Nanostructured Carbon Materials as Supports in the Preparation of Direct Methanol Fuel Cell Electrocatalysts


 ,
, 



Abstract
:1. Introduction
2. Results and Discussion
2.1. Carbon Materials Characterization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SBET (m2 g−1) | Vtotal (cm3 g−1) | SMeso (m2 g−1) | VMeso (cm3 g−1) | |
|---|---|---|---|---|
| CNF | 101 | 0.38 | 91 | 0.38 |
| CNC | 124 | 0.16 | 124 | 0.16 |
| CXG | 441 | 1.62 | 251 | 1.53 |

| CO2 (mmol g−1) | CO (mmol g−1) | O content (wt.%) | |
|---|---|---|---|
| CNF | 0.32 | 1.07 | 2.7 |
| CNC | 0.85 | 2.34 | 6.5 |
| CXG | 0.99 | 4.45 | 10.3 |
| Pt content (wt.%) | Crystallite size (nm) | |
|---|---|---|
| Pt/CNF | 19.2 | 6.8 |
| Pt/CNC | 20.0 | 4.7 |
| Pt/CXG | 21.0 | 4.1 |
| PtRu content (wt.%) | Atomic ratio Pt:Ru | Crystallite size (nm) | |
|---|---|---|---|
| PtRu/CNF | 21.0 | 47:53 | 2.3 |
| PtRu/CNC | 17.3 | 66:34 | 3.9 |
| PtRu/CXG | 24.0 | 50:50 | 2.5 |
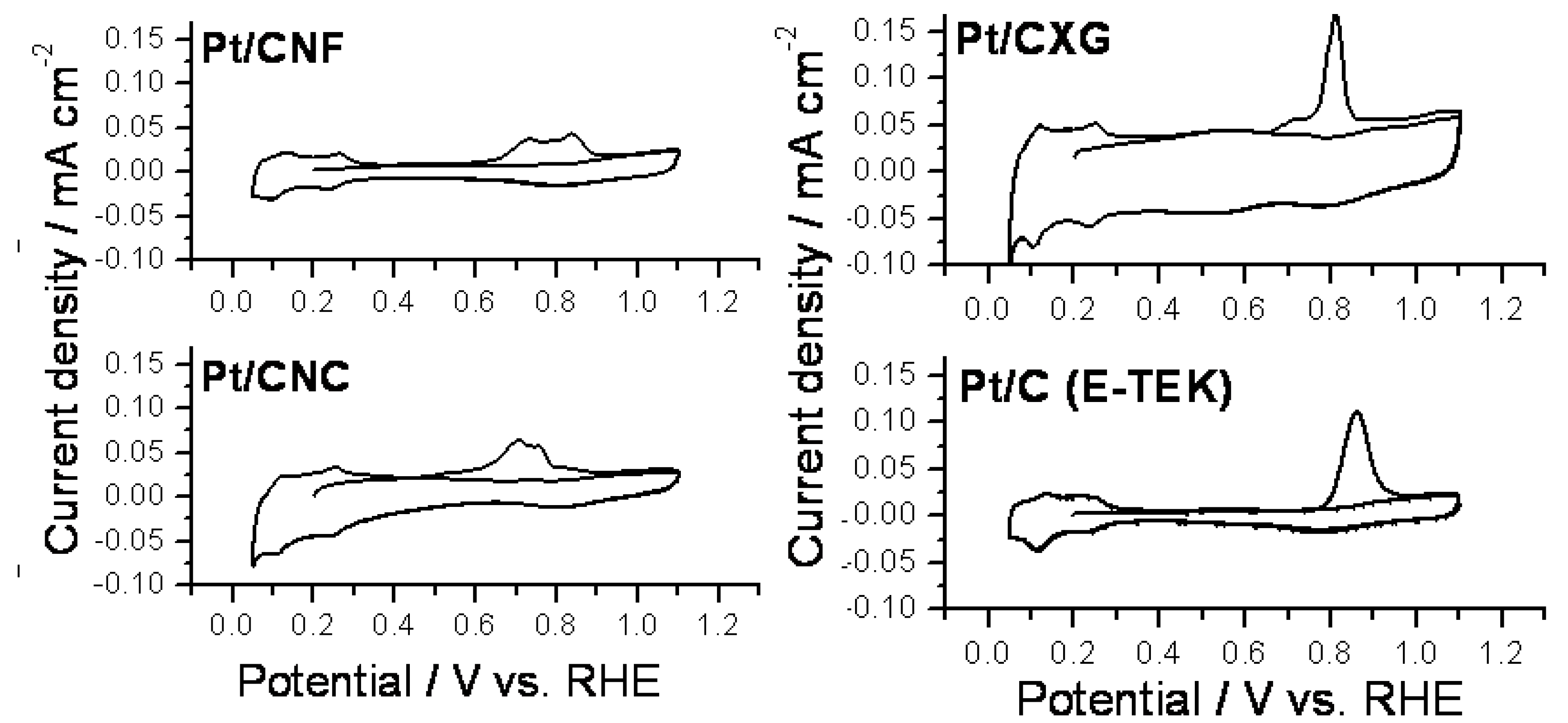
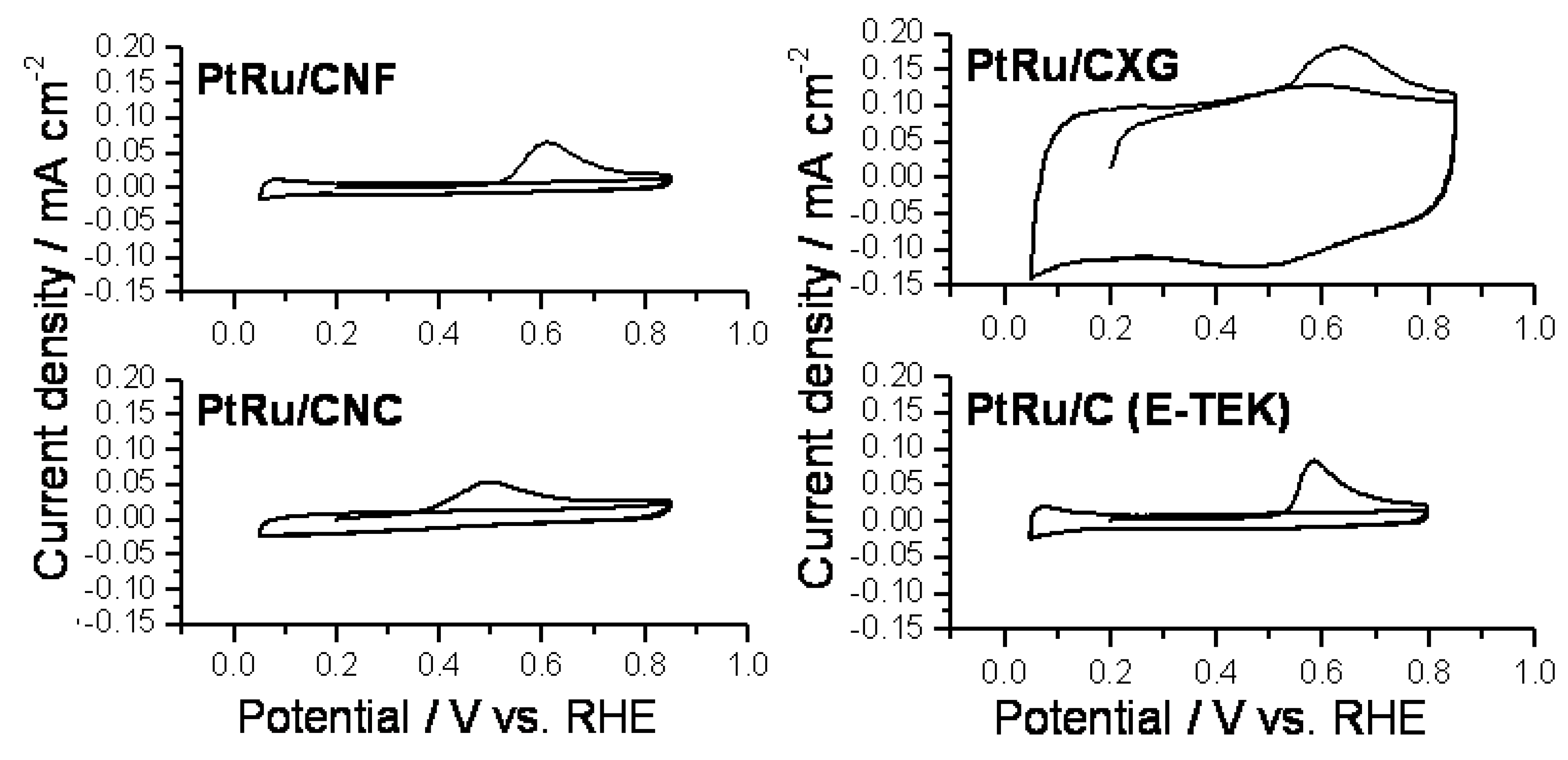
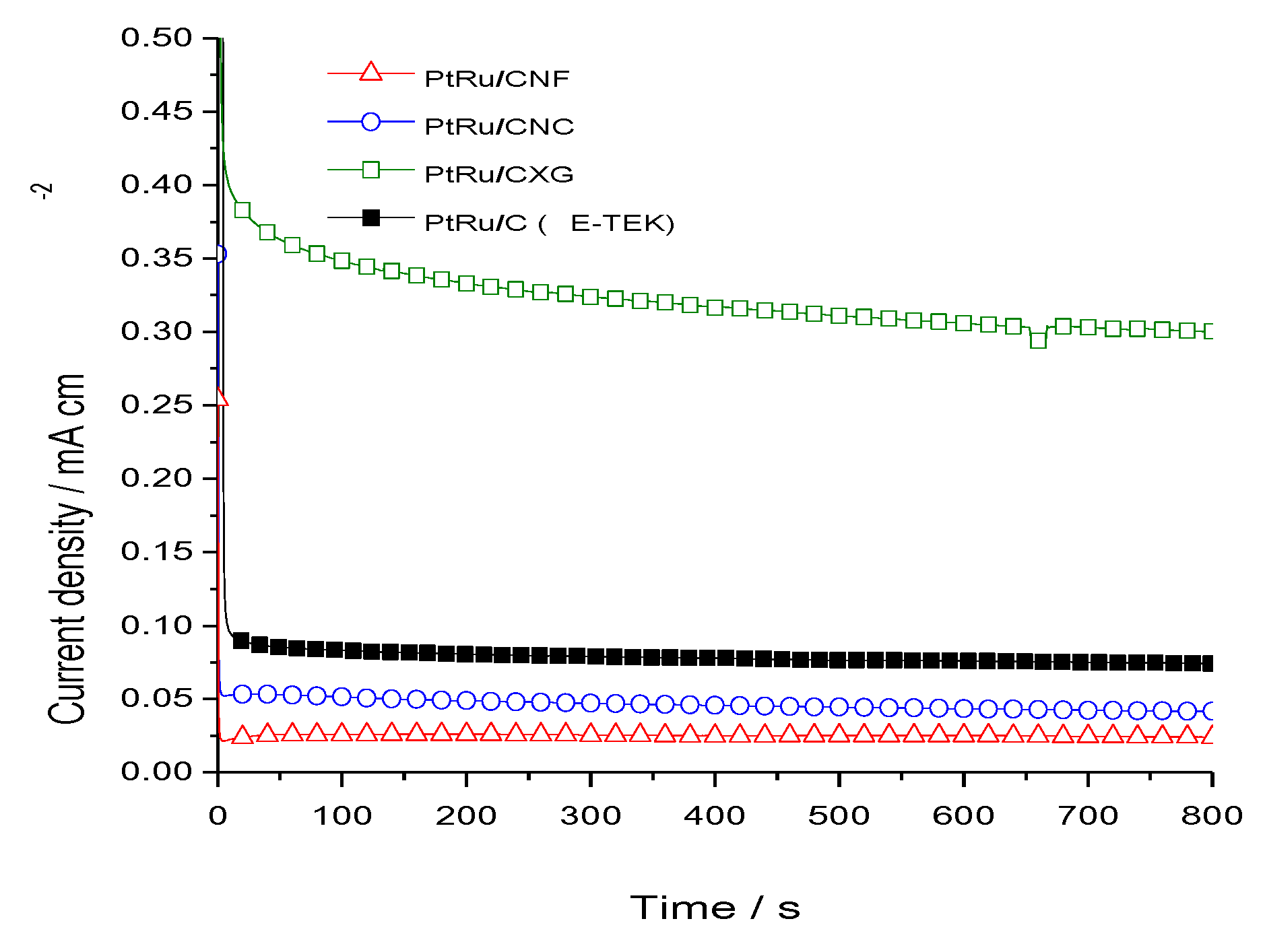
2.2. Electrochemical Characterization and Activity
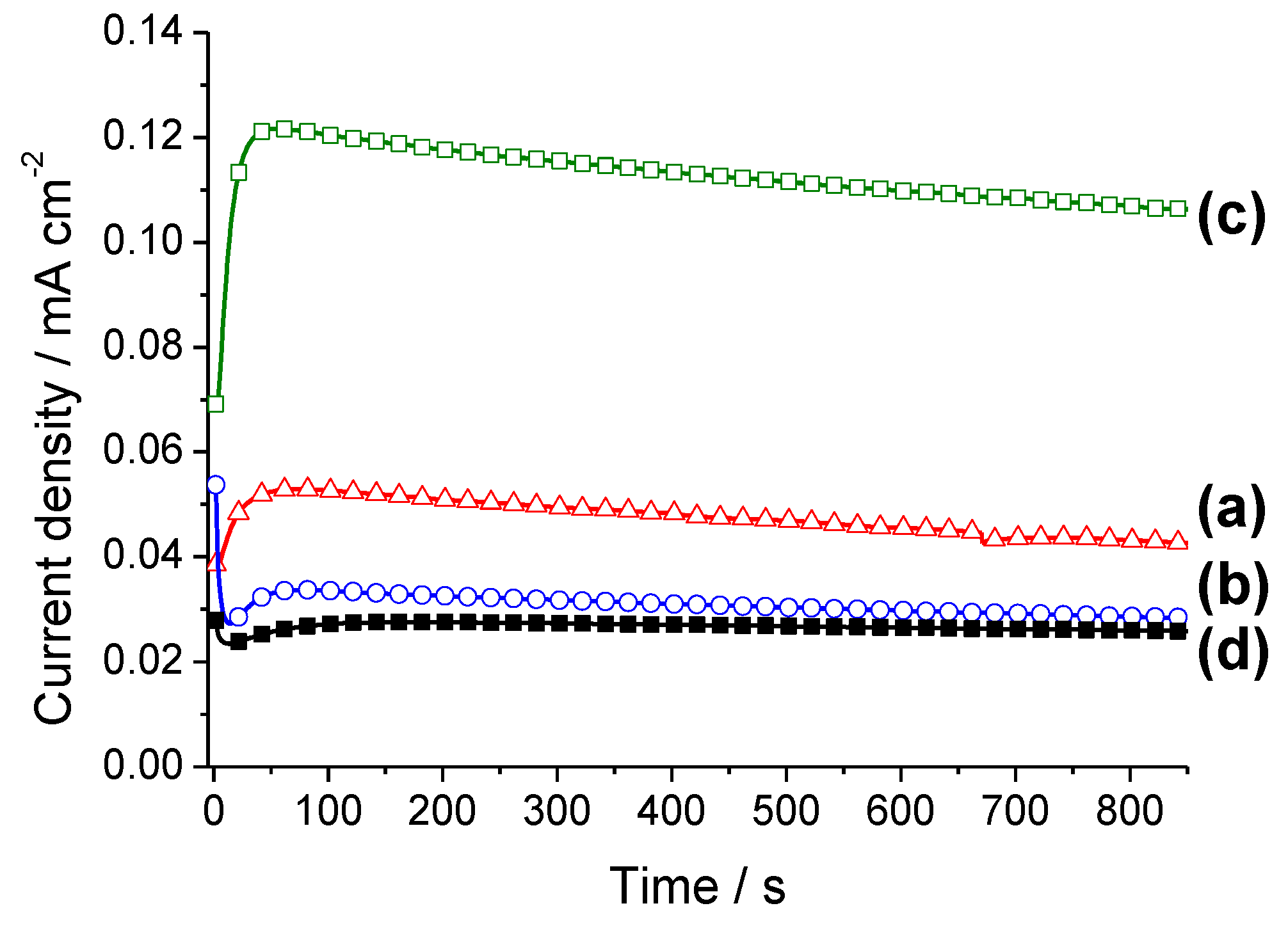
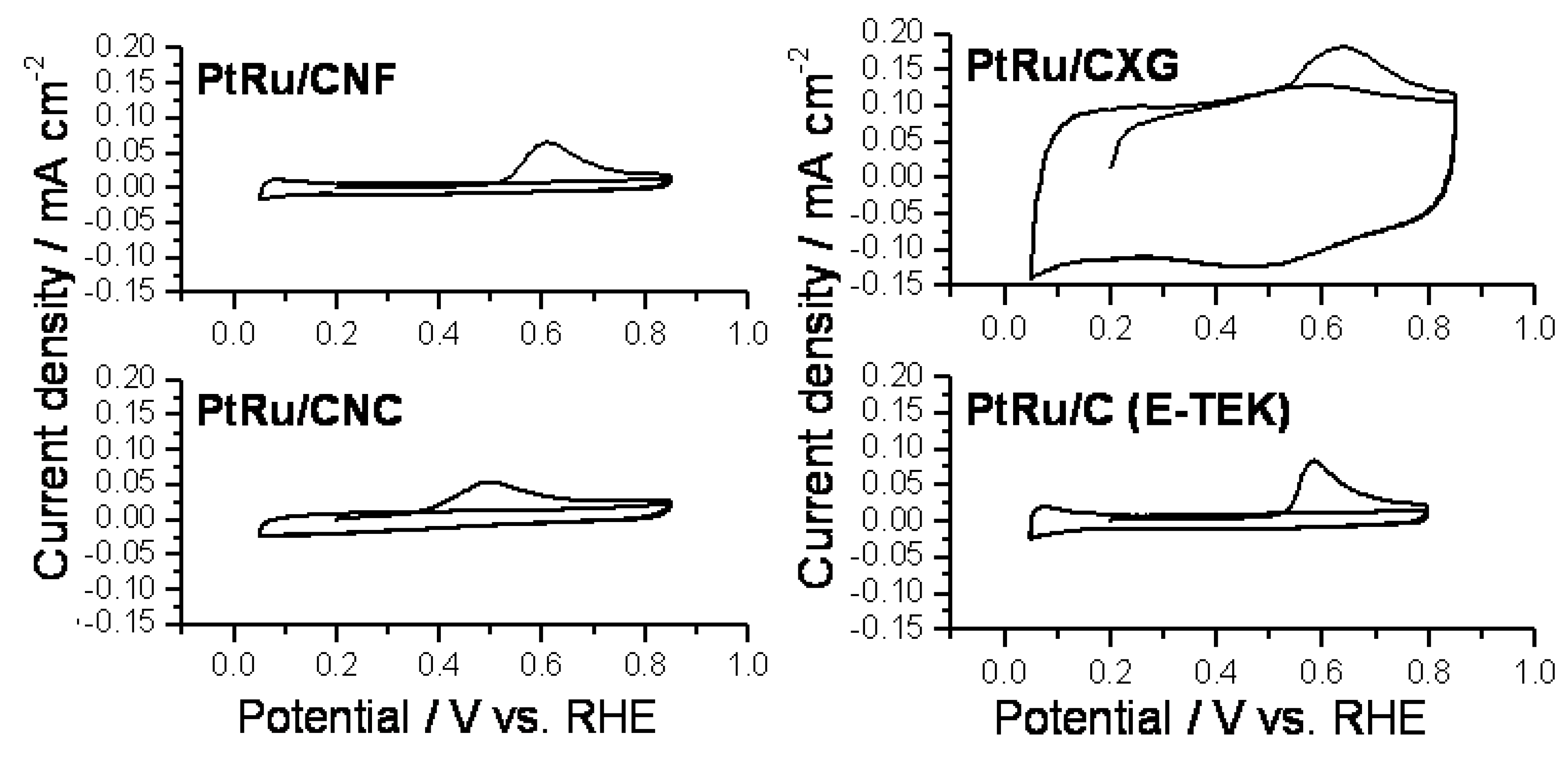
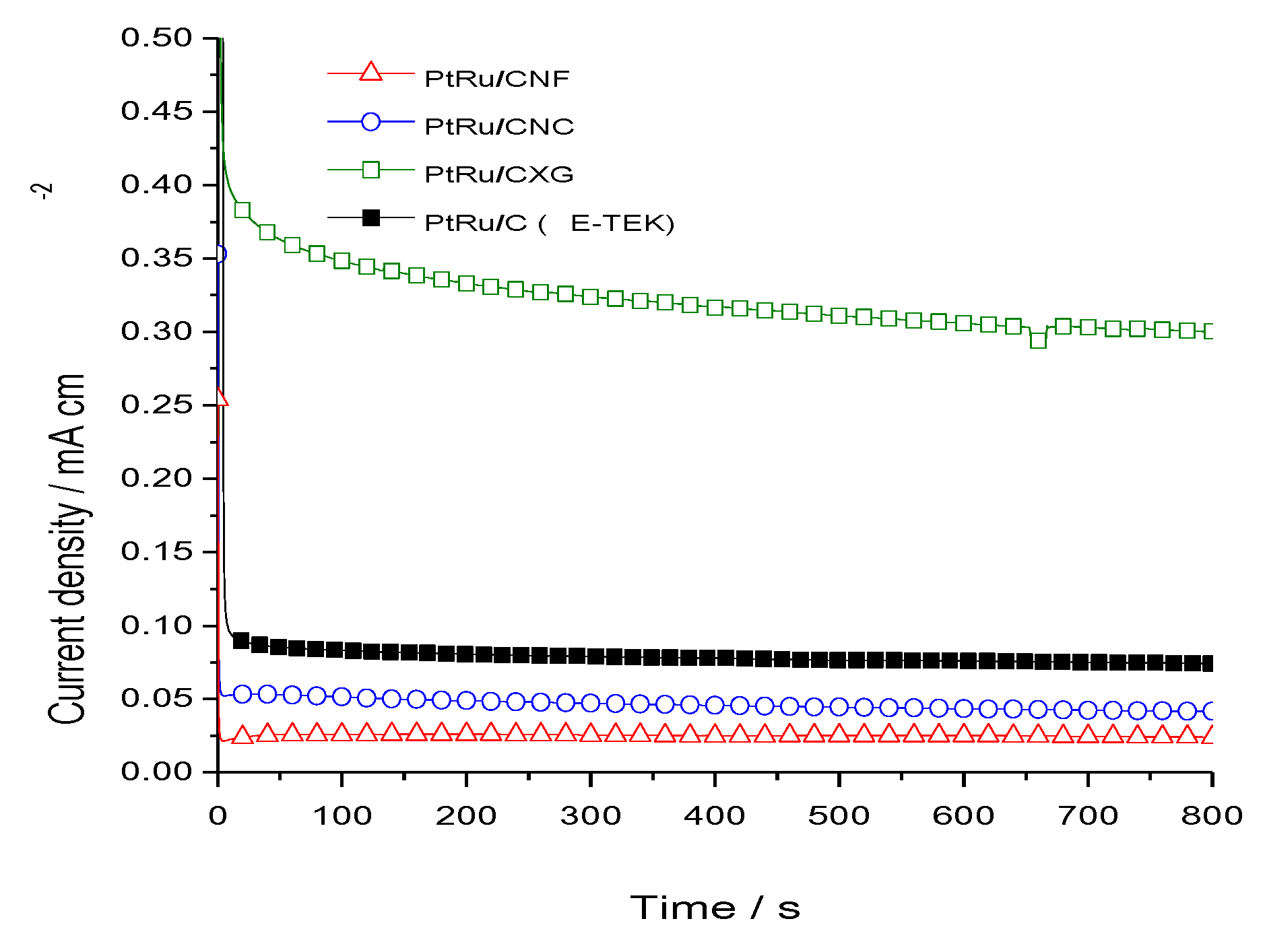
3. Experimental Section
3.1. Synthesis of the Carbon Materials
3.2. Catalysts Preparation
3.3. Physico-Chemical Characterization
3.4. Electrochemical Characterization and Activity
4. Conclusions
Acknowledgements
Conflicts of Interest
References
- Aricò, A.S.; Srinivasan, S.; Antonucci, V. DMFCs: From fundamentals aspects to technology development. Fuel Cells 2001, 1, 133–161. [Google Scholar] [CrossRef]
