3.2. Synthesis
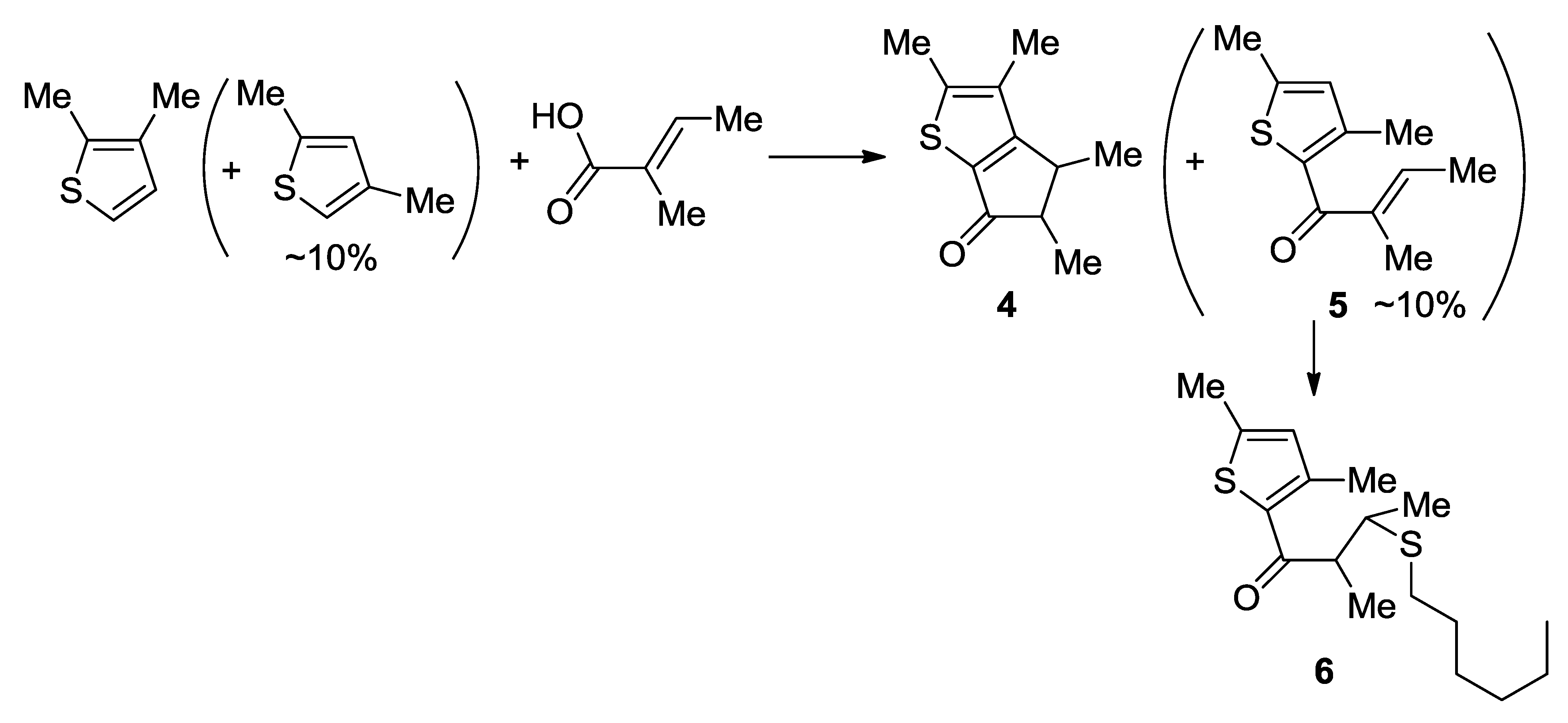
Large-Scale Synthesis of Compound 4: A CH2Cl2 solution (18 mL) containing dissolved 2,3-dimethylthiophene contaminated with 2,4-dimethylthiophene (30.0 g, 267 mmol) and tiglic acid (26.7 g, 267 mmol) was added dropwise to polyphosporic acid (220 g, 85%) with a syringe pump at 50 °C for 2 h, and then the mixture was stirred at 50 °C for 2 h. Ice (350 g) was added and the product was extracted with diethyl ether (4 × 100 mL). The combined extracts were washed with saturated aqueous Na2CO3 (160 mL) and dried over anhydrous MgSO4. The solvent was removed by using a rotary evaporator to give an oily residue (56 g), which was found by analysis of the 1H NMR spectrum to be a mixture of 4 and 5 in 6:1 ratio. Hexanethiol (l7.3 g, 600 mmol) and tetrabutylammonium fluoride (3.20 g, 12.2 mmol) were added to the crude mixture of 4 (~48.0 g, 247 mmol) and 5 (~8.0 g, 41.1 mmol). The neat mixture was stirred for 30 min at room temperature, and then, the flask was connected to a vacuum distillation set. Compound 4 was distilled out at 85 °C at reduced pressure (0.15 mmHg), whereas the thiol-added compound 6 remained in the distillation pot (39.0 g, 75%). 1H NMR (CDCl3): 3.32 (quintet, J = 7.2 Hz, 0.5H), 3.00 (quintet, J = 7.2 Hz, 0.5H), 2.80 (qd, J = 7.2, 3.2 Hz, 0.5H), 2.41 (qd, J = 7.2, 3.2 Hz, 0.5H), 2.40 (s, 3H), 2.12 (s, 1.5H), 2.11 (s, 1.5H), 1.34 (d, J = 7.2 Hz, 1.5H), 1.27 (d, J = 7.2 Hz, 1.5H), 1.17 (d, J = 7.2 Hz, 1.5H), 1.15 (d, J = 7.2 Hz, 3H) ppm. 13C{1H}17R (CDCl3): 199.04, 198.73, 172.94, 171.45, 150.83, 150.43, 134.35, 134.16, 130.06, 129.83, 55.70, 50.20, 40.71, 35.31, 18.86, 15.93, 15.89, 15.16, 15.08, 11.95, 11.80, 11.54 ppm.
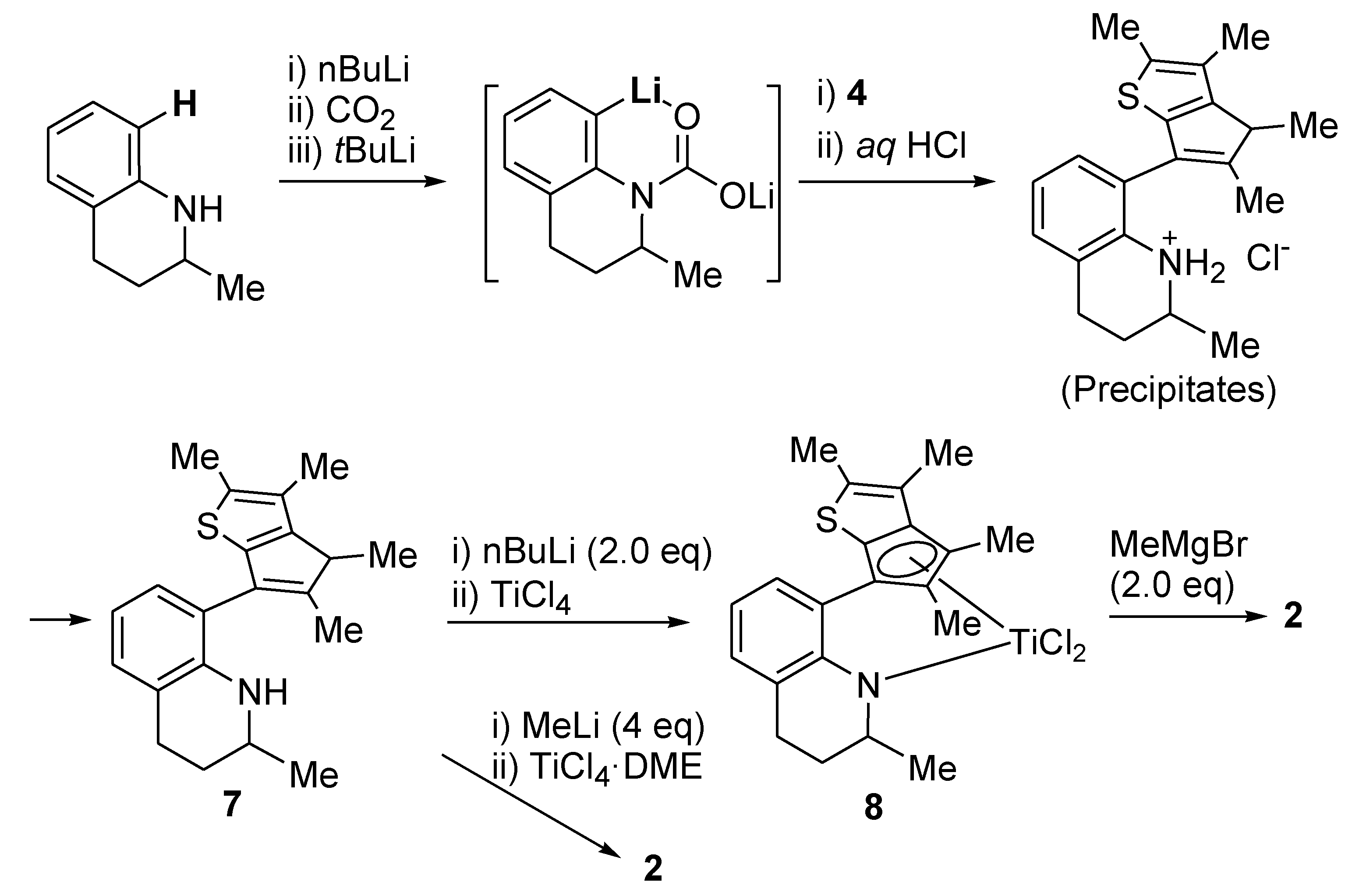
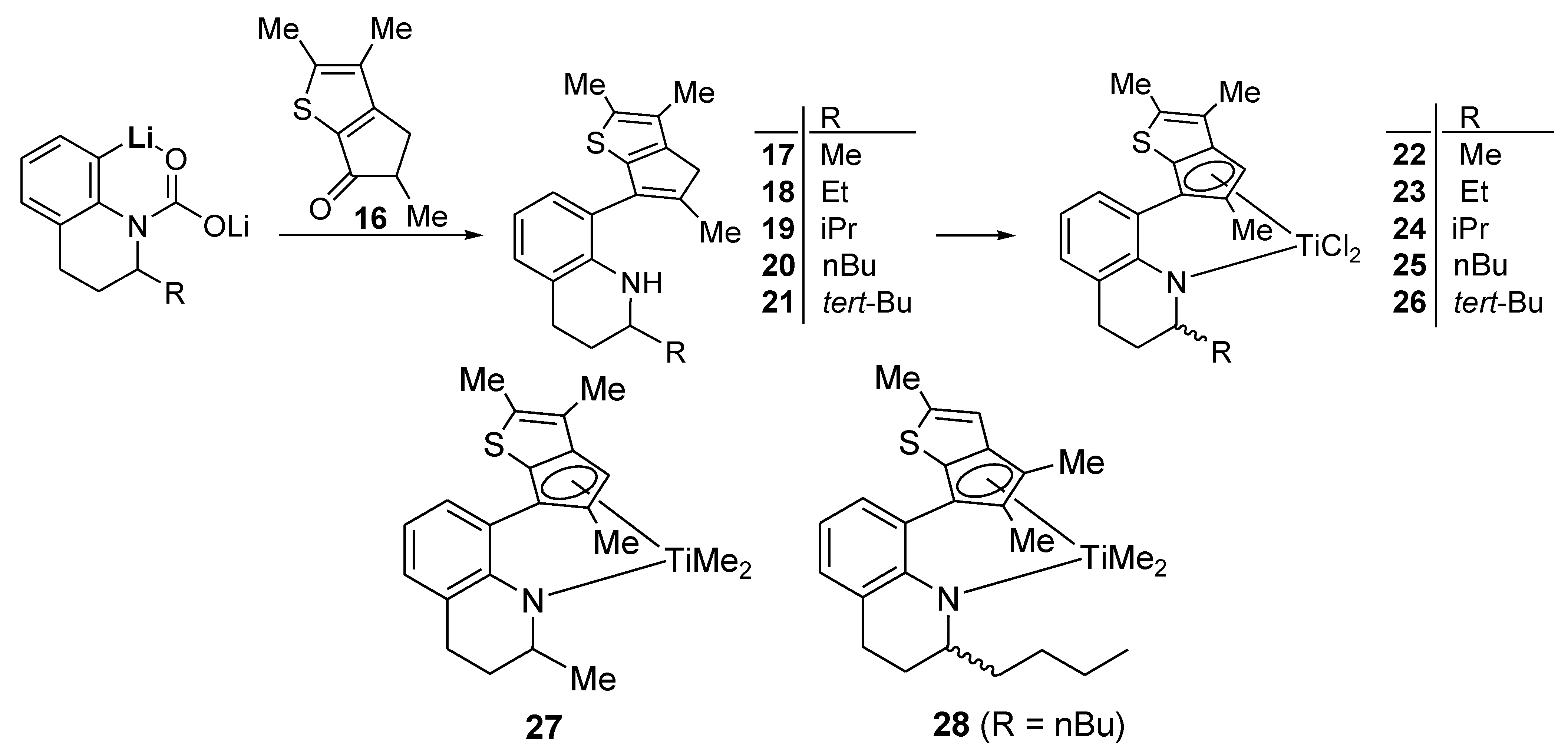
