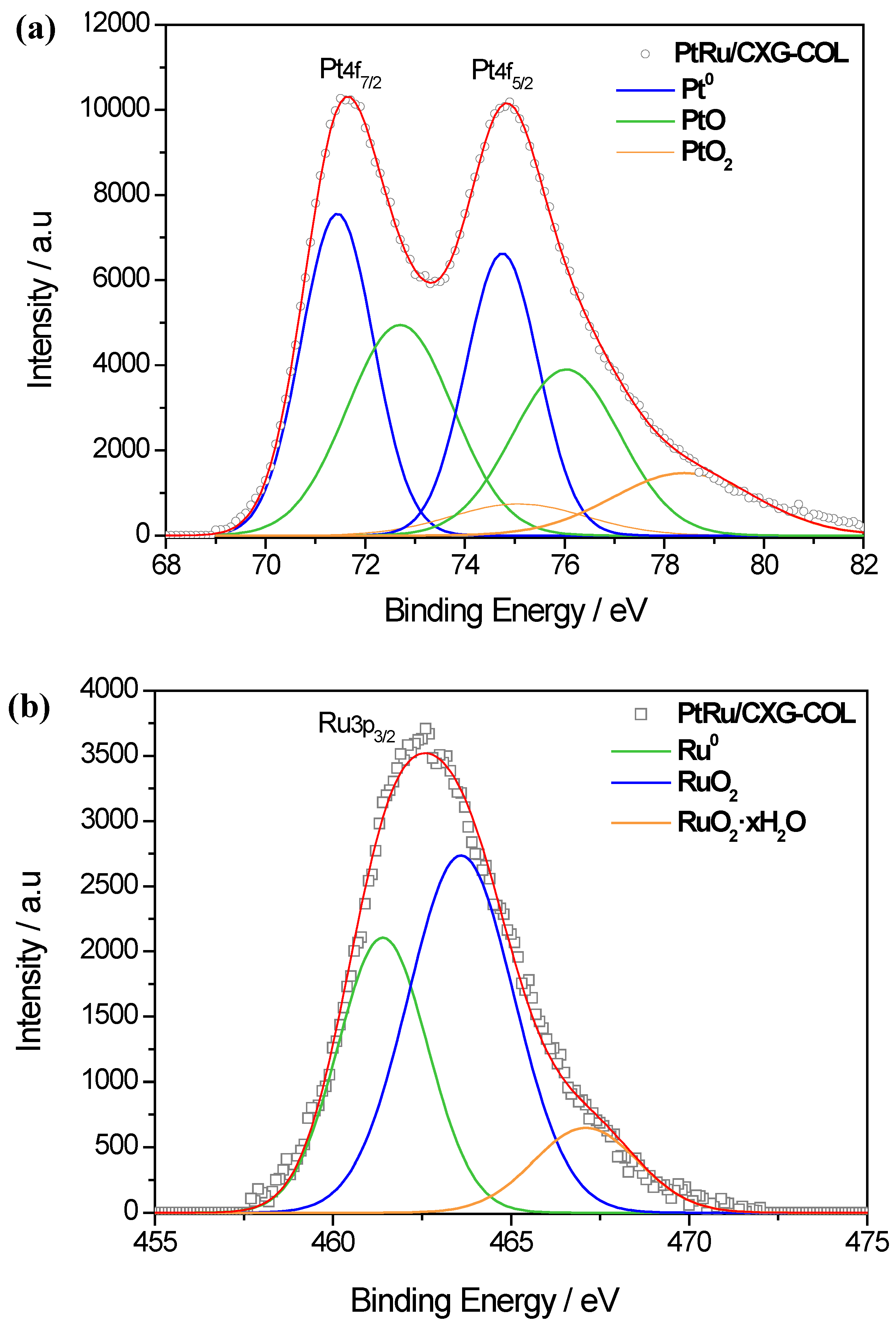
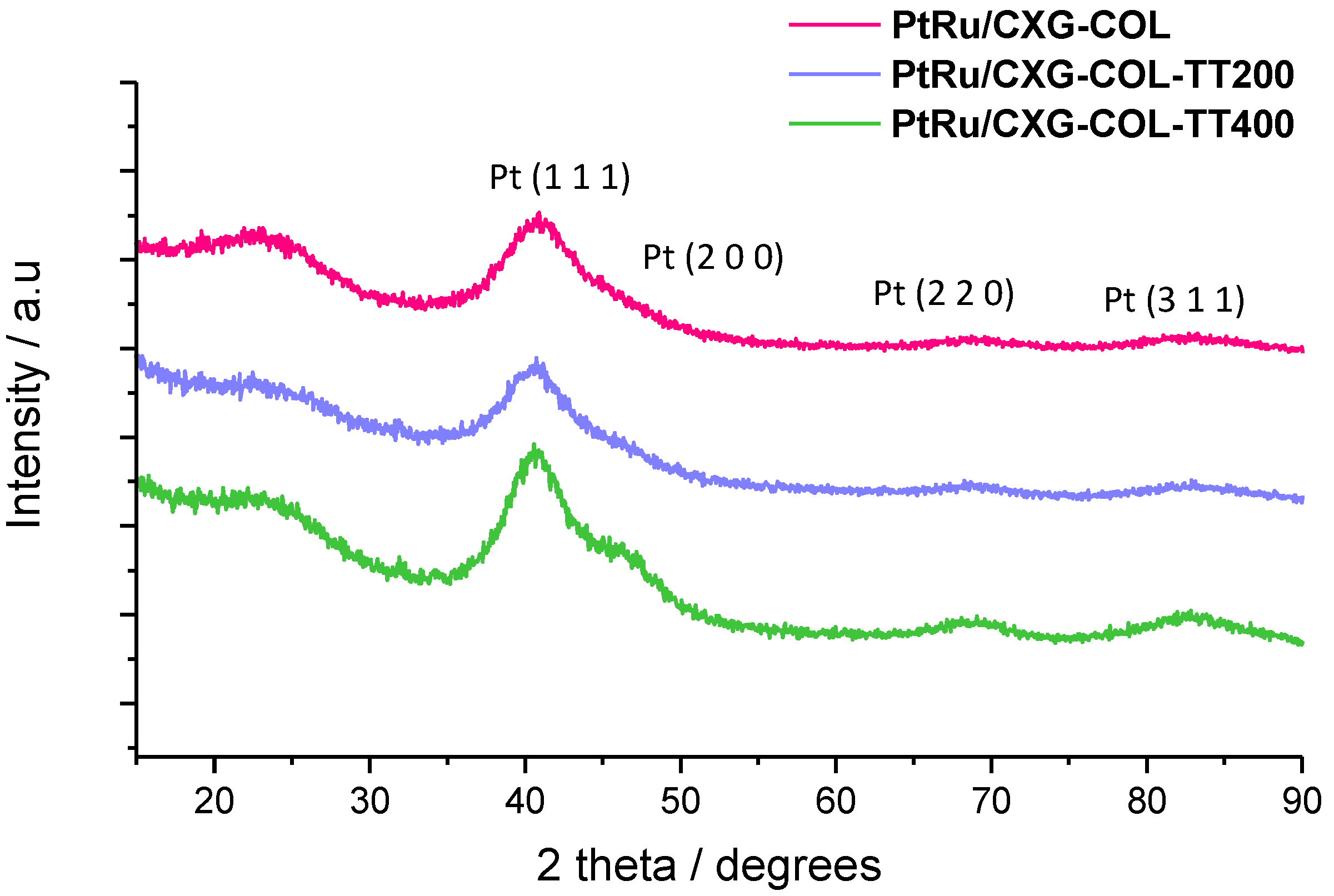
2.2. PtRu-Catalysts Characterization
| Sample | % w/w PtRu | Atomic ratio Pt:Ru | PtRu crystal size | Lattice parameter | XRu in PtRu alloy | |
|---|---|---|---|---|---|---|
| TGA | XRF | nm | nm | Vegard’s law | Antolini’s equation [22] | |
| PtRu/CXG-COL | 25 | 1.6 | 1.6 | 0.386 | 0.35 | 0.49 |
| PtRu/CXG-COLTT200 | 25 | 1.6 | 1.8 | 0.387 | 0.39 | 0.37 |
| PtRu/CXG-COLTT400 | 25 | 1.6 | 2.0 | 0.385 | 0.43 | 0.50 |

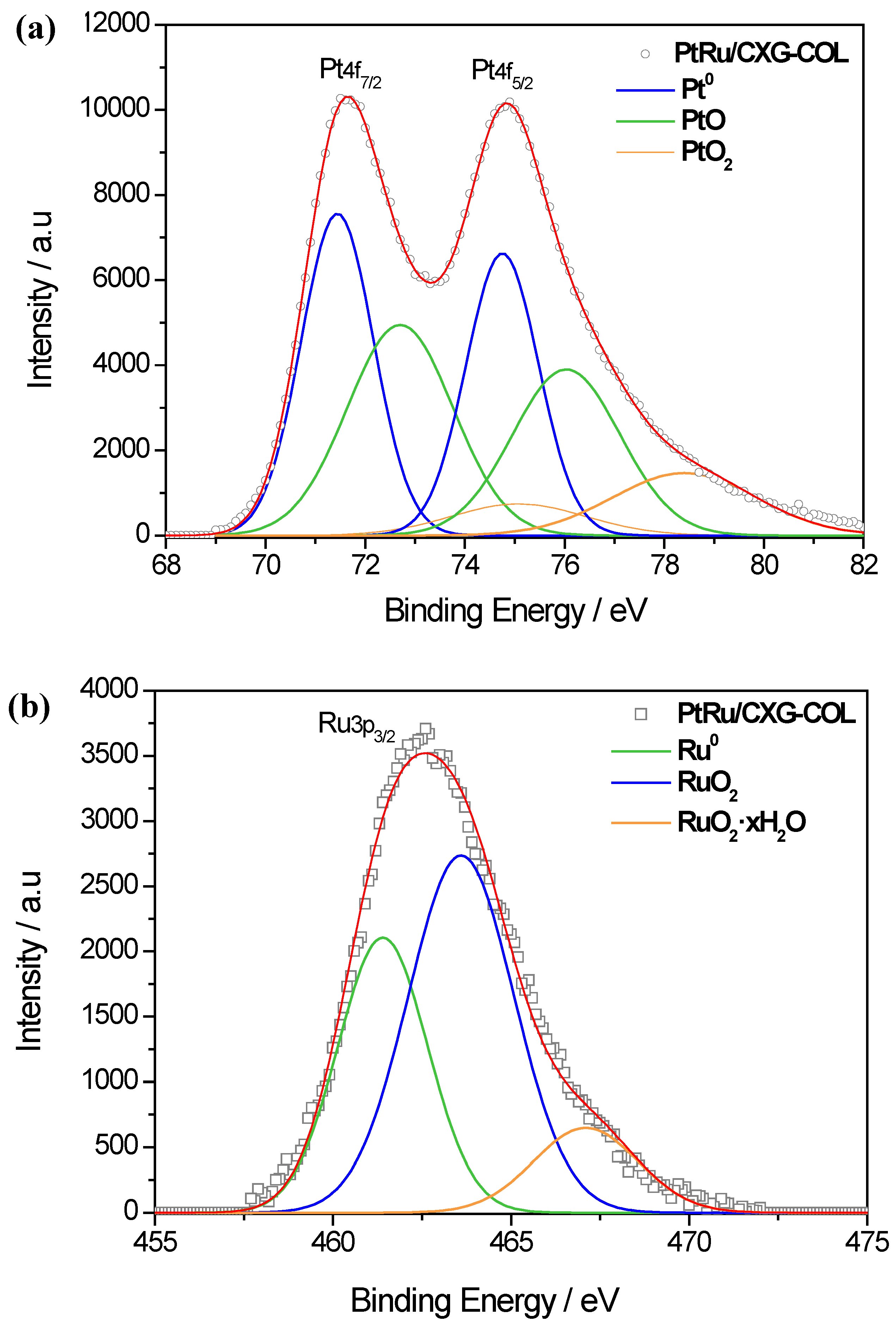
| Sample | Pt 4f7/2 | Ru 3p3/2 | Pt/Ru | ||
|---|---|---|---|---|---|
| Species | Intensity (%) | Species | Intensity (%) | ||
| PtRu/CXG- COL | Pt | 46.8 | Ru | 34.6 | 2.2 |
| PtO | 44.7 | RuO2 | 53.0 | ||
| PtO2 | 8.5 | RuO2·xH2O | 12.4 | ||
| PtRu/CXG-COL-TT200 | Pt | 54.2 | Ru | 38.2 | 2.2 |
| PtO | 36.6 | RuO2 | 50.2 | ||
| PtO2 | 9.2 | RuO2·xH2O | 11.6 | ||
| PtRu/CXG-COL-TT400 | Pt | 63.9 | Ru | 48.1 | 1.9 |
| PtO | 21.1 | RuO2 | 44 | ||
| PtO2 | 15 | RuO2·xH2O | 7.9 | ||
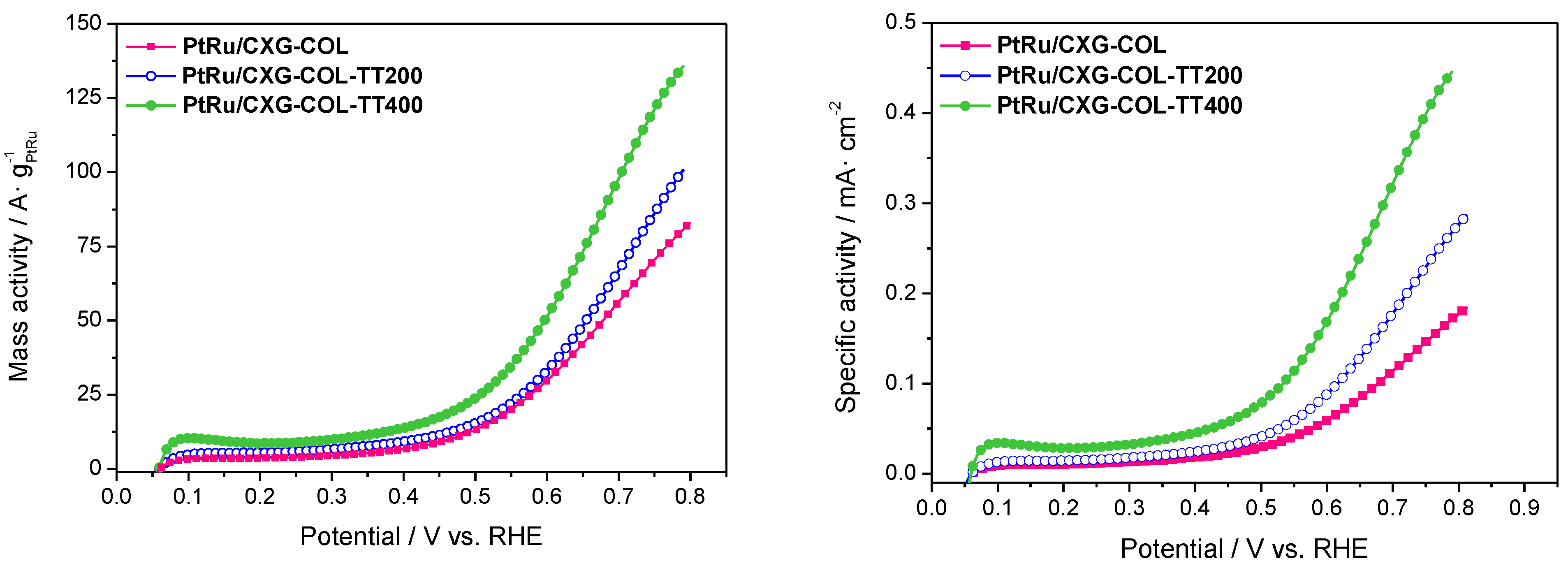
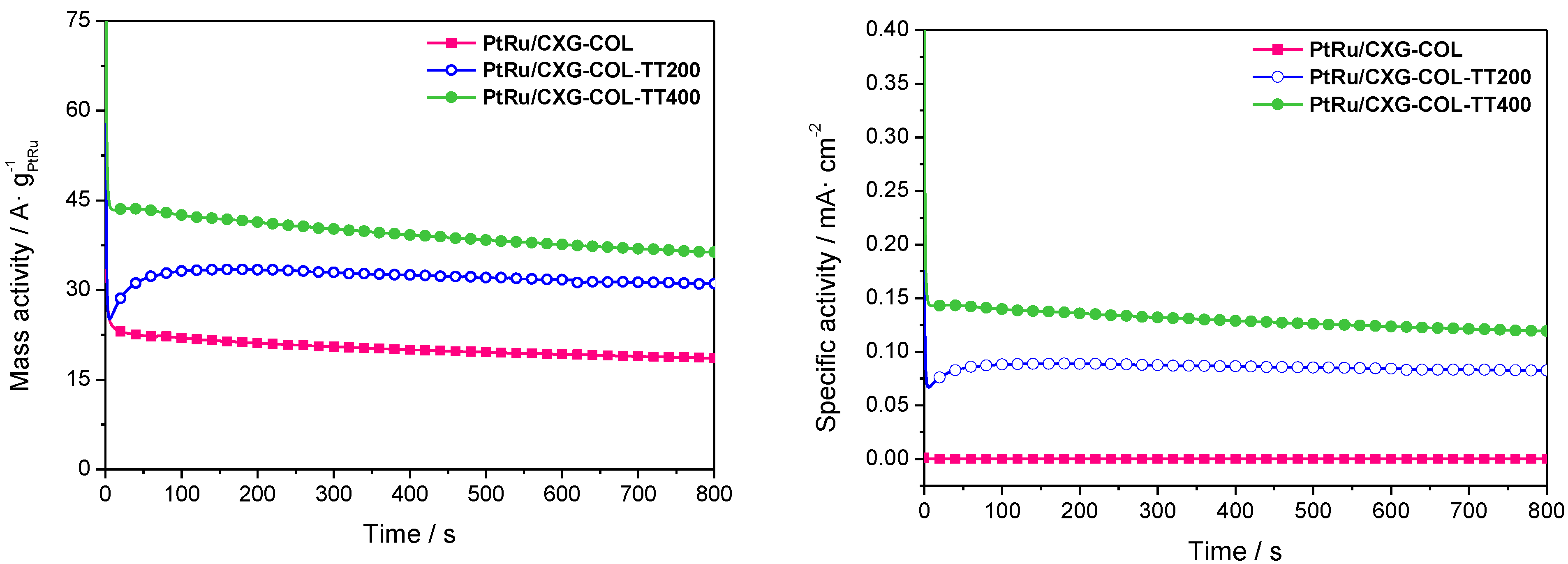
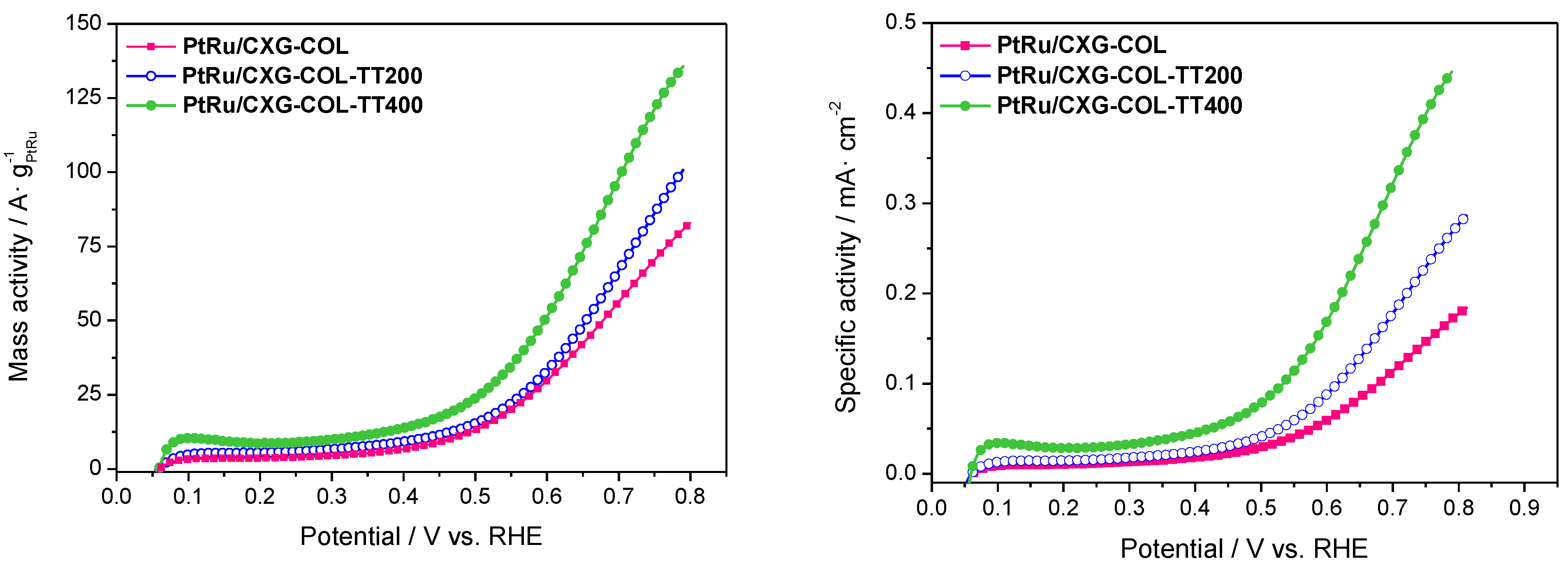
