1. Introduction
Biotransformations are chemical processes which occur under the influence of biological materials such as peptides and proteins. Amongst the myriad examples of bio-mediated transformations we have focused our attention on enzyme-catalyzed reactions at a silicon centre.
In the literature, there are several examples of organo-silicon biotransformations, such as the selective synthesis of organosilicon esters under mild reaction conditions [
1], enzymatic silicone oligomerization catalyzed by a lipid-coated lipase [
2], and the hydrolysis of silatranes catalyzed by an esterase obtained from the yeast
Rhodotorula mucilaginosa [
3]. In addition, nature provides many examples of reactions from simple to very complex with Si-substrates, where peptides and proteins are generally considered to be the undisputed arbiters [
4,
5]. Examples include silica formation in diatoms and other silica-forming organisms [
4,
5].
Our group has extensively studied silica precipitation [
6] and the enzyme-catalyzed hydrolysis and condensation of alxokysilanes [
7,
8,
9] under mild conditions. We have discovered several enzymatic candidates which were able to perform such reactions at room temperature and neutral pH and have investigated the potential involvement of their respective active sites in the biocatalyzed organo-silicon transformations.
In this work, we report a new enzyme-mediated reaction, namely the transetherification of alkoxysilanes, under mild conditions. Normally, the transetherification of alkoxysilanes requires extreme reaction conditions, including high temperatures, and the use of toxic chemicals. The use of mild (bio)-catalysts would therefore open a new way of performing such useful transformations, which is the subject of the present contribution.
2. Results and Discussion
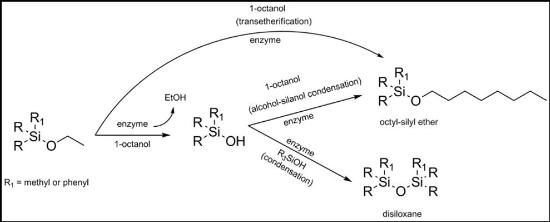
During our previous work on enzyme-catalyzed organo-silicon transformations, we were interested in siloxane-bond formation in the presence of biocatalysts both in aqueous and aqueous-organic media. Several monophasic and biphasic aqueous-organic systems were investigated as reaction solvents. One of the biphasic-aqueous organic systems employed during the alkoxysilane studies consisted of 1-octanol saturated with tris-buffered water. In addition to the previously observed enzyme-catalyzed hydrolysis and condensation of alkoxysilanes [
9], the formation of the octylsilyl ethers as a result of transetherification and/or silanol-alcohol condensation/exchange was also detected in this solvent whereas no equivalent reaction was seen in the negative control reactions (
Scheme 1). The “side-product” octyl-silyl ether was identified as a new peak which appeared in the gas-chromatogram following reaction and work-up when compared to our standard set of peaks which arise from solvents (THF, ethanol and 1-octanol), unreacted starting material, hydrolyzed silanol, and the disiloxane condensation product.
Scheme 1.
Enzyme-catalyzed transetherification and/or silanol-alcohol condensation.
Scheme 1.
Enzyme-catalyzed transetherification and/or silanol-alcohol condensation.
The identity of the structure was further investigated by means of GC-MS (not shown) and was confirmed to be the alkoxyoctylsilyl ether.
The reactions were formulated with approximately 5:1 alkoxysilane to enzyme weight ratio in wet (water-saturated) 1-octanol (5:1 solvent to alkoxysilane weight ratio) and conducted in inert glass vials. After 24 h of stirring at room temperature, the reactions were filtered and quantitatively analyzed by GC-FID. The gas chromatography analysis was performed with an Agilent 6890 Series injector on an Agilent 6890 plus gas chromatograph with a flame ionization detector.
Dodecane was used as an internal standard to quantitate the chromatographic analyses. The samples were prepared at ~1% (w/w) product in a THF solution containing 1% (w/w) dodecane.