Large-Scale Synthesis of Compound 7: nBuLi (54.3 mL, 136 mmol, 2.5 M solution in hexane) was added dropwise to a solution of 1,2,3,4-tetrahydroquinaline (20.0 g, 136 mmol) in hexane (280 mL) at room temperature. When the solution was stirred at room temperature overnight, a white solid precipitated, which was filtered and washed with hexane. The lithium amide compound was formed in quantitative yield (21.0 g). A diethyl ether solution (200 mL) containing the lithium amide compound (22.4 g, 115 mmol) was stirred at −78 °C and CO2 gas was added. The white solid disappeared immediately. After stirring of the solution for 40 min at −78 °C, the temperature was raised slowly to room temperature while excess CO2 gas was removed through a bubbler, and the solution was stirred overnight. A white solid precipitated again. THF (9.80 g, 136 mmol) and tert-BuLi (80.0 mL, 136 mmol, 1.7 M solution in pentane) were added successively to the slurry at −20 °C, and the solution was stirred for 2 h at this temperature. A diethyl ether solution (200 mL) with dissolved 4 (22.4 g, 115 mmol) was added to the ortho-lithiated compound using a syringe at −20 °C. The solution was stirred for 1 h at −20 °C and subsequently warmed slowly to room temperature. After stirring the solution overnight, aqueous 2M HCl (250 mL) was added at 0 °C and the mixture was stirred at room temperature for 30 min. The product was extracted with diethyl ether (3 × 100 mL). The organic phases were collected, and the solvent was removed with a rotary evaporator. Aqueous HCl solution (108 mL, 6 N) was added to the solution of crude products (~60 g) in hexane (600 mL) under vigorous stirring. The precipitated white solid was collected by filtration and washed with hexane (~300 mL). The solid was suspended in ethyl acetate (500 mL), and neutralized with saturated aqueous Na2CO3 solution (300 mL). The organic phase was collected and dried over anhydrous MgSO4. Removal of the solvent with a rotary evaporator gave a yellow viscous oil (52 g, 65%), which was pure enough to be used for the metallation without further purification.
