Oligomerization of Ethylene to Produce Linear α-Olefins Using Heterogeneous Catalyst Prepared by Immobilization of α-Diiminenickel(II) Complex into Fluorotetrasilicic Mica Interlayer

Abstract
:1. Introduction
2. Results and Discussion
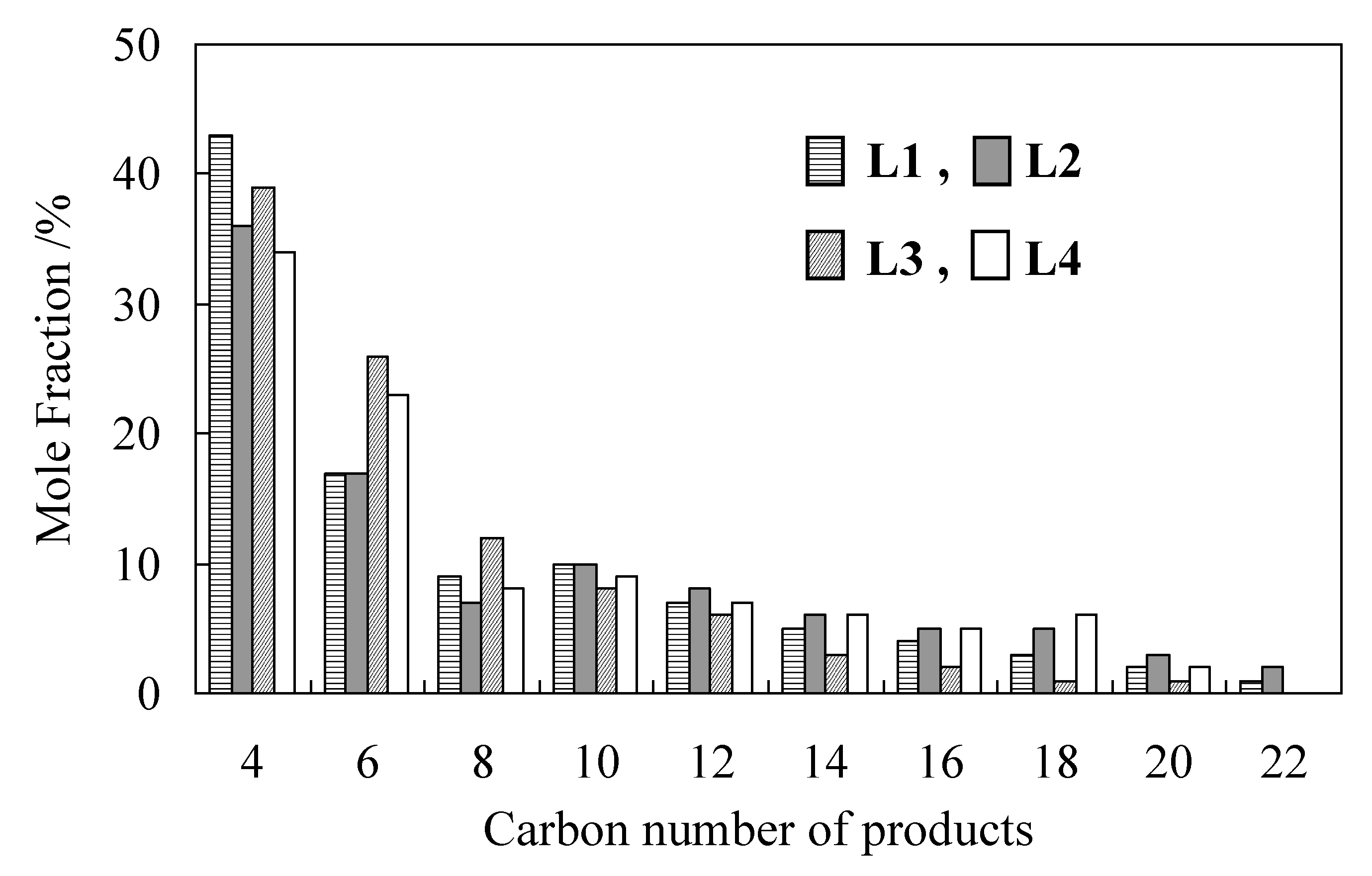
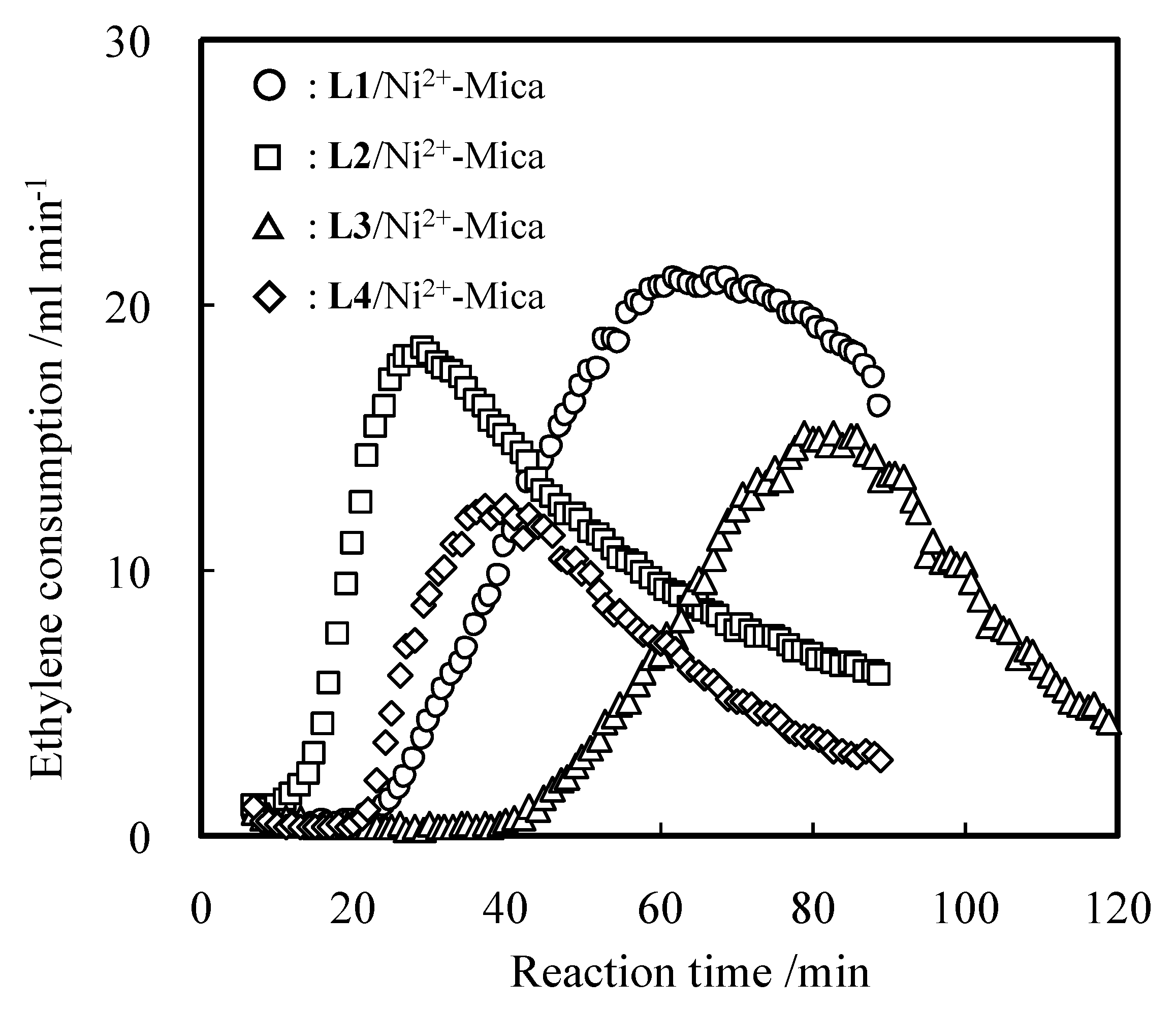
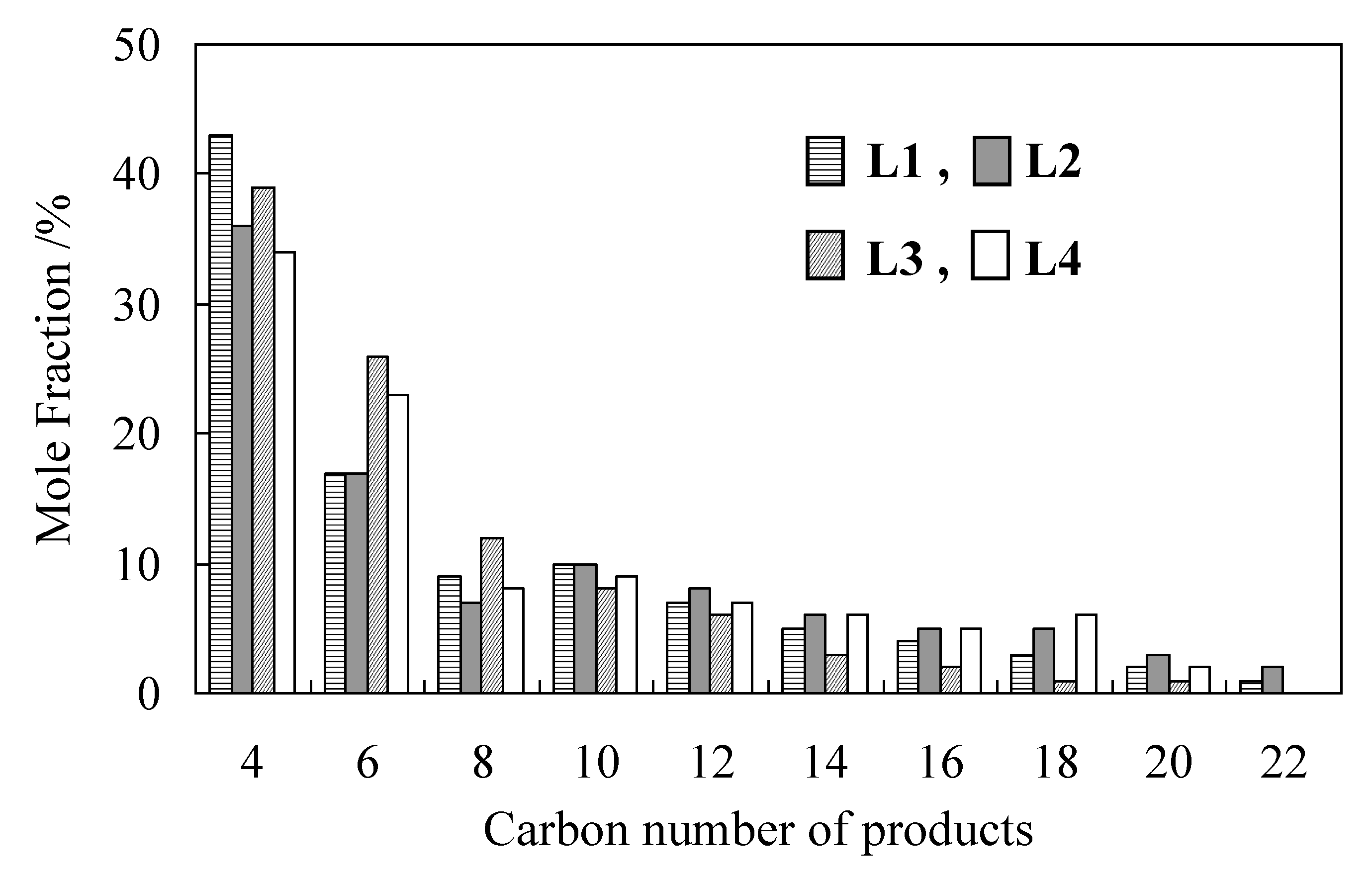
2.1. Oligomerization of Ethylene Using L1–4/Ni2+-Mica Procatalysts
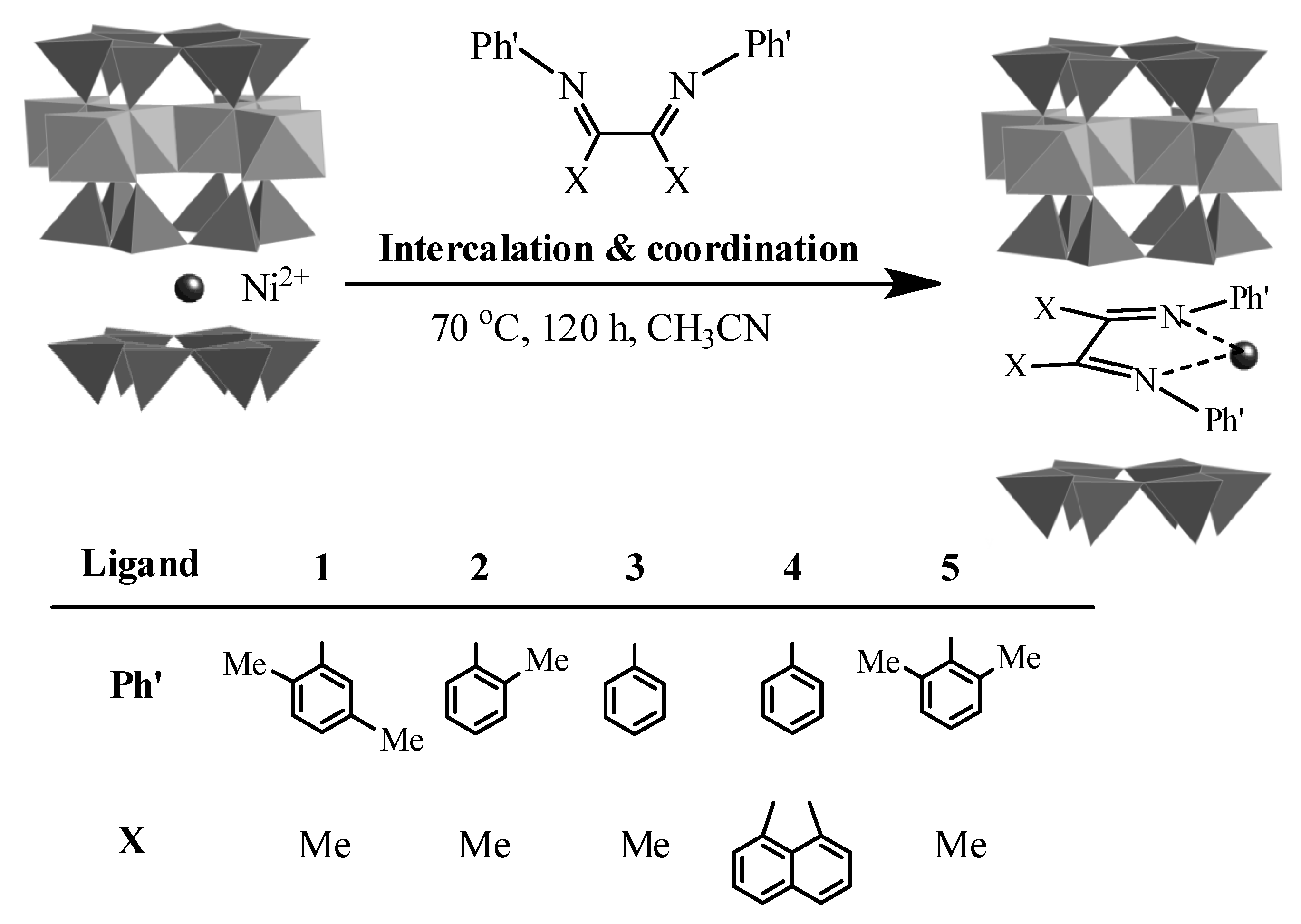

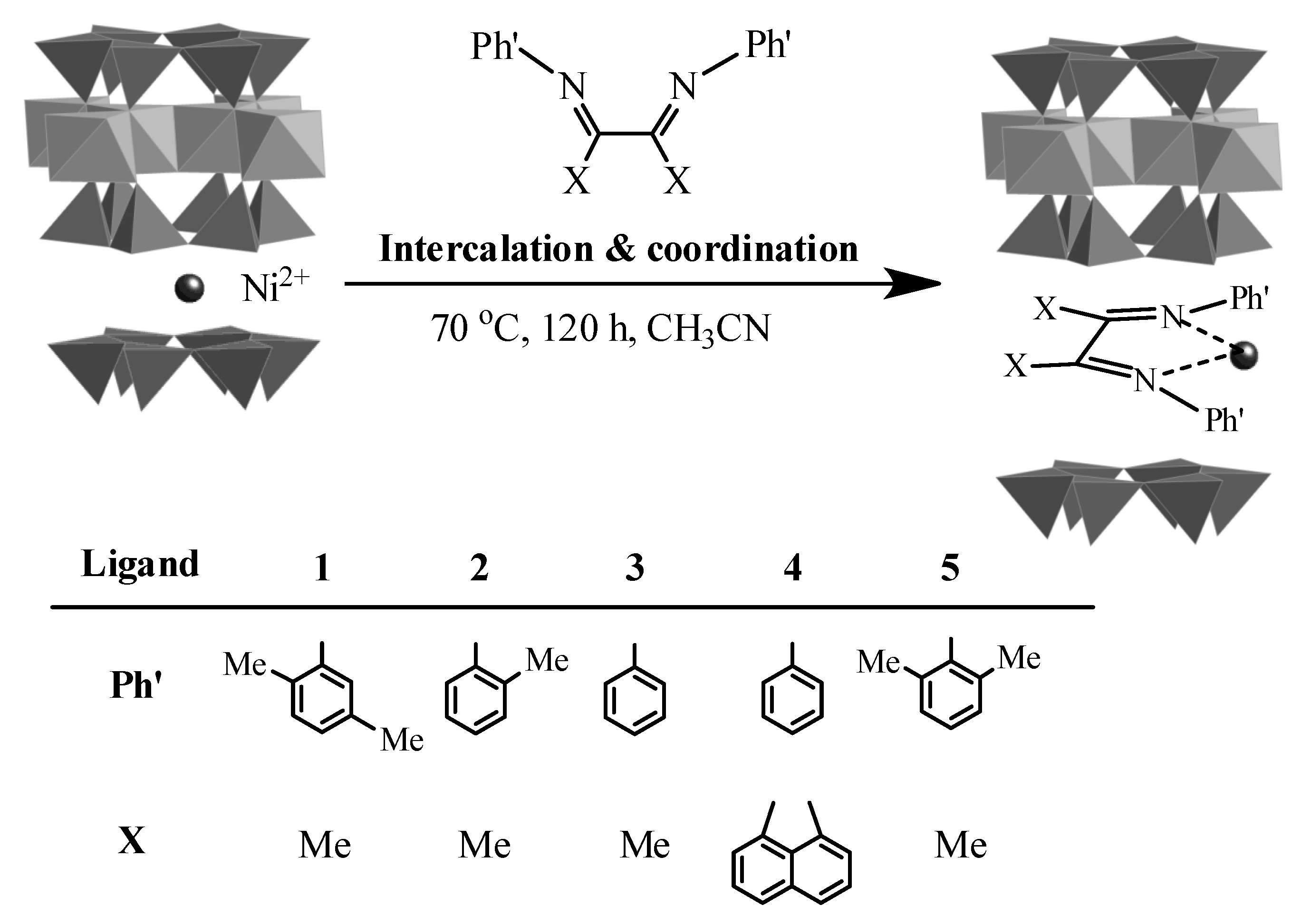{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Run | L | Time | Activity | Product/wt% 2 | Sα 3 | Tm 4 | |
|---|---|---|---|---|---|---|---|
| /h | /g-C2 g-cat−1 h−1 | Solid | C4–C22 | /% | /ºC | ||
| 1 | 1 | 1.5 | 193 | 94.9 | 4.4 | 72.1 | 128 |
| 2 | 2 | 1.5 | 188 | 87.9 | 6.2 | 74.4 | 127 |
| 3 | 3 | 2.0 | 101 | 22.1 | 53.9 | 91.2 | 125 |
| 4 | 4 | 1.5 | 96 | 71.7 | 12.6 | 92.5 | 128 |
| Ref. [15] | 5 | 1.0 | 1804 | 100 | 0 | - | 120 |
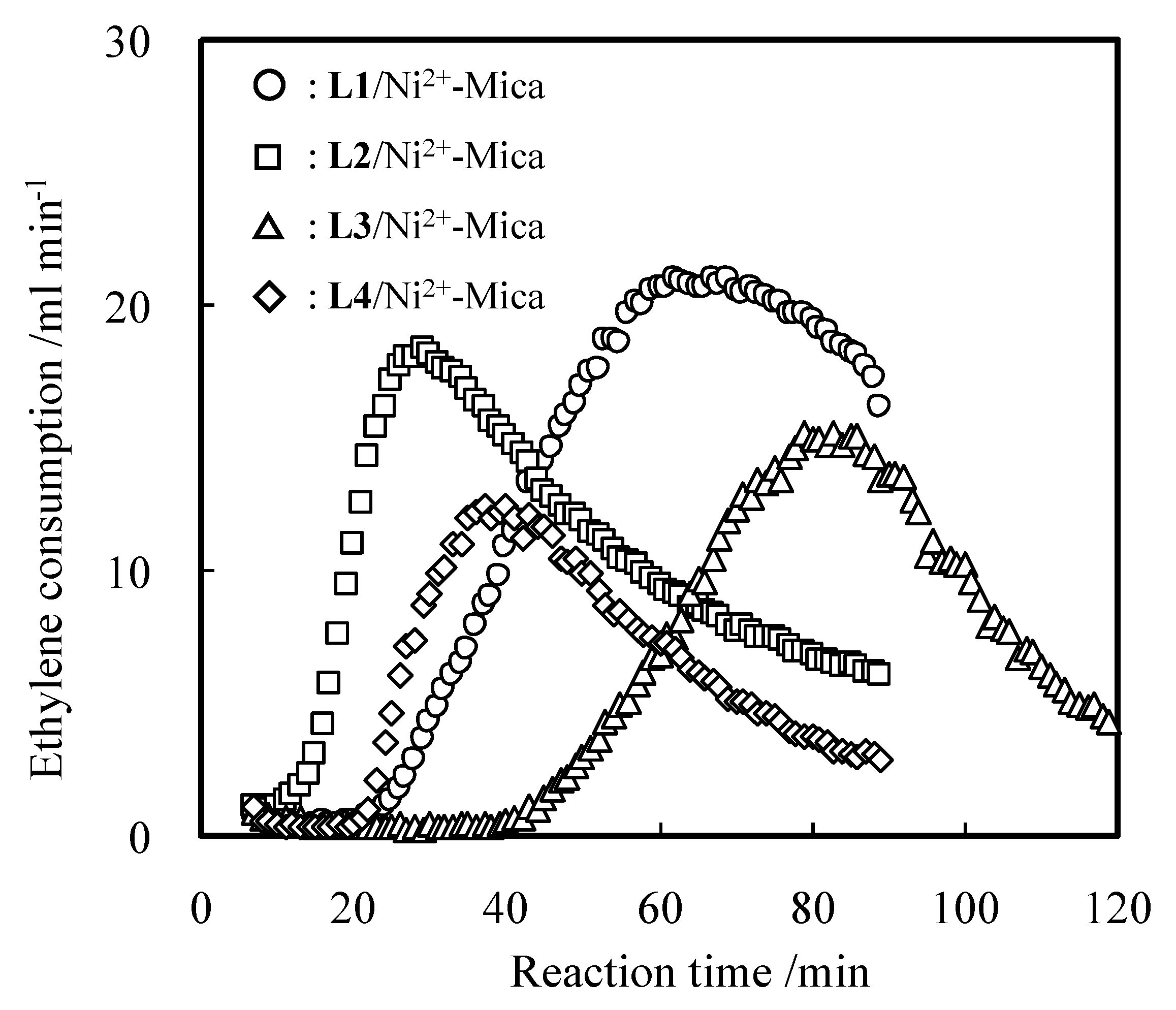
