1. Introduction
Synthetic macrocycles that are capable of interacting with small anionic species present a new area of study in organic chemistry, and the calixpyrroles are an important supramolecular subclass of this field. Calixpyrroles (
Figure 1) are interesting from a synthetic perspective for their use in the selective recognition of anions [
1]. Although the structural features of these compounds are best known for their ability to specifically recognize anions, appropriately functionalized calixpyrroles can recognize ionic pairs by binding to both the anion and the counter cation [
2].
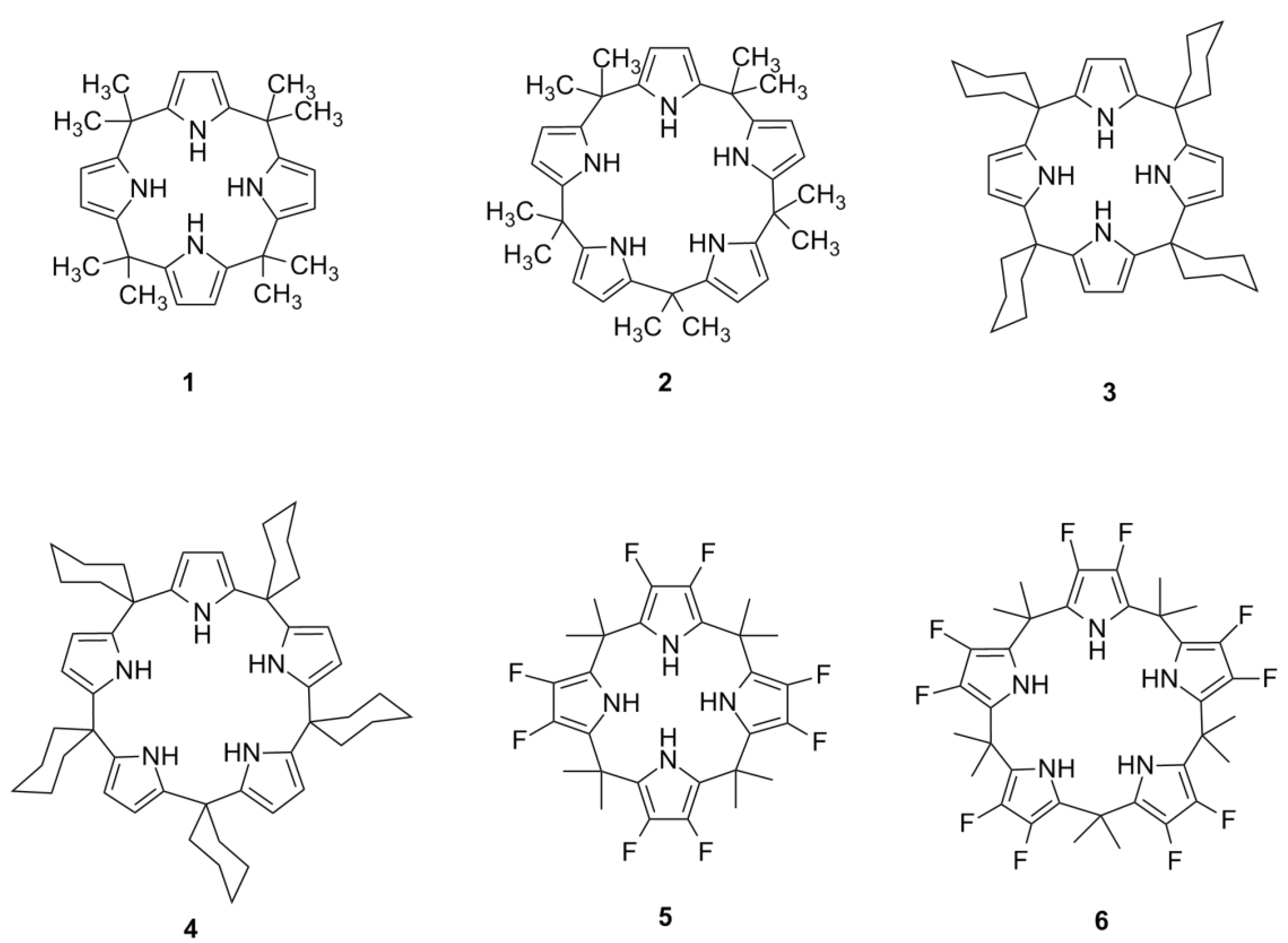
Figure 1.
Example of calix[4]pyrroles and calix[5]pyrroles.
Figure 1.
Example of calix[4]pyrroles and calix[5]pyrroles.
Calixpyrroles can act as hydrogen bond-donating organocatalysts through a mechanism similar to that used by other hydrogen bond-donating compounds, such as TADDOLs [
3]. Hydrogen bond-donating catalysts are used in a variety of synthetic transformations, including hetero-Diels–Alder, regioselective alkylation and acylation reactions, as well as vinylogous addition reactions of 2-trimethylsilyloxyfuran to aldehydes to afford γ-hydroxybutenolide products [
4].
Calixpyrroles are classified according to the number of pyrrolic subunits in the macrocycle. The most widely studied are the calix[4]pyrroles, largely due to their ease of synthesis [
5]. During the condensation of ketones with pyrrole, expanded calixpyrroles are also formed [
6]. Among the calixpyrroles that have been explored previously, the calix[5]pyrroles are the most elusive from a synthetic point of view and, therefore, the least explored in terms of anion recognition and other applications [
7,
8,
9,
10,
11].
The first synthetic calix[5]pyrrole was obtained by covalently binding a macrocycle to a calixarene using the product calix[5]arene as a template [
12]. A later approach described the synthesis of a calix[5]pyrrole by the reaction of 3,4-difluoropyrrole with acetone, leading to decafluorocalix[5]pyrrole
6 (35% yield), which was stable under the reaction conditions [
11]. The authors argued that
6 was difficult to obtain because the parent octafluorocalix[4]pyrrole
5 was the thermodynamic (stable) product and
6 was the kinetic (unstable) product. In 2002, Kohnke
et al. reported the first synthesis (an indirect synthesis) of a β-unsubstituted calix[5]pyrrole
2 from a calix[5] furan in a 1% yield [
9]. Recently, we described the first synthesis of
2 by a direct condensation of pyrrole with acetone [
7]. This surprising approach was stumbled upon by chance during an exploration of optimal conditions for the synthesis of calix[4]pyrroles using Lewis acids, particularly Bi(NO
3)
3 [
13]. By decreasing the concentration of the Lewis acid, the yield of
2 was found to increase with respect to the yield of
1, to a maximum ratio of 3:1. The same methodology was later applied to the condensation of pyrrole with cyclohexanone, which led to a new calix[5]pyrrole, pentaspirocyclohexyl calix[5]pyrrole
4, demonstrating that calix[5]pyrroles could be obtained by a direct condensation [
8].