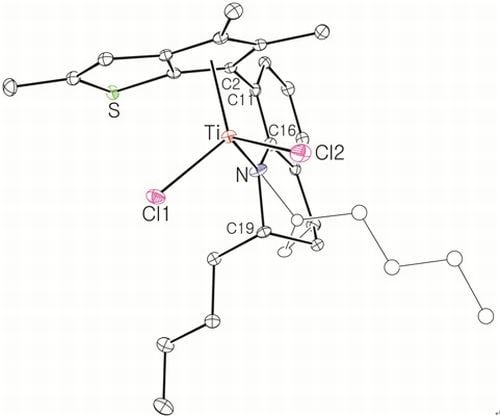
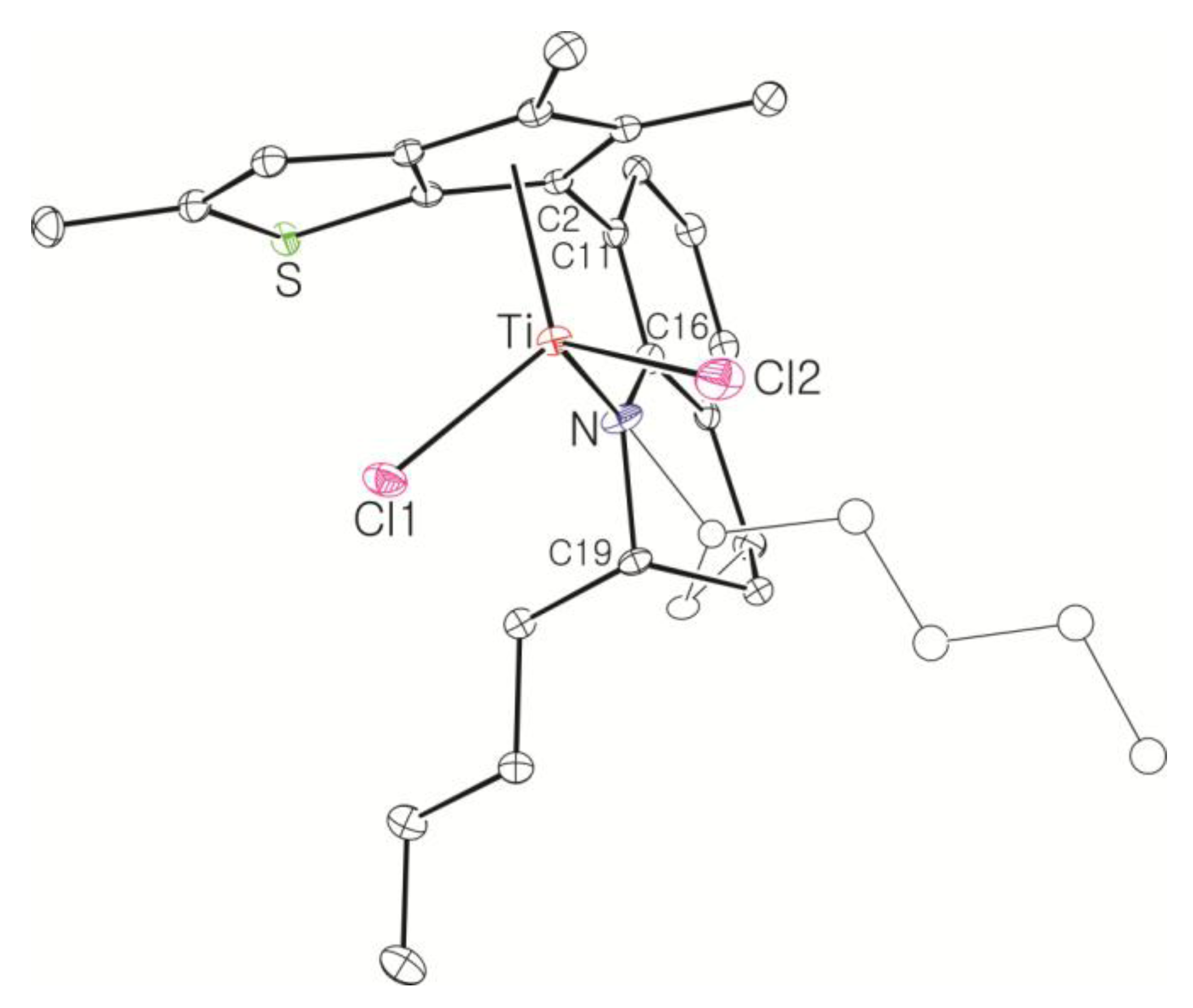
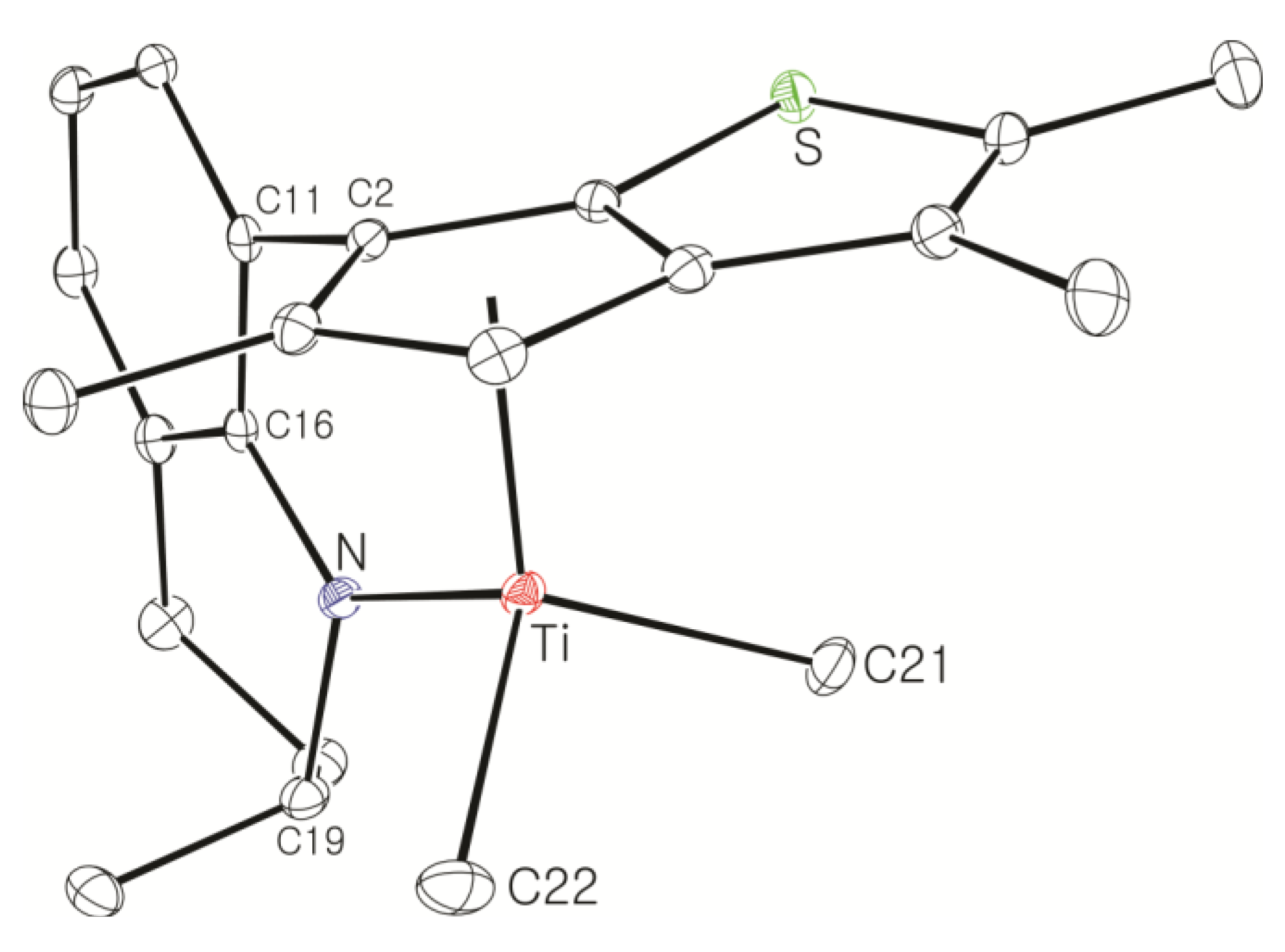
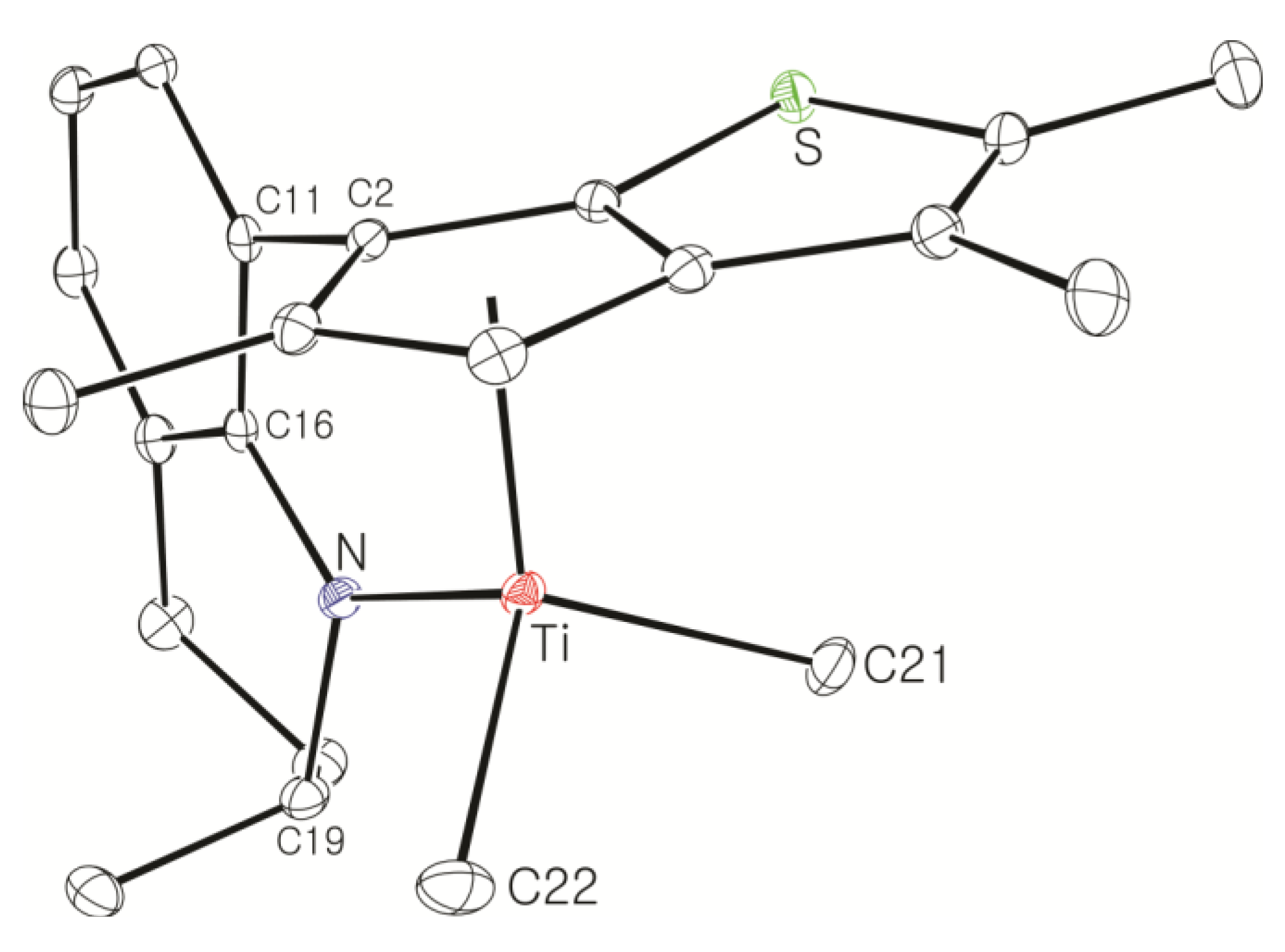
Large-Scale Synthesis of Complex 8: nBuLi (49.5 mL, 2.5 M in hexane, 120 mmol) was added dropwise to a stirred solution of 7 (20.0 g, 61.6 mmol) in hexane (100 mL), and then the solution was stirred overnight at room temperature. A white solid was deposited. Diethyl ether (54 mL) was added at −30 °C to dissolve the deposited solid. Diethyl ether (240 mL) was slowly added to another flask containing TiCl4 in toluene solution (11.7 g, 61.6 mmol) at −30 °C and the resulting solution was stirred for 1 h at room temperature to form slurry. The ether solution of the dilithiated compound was added in one portion to the flask containing the TiCl4 slurry at −30 °C. After stirring of the solution for 6 h at room temperature, the solution was filtered through Celite. The solvent was removed under vacuum to give a brown solid, which was triturated in hexane. The red powder was isolated by filtration (21.0 g, 78%). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.8 ratio. 1H NMR (C6D6): δ 7.10 (t, J = 4.4 Hz, 1H), 6.90 (d, J = 4.4 Hz, 2H), 5.27 and 5.22 (m, 1H, NCH), 2.54–2.38 (m, 1H, CH2), 2.20–2.08 (m, 1H, CH2), 2.36 and 2.35 (s, 3H), 2.05 and 2.03 (s, 3H), 1.94 and 1.93 (s, 3H), 1.89 and 1.84 (s, 3H), 1.72–1.58 (m, 2H, CH2), 1.36–1.28 (m, 2H, CH2), 1.17 and 1.14 (d, J = 6.4, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 162.78, 147.91, 142.45, 142.03, 136.91, 131.12, 130.70, 130.10, 128.90, 127.17, 123.39, 121.33, 119.87, 54.18, 26.48, 21.74, 17.28, 14.46, 14.28, 13.80, 13.27 ppm. Anal. Calcd. (C21H23Cl2NSTi): C, 57.29; H, 5.27; N, 3.18%. Found: C, 57.42; H, 5.51; N, 3.46%.
