Partial Oxidation of n-Butane over a Sol-Gel Prepared Vanadium Phosphorous Oxide



Abstract
:1. Introduction
2. Results and Discussion
2.1. Sol-Gel Synthesis in Tetrahydrofuran (THF)
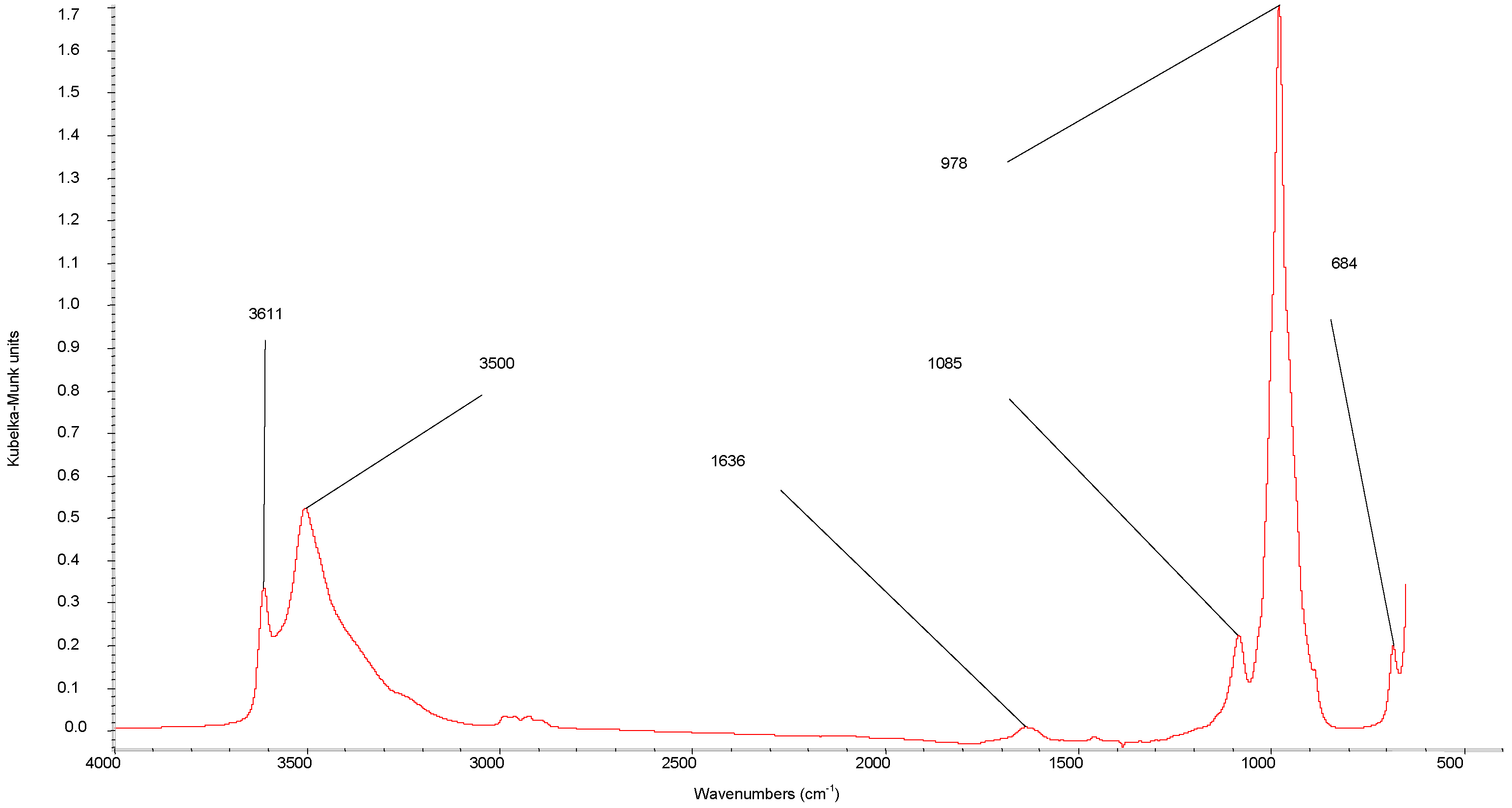

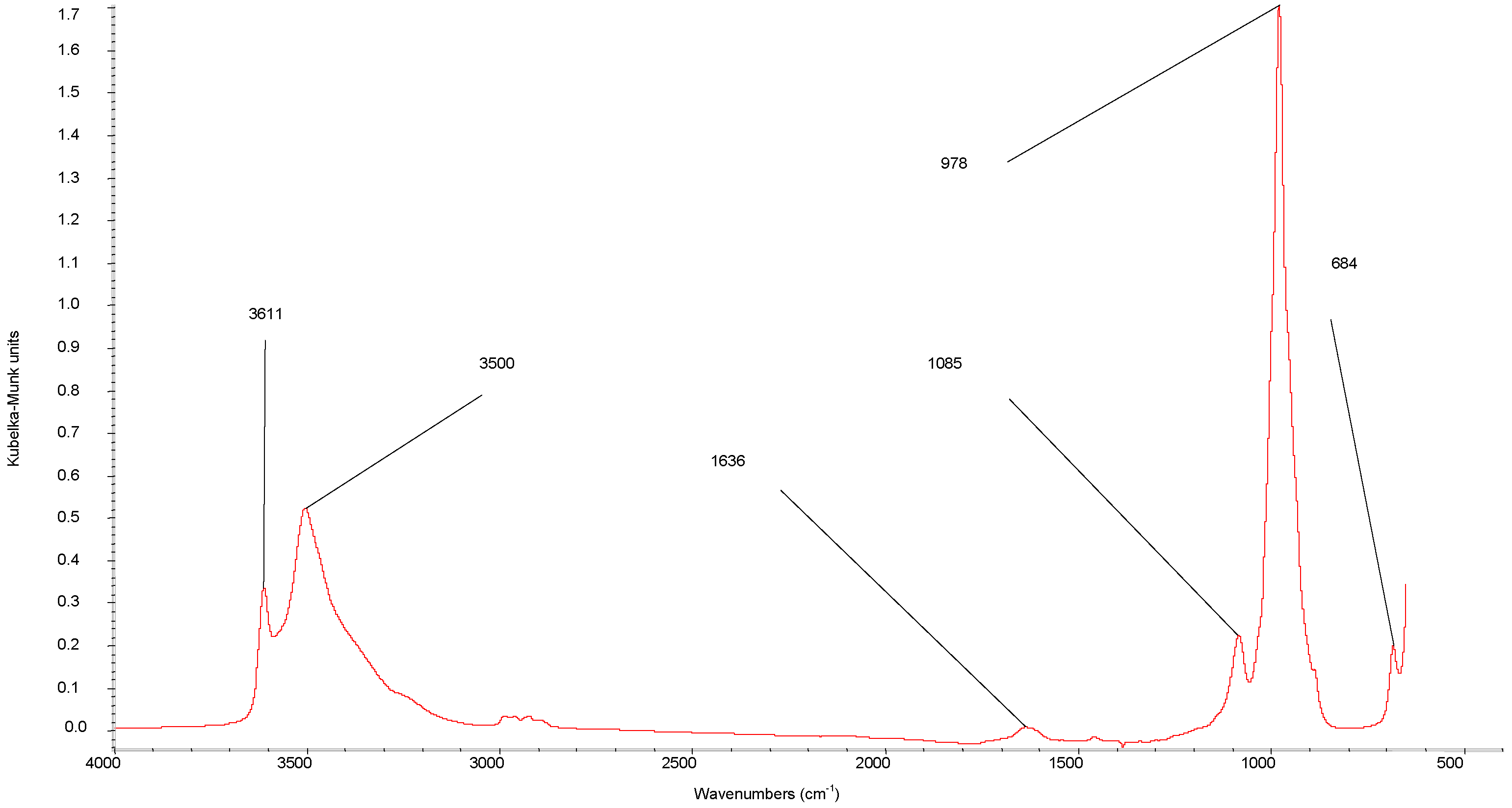{kind=link}
{kind=link}
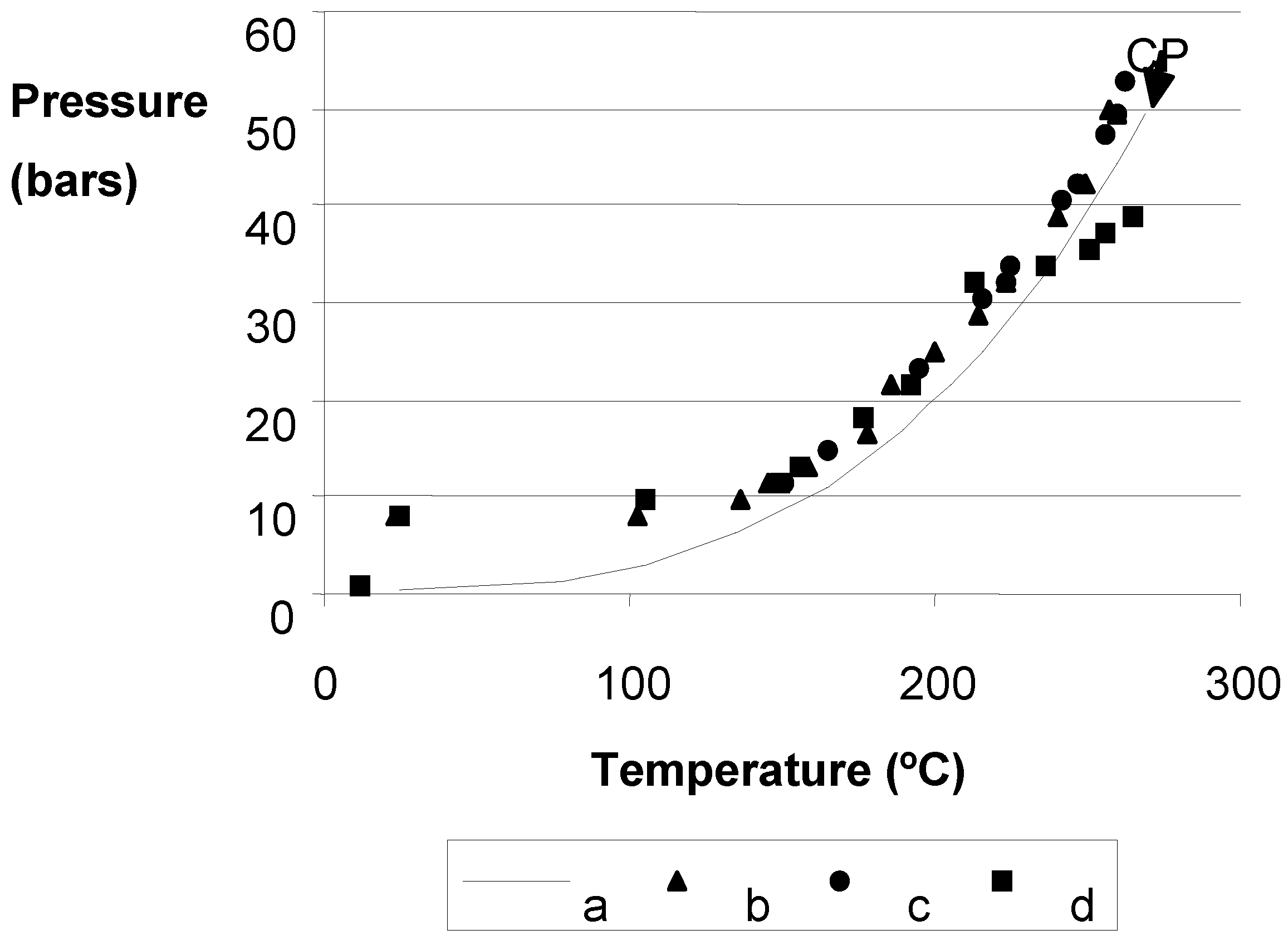{kind=link}
{kind=link}
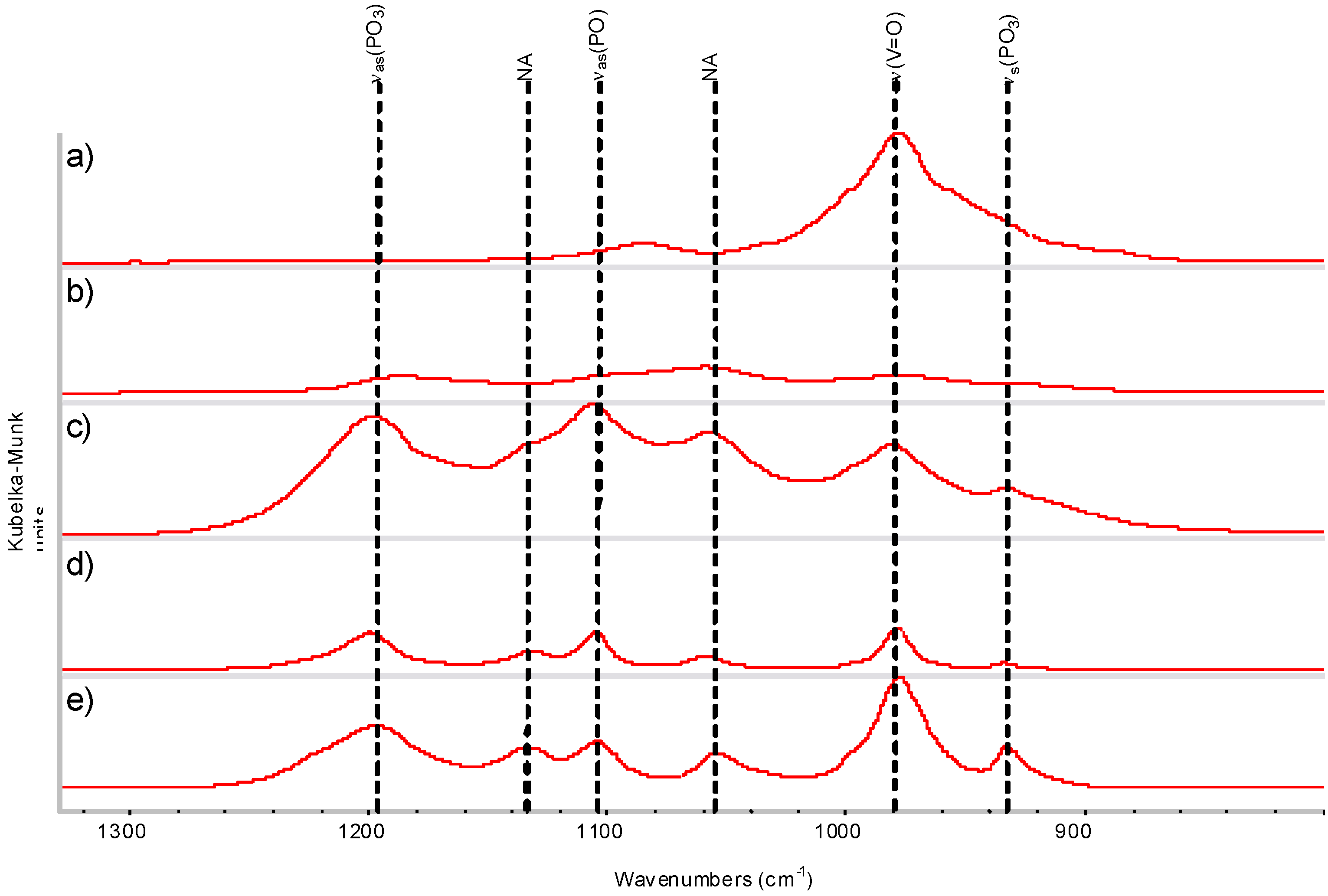{kind=link}
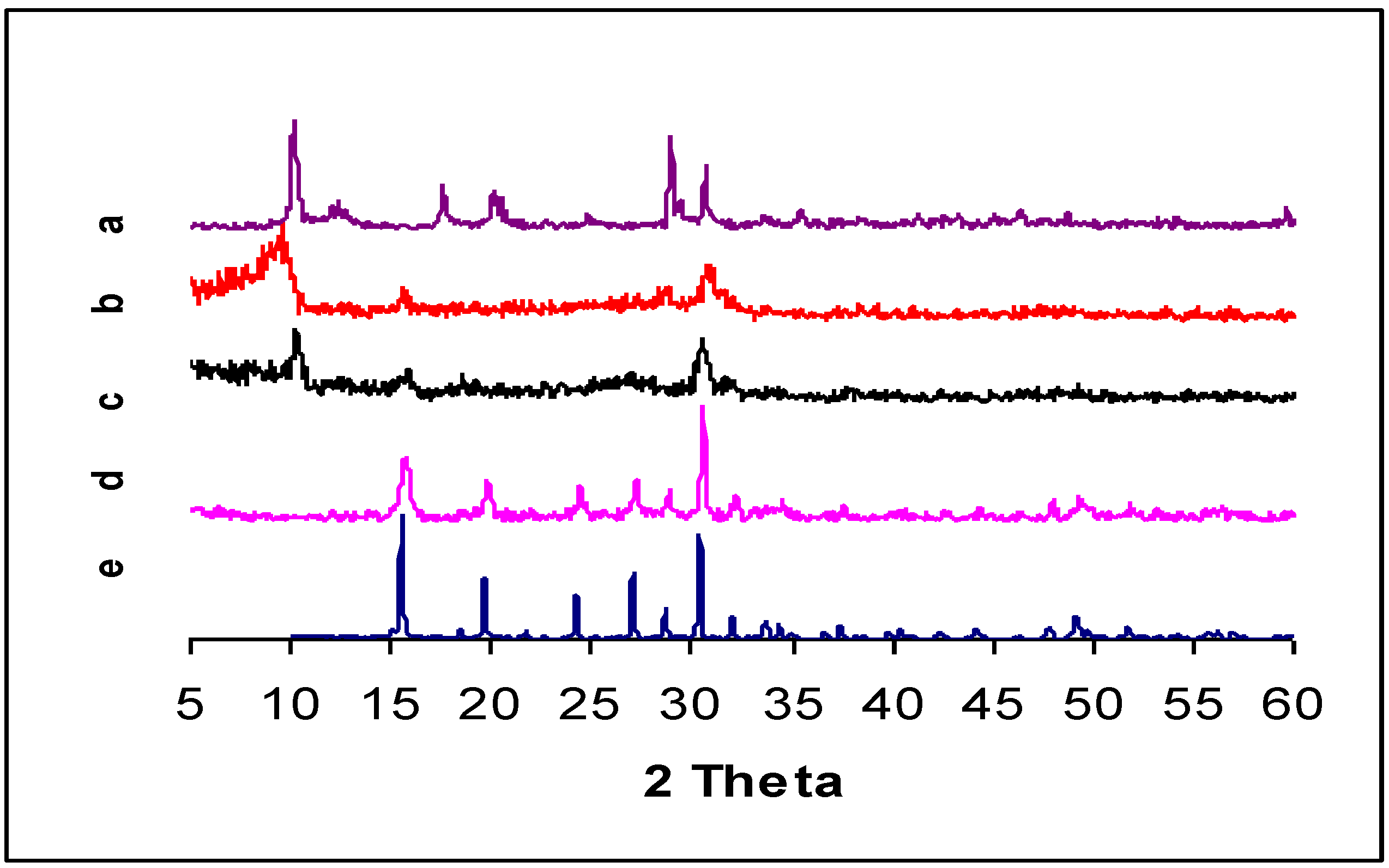{kind=link}
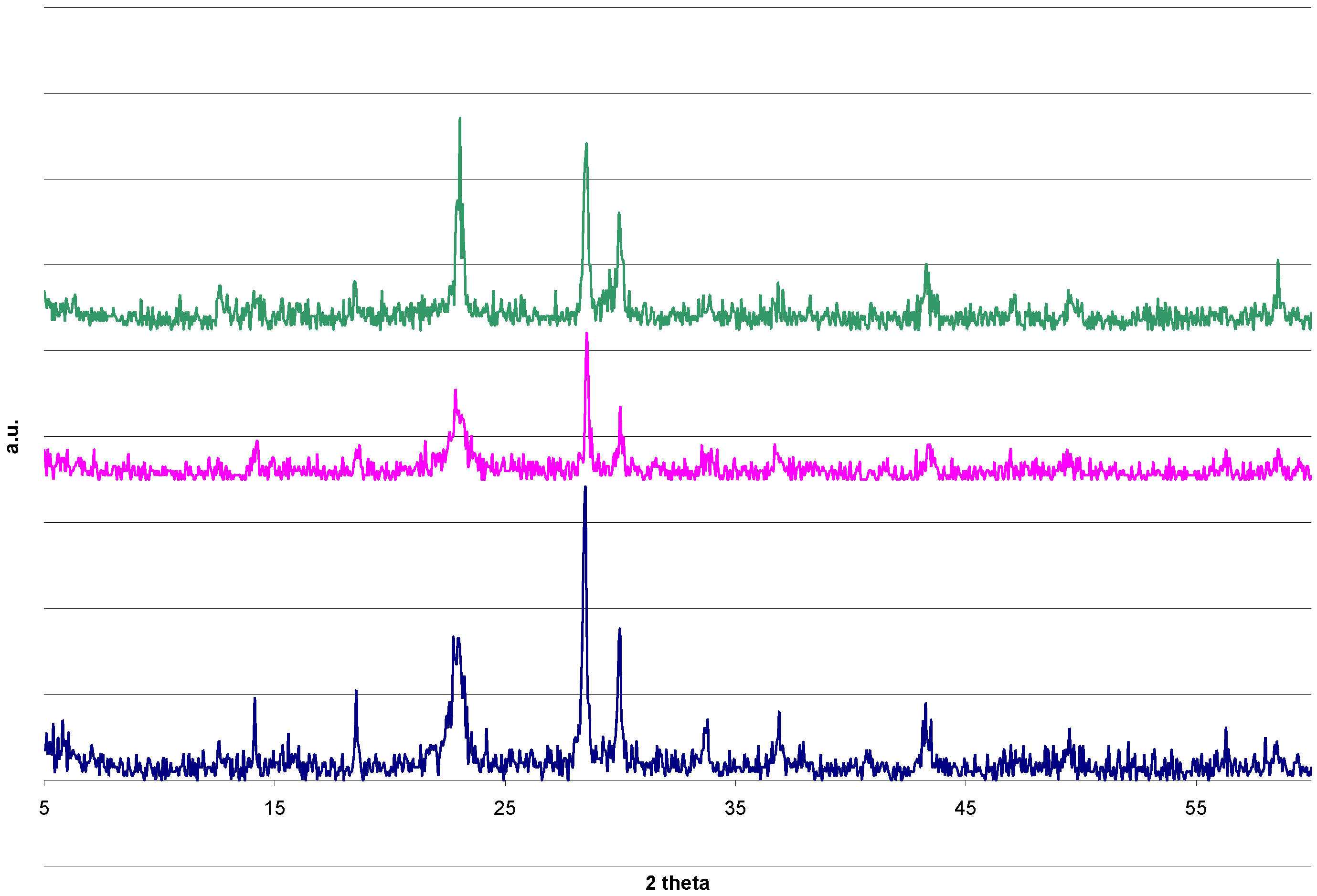{kind=link}
{kind=link}
| DRIFTS wavenumbers in cm−1 | XRD spacing in Å (relative intensity) | ||||||
|---|---|---|---|---|---|---|---|
| Reference Compounds | This work | Assignment [21,38] | Reference Compounds [13] | This work | |||
| R1 a | THF | R2 b | AD | R3c | R2 b | AD | |
| 3620 | 3611 | ν(OH) | 8.71 (100) | ||||
| 3580 | 3500 | ν(OH) | 7.45 (vS d) | 7.32 (21.5) | |||
| 2993 | 2993 | 2993 | ν(C-H) | 5.03 (40.9) | |||
| 2893 | 2893 | 2893 | ν(C-H) | 4.43 (w g) | 4.41 (35.5) | ||
| 1464 | 1460 | 1460 | δ(C-H) | 3.57 (S e) | 3.59 (11.8) | ||
| 1625 | 1636 | δ(H-O-H) | 3.07 (vS d) | 3.1 (M e) | 3.07 (83.9) | ||
| 1364 | 1346 | ν (C-O) | 2.91 (58.1) | ||||