- Liu, H.; Song, C.; Zhang, L.; Zhang, J.; Wang, H.; Wilkinson, D.P. A review of anode catalysis in the direct methanol fuel cell. J. Power Sources 2006, 155, 95–110. [Google Scholar] [CrossRef]
- Stassi, A.; Gatto, I.; Baglio, V.; Passalacqua, E.; Aricò, A.S. Oxide-supported PtCo alloy catalyst for intermediate temperature polymer electrolyte fuel cells. Appl. Catal. B 2013, 142–143, 15–24. [Google Scholar] [CrossRef]
- Zeng, J.; Francia, C.; Gerbaldi, C.; Baglio, V.; Specchia, S.; Aricò, A.S.; Spinelli, P. Hybrid ordered mesoporous carbons doped with tungsten trioxide as supports for Pt electrocatalysts for methanol oxidation reaction. Electrochim. Acta 2013, 94, 80–91. [Google Scholar] [CrossRef]
- Acres, G.J.K.; Frost, J.C.; Hards, G.A.; Potter, G.A.; Ralph, T.R.; Thompsett, D.; Burstein, G.T.; Hutchings, G.J. Electrocatalysts for fuel cells. Catal. Today 1997, 38, 393–400. [Google Scholar] [CrossRef]
- Sieben, J.M.; Duarte, M.M.E. Methanol, ethanol and ethylene glycol electro-oxidation at Pt and Pt-Ru catalysts electrodeposited over oxidized carbon nanotubes. Int. J. Hydrogen Energy 2012, 37, 9941–9947. [Google Scholar] [CrossRef]
- Wang, Z.B.; Yin, G.P.; Zhang, J.; Sun, Y.C.; Shi, P.F. Co-catalytic effect of Ni in the methanol electro-oxidation on Pt-Ru/C catalyst for direct methanol fuel cell. Electrochim. Acta 2006, 51, 5691–5697. [Google Scholar] [CrossRef]
- Liu, Z.; Ling, X.Y.; Su, X.; Lee, J.Y. Carbon-supported Pt and PtRu nanoparticles as catalysts for a direct methanol fuel cell. J. Phys. Chem. B 2004, 108, 8234–8240. [Google Scholar] [CrossRef]
- Chang, W.-C.; Nguyen, M.T. Investigations of a platinum–ruthenium/carbon nanotube catalyst formed by a two-step spontaneous deposition method. J. Power Sources 2011, 196, 5811–5816. [Google Scholar] [CrossRef]