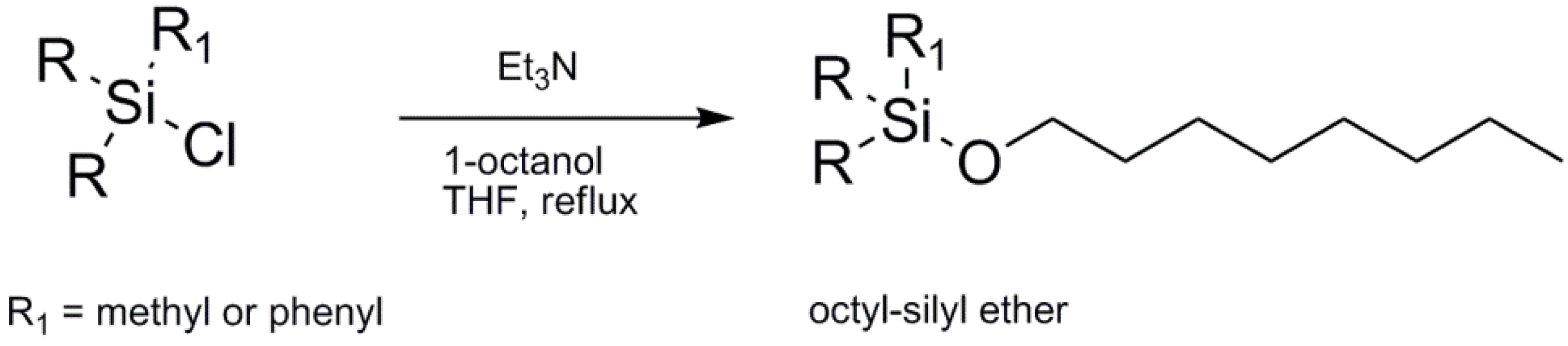
In order to chromatographically quantify the trimethyloctyloxysilane and phenyldimethyloctyloxysilane products, the two compounds had to be synthesized, as they were not commercially available. The synthetic procedures are detailed in the Experimental Section. In general, the appropriate chlorosilane was refluxed with 1-octanol in THF in the presence of triethylamine (
Scheme 2). The products were subsequently purified by vacuum distillation, characterized and used as standard references in the gas-chromatography analyses.
Scheme 2.
Chemical synthesis of the octyl-silylethers.
Scheme 2.
Chemical synthesis of the octyl-silylethers.
As shown in
Figure 1 and detailed in
Table 1,
Table 2, selected enzymes such as trypsin,
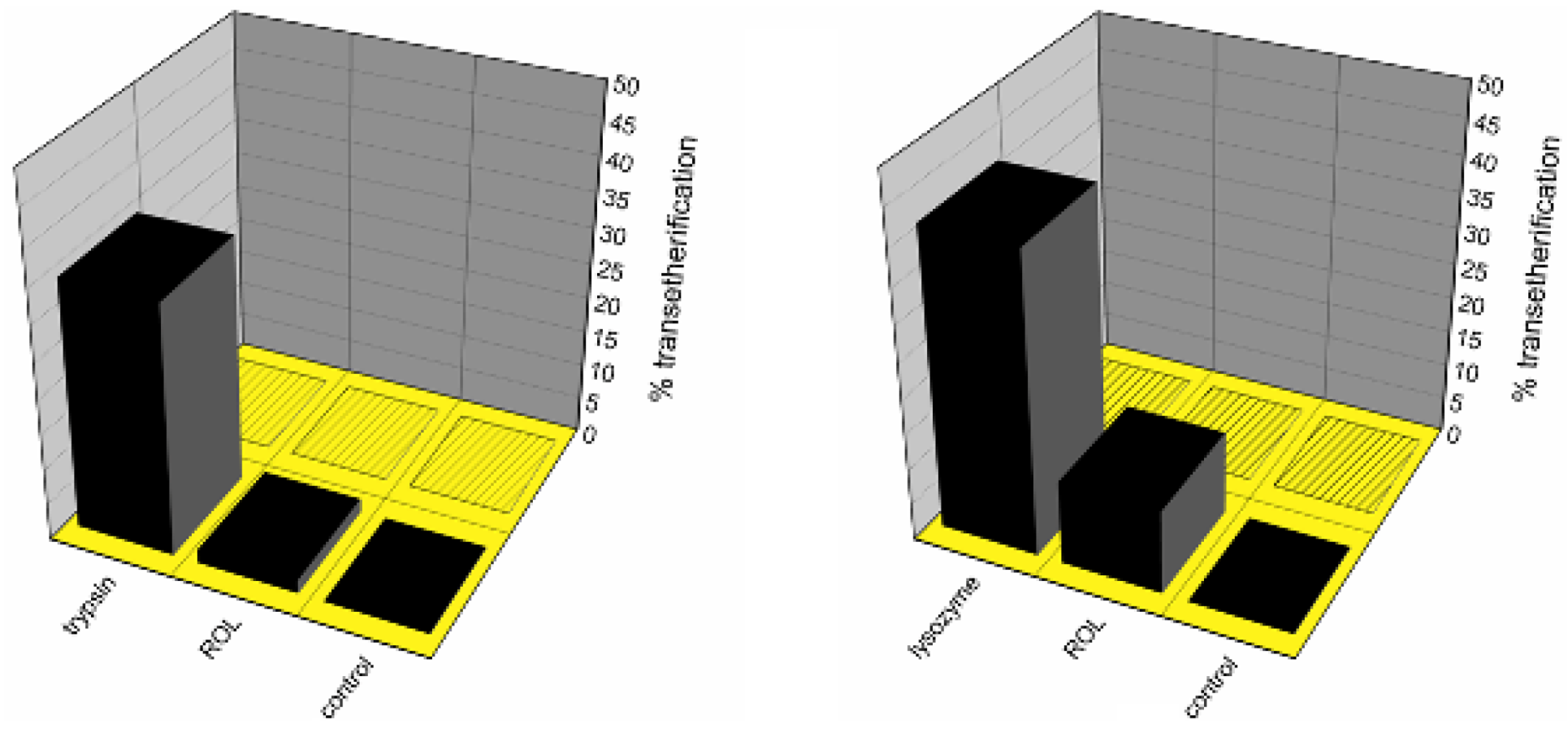
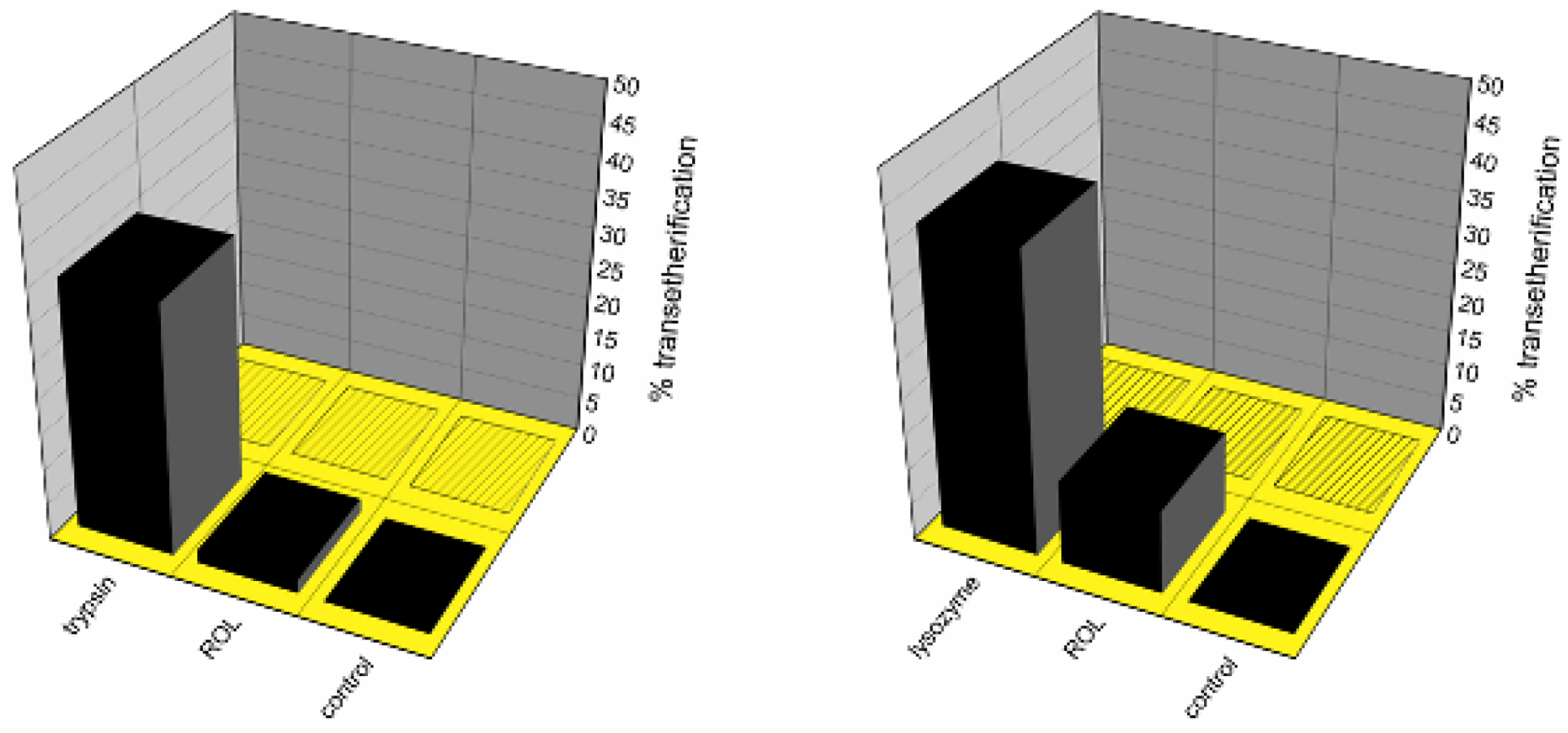
Rhizopus oryzae lipase (ROL) and lysozyme were able to catalyze the formation of the octyltrimethyl-silyl ether (
Figure 1, left) and/or the octylphenyldimethylsilyl ether (
Figure 1, right) after 24 h at room temperature. In similar conditions, no condensation was observed in the negative control reactions nor in the presence of all the other enzymes tested in the Si–O bond formation screening [
9].
Figure 1.