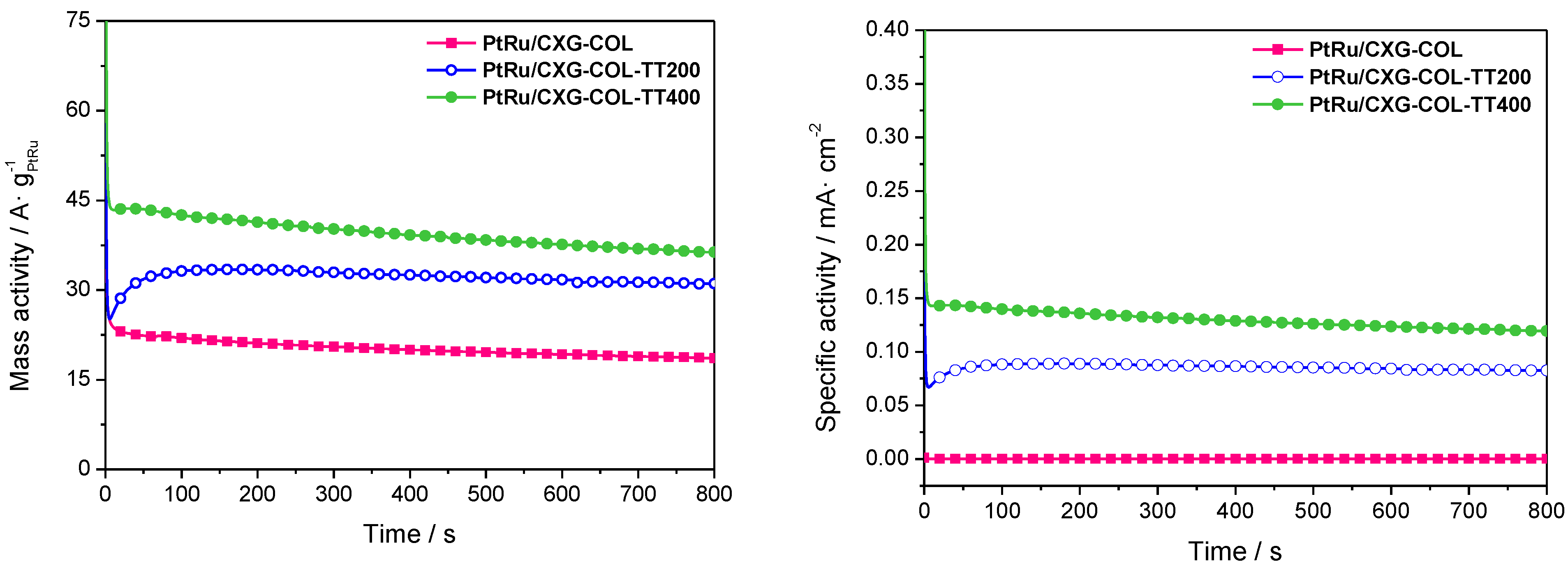
2.3. Catalytic Activity towards MOR
| Sample | ECSA/m2·g−1 PtRu |
|---|---|
| PtRu/CXG-COL | 56.9 |
| PtRu/CXG-COLTT200 | 35.9 |
| PtRu/CXG-COLTT400 | 30.4 |
3. Experimental Section
3.1. Carbon Xerogel Synthesis
3.2. Catalysts Preparation
3.3. Physico-Chemical Characterization
3.4. Electrochemical Experiments
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Aricó, A.S.; Baglio, V.; Antonucci, V. Direct Methanol Fuel Cells; Nova Publishers: New York, NY, USA, 2010. [Google Scholar]
- Antolini, E. Effect of the Structural Characteristics of Binary Pt–Ru and Ternary Pt–Ru–M Fuel Cell Catalysts on the Activity of Ethanol Electrooxidation in Acid Medium. ChemSusChem. [CrossRef]
- Petrii, O.A. Pt-Ru electrocatalysts for fuel cells: A representative review. J. Solid State Electrochem. 2008, 12, 609–642. [Google Scholar] [CrossRef]
- Antolini, E. Formation of carbon-supported PtM alloys for low temperature fuel cells: A review. Mater. Chem. Phys. 2003, 78, 563–573. [Google Scholar] [CrossRef]