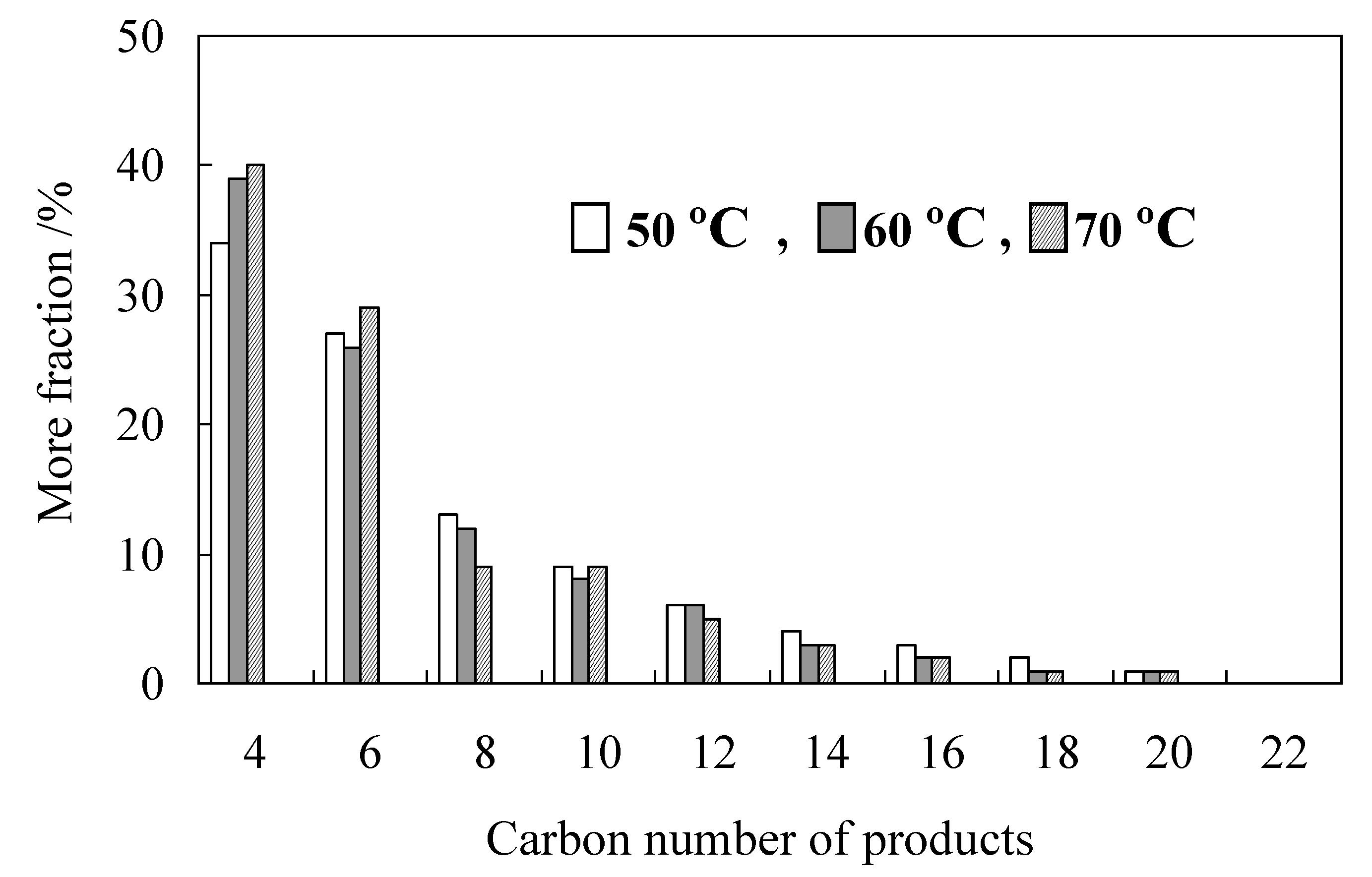
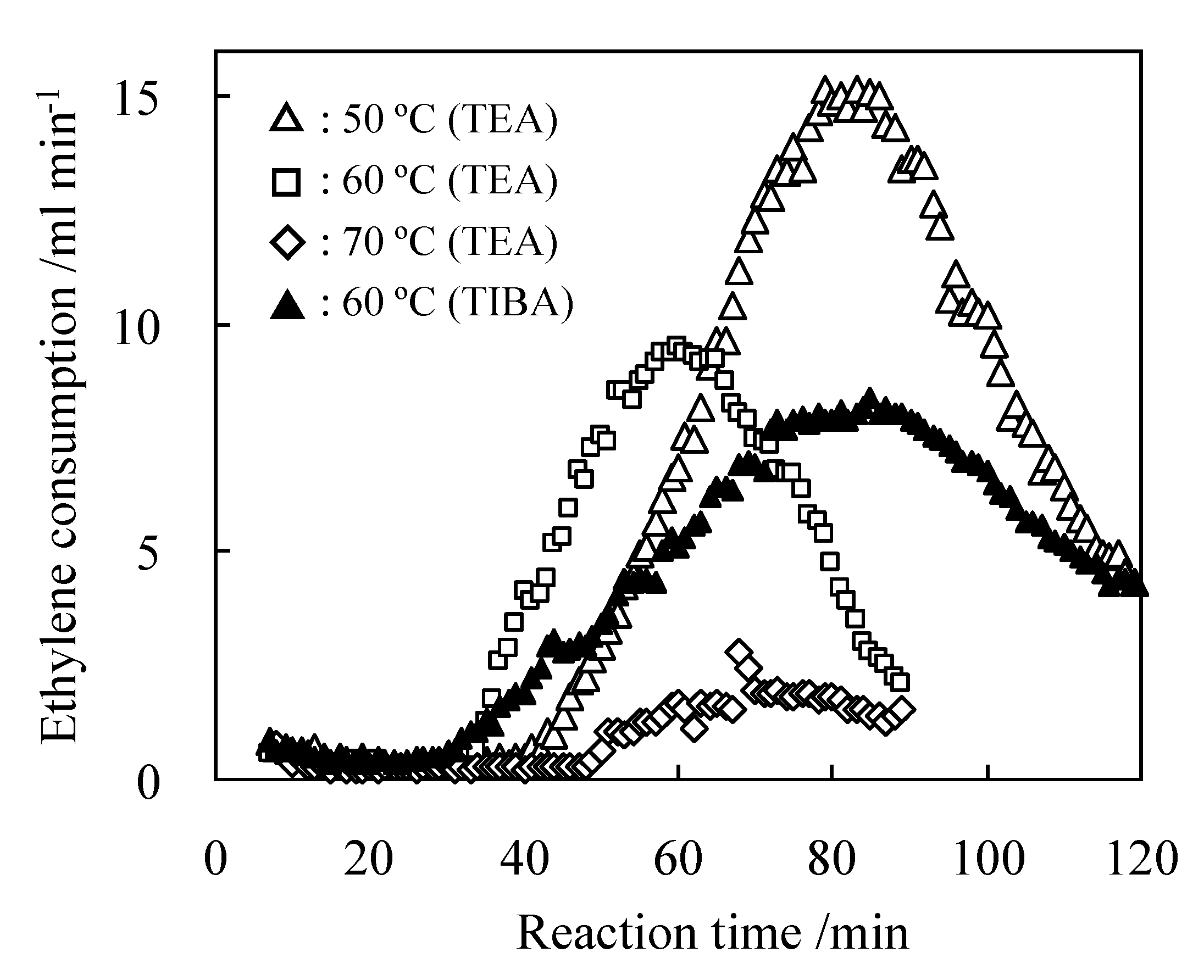
2.2. Detailed Evaluation of L3/Ni2+-Mica Procatalysts
| Run | To 2 | P 3 | Time | Productivity | Product/wt% 4 | Sα 5 | Tm 6 | |
|---|---|---|---|---|---|---|---|---|
| /°C | /bar | /h | /g-C2 g-cat−1 h−1 | Solid | C4–C22 | /% | /°C | |
| 3 | 50 | 0.7 | 2.0 | 101 | 22.1 | 53.9 | 91.2 | 124.5 |
| 5 | 60 | 0.7 | 1.5 | 67 | 15.3 | 61.2 | 88.2 | 122.0 |
| 6 | 70 | 0.7 | 1.5 | 14 | 6.9 | 71.1 | 78.8 | - |
| 7 | 50 | 1.0 | 1.5 | 165 | 25.3 | 54.5 | 93.8 | 124.5 |
| 8 | 50 | 0.4 | 1.5 | 88 | 19.2 | 53.1 | 88.1 | 123.0 |
| 9 7 | 60 | 0.7 | 2.0 | 71 | 9.2 | 83.5 | 87.8 | 123.1 |
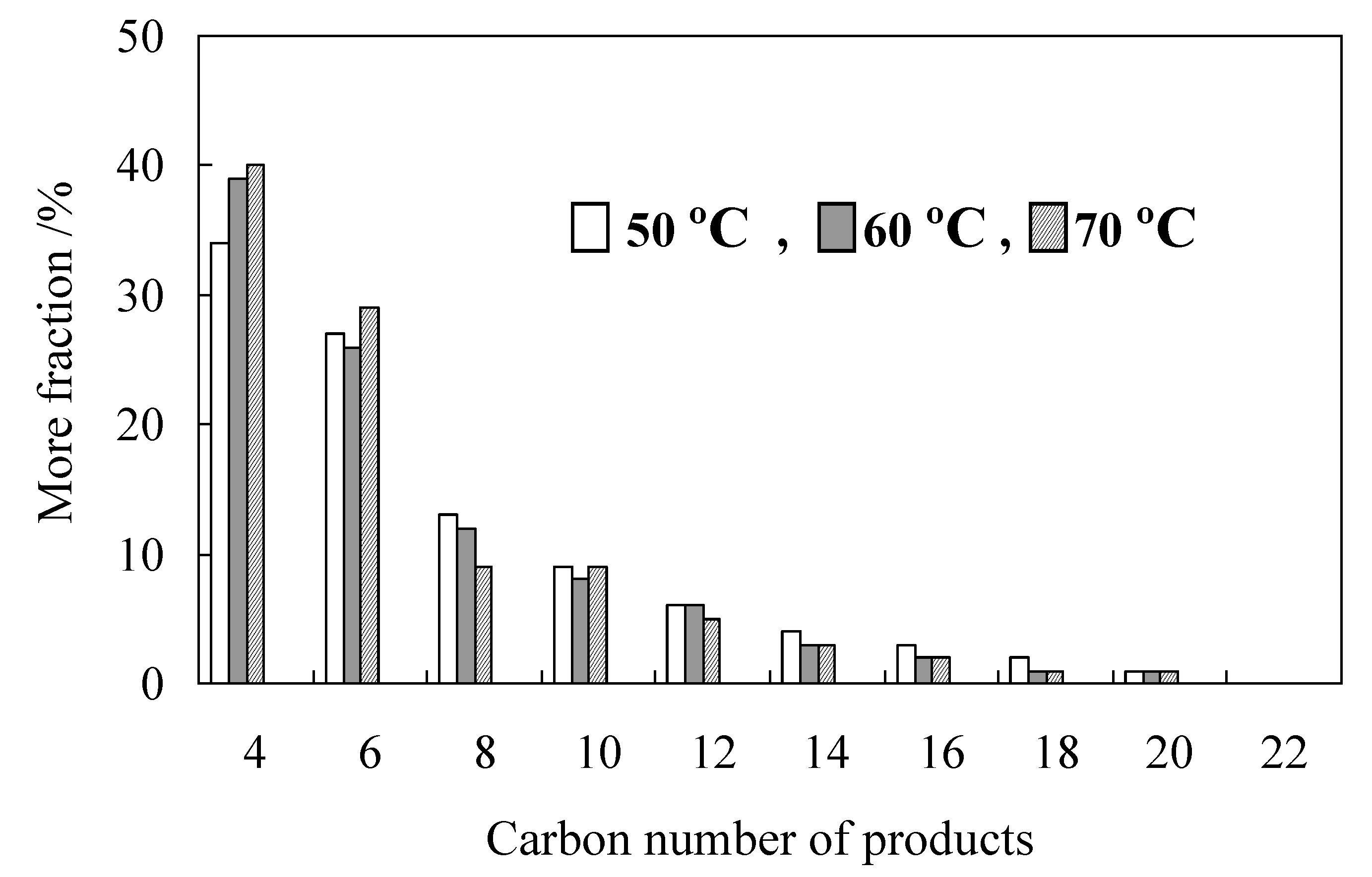
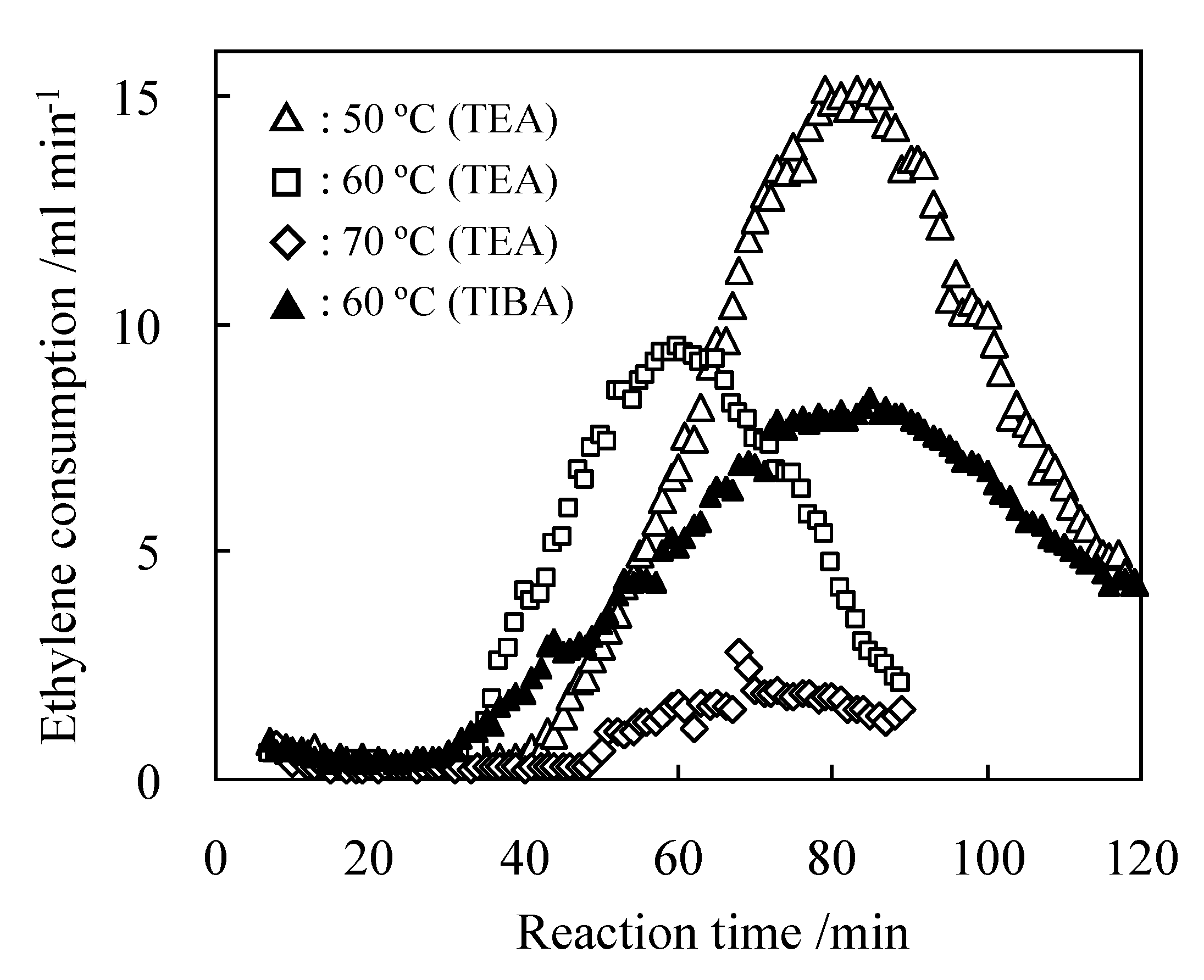
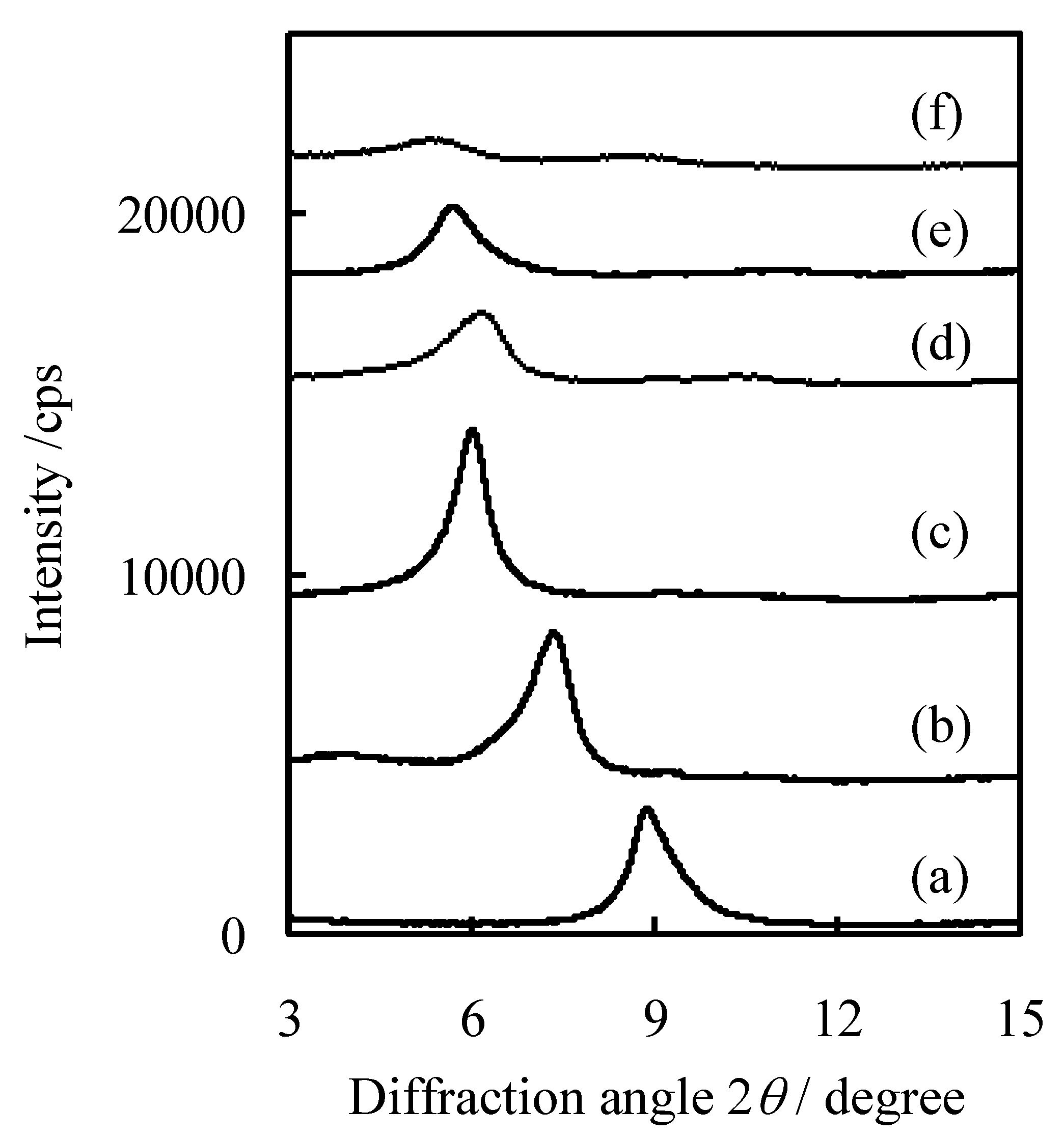
2.3. Characterization of Procatalysts
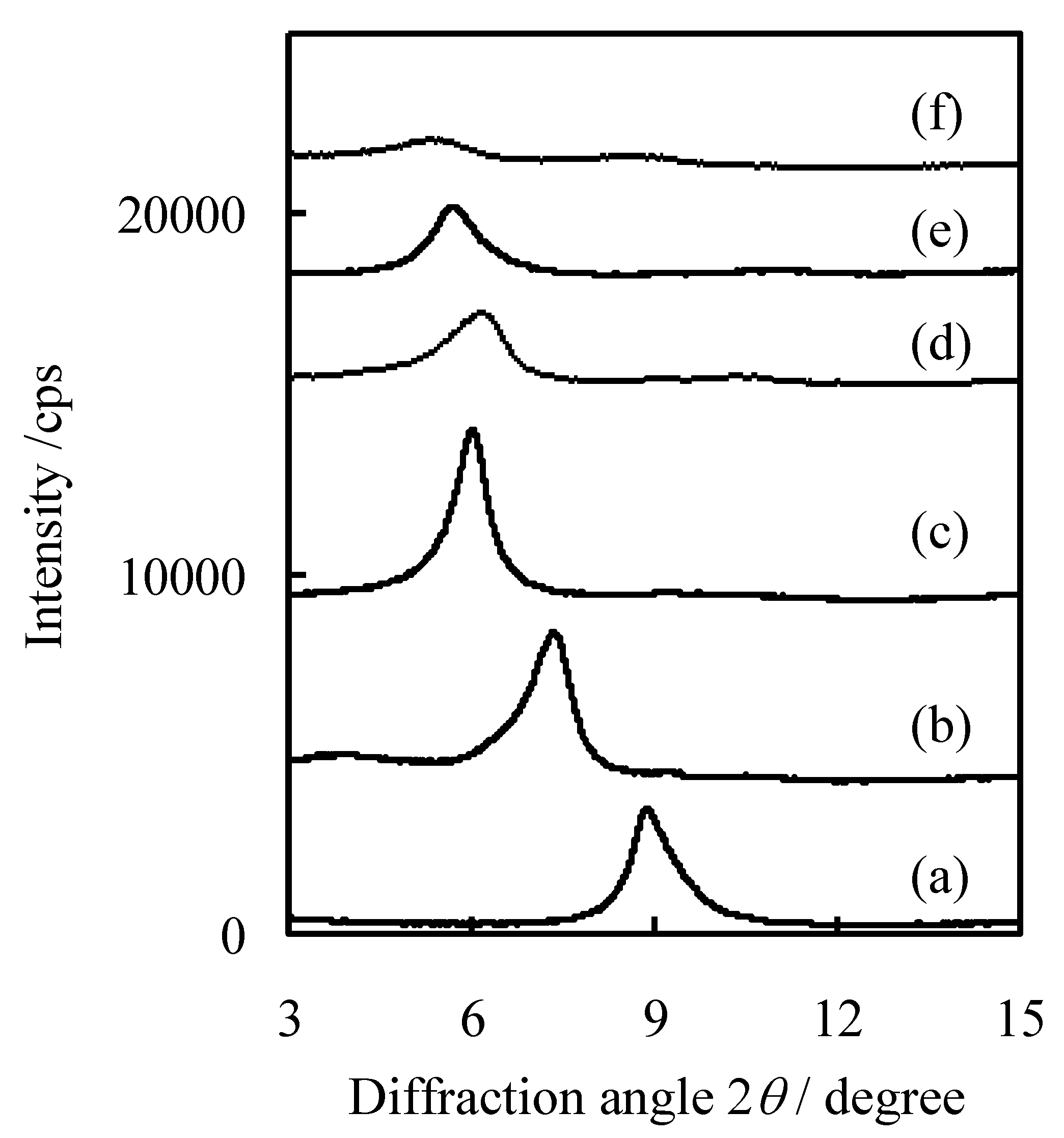
3. Experimental Section
3.1. Ligand Synthesis
3.2. Procatalyst Preparation
3.3. Characterization of Procatalyst