Enzyme-catalyzed transetherification and/or silanol alcohol condensation/exchange study between trimethylethoxysilane (left) or phenyldimethylethoxysilane (right) with 1-octanol (filled bars) or tert-butanol (bars with patterns), after 24 h at 25 °C.
Figure 1.
Enzyme-catalyzed transetherification and/or silanol alcohol condensation/exchange study between trimethylethoxysilane (left) or phenyldimethylethoxysilane (right) with 1-octanol (filled bars) or tert-butanol (bars with patterns), after 24 h at 25 °C.
Table 1.
Enzyme-catalyzed transetherification reactions between trimethylethoxysilane and 1-octanol or tert-butanol after 24 h at 25 °C.
Table 1.
Enzyme-catalyzed transetherification reactions between trimethylethoxysilane and 1-octanol or tert-butanol after 24 h at 25 °C.
| | % Yield (Normalized) |
|---|
| Reaction | Me3SiOEt 1 | Me3SiOH 2 | HMDS 3 | Me3SiOR 4 |
|---|
| negative control, 1-octanol | 91.3 | 5.3 | 3.4 | 0.0 |
| negative control, 5% water in tert-butanol | 34.7 | 64.4 | 1.0 | 0.0 |
| Rhizopus oryzae lipase, 1-octanol | 81.8 | 13.0 | 3.4 | 1.8 |
| Rhizopus oryzae lipase, 5% water in tert-butanol | 77.0 | 23.0 | 0.0 | 0.0 |
| bovine pancreatic trypsin, 1-octanol | 7.0 | 52.9 | 2.3 | 34.9 |
| bovine pancreatic trypsin, 5% water in tert-butanol | 3.8 | 92.1 | 4.1 | 0.0 |
Table 2.