- Viva, F.A.; Bruno, M.M.; Jobbagy, M.; Corti, H.R. Electrochemical Characterization of PtRu Nanoparticles Supported on Mesoporous Carbon for Methanol Electrooxidation. J. Phys. Chem. C 2012, 116, 4097–4104. [Google Scholar]
- Sharma, S.; Pollet, B.G. Support materials for PEMFC and DMFC electrocatalysts—A review. J. Power Sources 2012, 208, 96–119. [Google Scholar] [CrossRef]
- Monteverde Videla, A.H.A.; Zhang, L.; Kim, J.; Zeng, J.; Francia, C.; Zhang, J.; Specchia, S. Mesoporous carbons supported non-noble metal Fe-N X electrocatalysts for PEM fuel cell oxygen reduction reaction. J. Appl. Electrochem. 2013, 43, 159–169. [Google Scholar] [CrossRef]
- Baglio, V.; di Blasi, A.; D’Urso, C.; Antonucci, V.; Aricò, A.S.; Ornelas, R.; Morales-Acosta, D.; Ledesma-Garcia, J.; Godinez, L.A.; Arriaga, L.G.; et al. Development of Pt and Pt -Fe Catalysts Supported on Multiwalled Carbon Nanotubes for Oxygen Reduction in Direct Methanol Fuel Cells. J. Electrochem. Soc. 2008, 155, B829–B833. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, B.J.; Lee, S.Y. Effect of surface modification of mesoporous carbon supports on the electrochemical activity of fuel cells. J. Colloid Interface Sci. 2013, 405, 150–156. [Google Scholar] [CrossRef]