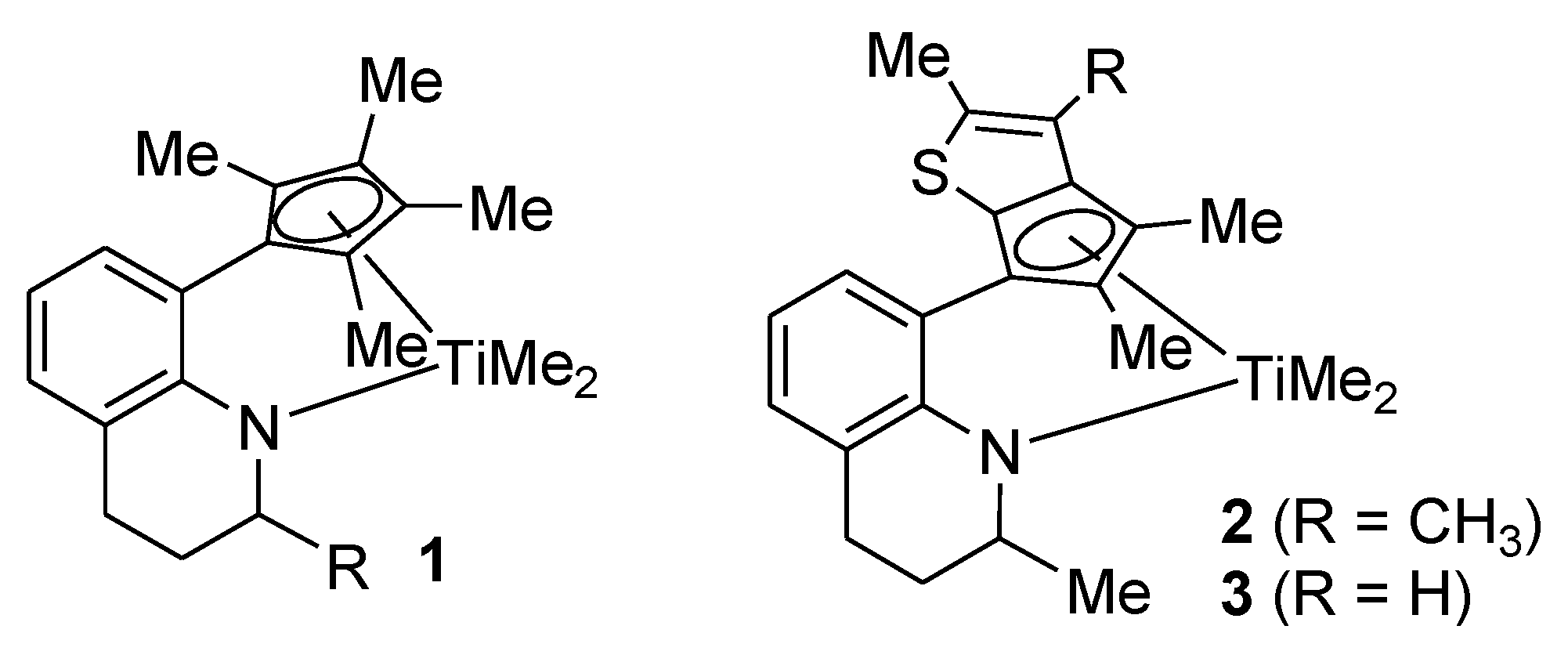
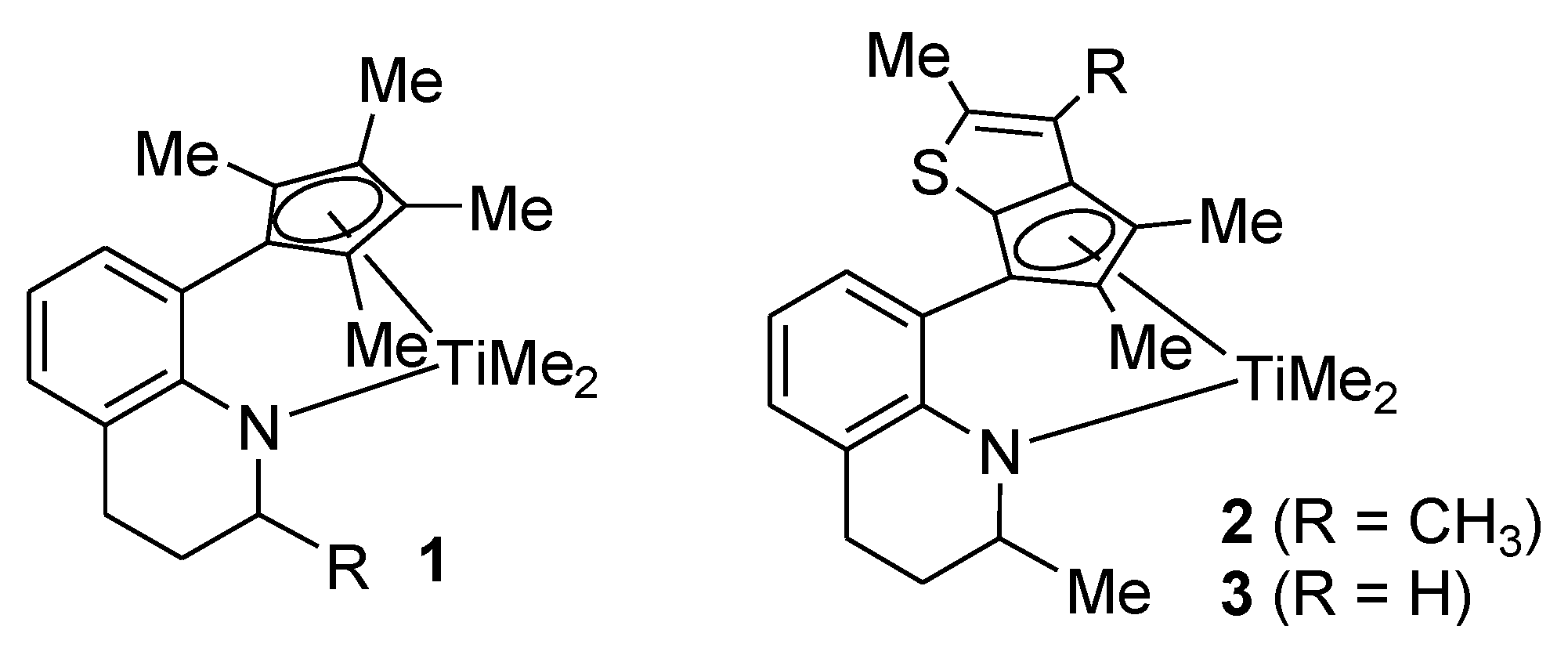
Complex 2: MeMgBr (0.235 g, 0.680 mmol, 3.0 M in diethyl ether) was added dropwise to a stirred solution of 8 (0.150 g, 0.340 mmol) in diethyl ether (2 mL) at −30 °C. After stirring of the solution for 4 h at room temperature, the solvent was removed under vacuum. The resulting residue was dissolved in hexane (5 mL) and filtered through Celite. After all the volatiles were removed under vacuum, the residue was triturated in hexane (~1 mL). The red solid was isolated by decantation (0.12 g, 87%). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.8 ratio. 1H NMR (C6D6): δ 7.13 and 7.10 (d, J = 7.2 Hz, 1H), 6.96 and 6.94 (d, J = 7.2 Hz, 1H), 6.82 and 6.81 (t, J = 7.2 Hz, 1H), 5.45 (m, 1H, NCH), 2.75–2.60 (m, 1H, CH2), 2.45–2.20 (m, 1H, CH2), 2.34 and 2.30 (s, 3H), 2.10 (s, 3H), 1.97 (s, 3H), 1.75 and 1.66 (s, 3H), 1.85–1.50 (m, 2H, CH2), 1.20 (d, J = 6.8 Hz, 3H), 0.76 and 0.72 (s, 3H, TiMe), 0.44 and 0.35 (s, 3H, TiMe) ppm. 13C{1H} NMR (C6D6): 160.13, 159.86, 141.33, 140.46, 138.39, 137.67, 136.74, 134.83, 131.48, 129.90, 129.78, 127.69, 127.65, 127.60, 127.45, 126.87, 126.81, 121.34, 121.23, 120.21, 120.15, 119.15, 118.93, 114.77, 111.60, 57.54, 55.55, 55.23, 51.73, 50.43, 50.36, 27.83, 27.67, 22.37, 22.31, 20.53, 20.26, 14.29, 13.51, 13.42, 13.06, 12.80 ppm.
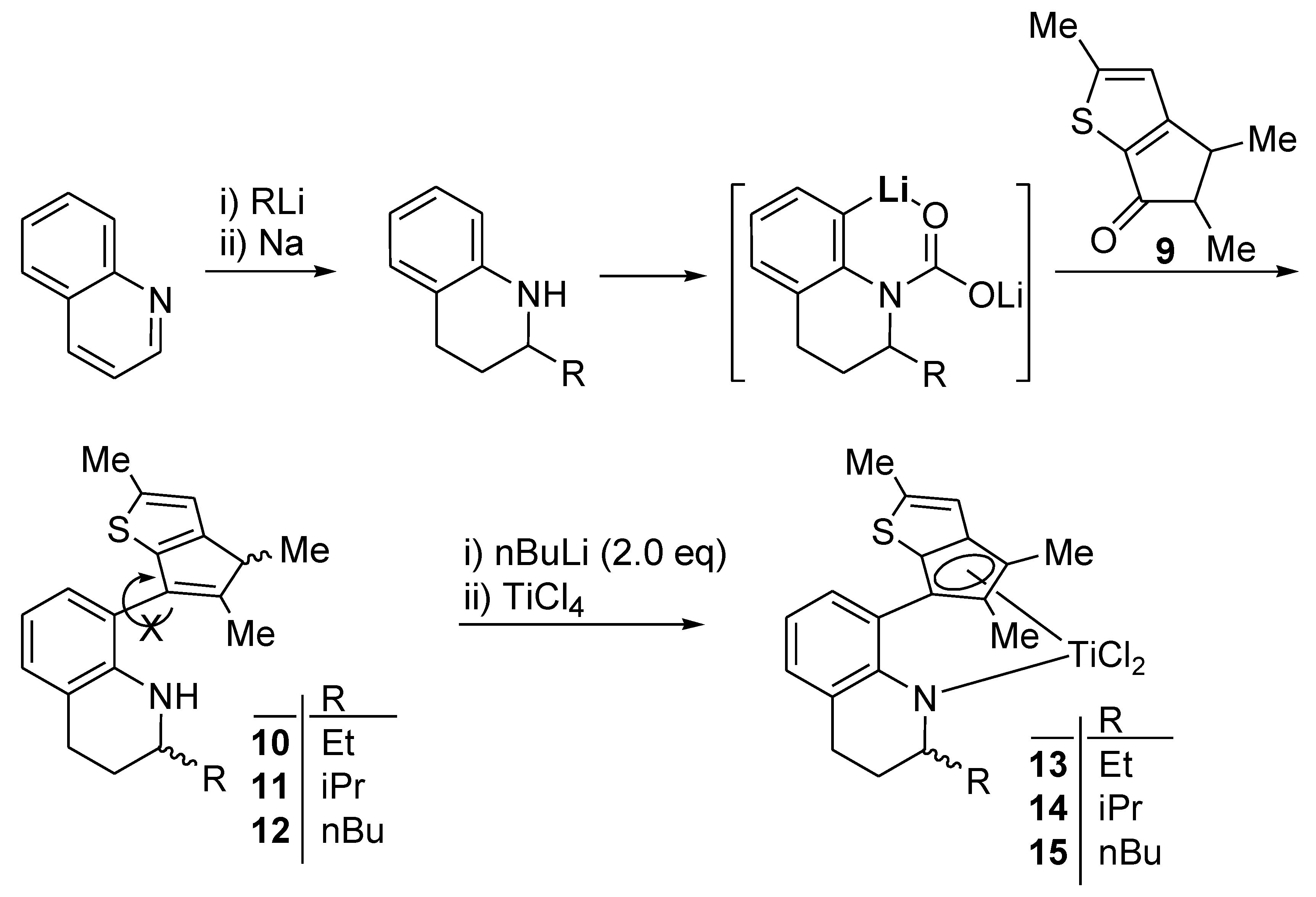
Compound 10: nBuLi (17.0 mL, 27.3 mmol, 1.6 M solution in hexane) was added dropwise to a solution of 2-ethyl-1,2,3,4-tetrahydroquinoline (4.00 g, 24.8 mmol) in hexane (34 mL) at room temperature. When the solution was stirred at room temperature overnight, a white solid precipitated, which was filtered and washed with hexane. The lithium amide compound was formed in quantitative yield (4.02 g). THF (6 mL) was added dropwise to a flask containing the lithium amide compound (0.440 g, 2.63 mmol) at −78 °C. A white suspension was formed, to which CO2 gas was added at −78 °C. The white solid was dissolved immediately by the addition of CO2 gas. After stirring for 1 h at −78 °C, the temperature was slowly raised to 0 °C while excess CO2 gas was removed through a bubbler. The solvent was removed by vacuum, and then diethyl ether (6 mL) was added. After being cooled to −20 °C, THF (0.40 mL) and tert-BuLi (1.70 mL, 2.89 mmol, 1.7 M solution in pentane) were added successively to the slurry. The solid was dissolved through treatment with tert-BuLi, and the resulting solution was stirred for 2 h at −20 °C. Compound 9 (0.403 g, 2.24 mmol) dissolved in diethyl ether (6 mL) was added dropwise, and the resulting solution was stirred for 1 h at −20 °C. After stirring of the solution overnight, aqueous HCl (2 N, 13 mL) was added to the solution at 0 °C. The two-phase solution was stirred for 30 min at room temperature. The organic phase was collected and the water phase was extracted further with ethyl acetate (3 × 15 mL).The collected organic phases were washed with aqueous saturated NaHCO3 (20 mL) and dried over anhydrous MgSO4. The solvent was removed with a rotary evaporator to give a residue which was purified by column chromatography on silica gel, eluting with hexane and ethyl acetate (v/v, 50:1). The product was obtained as a pale yellow viscous oil (0.41 g, 48%). 