3.4. Ethylene Oligomerization
4. Conclusions
Acknowledgment
Conflict of Interest
References
- Lappin, G.R.; Nemec, L.H.; Sauer, J.D.; Wagner, J.D. Higher Olefins; Kirk-Othmer Encyclopedia of Chemical Technology; Wiley & Sons, Inc.: New York, NY, USA, 2000; Volume 17, pp. 709–728. [Google Scholar]
- Leeuwen, P.W.N.M.; Clément, N.D.; Tschan, M.J.-L. New processes for the selective production of 1-octene. Coordin. Chem. Rev. 2011, 255, 1499–1517. [Google Scholar] [CrossRef]
- Skupinska, J. Oligomerization of α-olefins to higher oligomers. Chem. Rev. 1991, 91, 613–648. [Google Scholar]
- Shiraki, Y.; Nakamoto, Y.; Souma, Y. ZrCl4-TEA-EASC three-component catalyst for the oligomerization of ethylene: the role of organoaluminum co-catalysts and additives. J. Mol. Catal. A 2002, 187, 283–294. [Google Scholar] [CrossRef]
- Killian, C.M.; Johnson, L.K.; Brookhart, M. Preparation of linear α-olefins using cationic nickel(II) α-diimine catalysts. Organometallics 1997, 16, 2005–2007. [Google Scholar] [CrossRef]