Several important observations were made regarding the synthesis of the calix[5]pyrroles: 1. The decamethyl and pentaspirocyclohexyl calix[5]pyrroles,
2 and
4 could be synthesized directly via condensation of pyrrole with the corresponding ketone using 0.3 mol% Bi(NO
3)
3 at room temperature [
7,
8]; 2. Increasing the concentration of a Lewis acid (>0.5 mol%) favored formation of only calix[4]pyrrole [
7,
13]; 3. At low concentrations of Lewis acid (<0.1 mol%), macrocycle formation was not detected [
7]; 4. Unlike
2, compound
6 could be obtained directly via condensation of 3,4-difluoropyrrole with acetone under the conditions used to obtain
1, that is, using MeSO
3H as a catalyst [
11]; 5. All of
2 [
7,
9],
4 [
8] and
6 [
11] were relatively stable at room temperature; however, all were unstable above 60 °C and led to the respective calix[4]pyrroles; 6. Compound
4 was more stable than
2 under the purification conditions involving SiO
2 and heat [
8]; 7. The relative proportions of the various calix[n]pyrroles (
n = 4, 5, 6,
etc.) formed during a reaction could not be explained in purely statistical terms. The above observations suggest that calix[4]pyrrole is in equilibrium with calix[5]pyrrole, wherein the latter is the kinetic product and the former is the thermodynamic product. However, there is no formal report to demonstrate the mechanism that may favor the formation of calix[5]pyrroles. We were interested in exploring the mechanism by which low Lewis acid concentrations yielded calix[5]pyrrole as the main product and high Lewis acid concentrations yielded the calix[4]pyrrole exclusively. An understanding of the mechanism by which these macrocycles form could aid the synthesis of otherwise inaccessible homocalix[n]heteroisopyrazoles or hybrid[n]calixarenes or β-unsubstituted calix[n]pyrroles, particularly calix[8]pyrroles or calix[5]pyrrole with meso-position substituents other than CH
3 or cyclohexyl.
2. Results and Discussion
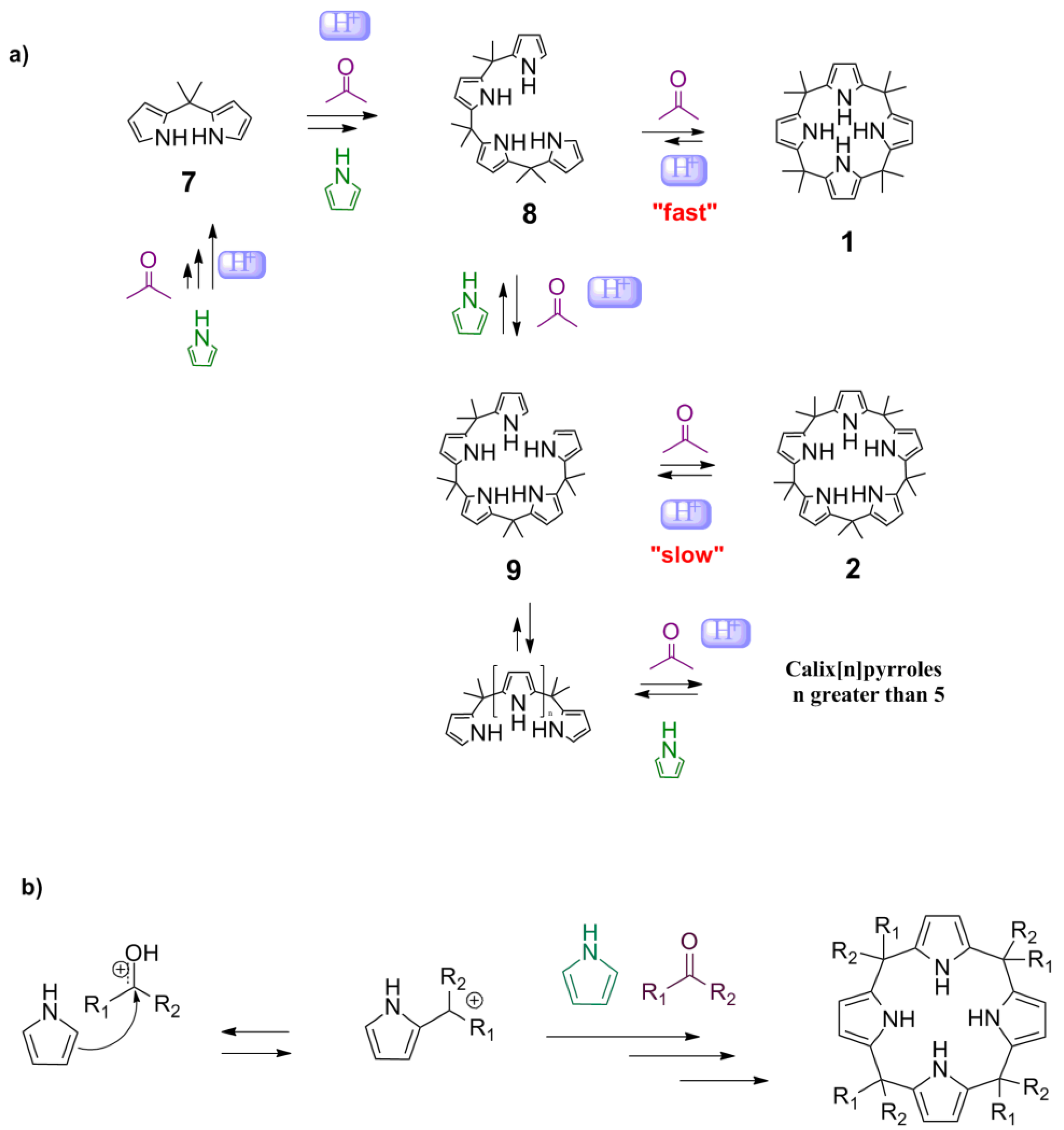
The mechanism by which the calixpyrroles are synthesized is assumed to be a condensation of pyrrole and acetone through the respective di-, tri-, tetra-, or pentameric intermediates (
Scheme 1a). The mechanism of the Brønsted–Lowry acid-catalyzed condensation reaction between pyrrole and ketone involves activation of the carbonyl by a proton, nucleophilic attack on carbon 2 of the pyrrole, and the subsequent elimination of water, to recover the aromaticity of the pyrrole (
Scheme 1b) [
14].