1H NMR (CDCl3): δ 7.05 and 7.01 (d, J = 8.0 Hz, 1H), 6.97 (d, J = 7.2 Hz, 1H), 6.75 (s, 1H), 6.68–6.58 (m, 1H), 4.20–3.90 (m, 1H, NH), 3.35–3.10 (m, 2H, NCHMe, CHMe), 3.00–2.75 (m, 2H, CH2), 2.52 (s, 3H, CH3), 1.97 and 1.96 (s, 3H, CH3), 1.75–1.55 (m, 2H, CH2), 1.55–1.40 (m, 2H, CH2), 1.37, 1.36, 1.35 and 1.33 (s, 3H, CH3), 0.91 (t, J = 7.2 Hz, 3H, CH3) ppm. 13C{1H} NMR (CDCl3): 150.80, 150.70, 145.83, 145.71, 145.30, 143.57, 143.31, 143.01, 141.95, 139.22, 139.08, 131.40, 131.18, 130.96, 130.69, 128.58, 127.83, 127.57, 127.46, 127.21, 121.30, 120.02, 119.99, 119.80, 119.71, 119.34, 115.91, 53.45, 53.39, 53.17, 53.08, 45.88, 45.74, 45.66, 45.56, 29.99, 29.86, 29.59, 28.10, 27.71, 27.34, 27.19, 27.13, 27.07, 26.94, 16.38, 16.35, 16.00, 14.34, 14.18, 13.87, 10.54, 10.39, 10.37 ppm. HRMS(EI): m/z Calcd. ([M+] C21H25NS) 323.1708. Found: 323.1709.
Compound 11: The compound was synthesized using the same conditions and procedure as those for 10 with 2-isopropyl-1,2,3,4-tetrahydroquinaline (0.480 g, 2.68 mmol) and 9 (0.410 g, 2.28 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). The light yellow viscous oil was obtained in 53% yield (0.41 g). 1H NMR (CDCl3): δ 7.07 and 7.03 (d, J = 7.2 Hz, 1H), 6.97 (d, J = 7.2 Hz, 1H), 6.75 (s, 1H), 6.68–6.58 (m, 1H), 4.20–3.83 (m, 1H, NH), 3.34–3.16 (m, 1H, NCHMe), 3.12–2.96 (m, 1H, CHMe), 2.96–2.76 (m, 2H, CH2), 2.53 (s, 3H, CH3), 1.99, 1.98, 1.97 and 1.96 (s, 3H, CH3), 2.04–1.92 (m, 1H, CH), 1.80–1.54 (m, 2H, CH2), 1.37, 1.36, 1.35 and 1.34 (s, 3H, CH3), 1.00–0.80 (m, 6H, CH3) ppm. 13C{1H} NMR (CDCl3): 150.80, 150.66, 150.60, 145.96, 145.76, 145.21, 143.75, 143.51, 143.48, 143.21, 142.35, 142.22, 139.19, 139.06, 139.02, 131.23, 130.90, 130.66, 128.52, 128.43, 127.72, 127.48, 127.28, 127.10, 121.32, 121.09, 120.02, 120.00, 119.76, 119.66, 119.49, 119.12, 115.88, 115.63, 57.82, 57.43, 45.87, 45.74, 45.67, 45.53, 33.23, 33.07, 33.05, 32.96, 27.59, 27.50, 27.44, 25.32, 24.73, 24.70, 19.21, 19.11, 18.90, 18.83, 18.74, 18.56, 16.54, 16.48, 16.40, 16.02, 14.47, 14.31, 13.90, 13.85 ppm. HRMS(EI): m/z Calcd. ([M+] C22H27NS) 337.1864. Found: 337.1863.
Compound 12: The compound was synthesized using the same conditions and procedure as those for compound 10 with 2-n-butyl-1,2,3,4-tetrahydroquinaline (1.50 g, 7.68 mmol) and 9 (1.18 g, 6.53 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). The light yellow viscous oil was obtained in 40% yield (1.1 g). 1H NMR (CDCl3): δ 7.06 and 7.02 (d, J = 7.2 Hz, 1H), 6.97 (d, J = 7.2 Hz, 1H), 6.80–6.70 (m, 1H), 6.70–6.58 (m, 1H, CH), 4.20–3.94 (m, 1H, NH), 3.38–3.16 (m, 2H, NCHMe, CHMe), 3.00–2.74 (m, 2H, CH2), 2.53 (s, 3H, CH3), 1.98 and 1.97 (s, 3H, CH3), 1.76–1.56 (m, 2H, CH2), 1.56–1.18 (m, 6H, CH2), 1.38, 1.37, 1.36 and 1.34 (s, 3H, CH3), 0.89 (t, J = 5.6 Hz, 3H, CH3) ppm. 13C{1H} NMR (CDCl3): 150.80, 150.71, 145.80, 145.75, 145.33, 145.26, 143.54, 143.30, 143.23, 142.97, 141.95, 139.21, 139.08, 131.38, 131.17, 130.98, 130.71, 128.57, 127.82, 127.58, 127.38, 127.18, 121.28, 120.00, 119.80, 119.70, 119.30, 115.89, 52.00, 51.82, 51.72, 51.56, 45.87, 45.72, 45.67, 45.58, 36.90, 36.80, 36.48, 28.66, 28.41, 28.29, 28.23, 27.88, 27.24, 27.19, 26.96, 23.13, 23.04, 16.37, 15.99, 14.47, 14.34, 14.18, 13.87 ppm. HRMS(EI): m/z Calcd. ([M+] C23H29NS) 351.2021. Found: 351.2021.