Enzyme-catalyzed transetherification reactions between phenyldimethyethoxysilane and 1-octanol or tert-butanol after 24 h at 25 °C.
Table 2.
Enzyme-catalyzed transetherification reactions between phenyldimethyethoxysilane and 1-octanol or tert-butanol after 24 h at 25 °C.
| | % Yield 1 |
|---|
| Reaction | PhMe2SiOEt 2 | PhMe2SiOH 3 | (PhMe2OSi)2-O 4 | PhMe2SiOR 5 |
|---|
| negative control, 1-octanol | 100.0 | 0.0 | 0.0 | 0.0 |
| negative control, 5% water in tert-butanol | 88.8 | 11.2 | 0.0 | 0.0 |
| Rhizopus oryzae lipase, 1-octanol | 66.8 | 21.5 | 0.0 | 11.7 |
| Rhizopus oryzae lipase, 5% water in tert-butanol | 95.8 | 4.2 | 0.0 | 0.0 |
| chicken egg white lysozyme, 1-octanol | 3.2 | 55.0 | 0.0 | 41.8 |
| chicken egg white lysozyme, 5% water in tert-butanol | 71.5 | 28.5 | 0.0 | 0.0 |
Figure 1 shows percentage yield of octyl-ether formation based on the quantitative chromatographic data, the mass balance being completed by either unreacted alkoxysilane, silanol formed by simple hydrolysis, or the corresponding disiloxane from the condensation product with another molecule of silanol (see
Scheme 1). Notably, in the absence of any biocatalyst (negative control), no octyl-silyl ether was observed, denoting the critical role of the enzyme in the alkoxysilane transetherification transformation.