| 1088 | 1085 | νas(P-O) | 2.19 (w g) | 2.18 (15.1) | |||
| 1000 | 978 | ν(V=O) | 1.9 (S e) | 1.95 (12.9) | |||
| 913 | 911 | ν(P-O) | 1.83 (M f) | 1.87 (14) | |||
| 685 | 684 | δ(V-OH)or δ(P-OH) | 1.56 (M f) | 1.55 (S e) | 1.55 (20.4) | ||
| 1.5 (Mf) | 1.52 (S e) | 1.52 (13.4) | |||||
| 1.43 (Mf) | 1.46 (M f) | 1.46 (13.8) | |||||
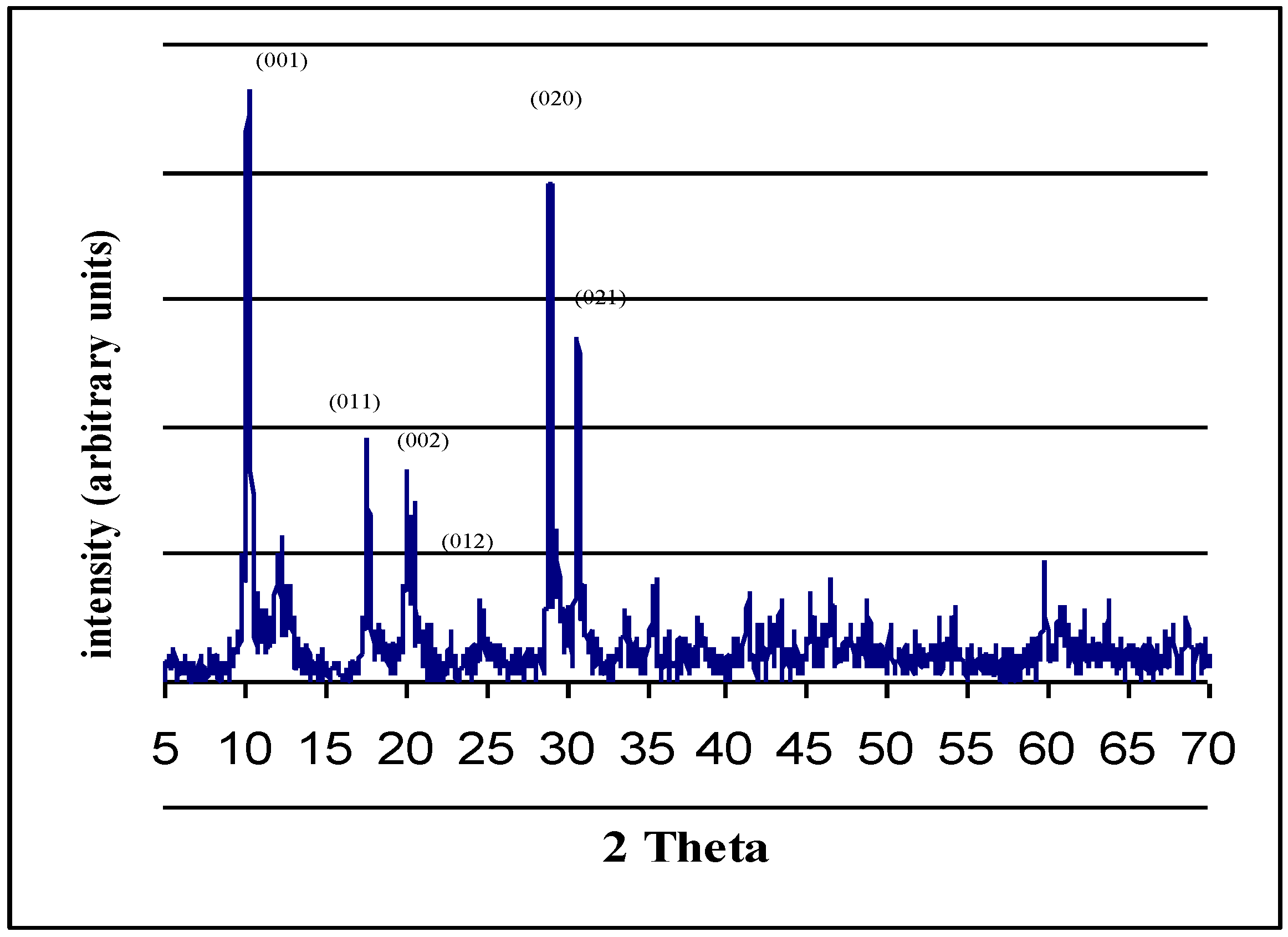
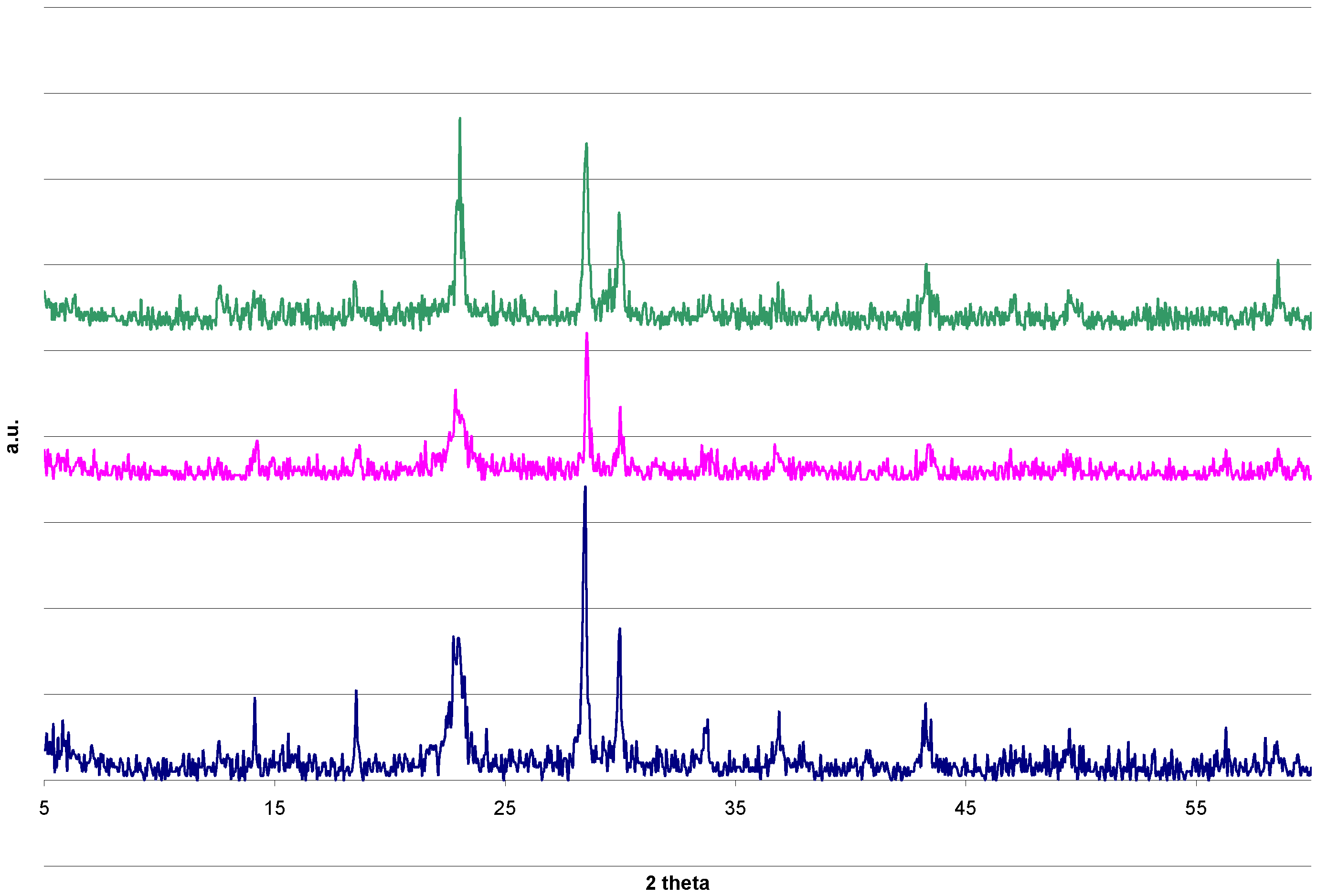
2.2. Autoclave Drying
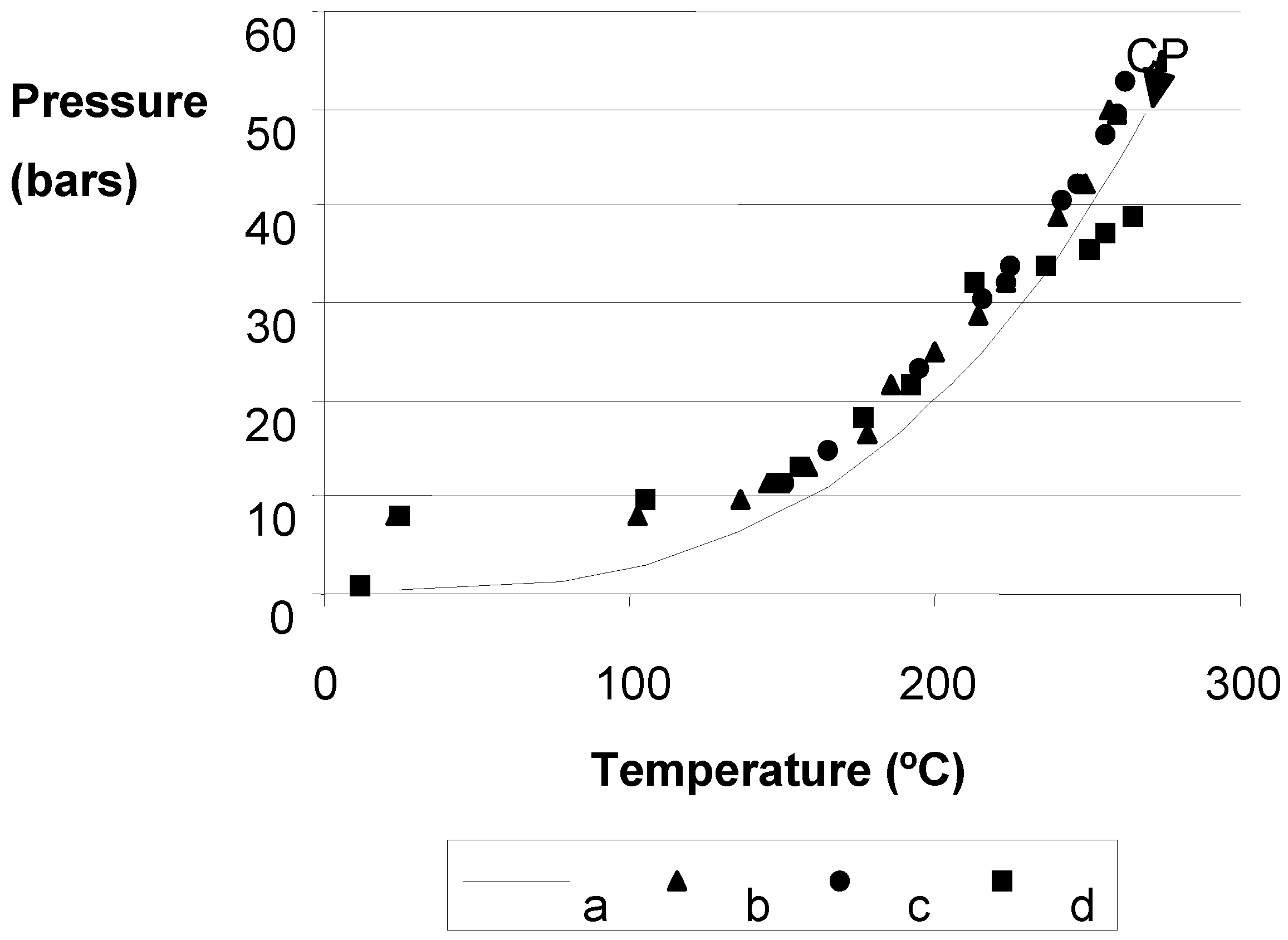

2.3. Surface Area Measurements of Precursors
| Precursor | Average Surface Area (m2/g) |
|---|---|
| LS | 65 |
| MS | 102 |
| HS | 121 |
2.4. DRIFTS and XRD Analysis
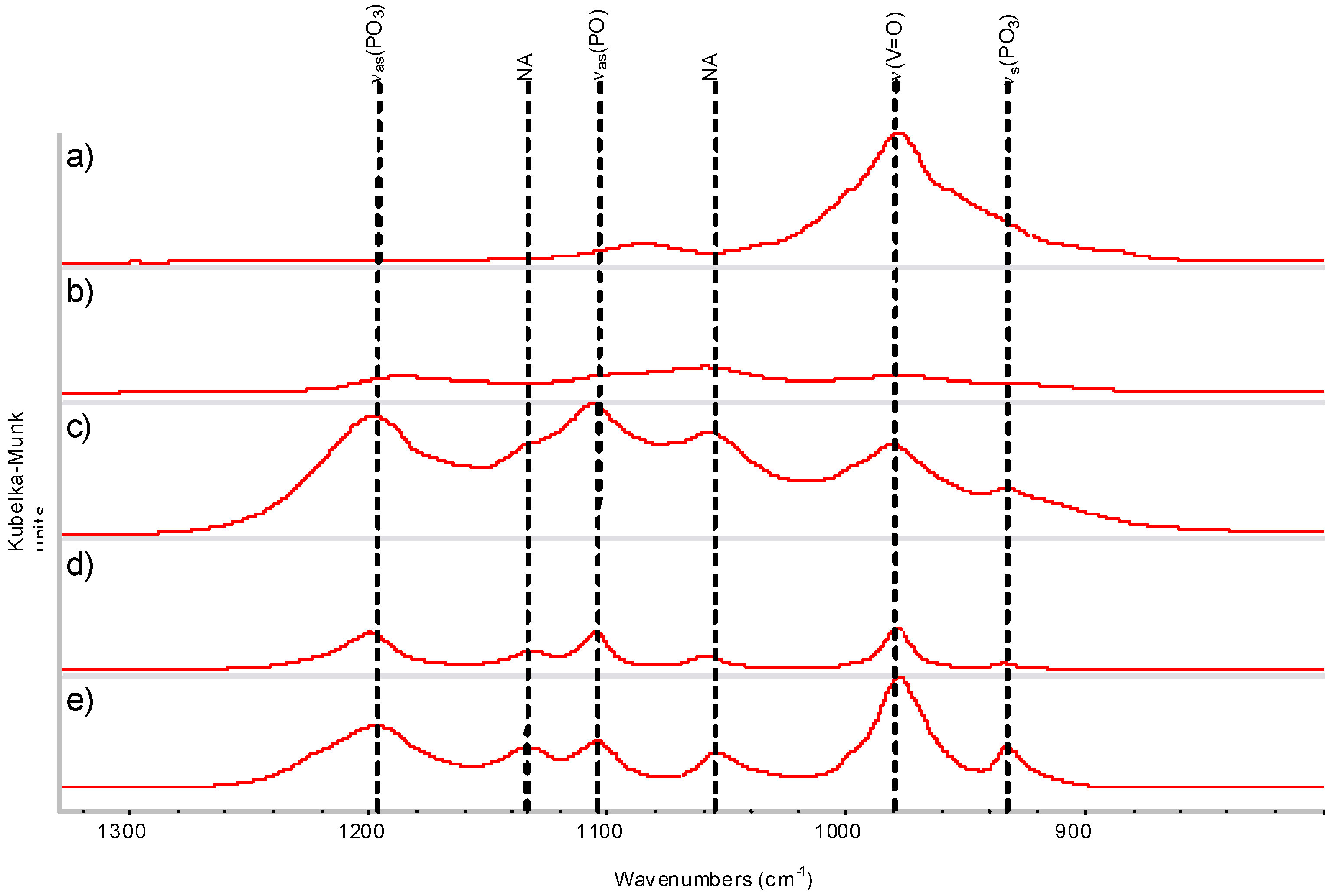
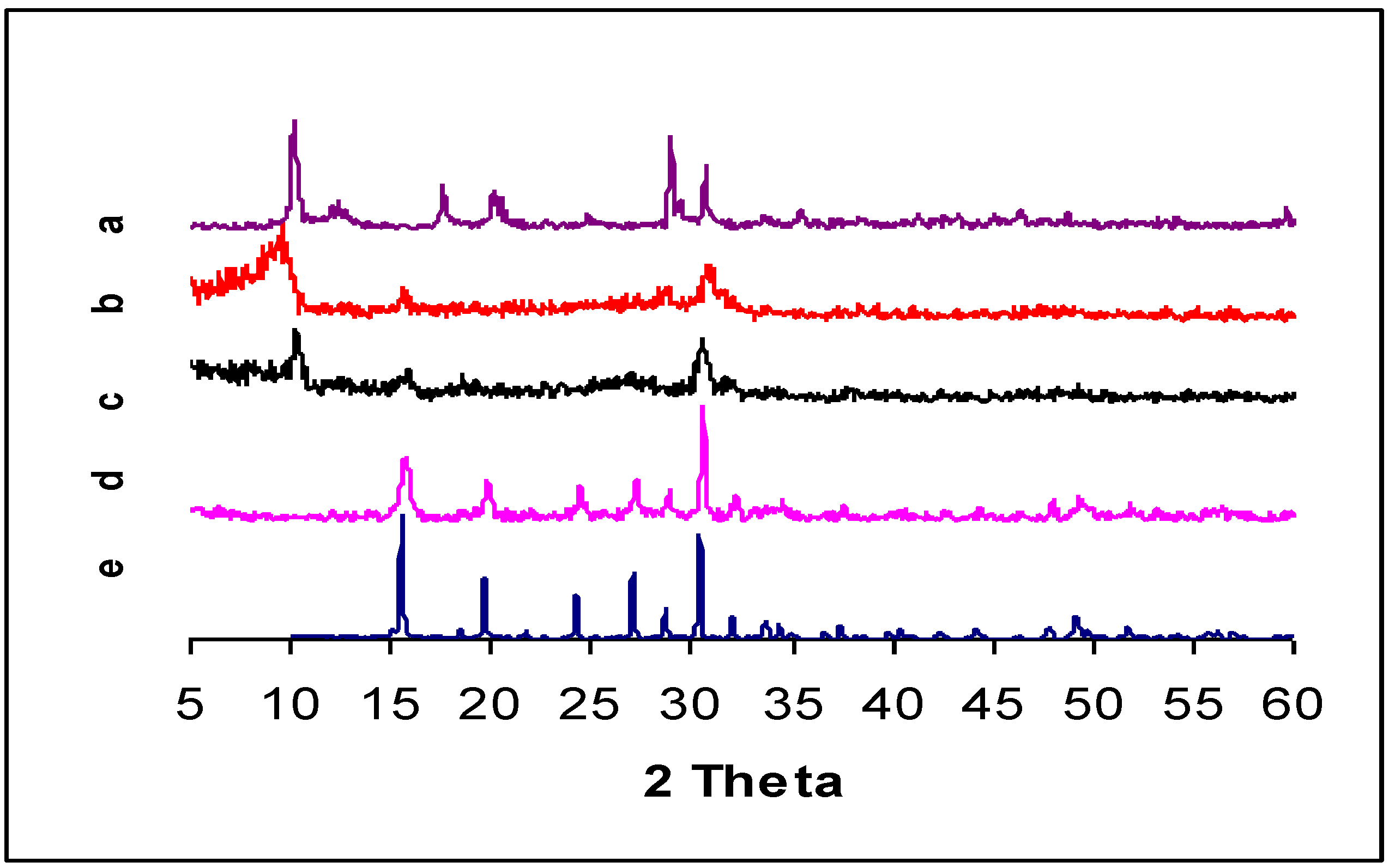
2.5. Reduction Process inside the Autoclave
2.6. Catalytic Evaluation
| Treatment | Butane conversion | 95% Confidence interval | Maleic anhydride selectivity | 95% confidence interval | Intrinsic activity 10−5 mol MA/m2/h |
|---|---|---|---|---|---|
| Traditional | 37.4 | (29.2, 41.7) | 28.7 | (22.6, 34.7) | 2.11 |
| Low solvent | 31.1 | (29.8, 39.7) | 41.8 | (35.7, 47.8) | 1.13 |
| Medium Solvent | 20.6 | (12.3, 29.0) | 28.0 | (22.0, 34.0) | 1.8 |