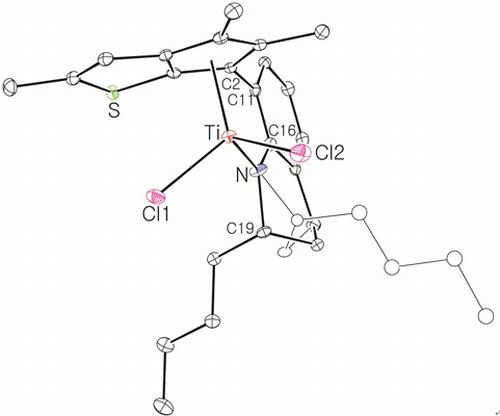
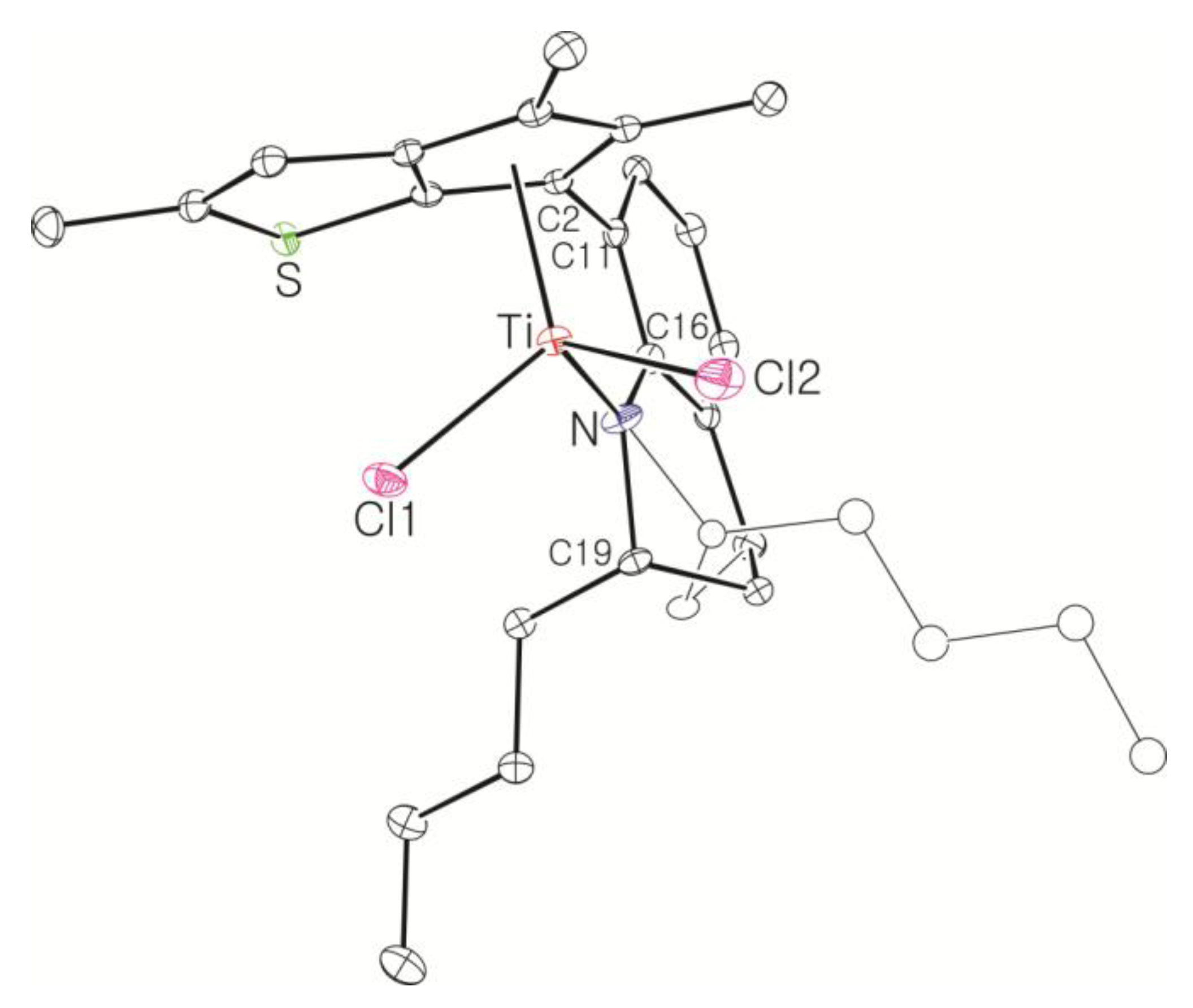
Complex 13: The complex was synthesized using the same conditions and procedure as those for 8 with 10 (1.00 g, 3.09 mmol). It was obtained as a red solid in 70% yield (1.12 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.5 ratio. 1H NMR (C6D6): δ 7.08 (t, J = 4.0 Hz, 1H), 6.94–6.84 (m, 2H), 6.41 and 6.29 (s, 1H), 5.07–4.83 (m, 1H, NCH), 2.54–2.36 (m, 1H, CH2), 2.28 and 2.27 (s, 3H, CH3), 2.25–2.15 (m, 1H, CH2), 2.04 and 2.02 (s, 3H, CH3), 1.76 (s, 3H, CH3), 1.74–1.65 (m, 2H, CH2), 1.10–0.85 (m, 2H, CH2), 0.78 and 0.75 (t, J = 6.8 Hz, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 163.17, 151.31, 149.74, 147.16, 146.21, 142.20, 140.41, 137.21, 129.90, 129.70, 129.43, 126.70, 126.54, 123.00, 122.89, 121.49, 120.33, 119.93, 118.94, 118.64, 118.10, 63.62, 63.19, 32.09, 23.04, 22.55, 21.06, 20.84, 20.44, 19.98, 16.72, 16.63, 14.65, 13.84 ppm. Anal. Calcd. (C21H23Cl2NSTi): C, 57.29; H, 5.27; N, 3.18%. Found: C, 57.56; H, 5.49; N, 3.32%.
Complex 14: The complex was synthesized using the same conditions and procedure as those for 8 with 11 (0.185 g, 0.550 mmol). It was obtained as a red solid in 86% yield (0.22 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.5 ratio. 1H NMR (C6D6): δ 7.08 (t, J = 4.4 Hz, 1H), 6.92–6.82 (m, 2H), 6.39 and 6.27 (s, 1H), 5.10–5.34 (m, 1H, NCH), 2.56–2.34 (m, 2H, CH2), 2.28 and 2.27 (s, 3H, CH3), 2.30–2.18 (m, 1H, NCH) 2.12–2.04 (m, 1H, CH), 2.02 and 2.01 (s, 3H, CH3), 1.96 and 1.76 (s, 3H, CH3), 1.74–1.58 (m, 2H, CH2), 0.99 and 0.92 (d, J = 6.8 Hz, 3H, CH3), 0.76 and 0.75 (d, J = 6.8 Hz, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 162.68, 162.63, 148.09, 146.90, 144.40, 143.25, 142.41, 142.15, 140.73, 138.82, 132.45, 132.32, 129.93, 129.91, 129.80, 127.13, 126.70, 126.60, 126.49, 123.31, 123.23, 120.26, 119.90, 106.32, 104.54, 60.42, 60.07, 32.20, 23.32, 23.11, 22.55, 21.65, 21.57, 21.12, 16.70, 16.19, 14.66, 14.26, 12.34, 12.22, 11.21 ppm. Anal. Calcd. (C22H25Cl2NSTi): C, 58.17; H, 5.55; N, 3.08%. Found: C, 58.34; H, 5.72; N, 3.29%.
Complex 15: The complex was synthesized using the same conditions and procedure as those for 8 with 12 (0.681 g, 1.94 mmol). It was obtained as a brown solid in 75% yield (0.68 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.6 ratio. 1H NMR (C6D6): δ 7.15–7.06 (m, 1H), 6.96–6.84 (m, 2H), 6.39 and 6.32 (s, 1H), 5.26–5.00 (m, 1H, NH), 2.53–2.36 (m, 1H, CH2), 2.32–2.24 (m, 1H, CH2) 2.28 and 2.27 (s, 3H, CH3), 2.18–2.06 (m, 1H, NCHMe), 2.04 and 2.02 (s, 3H, CH3), 1.88 and 1.82 (s, 3H, CH3), 1.72–1.54 (m, 2H, CH2), 1.44–1.00 (m, 6H, CH2), 0.85 and 0.79 (t, J = 6.8 Hz, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 162.77, 162.58, 150.94, 150.11, 147.23, 146.42, 145.33, 143.20, 139.52, 137.21, 130.87, 130.33, 129.92, 129.80, 126.81, 126.67, 123.15, 123.07, 120.48, 119.95, 119.75, 118.88, 118.64, 118.13, 58.48, 58.39, 29.45, 29.26, 28.95, 28.92, 23.01, 22.96, 22.28, 22.08, 21.36, 16.77, 16.68, 14.64, 14.34, 14.29, 13.88, 13.72 ppm. Anal. Calcd. (C23H27Cl2NSTi): C, 58.99; H, 5.81; N, 2.99%. Found: C, 58.78; H, 5.67; N, 2.88%.
Compound 16: Methacrylic acid (22.7 g, 264 mmol) and 2,3-dimethylthiophene (24.7 g, 220 mmol) were mixed and the mixture was added dropwise to Eaton’s reagent (220 mL) for 3 h at 80 °C. The mixture was then stirred for 30 min at 80 °C. The resulting solution was poured slowly into a flask containing a two-phase mixture of water (440 mL) and diethyl ether (75 mL) under vigorous stirring. The organic phase was collected, and the water phase was further extracted with additional diethyl ether (3 × 80 mL). The collected organic phases were combined and washed with saturated aqueous NaHCO3 (200 mL). The combined organic phases were dried over anhydrous MgSO4 and the solvent was removed by using a rotary evaporator. The oily residue was purified by column chromatography on silica gel, eluting with hexane and ethyl acetate (v/v, 20:1). The product was obtained as a mixture of two diastereomers in a 1:1 ratio (14.3 g, 36%). 1H NMR (C6D6): 2.58 (qd, J = 7.6, 3.2 Hz, 1H), 2.48 and 2.44 (d, J = 7.2 Hz, 1H), 1.90 (s, 3H, CH3), 1.62 (s, 3H, CH3), 1.16 (d, J = 7.6 Hz, 3H, CH3) ppm. 13C{1H} NMR (CDCl3): 199.52, 168.13, 150.54, 134.95, 130.04, 46.23, 46.14, 32.54, 32.31, 17.35, 17.13, 15.24, 15.05, 11.50, 11.29 ppm. HRMS(EI): m/z Calcd. ([M+] C10H12OS) 181.0609. Found: 181.0608.
Compound 17: The compound was synthesized using the same conditions and procedure as those for 7 with 1,2,3,4-tetrahydroquinaldine (20.0 g, 139 mmol) and 16 (1.50 g, 8.33 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). The light yellow viscous oil was obtained in 48% yield (1.50 g). 1H NMR (CDCl3): δ 7.14 (d, J = 7.2 Hz, 1H), 7.07 (d, J = 7.2 Hz, 1H), 6.75 (t, J = 7.2 Hz, 1H), 4.14–3.92 (m, 1H, NH), 3.62–3.44 (m, 1H, NCHMe), 3.31 (d, J = 5.2 Hz, 2H, CH2), 3.10–2.86 (m, 2H, CH2), 2.50 (s, 3H, CH3), 2.26 (s, 3H, CH3), 2.19 (s, 3H, CH3), 2.10–2.00 (m, 1H, CH2), 1.82–1.68 (m, 1H, CH2), 1.29, 1.28 and 1.26 (s, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 145.65, 142.30, 142.11, 141.70, 141.35, 139.96, 139.56, 133.17, 132.65, 132.30, 132.12, 128.59, 128.29, 127.97, 127.75, 127.39, 120.87, 120.50, 119.15, 118.96, 115.84, 115.50, 47.34, 47.20, 40.07, 39.97, 30.39, 29.89, 27.25, 26.93, 23.06, 22.78, 16.26, 15.90, 14.09, 12.44 ppm. HRMS(EI): m/z Calcd. ([M+] C20H23NS) 309.1551. Found: 309.1551.