- Tsuji, M.; Kubokawa, M.; Yano, R.; Miyamae, N.; Tsuji, T.; Jun, M.S.; Hong, S.; Lim, S.; Yoon, S.H.; Mochida, I. Fast preparation of PtRu catalysts supported on carbon nanofibers by the microwave-polyol method and their application to fuel cells. Langmuir 2007, 23, 387–390. [Google Scholar] [CrossRef]
- Lázaro, M.J.; Celorrio, V.; Calvillo, L.; Pastor, E.; Moliner, R. Influence of the synthesis method on the properties of Pt catalysts supported on carbon nanocoils for ethanol oxidation. J. Power Sources 2011, 196, 4236–4241. [Google Scholar] [CrossRef]
- Suelves, I.; Lázaro, M.J.; Moliner, R.; Echegoyen, Y.; Palacios, J.M. Characterization of NiAl and NiCuAl catalysts prepared by different methods for hydrogen production by thermo catalytic decomposition of methane. Catal. Today 2006, 116, 271–280. [Google Scholar] [CrossRef]
- Lázaro, M.J.; Sebastián, D.; Suelves, I.; Moliner, R. Carbon nanofiber growth optimization for their use as electrocatalyst support in proton exchange membrane (PEM) fuel cells. J. Nanosci. Nanotechnol. 2009, 9, 4353–4359. [Google Scholar] [CrossRef]
- Sebastián, D.; Calderón, J.C.; González-Expósito, J.A.; Pastor, E.; Martínez-Huerta, M.V.; Suelves, I.; Moliner, R.; Lázaro, M.J. Influence of carbon nanofibers properties as electrocatalyst support on the electrochemical performance for PEM fuel cells. Int. J. Hydrogen Energy 2010, 35, 9934–9942. [Google Scholar] [CrossRef]
- Celorrio, V.; Calvillo, L.; Martínez-Huerta, M.V.; Moliner, R.; Lázaro, M.J. Study of the Synthesis Conditions of Carbon Nanocoils for Energetic Applications. Energy Fuels 2010, 24, 3361–3365. [Google Scholar] [CrossRef]