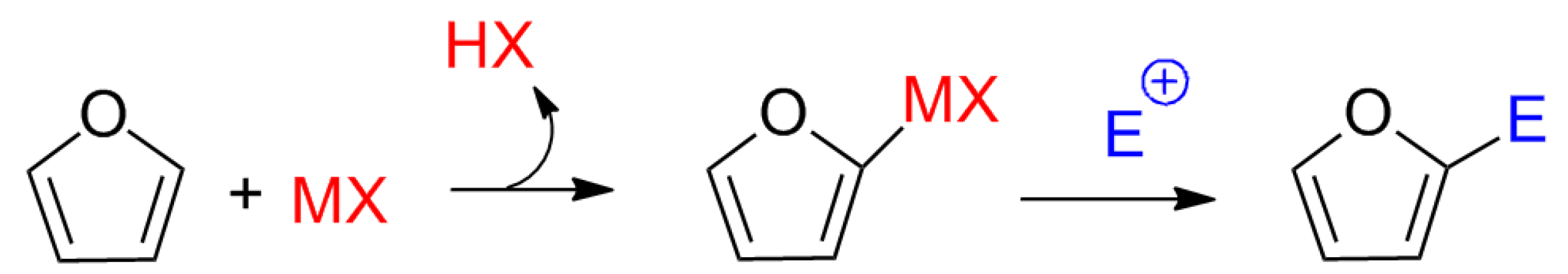
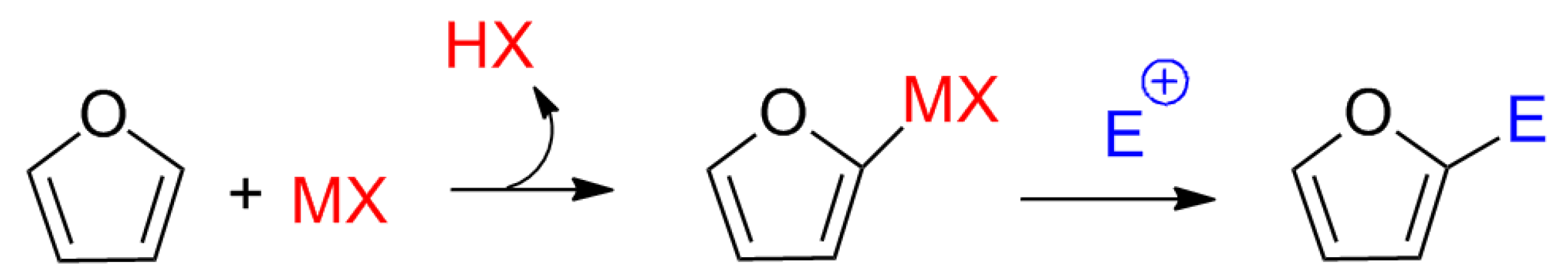
Although no previous studies suggested a mechanism involving Lewis acids for the synthesis of calixpyrroles, the electron pair of the carbonyl oxygen appeared to interact with the metal to activate the carbonyl. Other mechanisms cannot be ruled out, such as activation by a pyrrole-metal complex, as proposed for the addition of electrophiles to furans (
Scheme 2) [
15]. The mechanism can be described as an equilibrium between
1 and
2 through
8 and
9 (
Scheme 1a). Note that
1 and
2 may possibly be generated independently without requiring an equilibrium between the two species.
Scheme 1.
Reaction mechanism for the formation of calixpyrroles. (a) General mechanism; (b) Electrophilic substitution of ketones to yield the calixpyrrole.
Scheme 1.
Reaction mechanism for the formation of calixpyrroles. (a) General mechanism; (b) Electrophilic substitution of ketones to yield the calixpyrrole.
Scheme 2.
Proposed mechanism for the electrophilic substitution of furans via Lewis acid catalysis.
Scheme 2.
Proposed mechanism for the electrophilic substitution of furans via Lewis acid catalysis.
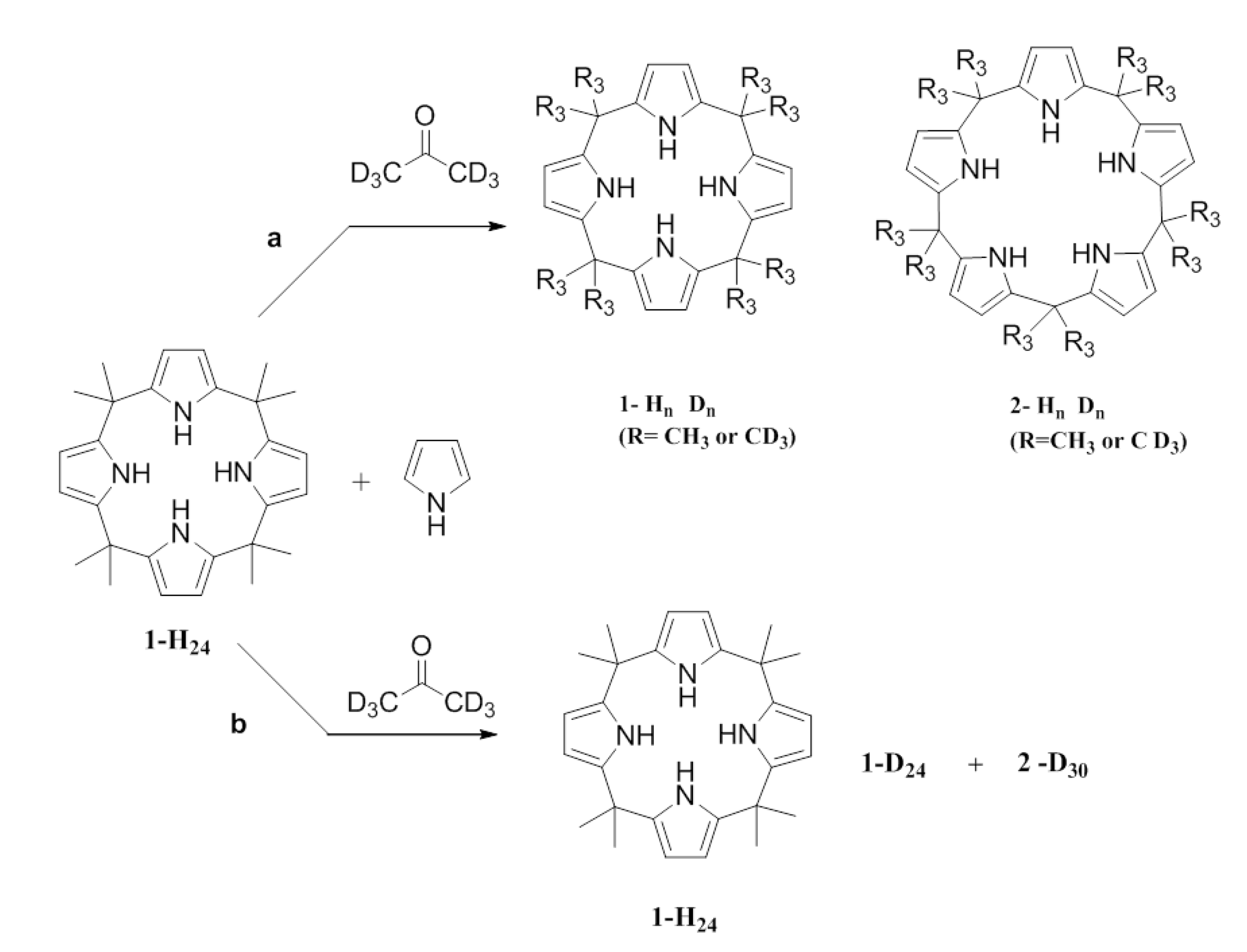
To demonstrate whether
1 and
2 interconverted, an isotopic labeling experiment was conducted to test the reversibility of the process. Distilled pyrrole and acetone-d
6 were added to a reaction mixture containing 0.1 mmol of the fully protonated
1 (
1-
1H
24). The composition of products was expected to be either: (a) a mixture of
1-H
n D
n (mainly meso-octamethyl deuterated) and
2-H
n D
n (mainly meso-decamethyl deuterated), or (b) a mixture of
1-H
24 (starting material),
1-D
24 (fully meso-octamethyl deuterated), and
2-D
30 (fully meso-decamethyl deuterated). This is so if the composition of the reaction mixture is that indicated for case (a) the equilibria 8-1 and 9-2 are occurring; if the composition is that indicated for case (b) then 1 is not in equilibrium with the linear tetramer 8 (
Scheme 3).