To our knowledge, this is the first case of an enzyme-mediated transetherification reaction of organo-silicon substrate under mild conditions. It is apparent that there is advantage in using an enzyme at room temperature over the conventional synthetic procedure the synthesis of the octyl-silyl products by avoiding the use of harsh chemicals and elevated reaction temperature.
Interestingly, the enzymatic screening conducted during the hydrolysis and condensation study of monoalkoxysilanes in wet
tert-butanol (see [
9],
Figure 1,
Table 1,
Table 2) did not lead to any
tert-butylsilyl ether product formation. This may be due to the steric hindrance of
tert-butyl groups, as opposed to the longer but more flexible octyl chains, which may be more accessible to the enzyme cavities.
Rhizopus oryzae lipase (ROL) is a small globular protein of approximately 32 kDa that exists as a monomer in solution. This enzyme catalyzes the hydrolysis of triglycerides to yield the free fatty acid. It hydrolyzes triglycerides with alkyl chains in the region of 8 to 18 carbons long, whereas poor activity was observed with short-chain fatty esters. Notably, ROL was observed to catalyze the formation of both trimethyloctyloxysilane and phenyldimethyloctyloxysilane. Conversely, the lipase was not able to catalyze the formation of
tert-butyl silyl ethers. This is in agreement with the natural substrate-selectivity of the ROL, and suggests the involvement of the active site during the catalysis. Trypsin, a serine-protease that cleaves peptide chains mainly at the carboxyl side of the amino acids lysine or arginine, was a good biocatalyst for trimethyloctyl silyl ether formation, and this is in line with our previous studies on the alkoxysilane hydrolysis and condensation reactions [
7,
9]. Lysozyme is a small (14.3 kDa) but robust protein consisting of a single 129 amino acid polypeptide chain , and it was already observed to be a good siloxane-bond biocatalyst [
8]. This enzyme produced the highest yield of the phenyldimethyloctyl-silyl ether in this study. The reason for the unusual selectivity of this glycoside hydrolase is not yet understood and will be the subject of further investigations. The work proves the potential of the use of (bio)-macromolecules as catalytic aids on unusual substrates under facile and mild reaction conditions and justifies further exploration for biotransformation in silicon chemistry.
3. Experimental Section
3.1. Materials
Trimethylsilanol (Me3SiOH) was obtained from the Dow Corning Corporation (Midland, MI, USA). Enzymes and all the other chemicals were purchased from Sigma-Aldrich (Poole, UK). All the materials were used as received without further modification. Ultra high purity water (UHP) was obtained from a f Milli-Q system at The Open University (Milton Keynes, UK).
3.1.1. Synthesis of Trimethyloctyloxysilane
1-Octanol (31.25 g, 0.24 M) and triethylamine (24.28 g, 0.24 M) were dissolved in anhydrous THF (300 mL) under nitrogen in a 3-necked round-bottomed flask, and a solution of chlorotrimethylsilane (15.21 g, 0.14 M) in anhydrous THF (100 mL) was added dropwise to the mixture over one hour. After gently refluxing at approximately 70 °C, a white solid precipitated (triethylammonium chloride). The solid was filtered off and the filtrate recovered. The solvent and excess triethylamine were removed using a rotary evaporator, and the transparent liquid purified by distillation at 120 °C and approximately 85 mbar. The product was characterized by NMR spectroscopy and GC-MS, and it was used as a GC-standard in order to understand the product distribution during the enzyme-catalyzed transetherification studies. Yield 67%.