| High Solvent | 28.4 | (20.1, 36.6) | 12.1 | (6.1, 18.1) | 1.17 |

3. Experimental Section
3.1. Sol-Gel Synthesis in THF
3.2. Drying Process
3.3. Catalytic Evaluation
3.4. Characterization
4. Conclusions
Conflict of Interest
References
- Hodnett, B.K. Vanadium-Phosphorous Oxide Catalysts for the Selective Oxidation of C-4 Hydrocarbons to Maleic-Anhydride. Catal. Rev. Sci. Eng. 1985, 27, 373–424. [Google Scholar] [CrossRef]
- Centi, G.; Cavani, F.; Trifiro, F. Selective Oxidation by Heterogeneous Catalysis; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2001; p. 143. [Google Scholar]
- Hutchings, G.J. Vanadium phosphate: A new look at the active components of catalysts for the oxidation of butane to maleic anhydride. Mater. Chem. 2004, 14, 3385–3395. [Google Scholar] [CrossRef]
- Ballarini, N.; Cavani, F.; Cortelli, C.; Ligi, S.; Pierelli, F.; Trifiro, F.; Fumagalli, C.; Mazzoni, G.; Monti, T. VPO catalyst for n-butane oxidation to maleic anhydride: A goal achieved, or a still open challenge? Topics Catal. 2006, 38, 147–156. [Google Scholar]