Scheme 3.
Two possible routes for the condensation of pyrrole with deuterated acetone.
Scheme 3.
Two possible routes for the condensation of pyrrole with deuterated acetone.
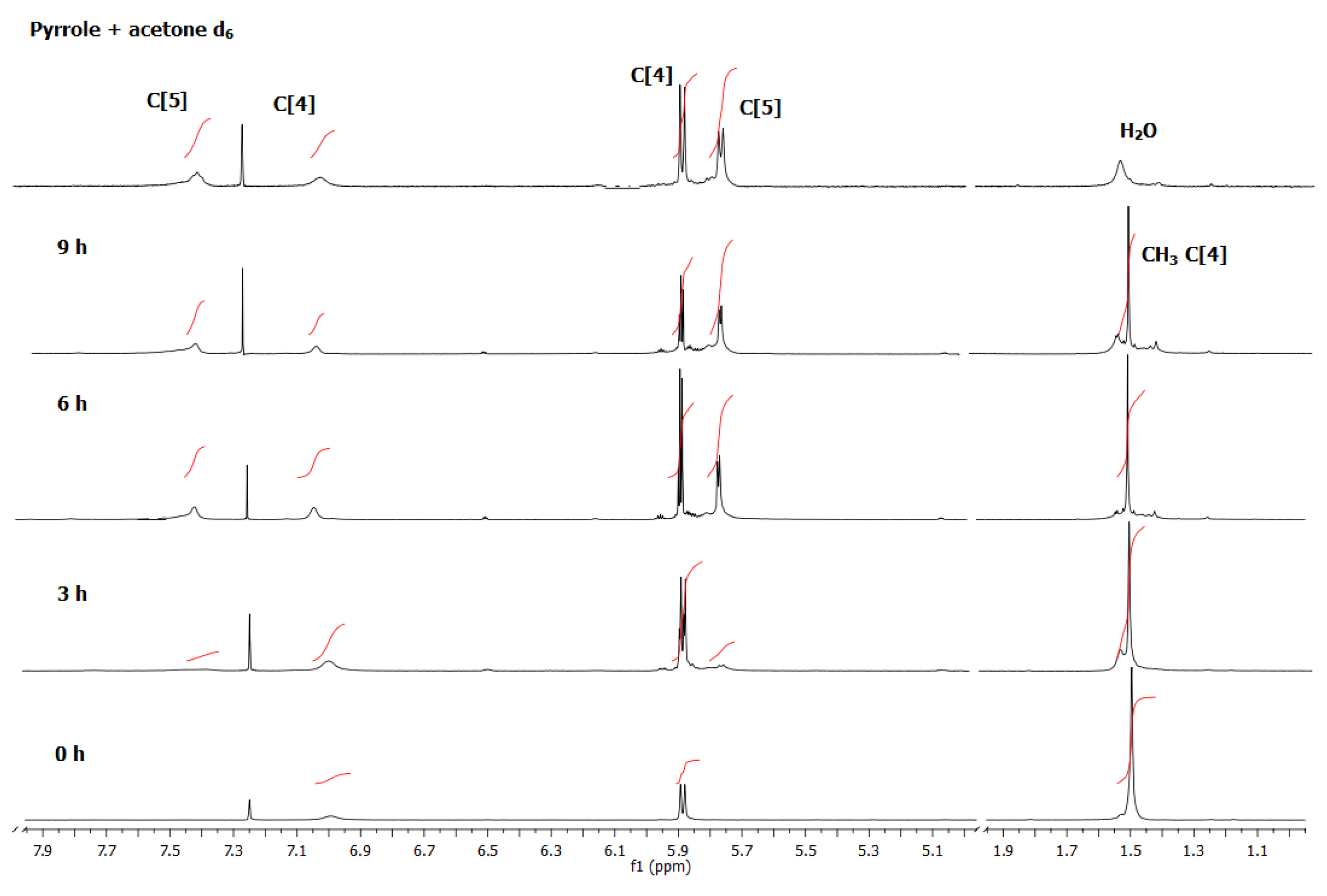
The
1H NMR peak integrals corresponding to the protons β (5.77 ppm) and the absence of the methyl protons at 1.51 ppm, as illustrated in
Figure 2, showed that after 3 hr,
2-D
30 (full meso-decamethyl deuterated) appeared, and no
2-CH
3 was observed until the end of the reaction sequence. As in the case of
2, compound
1 was not found to be partially deuterated, indicating that no interconversion between
1 and
2 occurred after 9 hr reaction under these conditions. Both macrocycles followed their respective reaction courses, from the common intermediate
8 (
Scheme 1), without significant reversibility. Compounds
8 and
9 must presumably form, but their concentrations is marginal, indicating that the equilibria 8-1 and 9-2 shown in (
Scheme 1) are largely shifted towards the formation of the C4 and C5 respectively.
Figure 2.
1H NMR spectra over the course of the reaction of pyrrole with acetone d6 at room temperature after work up. Spectra were collected at different reaction times in CD3Cl. At the top a mixture of the two calix compounds deuterated at all of their CD3 positions is shown.
Figure 2.
1H NMR spectra over the course of the reaction of pyrrole with acetone d6 at room temperature after work up. Spectra were collected at different reaction times in CD3Cl. At the top a mixture of the two calix compounds deuterated at all of their CD3 positions is shown.