Compound 18: The compound was synthesized using the same conditions and procedure as those for 10 with 2-ethyl-1,2,3,4-tetrahydroquinoline (0.500 g, 2.99 mmol) and 16 (0.458 g, 2.54 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). The light yellow viscous oil was obtained in 44% yield (0.43 g). 1H NMR (CDCl3): δ 7.03 (d, J = 7.2 Hz, 1H), 6.97 (d, J = 6.8 Hz, 1H), 6.68–6.58 (m, 1H, CH), 4.12–3.90 (m, 1H, NH), 3.30–3.10 (m, 3H, NCH, CH2), 2.97–2.75 (m, 2H, CH2), 2.39 (s, 3H, CH3), 2.15 (s, 3H, CH3), 2.08 (s, 3H, CH3), 2.05–1.94 (m, 1H, CH2), 1.72–1.57 (m, 1H, CH2), 1.57–1.41 (m, 2H, CH2), 0.91 (t, J = 7.2 Hz, 3H, CH3) ppm. 13C{1H} NMR (CDCl3): 145.61, 142.11, 141.95, 141.71, 141.44, 139.96, 139.47, 132.98, 132.50, 132.25, 132.08, 128.48, 128.27, 127.99, 127.58, 127.34, 121.17, 120.87, 119.17, 115.68, 53.28, 52.98, 40.06, 39.94, 29.83, 29.48, 27.61, 27.21, 27.07, 26.88, 16.30, 15.91, 14.07, 12.42, 10.49, 10.31 ppm. HRMS(EI): m/z Calcd. ([M+] C21H25NS) 323.1708. Found: 323.1709.
Compound 19: The compound was synthesized using the same conditions and procedure as those for 10 with 2-isopropyl-1,2,3,4-tetrahydroquinaline (0.500 g, 2.76 mmol) and 16 (0.423 g, 2.35 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). The light yellow viscous oil was obtained in 40% yield (0.34 g). 1H NMR (C6D6): δ 7.33 and 7.25 (d, J = 6.8 Hz, 1H), 6.99 (d, J = 7.6 Hz, 1H), 6.88–6.70 (m, 1H), 4.22–4.00 (m, 1H, NH), 3.05–2.50 (m, 5H, NCH, CH2), 2.15 (s, 3H, CH3), 2.01 (d, J = 7.2 Hz, 3H, CH3), 1.93 (s, 3H, CH3), 1.70–1.45 (m, 2H, CH2), 1.44–1.30 (m, 1H, CH), 0.73 (d, J = 6.8 Hz, 6H, CH3) ppm. 13C{1H} NMR (C6D6): 146.07, 145.97, 143.03, 142.89, 142.35, 142.11, 139.64, 139.17, 134.30, 133.76, 132.51, 132.29, 129.10, 128.15, 127.94, 121.70, 121.43, 119.98, 119.66, 116.71, 116.43, 57.98, 57.49, 40.07, 39.93, 33.26, 33.14, 27.85, 25.06, 24.98, 19.11, 18.96, 18.72, 18.44, 16.32, 15.83, 13.98, 12.35 ppm. HRMS(EI): m/z Calcd. ([M+] C22H27NS) 337.1864. Found: 337.1865.
Compound 20: The compound was synthesized using the same conditions and procedure as those for 10 with 2-n-butyl-1,2,3,4-tetrahydroquinaline (1.00 g, 5.12 mmol) and 16 (0.785 g, 4.35 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). The light yellow viscous oil was obtained in 42% yield (0.76 g). 1H NMR (C6D6): δ 7.32 and 7.26 (d, J = 7.6 Hz, 1H), 7.20 (d, J = 7.2 Hz, 1H), 6.86–6.74 (m, 1H), 4.24–4.06 (m, 1H, NH), 3.14–2.98 (m, 1H, NCH), 2.56–2.96 (m, 4H, CH2), 2.15 (s, 3H, CH3), 2.01 (s, 3H, CH3), 1.93 (s, 3H, CH3), 1.74–1.62 (m, 1H, CH2), 1.56–1.42 (m, 1H, CH2), 1.34–1.00 (m, 6H, CH2), 0.78 (t, J = 6.8 Hz, 3H, CH3) ppm. 13C{1H} NMR (CDCl3): 145.61, 142.13, 141.87, 141.65, 141.43, 139.94, 139.46, 132.99, 132.53, 132.23, 132.09, 128.49, 128.25, 127.96, 127.58, 127.35, 121.17, 120.87, 119.19, 118.92, 115.68, 115.41, 51.83, 51.56, 40.07, 39.96, 36.81, 36.45, 28.25, 28.20, 28.13, 27.78, 27.12, 26.91, 23.05, 16.29, 15.91, 14.37, 14.05, 12.42 ppm. HRMS(EI): m/z Calcd. ([M+] C23H29NS) 351.2021. Found: 351.2021.
Compound 21: The compound was synthesized using the same conditions and procedure as those for 10 with 2-tert-butyl-1,2,3,4-tetrahydroquinaline (1.00 g, 5.12 mmol) and 16 (0.785 g, 4.35 mmol). It was purified by column chromatography on silica gel eluting with hexane and ethyl acetate (v/v, 50:1). A light yellow viscous oil was obtained in 31% yield (0.56 g). 1H NMR (C6D6): δ 7.35 and 7.28 (d, J = 7.6 Hz, 1H), 7.00 (d, J = 7.2 Hz, 1H), 6.85–6.74 (m, 1H) 4.40–4.16 (m, 1H, NH), 3.06–2.54 (m, 5H, NCH, CH2), 2.14 and 2.12 (s, 3H, CH3), 2.04 and 1.99 (s, 3H, CH3), 1.91 and 1.90 (s, 3H, CH3), 1.70–1.60 (m, 1H, CH2), 1.58–1.40 (m, 1H, CH2), 0.76 (s, 9H, CH3) ppm. 13C{1H} NMR (C6D6): 146.07, 145.95, 143.32, 143.22, 142.37, 142.16, 139.65, 138.97, 134.29, 133.64, 132.52, 132.23, 128.97, 128.89, 121.79, 121.51, 120.27, 119.77, 116.86, 116.52, 61.47, 60.95, 40.08, 39.89, 33.79, 33.58, 28.46, 28.41, 26.32, 23.69, 23.53, 16.49, 15.81, 13.98, 12.35 ppm. HRMS(EI): m/z Calcd ([M+] C23H29NS) 351.2021. Found: 351.2020.
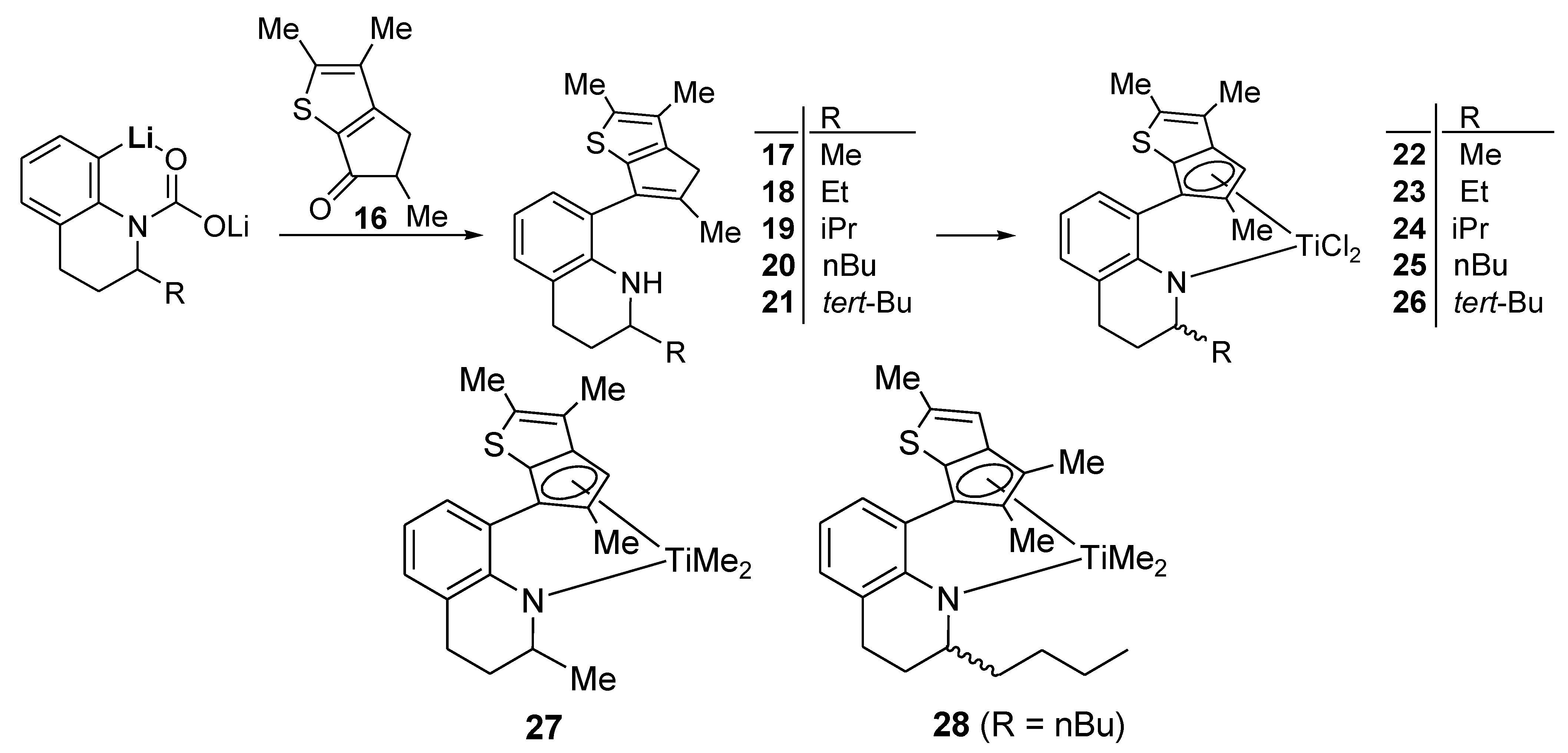
Complex 22: The complex was synthesized using the same conditions and procedure as those for 8 with 17 (0.500 g, 1.62 mmol). It was obtained as a brown solid in 73% yield (0.50 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.4 ratio. 1H NMR (C6D6): δ 7.25–6.98 (m, 1H), 6.94–6.78 (m, 2H), 6.15 and 6.09 (s, 1H), 5.44–5.26 (m, 1H, NCH), 2.52–2.32 (m, 1H, CH2), 2.16–1.98 (m, 1H, CH2), 1.95 (s, 3H, CH3), 1.90 (s, 3H, CH3), 1.88 (s, 3H, CH3), 1.70–1.50 (m, 2H, CH2), 1.14 and 1.12 (d, J = 6.8, 3H, CH3) ppm. 13C{1H} NMR (CDCl3): 161.99, 161.92, 148.63, 147.61, 144.25, 143.22, 142.63, 142.32, 140.58, 138.83, 132.54, 132.40, 129.91, 129.88, 129.57, 129.49, 126.93, 126.61, 126.52, 123.26, 123.24, 119.78, 119.54, 106.07, 106.57, 54.30, 59.96, 26.25, 21.33, 16.78, 16.75, 16.46, 16.41, 14.78, 14.75, 12.64, 12.58 ppm. Anal. Calcd. (C20H21Cl2NSTi): C, 56.36; H, 4.97; N, 3.29%. Found: C, 56.23; H, 4.79; N, 3.02%.