- Hutchings, G.J.; Sananes, M.T.; Sajip, S.; Kiely, C.J.; Burrows, A.; Ellison, I.J.; Volta, J.C. Improved method of preparation of vanadium phosphate catalysts. Catal. Today 1997, 33, 161–171. [Google Scholar]
- Higgins, R.; Hutchings, G.J. To Imperial Chemical Industries Limited. US Patent 4 317 777, 2 March 1982. [Google Scholar]
- Kamiya, Y.; Ueki, S.; Hiyoshi, N.; Yamamoto, N.; Okuhara, T. Preparation of catalyst precursors for selective oxidation of n-butane by exfoliation-reduction of VOPO4·2H2O) in primary alcohol. Catal. Today 2003, 78, 281–290. [Google Scholar]
- Kamiya, Y.; Imai, H.; Okohura, T. Selective Oxidation of n-Butane over Highly Crystalline Vanadyl Pyrophosphate Catalyst Synthesized by Intercalation-exfoliation-reduction of Layered Vanadyl Phosphate Dihydrate. J. Jpn. Petrol. Inst. 2009, 52, 81–89. [Google Scholar] [CrossRef]
- Carreon, M.A.; Guliants, V.V.; Pierelli, F.; Cavani, F. Ordered mesostructured mixed metal oxides: microporous VPO phases for n-butane oxidation to maleic anhydride. Cataly. Lett. 2004, 92, 11–16. [Google Scholar] [CrossRef]
- Carreon, M.A.; Guliants, V.V. Macroporous vanadium phosphorous oxide phases displaying three-dimensional arrays of spherical voids. Chem. Mater. 2002, 14, 2670–2675. [Google Scholar] [CrossRef]