- Small, B.L.; Brookhart, M. Iron-based catalysts with exceptionally high activities and selectivities for oligomerization of ethylene to linear α-olefins. J. Am. Chem. Soc. 1998, 120, 7143–7144. [Google Scholar] [CrossRef]
- Svejda, S.A.; Brookhart, M. Ethylene oligomerization and propylene dimerization using cationic (α-diimine)nickel(II) catalysts. Organometallics 1999, 18, 65–74. [Google Scholar]
- Britovsek, G.J.P.; Mastroianni, S.; Solan, G.A.; Baugh, S.P.D.; Redshaw, C.; Gibson, V.C.; White, A.J.P.; Williams, D.J.; Elsegood, M.R.J. Oligomerization of ethylene by bis(imino)pyridyliron and -cobalt complexes. Chem. Eur. J. 2000, 6, 2221–2231. [Google Scholar] [CrossRef]
- Ittel, S.D.; Johnson, L.K.; Brookhart, M. Late-metal catalysts for ethylene homo- and copolymerization. Chem. Rev. 2000, 100, 1169–1203. [Google Scholar] [CrossRef]
- Mecking, S. Olefin polymerization by late transition metal complexes: A root of Ziegler catalysts gains new ground. Angew. Chem. Int. Ed. 2001, 40, 534–540. [Google Scholar] [CrossRef]