1H NMR (CDCl
3, 300 MHz): δ 3.45 (
t, 2H,
J 7.4, O-C
H2), 1.41 (
m, 2H, OCH
2-C
H2), 1.16 (
m, 10H, OCH
2CH
2-(CH
2)
5), 0.77 (3H,
t,
J 7.4, O(CH
2)
7-C
H3) and 0.0 (
s, 9H, Si(C
H3)
3) ppm.
13C NMR (CDCl
3, 75.45 MHz) δ 63.2 (O-
CH
2), 33.2 (OCH
2-CH
2), 32.3 (OCH
2CH
2-
CH
2), 29.8 (O(CH
2)
3-CH
2), 29.7 (O(CH
2)
4-
CH
2), 26.3 (O(CH
2)
5-
CH
2), 23.1 (O(CH
2)
6-
CH
2), 14.5 (O(CH
2)
7-CH
3) and 0.0 (Si-(
CH
3)
3).
29Si NMR (CDCl
3, 79.3 MHz): δ 17.5 ppm. Mass
m/
z (EI) 202 (M
+), 187 (M
+-CH
3). NMR data were consistent with those reported [
10].
3.1.2. Synthesis of Phenyldimethyloctyloxysilane
1-Octanol (28.65 g, 0.22 M) and triethylamine (22.26 g, 0.22 M) were dissolved in anhydrous THF (250 mL) under nitrogen in a 3-necked round-bottomed flask and a solution of chlorodimethylphenylsilane (20.48 g, 0.12 mM) in anhydrous THF (100 mL) added dropwise to the mixture over one hour. After gently refluxing at approximately 70 °C for 2 h, the triethylammonium chloride was filtered off and the filtrate collected. After removing the solvent and excess of triethylamine using a rotary evaporator, the product was purified by distillation at 142 °C and approximately 85 mbar. The product was characterized by NMR spectroscopy and GC-MS and it was used as a GC-standard in order to understand the product distribution during the enzyme-catalyzed transetherification studies. Yield 44%.
1H NMR (CDCl
3, 300 MHz): δ 7.22-7.02-7.01 (
m, 5H, aromatic H), 3.26 (
t, 2H,
J 6.6, O-C
H2), 1.17 (
m, 2H, OCH
2-C
H2), 0.90 (
m, 10H, OCH
2CH
2-(C
H2)
5), 0.51 (
t, 3H,
J 3.0, O(CH
2)
7-C
H3) and 0.0 (
s, 6H, Si(C
H3)
2) ppm.
13C NMR (CDCl
3, 75.45 MHz): δ 138.1-133.4-129.5-127.7 (aromatic C), 63.0 (O-CH
2), 32.6 (OCH
2-CH
2), 31.8 (O(CH
2)
2-CH
2), 29.4 (O(CH
2)
3-
CH
2), 29.2 (O(CH
2)
4-
CH
2), 25.7 (O(CH
2)
5-
CH
2), 22.6 (O(CH
2)
6-
CH
2), 14.0 (O(CH
2)
7-
CH
3) and -1.7 (Si-(
CH
3)
3).
29Si NMR (CDCl
3, 79.3 MHz): δ 7.5 ppm. Mass
m/
z (EI) 264 (M
+), 249 (M
+-CH
3). NMR data were consistent with those reported [
11].
3.2. Enzyme-Catalyzed Transetherification Reactions
The reactions were formulated with a 5:1 alkoxysilane (100 mg) to enzyme (20 mg) weight ratio in 0.5 g of alcohol (water-equilibrated 1-octanol or tert-butanol containing 5% (v/v) buffered water). Prior to analysis, the reactions were filtered through a Whatman Autovial® 5 0.45-μm Teflon® filter (Fisher Scientific Ltd., Loughborough, UK). The closed (screw capped) two-phase reactions were conducted in inert glass vials at 25 °C with magnetic stirring for 24 h. The reaction products were isolated and analyzed by GC-FID (quantitative, Agilent, Wokingham, UK) and GC-MS (qualitative, Agilent, Wokingham, UK).
Control reactions are defined as non-enzymatic reactions. Specifically, experiments conducted in the absence of a protein are defined as negative control reactions.
3.3. Gas Chromatography-Flame Ionization Detection
The gas chromatography (GC) analyses were performed with an Agilent 6890 Series injector on an Agilent 6890 plus gas chromatograph (GC) with a flame-ionization detector (FID).