- Rownaghi, A.A.; Taufig-Yup, Y.H. Novel Synthesis Technique for Preparation of Ultrahigh-Crystalline Vanadyl Pyrophosphate as a Highly Selective Catalyst for n-Butane Oxidation. Ind. Eng. Chem. Res. 2010, 49, 2135–2143. [Google Scholar]
- Shpeizer, B.G.; Ouyang, X.; Heising, J.M.; Clearfield, A. Synthesis and crystal structure of a new vanadyl phosphate [H-0.6(VO)3(PO4)(3)(H2O)(3)]·4H(2)O and its conversion to porous products. Chem. Mater. 2001, 13, 2288–2296. [Google Scholar] [CrossRef]
- Centi, G.; Trifiro, F.; Ebner, J.R.; Franchetti, V.M. Mechanistic Aspects of Maleic-Anhydride Synthesis from C4-hydrocarbons over Phosphorus Vanadium-Oxide. Chem. Rev. 1988, 88, 55–80. [Google Scholar]
- Horowitz, H.S.; Blackstone, C.M.; Sleight, A.W.; Teufer, G. V-P-O Catalysts for Oxidation of Butane to Maleic Anhydride–Influence of (VO)2H4P2O9 Precursor Morphology on Catalytic Properties. Appl. Catal. 1988, 38, 193–210. [Google Scholar] [CrossRef]
- Bordes, E. Crystallochemistry of V-P-O Phases and Application to Catalysis. Catal. Today 1987, 1, 499–526. [Google Scholar] [CrossRef]