- Cui, Z.; Liu, C.; Liao, J.; Xing, W. Highly active PtRu catalysts supported on carbon nanotubes prepared by modified impregnation method for methanol electro-oxidation. Electrochim. Acta 2008, 53, 7807–7811. [Google Scholar] [CrossRef]
- Zhou, W.J.; Li, W.Z.; Song, S.Q.; Zhou, Z.H.; Jiang, L.H.; Sun, G.Q.; Xin, Q.; Poulianitis, K.; Kontou, S.; Tsiakaras, P. Bi- and tri-metallic Pt-based anode catalysts for direct ethanol fuel cells. J. Power Sources 2004, 131, 217–223. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Serp, P.; Kalck, P.; Samant, P.V.; Fernandes, J.B. Development of carbon nanotube and carbon xerogel supported catalysts for the electro-oxidation of methanol in fuel cells. Carbon 2006, 44, 2516–2522. [Google Scholar] [CrossRef]
- Yu, X.; Ye, S. Recent advances in activity and durability enhancement of Pt/C catalytic cathode in PEMFC: Part I. Physico-chemical and electronic interaction between Pt and carbon support, and activity enhancement of Pt/C catalyst. J. Power Sources 2007, 172, 133–144. [Google Scholar] [CrossRef]
- Kim, M.; Park, J.N.; Kim, H.; Song, S.; Lee, W.H. The preparation of Pt/C catalysts using various carbon materials for the cathode of PEMFC. J. Power Sources 2006, 163, 93–97. [Google Scholar] [CrossRef]
- Job, N.; Chatenet, M.; Berthon-Fabry, S.; Hermans, S.; Maillard, F. Efficient Pt/carbon electrocatalysts for proton exchange membrane fuel cells: Avoid chloride-based Pt salts! J. Power Sources 2013, 240, 294–305. [Google Scholar] [CrossRef]
- Arbizzani, C.; Beninati, S.; Soavi, F.; Varzi, A.; Mastragostino, M. Supported PtRu on mesoporous carbons for direct methanol fuel cells. J. Power Sources 2008, 185, 615–620. [Google Scholar] [CrossRef]
- Job, N.; Lambert, S.; Chatenet, M.; Gommes, C.J.; Maillard, F.; Berthon-Fabry, S.; Regalbuto, J.R.; Pirard, J.P. Preparation of highly loaded Pt/carbon xerogel catalysts for Proton Exchange Membrane fuel cells by the Strong Electrostatic Adsorption method. Catal. Today 2010, 150, 119–127. [Google Scholar] [CrossRef]
- Arbizzani, C.; Beninati, S.; Manferrari, E.; Soavi, F.; Mastragostino, M. Electrodeposited PtRu on cryogel carbon–Nafion supports for DMFC anode. J. Power Sources 2006, 161, 826–830. [Google Scholar] [CrossRef]