- Gibson, B.C.; Spitzmesser, S.K. Advances in non-metallocene olefin polymerization catalysis. Chem. Rev. 2003, 103, 283–315. [Google Scholar]
- Bianchini, C.; Giambastiani, G.; Rios, I.G.; Mantovani, G.; Meli, A.; Segarra, A.M. Olefin oligomerization, homopolymerization and copolymerization by late transition metals supported by (imino)pyridine ligands. Coord. Chem. Rev. 2010, 254, 431–455. [Google Scholar] [CrossRef]
- Song, S.; Xiao, T.; Wang, L.; Redshaw, C.; Wang, F.; Sun, W.-H. 2-(1-Arylimino)quinolylnickel halides: synthesis, characterization and catalytic behavior towards ethylene. J. Organomet. Chem. 2012, 699, 18–25. [Google Scholar]
- Kurokawa, H.; Matsuda, M.; Fujii, K.; Ishihama, Y.; Sakuragi, T.; Ohshima, M.; Miura, H. Bis(imino)pyridine iron and cobalt complexes immobilized into interlayer space of fluorotetrasilicic mica: highly active heterogeneous catalysts for polymerization of ethylene. Chem. Lett. 2007, 36, 1004–1005. [Google Scholar] [CrossRef]
- Fujii, K.; Ishihama, Y.; Sakuragi, T.; Ohshima, M.; Kurokawa, H.; Miura, H. Heterogeneous catalysts immobilizing α-diimine nickel complexes into fluorotetrasilicic mica interlayers to prepare branched polyethylene from only ethylene. Catal. Commun. 2008, 10, 183–186. [Google Scholar]
- Kondo, T.; Yamamoto, K.; Sakuragi, T.; Kurokawa, H.; Miura, H. Acetyliminopyridineiron(III) complexes immobilized in fluorotetrasilicic mica interlayer as efficient catalysts for oligomerization of ethylene. Chem. Lett. 2012, 43, 461–464. [Google Scholar]
- Helldörfer, M.; Backhaus, J.; Milius, W.; Alt, H.G. (α-Diimine)nickel(II) complexes containing chloro substituted ligands as catalyst precursors for the oligomerization and polymerization of ethylene. J. Mol. Catal. A 2003, 193, 59–70. [Google Scholar] [CrossRef]