- Kamiya, Y.; Hiyoshi, N.; Ryumon, N.; Okuhara, T. Microstructures of V-P-O catalysts derived from VOHPO4 center dot 0.5H(2)O of different crystallite sizes. J. Mol. Catal. A 2004, 220, 103–112. [Google Scholar] [CrossRef]
- Hiyoshi, N.; Yamamoto, N.; Ryumon, N.; Kamiya, Y.; Okuhara, T. Selective oxidation of n-butane in the presence of vanadyl pyrophosphates synthesized by intercalation–exfoliation–reduction of layered VOPO4·2H2O in 2-butanol. J. Catal. 2004, 221, 225. [Google Scholar]
- Kamiya, Y.; Ryumon, N.; Imai, H.; Okuhara, T. Nano-sized crystallites of vanadyl pyrophosphate as a highly selective catalyst for n-butane oxidation. Catal. Lett. 2006, 111, 159–163. [Google Scholar] [CrossRef]
- Ryumon, N.; Imai, H.; Kamiya, Y.; Okuhara, T. Effect of water vapor on the transformation of VOHPO4·0.5H(2)O into (VO)(2)P2O7. Appl. Catal. A 2006, 297, 73–80. [Google Scholar] [CrossRef]
- Yan, C.; Sun, L.; Cheng, F. Handbook of Nanophase and Nanostructured Materials; Wang, Z.L., Li, Y., Zhang, Z., Eds.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2003; Volume 1, p. 72. [Google Scholar]