- Alegre, C.; Calvillo, L.; Moliner, R.; González-Expósito, J.A.; Guillén-Villafuerte, O.; Huerta, M.V.M.; Pastor, E.; Lázaro, M.J. Pt and PtRu electrocatalysts supported on carbon xerogels for direct methanol fuel cells. J. Power Sources 2011, 196, 4226–4235. [Google Scholar] [CrossRef]
- Alegre, C.; Gálvez, M.E.; Baquedano, E.; Pastor, E.; Moliner, R.; Lázaro, M.J. Influence of support’s oxygen functionalization on the activity of Pt/carbon xerogels catalysts for methanol electro-oxidation. Int. J. Hydrogen Energy 2012, 37, 7180–7191. [Google Scholar]
- Alegre, C.; Gálvez, M.E.; Baquedano, E.; Moliner, R.; Pastor, E.; Lázaro, M.J. Oxygen-Functionalized Highly Mesoporous Carbon Xerogel Based Catalysts for Direct Methanol Fuel Cell Anodes. J. Phys. Chem. C 2013, 117, 13045–13058. [Google Scholar]
- Antolini, E.; Cardellini, F. Formation of carbon supported PtRu alloys: An XRD analysis. J. Alloys Compd. 2001, 315, 118–122. [Google Scholar] [CrossRef]
- De la Fuente, J.L.G.; Martínez-Huerta, M.V.; Rojas, S.; Fierro, J.L.G.; Peña, M.A. Methanol electrooxidation on PtRu nanoparticles supported on functionalised carbon black. Catal. Today 2006, 116, 422–432. [Google Scholar] [CrossRef]