The system was configured as detailed in
Table 3. Dodecane was used as an internal standard to gravimetrically quantitate the chromatographic analyses. The samples were prepared at ~1% (
w/
w) product in a THF solution containing 1% (
w/
w) dodecane. Based on triplicate measurements, the response factors for the analytes were calculated, Equation (1), and found to be linear as a function of concentration over four orders of magnitude (
i.e., 0.01%–10% (
w/
w) (
Table 4):
where RF
analyte = response factor for the analyte, [analyte] = concentration of the analyte, Area
analyte = peak area of the analyte, Area
IS = peak area of the internal standard, [IS] = concentration of the internal standard, RF
IS = response factor for the internal standard = 1. Equation (1) was then solved to quantitatively calculate the concentration of an analyte in the presence of an internal standard, Equation (2):
Table 3.
GC-FID experimental parameters.
Table 3.
GC-FID experimental parameters.
| Parameter | Setting |
|---|
| Carrier gas | 99.9995% Ultra high purity helium (UHP) |
| GC inlet, split | 250 °C, split ratio = 100:1, constant flow (rate = 1.0 mL/min) |
| Detector | Flame ionization detector at 275 °C, H2 = 40 mL/min, Make up N2 = 45 mL/min |
| GC column | HP-5MS crosslinked 5% phenylmethylsiloxane film (30 m × 0.25 mm, 0.25 μm film) |
| GC temperature program | 50 (2) → 250 (8) at 10 °C/min, 30 min total run time |
| Internal standard | ~1% (w/w) dodecane in THF |
| Data system | Agilent Technologies ChemStation |
Table 4.
GC-FID analyte retention times and response factors.
Table 4.
GC-FID analyte retention times and response factors.
| | Response Factor (RF) |
|---|
| Analyte 1 | Retention Time (min) | Average | Standard Deviation | RSD 2 |
|---|
| Me3SiOEt | 2.75 | 2.00 | 0.007 | 3.3% |
| Me3SiOH | 2.63 | 2.35 | 0.091 | 3.9% |
| HMDS | 3.22 | 2.01 | 0.052 | 2.6% |
| PhMe2SiOEt | 10.37 | 1.35 | 0.007 | 0.5% |
| PhMe2SiOH | 9.92 | ND | ND | ND |
| Phenyl disiloxane | 17.42 | ND | ND | ND |
| PhMe2SiO(CH2)7CH3 | 17.48 | 2.02 | 0.038 | 0.2% |
| Me3SiO(CH2)7CH3 | 10.97 | 2.15 | 0.027 | 1.3% |
| Dodecane | 11.11 | 1.00 | | |
3.4. Gas Chromatography-Mass Spectrometry
The gas chromatography-mass spectrometry (GC-MS) analyses were performed with an Agilent 6890 Series injector on an Agilent 6890 plus gas chromatograph with a 5973 MS detector. The MS detector was autotuned with perfluorotributylamine (PFTBA) prior to analysis. The system was configured as detailed in
Table 5.
Table 5.
GC-MS Experimental Parameters.
Table 5.
GC-MS Experimental Parameters.
| Parameter | Setting |
|---|
| Carrier gas | 99.999% high purity helium |
| GC inlet, split | 250 °C, split ratio = 50:1, constant flow (rate = 1.0 mL/min.) |
| GC column | HP-5MS crosslinked 5% phenyl methylsiloxane film (30 m × 0.25 mm, 0.25 μm film) |
| GC temperature program | 50 (2) → 250 (8) at 10 °C/min, 30 min total run time |
| GC-MS transfer line temperature | 350 °C |
| MS ionization | electron impact |
| MS full scanning mass range | 15–500 amu, 1 scan/s |
| Data system | Agilent Technologies ChemStation |
3.5. NMR Spectroscopy
NMR spectra were obtained using a JEOL ECX 400 NMR spectrometer (Jeol, Watchmead, UK) fitted with multinuclear probes. 13C spectra were broad-band decoupled. The pulse delay for 29Si spectra was standardized at 15 s. All spectra were recorded at room temperature (20 °C) using deuterated solvents. The internal NMR reference compound was TMS in all 1H, 13C and 29Si NMR spectra, unless it interfered with the product peaks; where this was the case, the residual solvent peak was used as standard.




{kind=link}
{kind=link}
{kind=link}
{kind=link}