- nnaciri, S.A.; Bardoux, P. Solution Synthesis of Vanadium and Titanium Phosphates. Mater. Sci. Forum 1994, 152–153, 331–334. [Google Scholar]
- Farrusseng, D.; Julbe, A.; Lopez, M.; Guizard, C. Investigation of sol-gel methods for the synthesis of VPO membrane materails adapted to the partial oxidation of n-butane. Catal. Today 2000, 56, 211–220. [Google Scholar]
- R'kha, C.; Vandenborre, M.T.; Livage, J.; Prost, R.; Huard, E. Spectroscopic Study of Colloidal VOPO4. 2H2O. J. Solid State Chem. 1986, 63, 202–215. [Google Scholar]
- Pierre, A.C. Introduction to Sol-Gel Processing; Kluwer Academic Publishers: Norwell, MA, USA, 1998; p. 242. [Google Scholar]
- Teichner, S.J. Aerogels; Fricke, J., Ed.; Sringer-Verlag: Berlin, Germany, 1986; p. 22. [Google Scholar]
- Griesel, L.; Bartley, J.K.; Wells, R.P.K.; Hutchings, G.J. Preparation of vanadium phosphate catalyst precursors using a high pressure method. Catal. Today 2005, 99, 131–136. [Google Scholar]
- Dong, W.; Bartley, J.K.; Dummer, N.F.; Girgsdies, F.; Su, D.; Schloegl, R.; Volta, J.; Hutchings, G.J. Reaction of vanadium phosphates with alcohols at elevated temperature and pressure. J. Mater. Chem. 2005, 15, 3214–3220. [Google Scholar] [CrossRef]