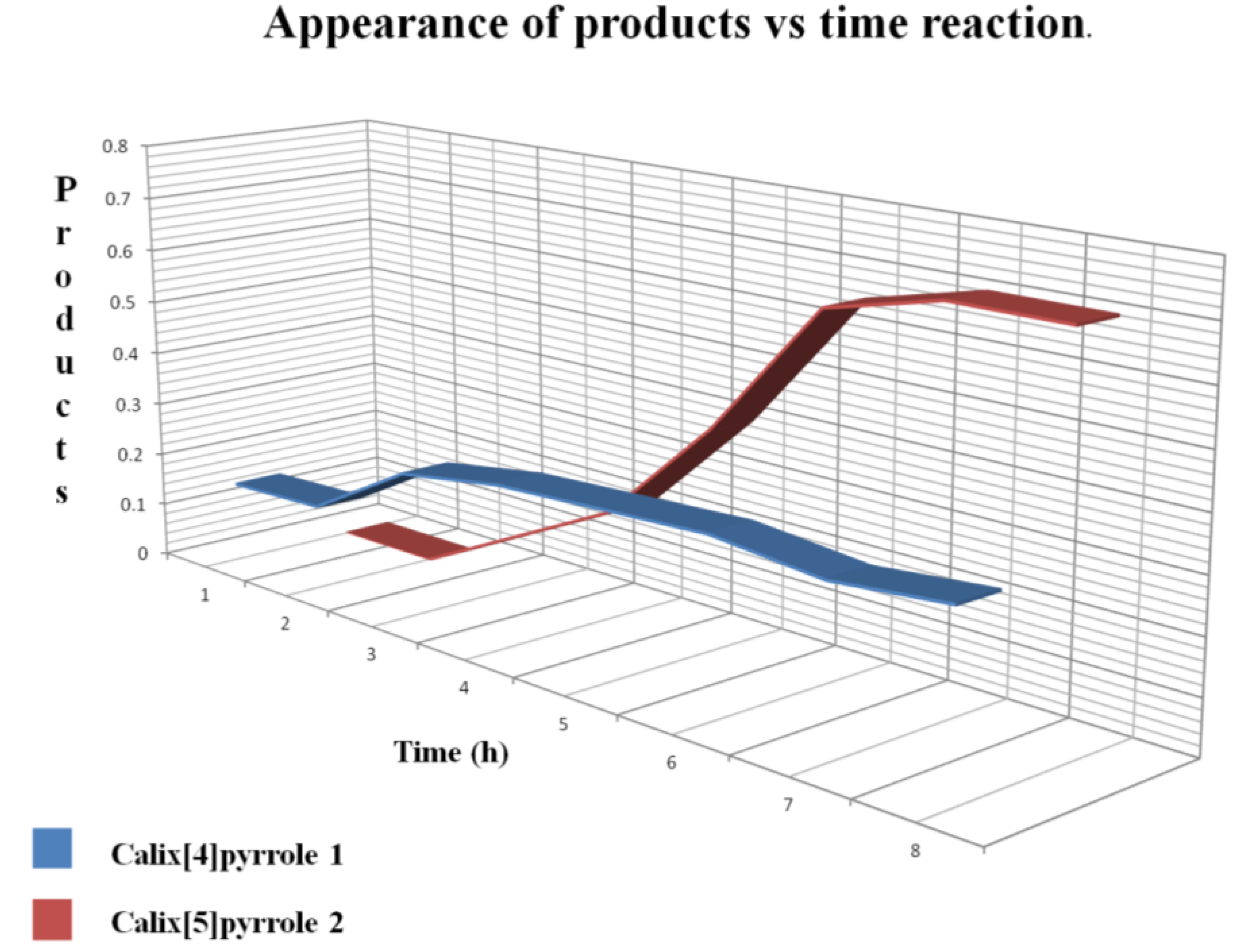
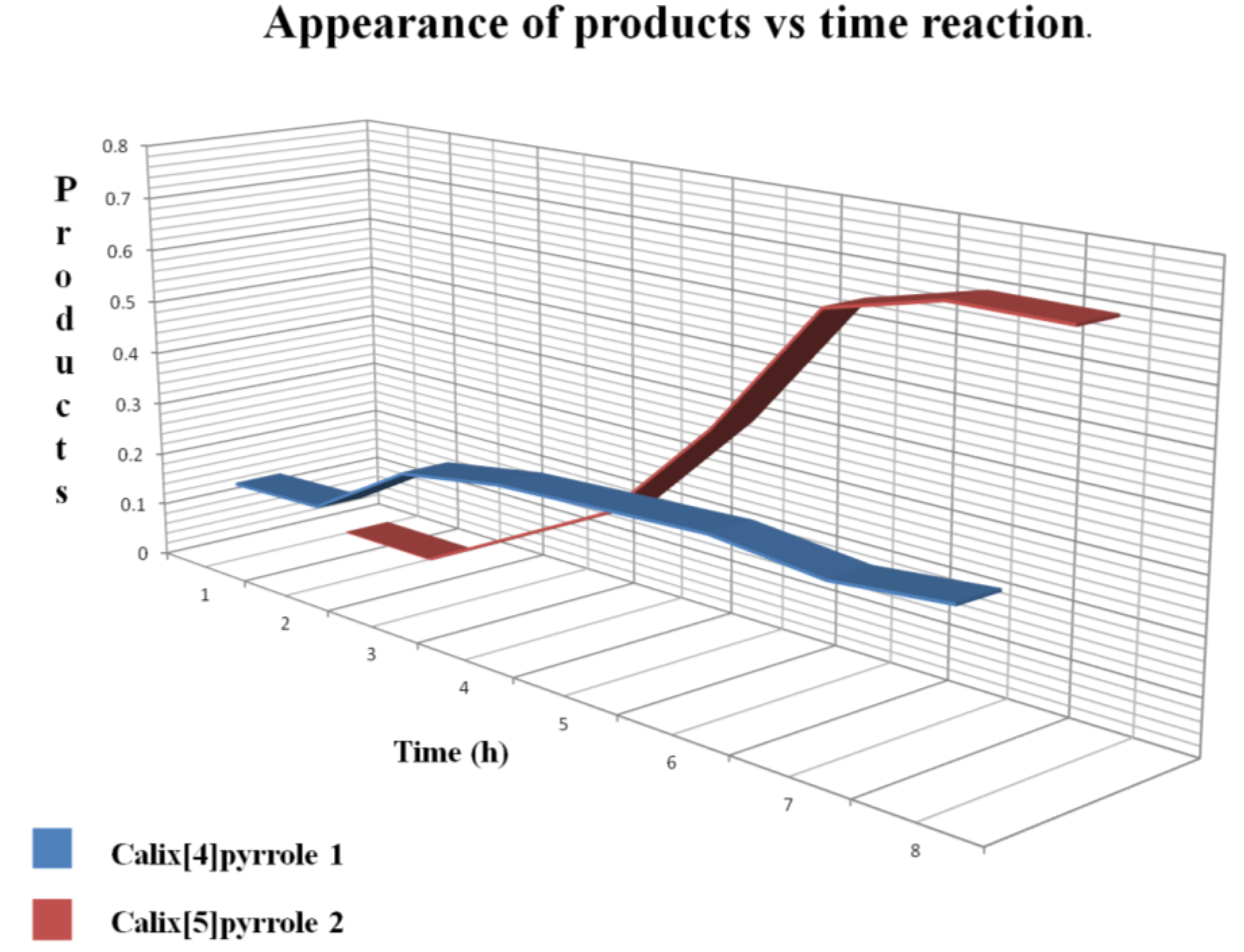
Figure 3 shows that the first product formed was
1 after 2 h, and
2 began to appear after 4 h. After 9 h reaction time the major product was
2, whereas
1 remained constant.
Figure 3.
Progress of the reaction, showing the ratio of
1 to
2 obtained from the integrals of protons β as the reaction progressed (see
Figure 2).
Figure 3.
Progress of the reaction, showing the ratio of
1 to
2 obtained from the integrals of protons β as the reaction progressed (see
Figure 2).
The reaction was also tested using acetone and deuterated pyrrole-d
5; however, the results were uninterpretable due to the emergence of hydrogen peaks instead of deuterium peaks in the pyrrole fragment of
1 and
2, in a much higher proportion than was present in the reactant. This hydrogen resulted from proton exchange in the β-pyrrole of the macrocycle. The result was interesting because it suggested the involvement of the Brønsted acid in the medium. The basicity of the 3- and 4-positions of pyrrole has been demonstrated in 2,5-disubstituted pyrroles (
Scheme 4) [
16].
Scheme 4.