- Helldörfer, M.; Milius, W.; Alt, H.G. The influence of halogen substituents at the ligand framework of (α-diimine)nickel(II) catalyst precursors on their behavior in ethylene oligomerization and polymerization. J. Mol. Catal. A 2003, 197, 1–13. [Google Scholar] [CrossRef]
- Benito, J.M.; de Jesús, E.; de la Mata, F.J.; Flores, J.C.; Gómez, R.; Cómez-Sal, P. Mononuclear and dendritic nickel(II) complexes containing N,N'-iminopyridine chelating ligands: generation effects on the catalytic oligomerization and polymerization of ethylene. Organometallics 2006, 25, 3876–3887. [Google Scholar] [CrossRef]
- Kurokawa, H.; Nakazato, Y.; Tahara, S.; Katakura, T.; Ishihama, Y.; Sakuragi, T.; Miura, H. Copolymerization of ethylene with vinyl monomers using heterogeneous catalysts consisting of α-diimine Ni (II) complexes immobilized into a fluorotetrasilicic mica interlayer in the presence of an alkylaluminum compound. Macromol. React. Eng 2013, in press. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kurokawa, H.; Miura, K.; Yamamoto, K.; Sakuragi, T.; Sugiyama, T.; Ohshima, M.-a.; Miura, H. Oligomerization of Ethylene to Produce Linear α-Olefins Using Heterogeneous Catalyst Prepared by Immobilization of α-Diiminenickel(II) Complex into Fluorotetrasilicic Mica Interlayer. Catalysts 2013, 3, 125-136. https://doi.org/10.3390/catal3010125
Kurokawa H, Miura K, Yamamoto K, Sakuragi T, Sugiyama T, Ohshima M-a, Miura H. Oligomerization of Ethylene to Produce Linear α-Olefins Using Heterogeneous Catalyst Prepared by Immobilization of α-Diiminenickel(II) Complex into Fluorotetrasilicic Mica Interlayer. Catalysts. 2013; 3(1):125-136. https://doi.org/10.3390/catal3010125
Chicago/Turabian StyleKurokawa, Hideki, Kazuki Miura, Kazuhiro Yamamoto, Tsutomu Sakuragi, Takao Sugiyama, Masa-aki Ohshima, and Hiroshi Miura. 2013. "Oligomerization of Ethylene to Produce Linear α-Olefins Using Heterogeneous Catalyst Prepared by Immobilization of α-Diiminenickel(II) Complex into Fluorotetrasilicic Mica Interlayer" Catalysts 3, no. 1: 125-136. https://doi.org/10.3390/catal3010125