- Celorrio, V.; Calvillo, L.; Pérez-Rodríguez, S.; Lázaro, M.J.; Moliner, R. Modification of the properties of carbon nanocoils by different treatments in liquid phase. Micropor. Mesopor. Mat. 2011, 142, 55–61. [Google Scholar] [CrossRef]
- Alegre, C.; Gálvez, M.E.; Baquedano, E.; Pastor, E.; Moliner, R.; Lázaro, M.J. Influence of support’s oxygen functionalization on the activity of Pt/Carbon xerogels catalysts for methanol electro-oxidation. Int. J. Hydrogen Energy 2012, 37, 7180–7191. [Google Scholar] [CrossRef]
- Salgado, J.R.C.; Antolini, E.; González, E.R. Structure and activity of carbon-supported Pt−Co electrocatalysts for oxygen reduction. J. Phys. Chem. B 2004, 108, 17767–17774. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gálvez, M.E.; Calvillo, L.; Alegre, C.; Sebastián, D.; Suelves, I.; Pérez-Rodríguez, S.; Celorrio, V.; Pastor, E.; Pardo, J.I.; Moliner, R.; et al. Nanostructured Carbon Materials as Supports in the Preparation of Direct Methanol Fuel Cell Electrocatalysts. Catalysts 2013, 3, 671-682. https://doi.org/10.3390/catal3030671
Gálvez ME, Calvillo L, Alegre C, Sebastián D, Suelves I, Pérez-Rodríguez S, Celorrio V, Pastor E, Pardo JI, Moliner R, et al. Nanostructured Carbon Materials as Supports in the Preparation of Direct Methanol Fuel Cell Electrocatalysts. Catalysts. 2013; 3(3):671-682. https://doi.org/10.3390/catal3030671
Chicago/Turabian StyleGálvez, María Elena, Laura Calvillo, Cinthia Alegre, David Sebastián, Isabel Suelves, Sara Pérez-Rodríguez, Verónica Celorrio, Elena Pastor, Juan Ignacio Pardo, Rafael Moliner, and et al. 2013. "Nanostructured Carbon Materials as Supports in the Preparation of Direct Methanol Fuel Cell Electrocatalysts" Catalysts 3, no. 3: 671-682. https://doi.org/10.3390/catal3030671