Deuteron exchange in pyrroles via Bronsted acid-catalyst.
Scheme 4.
Deuteron exchange in pyrroles via Bronsted acid-catalyst.
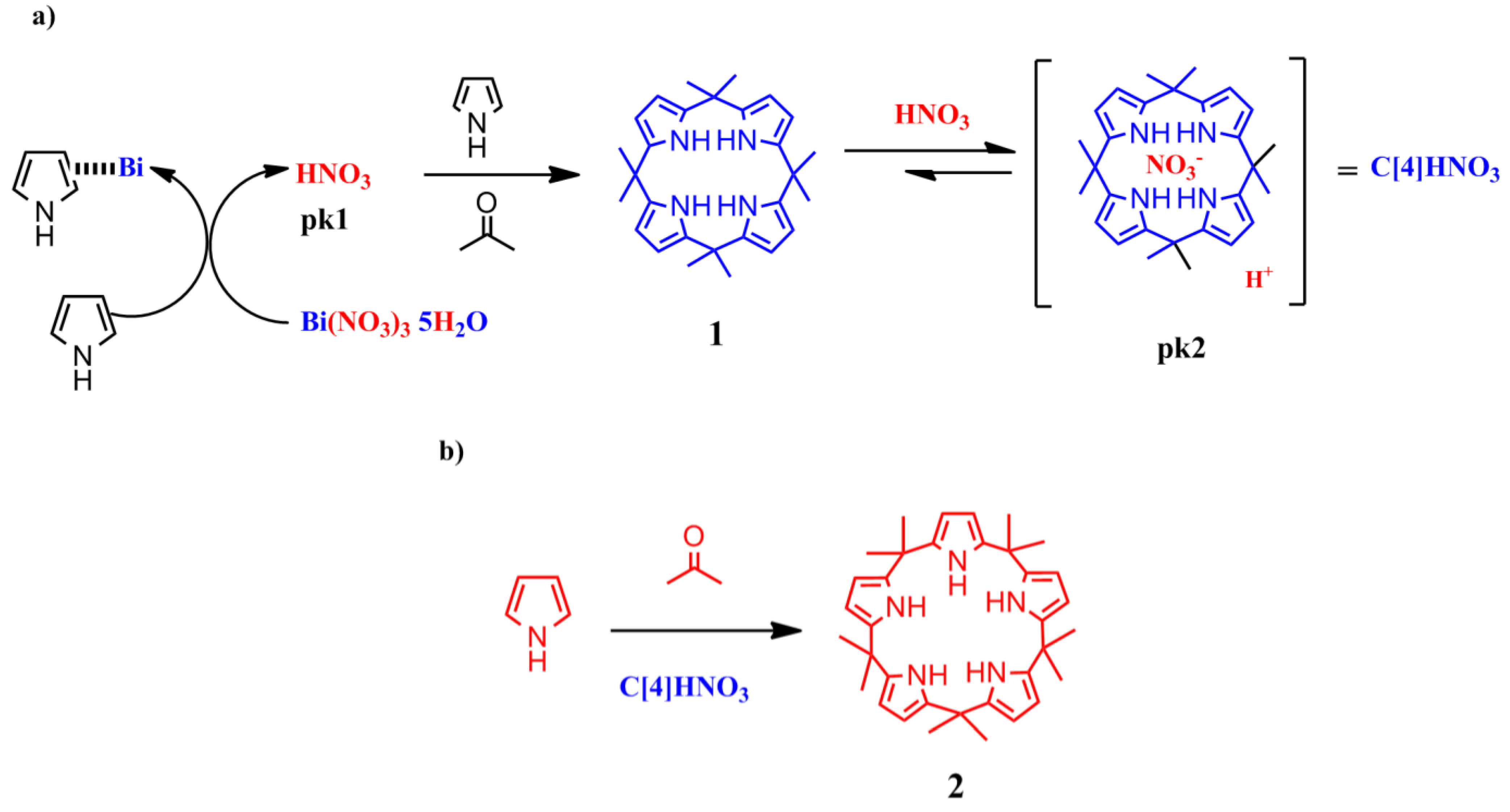
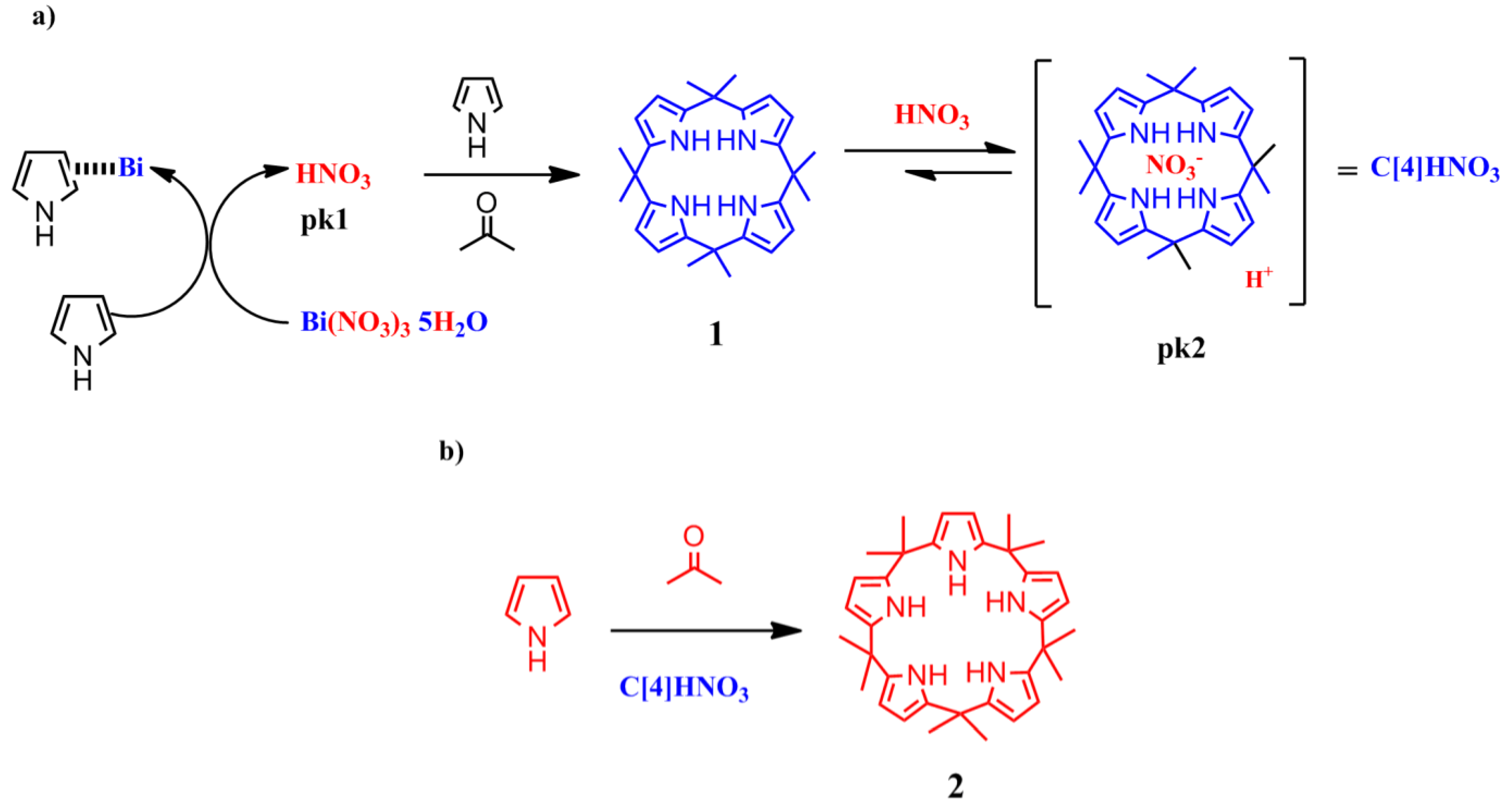
To explain the above results, we propose that a first step in the formation of
2 (
Scheme 5) involves the reaction of Bi(NO
3)
3 with pyrrole to release the Brønsted acid (nitric acid), as has been proposed for the addition of furans to vinyl ketones catalyzed by Lewis acids [
15].