- Frelink, T.; Visscher, W.; van Veen, J.A.R. Particle size effect of carbon-supported platinum catalysts for the electrooxidation of methanol. J. Electroanal. Chem. 1995, 382, 65–72. [Google Scholar] [CrossRef]
- Chrzanowski, W.; Wieckowski, A. Surface Structure Effects in Platinum/Ruthenium Methanol Oxidation Electrocatalysis. Langmuir 1998, 14, 1967–1970. [Google Scholar] [CrossRef]
- Garcia, G.; Baglio, V.; Stassi, A.; Pastor, E.; Antonucci, V.; Aricò, A.S. Investigation of Pt–Ru nanoparticle catalysts for low temperature methanol electro-oxidation. J. Solid State Electrochem. 2007, 11, 1229–1238. [Google Scholar] [CrossRef]
- Alegre, C.; Sebastián, D.; Baquedano, E.; Gálvez, M.E.; Moliner, R.; Lázaro, M.J. Tailoring Synthesis Conditions of Carbon Xerogels towards Their Utilization as Pt-Catalyst Supports for Oxygen Reduction Reaction (ORR). Catalysts 2012, 2, 466–489. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Alegre, C.; Sebastián, D.; Gálvez, M.E.; Moliner, R.; Stassi, A.; Aricò, A.S.; Lázaro, M.J.; Baglio, V. PtRu Nanoparticles Deposited by the Sulfite Complex Method on Highly Porous Carbon Xerogels: Effect of the Thermal Treatment. Catalysts 2013, 3, 744-756. https://doi.org/10.3390/catal3030744
Alegre C, Sebastián D, Gálvez ME, Moliner R, Stassi A, Aricò AS, Lázaro MJ, Baglio V. PtRu Nanoparticles Deposited by the Sulfite Complex Method on Highly Porous Carbon Xerogels: Effect of the Thermal Treatment. Catalysts. 2013; 3(3):744-756. https://doi.org/10.3390/catal3030744
Chicago/Turabian StyleAlegre, Cinthia, David Sebastián, María Elena Gálvez, Rafael Moliner, Alessandro Stassi, Antonino Salvatore Aricò, María Jesús Lázaro, and Vincenzo Baglio. 2013. "PtRu Nanoparticles Deposited by the Sulfite Complex Method on Highly Porous Carbon Xerogels: Effect of the Thermal Treatment" Catalysts 3, no. 3: 744-756. https://doi.org/10.3390/catal3030744
APA StyleAlegre, C., Sebastián, D., Gálvez, M. E., Moliner, R., Stassi, A., Aricò, A. S., Lázaro, M. J., & Baglio, V. (2013). PtRu Nanoparticles Deposited by the Sulfite Complex Method on Highly Porous Carbon Xerogels: Effect of the Thermal Treatment. Catalysts, 3(3), 744-756. https://doi.org/10.3390/catal3030744