Complex 23: The complex was synthesized using the same conditions and procedure as those for 8 with 18 (0.271 g, 0.838 mmol). It was obtained as a brown solid in 80% yield (0.30 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.8 ratio. 1H NMR (C6D6): δ 7.13 and 7.09 (t, J = 3.2 Hz, 1H), 6.95–6.87 (m, 2H), 6.40 and 6.33 (s, 1H), 5.20–5.00 (m, 1H, NCH), 2.50–2.38 (m, 1H, CH2), 2.29 and 2.27 (s, 3H, CH3), 2.18–2.05 (m, 1H, CH2), 2.05 and 2.03 (s, 3H, CH3), 1.88 and 1.82 (s, 3H, CH3), 1.68–1.54 (m, 2H, CH2), 1.36–1.16 (m, 2H, CH2), 0.85 and 0.79 (t, J = 6.8 Hz, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 162.81, 162.61, 150.94, 150.11, 147.24, 146.42, 145.32, 143.21, 139.51, 137.19, 130.85, 130.37, 130.34, 129.92, 129.79, 126.80, 126.67, 123.12, 123.04, 120.48, 119.99, 119.78, 118.84, 118.62, 118.12, 58.47, 58.37, 29.46, 29.27, 28.94, 23.01, 22.96, 22.25, 22.05, 21.34, 16.73, 16.65, 14.64, 14.32, 13.85, 13.70 ppm. Anal. Calcd. (C21H23Cl2NSTi): C, 57.29; H, 5.27; N, 3.18%. Found: C, 57.42; H, 5.49; N, 3.33%.
Complex 24: The complex was synthesized using the same conditions and procedure as those for 8 with 19 (0.050 g, 0.15 mmol). It was obtained as a brown solid in 81% yield (0.06 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.8 ratio. 1H NMR (C6D6): δ 7.05 (t, J = 4.0 Hz, 1H), 6.94–6.80 (m, 2H), 6.17 and 6.14 (s, 1H), 5.20–4.90 (m, 1H, NCH), 2.60–2.34 (m, 1H, CH2), 2.26–2.10 (m, 1H, CH2), 2.07 and 1.90 (s, 3H, CH3), 1.93 and 1.90 (s, 3H, CH3), 1.85 (s, 3H, CH3), 1.80–1.68 (m, 2H, CH2), 1.70–1.56 (m, 1H, CH), 0.97 and 0.90 (d, J = 6.8 Hz, 3H), 0.75 and 0.73 (d, J = 7.2 Hz, 3H) ppm. 13C{1H} NMR (C6D6): 163.37, 163.16, 148.24, 146.68, 144.82, 143.61, 142.15, 141.43, 141.15, 138.96, 131.53, 131.32, 129.90, 129.71, 129.15, 129.00, 127.01, 126.73, 126.54, 126.41, 123.24, 123.12, 120.59, 120.05, 107.22, 104.82, 64.12, 63.27, 32.12, 31.64, 22.99, 22.47, 22.38, 22.31, 21.23, 20.76, 20.45, 19.96, 17.05, 16.25, 14.26, 14.20, 12.41, 12.18 ppm. Anal. Calcd. (C22H25Cl2NSTi): C, 58.17; H, 5.55; N, 3.08%. Found: C, 58.37; H, 5.75; N, 3.29%.
Complex 25: The complex was synthesized using the same conditions and procedure as those for 8 with 20 (1.50 g, 9.79 mmol). The product was obtained as a brown solid in 88% yield (1.50 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.9 ratio. 1H NMR (C6D6): δ 7.11 (t, J = 4.4 Hz, 1H), 6.90 (d, J = 5.6 Hz, 2H), 6.19 and 6.12 (s, 1H), 5.26–5.10 (m, 1H, NCH), 2.52–2.36 (m, 1H, CH2), 2.18–2.02 (m, 1H, CH2), 1.97 (s, 3H, CH3), 1.92 (s, 3H, CH3), 1.91 (s, 3H, CH3), 1.70–1.54 (m, 2H, CH2), 1.36–1.00 (m, 6H, CH2), 0.84 and 0.77 (t, J = 7.6 Hz, 3H, CH3) ppm. 13C{1H} NMR (C6D6): 162.74, 162.68, 148.11, 146.92, 144.38, 143.21, 142.46, 142.17, 140.71, 138.84, 132.44, 132.29, 129.94, 129.87, 129.82, 127.08, 126.71, 126.65, 126.54, 123.37, 123.29, 120.20, 119.89, 106.30, 104.55, 58.96, 58.64, 29.41, 28.92, 23.00, 22.93, 22.21, 22.15, 21.32, 16.70, 16.24, 14.62, 14.30, 14.25, 12.35, 12.26 ppm. Anal. Calcd. (C23H27Cl2NSTi): C, 58.99; H, 5.81; N, 2.99%. Found: C, 58.73; H, 5.57; N, 2.69%.
Complex 26: The complex was synthesized using the same conditions and procedure as those for 8 with 21 (0.050 g, 0.142 mmol). It was obtained as a brown solid in 82% yield (0.050 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.4 ratio. 1H NMR (C6D6): δ 7.13 (t, J = 6.8 Hz, 1H), 6.90–6.70 (m, 2H), 6.18 and 6.16 (s, 1H), 5.30–4.86 (m, 1H, NCH), 2.58–2.42 (m, 1H, CH2), 2.16 (s, 3H, CH3), 2.08–1.94 (m, 1H, CH2), 1.89 (s, 6H, CH3), 1.90–1.74 (m, 2H, CH2), 0.92 (s, 9H, CH3) ppm. 13C{1H} NMR (C6D6): 163.57, 144.82, 144.69, 144.45, 142.69, 129.66, 129.43, 126.32, 126.19, 122.77, 120.35, 108.65, 105.06, 66.99, 41.29, 30.73, 30.63 23.29, 23.19, 16.12, 14.32, 12.00 ppm. Anal. Calcd. (C23H27Cl2NSTi): C, 58.99; H, 5.81; N, 2.99%. Found: C, 58.69; H, 5.51; N, 2.65%.
Complex 27: The complex was synthesized using the same conditions and procedure as those for 2 with 22 (0.500 g, 1.17 mmol). It was obtained as red viscous oil in 86% yield (0.40 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.7 ratio. 1H NMR (C6D6): δ 7.12 and 7.08 (d, J = 6.8 Hz, 1H), 6.94 and 6.92 (d, J = 7.6 Hz, 1H), 6.79 (t, J = 7.6 Hz, 1H), 6.31 and 6.12 (s, 1H), 5.60–5.44 (m, 1H, NCH), 2.75–2.60 (m, 1H, NCHMe), 2.00 and 1.99 (s, 3H, CH3), 1.95 (s, 3H, CH3), 1.84 and 1.73 (s, 3H, CH3), 1.72–1.64 (m, 1H, CH2), 1.60–1.50 (m, 1H, CH2), 1.18 and 1.17 (s, 3H, CH3), 0.83 and 0.80 (s, 3H, TiMe), 0.35 and 0.27 (s, 3H, TiMe) ppm. 13C{1H} NMR (C6D6): 159.47, 141.38, 138.38, 138.10, 135.95, 129.64, 129.51, 127.11, 127.02, 125.75, 122.82, 120.11, 119.98, 119.01, 102.05, 99.27, 58.55, 55.61, 52.30, 50.42, 50.12, 27.53, 27.28, 21.96, 20.15, 19.71, 15.31, 15.12, 14.01, 12.50, 12.37 ppm. Anal. Calcd. (C22H27Cl2NSTi): C, 68.56; H, 7.06; N, 3.63%. Found: C, 68.77; H, 7.24; N, 3.79%.
Complex 28: The complex was synthesized using the same conditions and procedure as those for 2 with 15 (0.500 g, 1.07 mmol). It was obtained as an red viscous oil in 85% yield (0.39 g). The 1H NMR spectrum indicated a mixture of two stereoisomers in 1:0.7 ratio. 1H NMR (C6D6): δ 7.11 and 7.08 (d, J = 7.2 Hz, 1H), 6.95 (t, J = 7.6 Hz, 1H), 6.81 and 6.79 (d, J = 7.6 Hz, 1H), 6.48 and 6.47 (s, 1H), 5.50–5.38 (m, 1H, NCH), 2.78–2.60 (m, 1H, CH2), 2.44–2.26 (m, 1H, CH2), 2.28 and 2.22 (s, 3H, CH3), 2.09 (s, 3H, CH3), 1.84–1.52 (m, 2H, CH2), 1.73 and 1.63 (s, 3H, CH3), 1.40–1.15 (m, 6H, CH2), 1.18 (t, J = 5.6 Hz, 3H, CH3), 0.74 and 0.68 (s, 3H, TiMe), 0.46 and 0.37 (s, 3H, TiMe) ppm. 13C{1H} NMR (C6D6): 159.73, 159.42, 145.84, 144.82, 140.65, 139.82, 139.13, 138.75, 135.18, 131.50, 129.60, 129.49, 127.41, 127.36, 127.28, 127.14, 121.22, 121.10, 119.96, 119.88, 118.80, 118.65, 117.91, 117.66, 113.77, 110.31, 57.83, 55.23, 54.84, 51.63, 50.20, 50.06, 27.54, 27.24, 22.01, 21.99, 20.24, 19.83, 16.61, 16.58, 13.04, 13.00, 12.91, 12.60 ppm. Anal. Calcd. (C25H33Cl2NSTi): C, 70.24; H, 7.78; N, 3.28%. Found: C, 70.46; H, 7.98; N, 3.53%.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}