Scheme 5.
Proposed mechanism for the formation of calix[5]pyrrole using Bi as catalyst (a) Acid activation; (b) Condensation reaction.
Scheme 5.
Proposed mechanism for the formation of calix[5]pyrrole using Bi as catalyst (a) Acid activation; (b) Condensation reaction.
In fact, we observed that the reaction of the pyrrole with the bismuth salt was exothermic in the absence of solvent. The addition of acetone to this mixture yielded the immediate formation of 1 along with several other products, presumably polypyrroles.
The acid was then released to catalyze the formation of
1, as expected in the presence of protic acids; however,
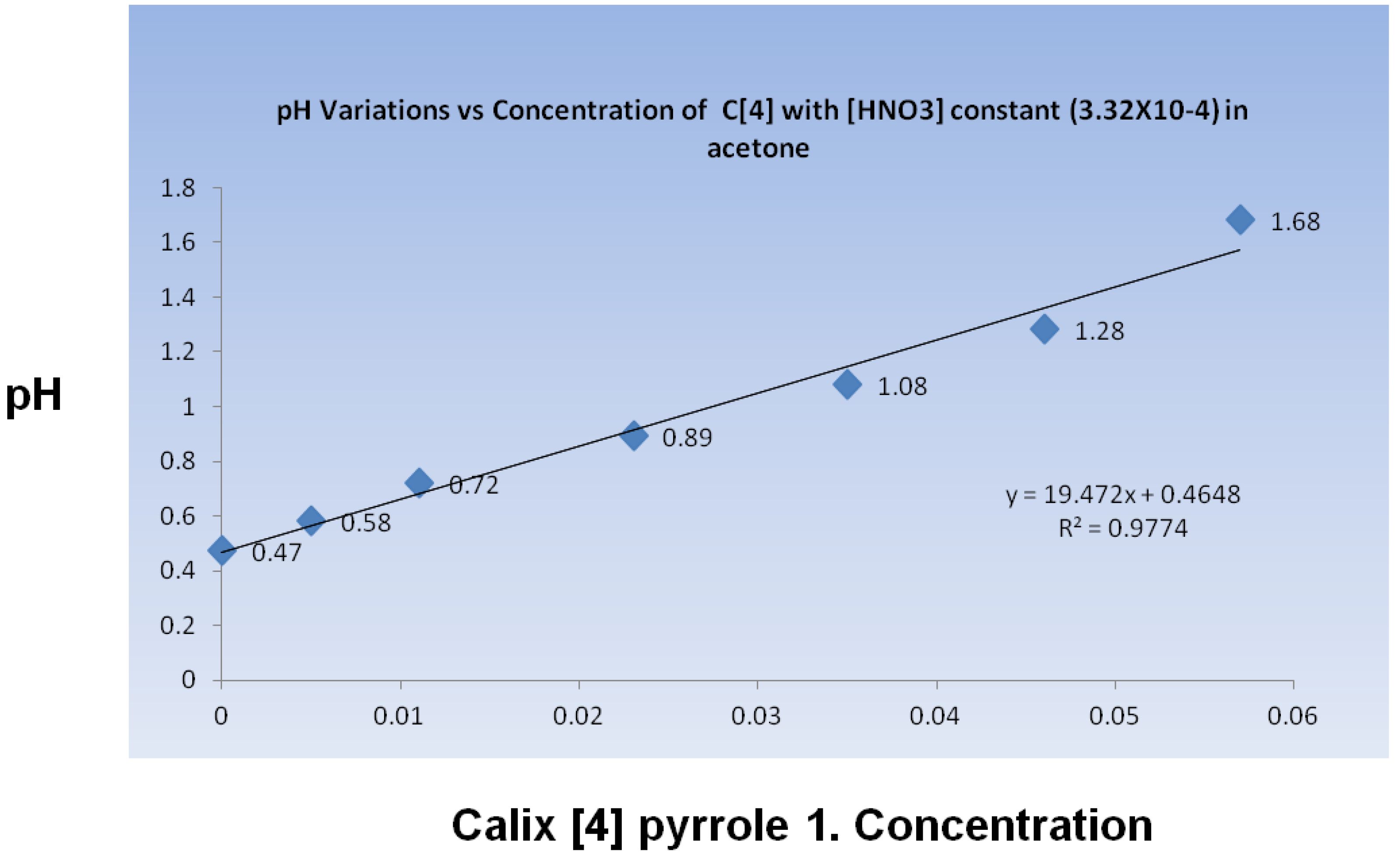
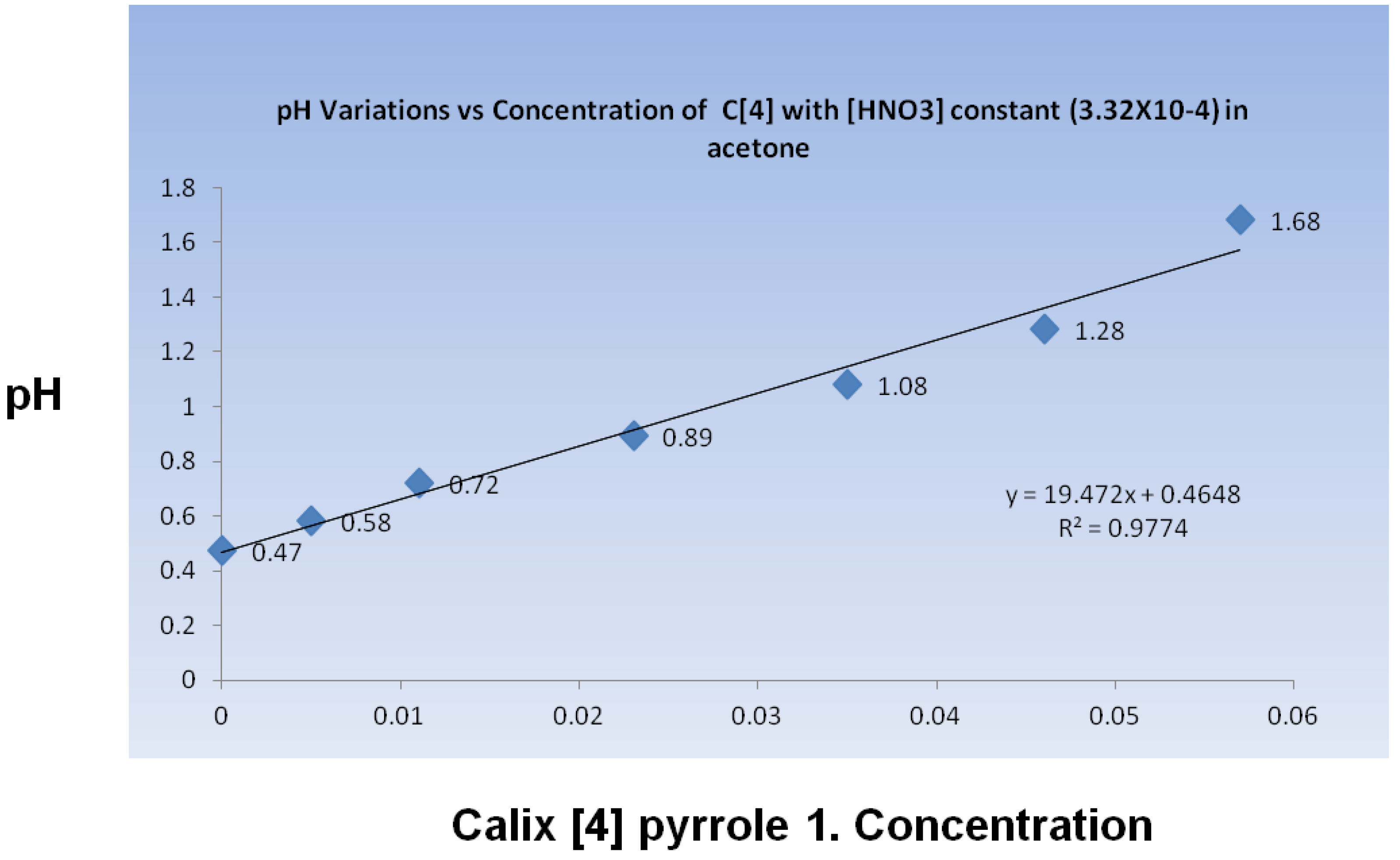
1 interacted with the protic acid in the reaction media to alter the acidity of the reactive environment. The pH was quantified through potentiometric measurements in acetone. It was monitored during a titration in which calixpyrrole
1 was added to an acetone solution containing HNO3. As compound 1 was added, the pH level increased linearly (
Figure 4). The pH change was not entirely unexpected, considering that
1 has been reported to recognize nitrate anions [
17].
In the presence of high concentrations of a weak acid (such as acetic acid, pKa = 4.5), the reaction did not proceed to the calixpyrroles, suggesting that a strong acid was essential. Carbonyls, which are relatively weak bases (carbonyl protonation pKa = −7), must be activated by a strong acid. Low concentrations of nitric acid (pKa = −1.5) react to induce cyclization of the tetrapyrrole 8 to give 1. This cyclization is slower than the formation of the pentapyrrole 9, which subsequently reacts to give 2. If the controlling step for these condensations were the addition of the pyrrole to the benzylic-like carbocation, the degrees of freedom would be expected to play an important role in the nucleophile-electrophile interaction geometry. Under these conditions (low pH and strong acid) the reaction is expected to favor 2 as the kinetic product.
Because strong acids govern the reversibility of the reaction via protonation of the pyrrole ring (which is a strong base relative to the carbonyl: pyrrole protonation p
Ka = −3.8), ring opening was favored for
2, due to strain within the macrocycle. A comparison of the bond angles of the calixpyrroles described by Sessler [
18] and Kohnke [
9], listed in
Table 1, indicates that strain is present in
2, in which the C1–C25–C24 bond angles differ by as much as 4° with respect to the parent compound
1.
Figure 4.
pH change in acetone containing HNO3 during the addition of calix[4]pyrrole 1.
Figure 4.
pH change in acetone containing HNO3 during the addition of calix[4]pyrrole 1.
Table 1.
Representative bond angles in
1 and
2 from the X-ray diffraction data [
9,
18].
![Catalysts 03 00588 i001]()
Table 1.
Representative bond angles in 1 and 2 from the X-ray diffraction data [9,18]. ![Catalysts 03 00588 i001]()
| Angle | X-Y-Z (1) | α1 | X-Y-Z (2) | α2 | α2 − α1 |
|---|
| a | N(1)-C(4)-C(5) | 121.6 | N(1)-C(4)-C(5) | 123.3 | 1.7 |
| b | C(4)-C(5)-C(6) | 109.8 | C(4)-C(5)-C(6) | 111.6 | 1.8 |
| c | C(5)-C(6)-C(7) | 131.5 | C(5)-C(6)-C(7) | 133.3 | 1.8 |
| d | N(4)-C(19)-C(20) | 121.3 | N(5)-C(24)-C(25) | 123.9 | 2.6 |
| e | C(18)-C(19)-C(20) | 131.8 | C(23)-C(24)-C(25) | 129.7 | −2.1 |
| f | C(19)-C(20)-C(1) | 109.9 | C(24)-C(25)-C(1) | 113.5 | 3.6 |
| g | C(19)-C(20)-C(20A) | 109.8 | C(24)-C(25)-C(25A) | 108.4 | −1.4 |
The strained geometry approached that of a carbocation (sp
2) intermediary and, therefore, a transition state. The activation energy for cyclization is low and the aperture of the macrocycle is relatively small, favoring the formation of
2 (the kinetic product) over
1 (the thermodynamic product). These results are consistent with the proposed mechanism of formation of
6 and the other higher calixpyrroles, such as the hexamethylmeso-hexaphenyl-calix[6]pyrrole [
19].
The formation of
6 was catalyzed in CH
3SO
4H, then the 3,4-difluoropyrrole slowed the reaction and facilitated the reverse reaction by destabilizing the carbocation pyrrole via electron-withdrawing effects. The formation of hexamethylmeso-hexaphenyl-calix[6]pyrrole proceeded in 2,2,2-trichloroethanol, then the equilibrium shifted to the right because the product was stabilized by interactions with the alcohol group [
19]. Cyclopentanone, a weaker base than cyclohexanone, required more strongly acidic conditions to achieve condensation, although the reverse process was also favorable [
8].
The proposed mechanism was tested by exploring the reaction at various HNO
3 concentrations. The results in
Table 2 show that
2 was formed at low acid concentrations (0.003 mol%), whereas at high concentrations, the only product formed was
1. At sufficiently low concentrations (0.0003 mol%), no macrocycles were observed to form, and only the open products or the starting materials were observed.
Table 2.
The ratios of 1 to 2 as a function of HNO3 concentration.
Table 2.
The ratios of 1 to 2 as a function of HNO3 concentration.
| HNO3 mMol | 1 | 2 |
|---|
| 0.7 | 100 | - |
| 0.014 | 64 | 36 |
| 0.006 | 44 | 56 |
| 0.003 | 25 | 75 |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}