- Griesel, L.; Bartley, J.K.; Wells, R.P.K.; Hutchings, G.J. Preparation of vanadium phosphate catalysts from VOPO4·2H(2)O: Effect of VOPO4·2H(2)O preparation on catalyst preparation. J. Mol. Catal. A 2004, 220, 113–119. [Google Scholar] [CrossRef]
- Taufiq-Yap, Y.H.; Hasbi, A.R.M.; Hussein, M.Z.; Hutchings, G.J.; Bartley, J.; Dummer, N. Synthesis of vanadium phosphate catalysts by hydrothermal method for selective oxidation of n-butane to maleic anhydride. Catal. Lett. 2006, 106, 177–181. [Google Scholar]
- Yamamoto, N.; Hiyoshi, N.; Okuhara, T. Thin-layered sheets of VOHPO4·0.5H2O prepared from VOPO4·2H2O by intercalation-exfoliation-reduction in alcohol. Chem. Mater. 2002, 14, 3882–3888. [Google Scholar] [CrossRef]
- Johnson, J.W.; Jacobson, A.J.; Brody, J.F.; Rich, S.M. Coordination Intercalation Reactions of the Layered Compounds VOPO4 and VOASO4 with Pyridine. Inorg. Chem. 1982, 21, 3820–3825. [Google Scholar] [CrossRef]
- Johnson, J.W.; Jacobson, A.J.; Brody, J.F.; Lewandowski, J.T. Layered Compounds with Alternating Organic and Inorganic Layers–Vandyl Organophophonates. Inorg. Chem. 1984, 23, 3842–3844. [Google Scholar] [CrossRef]
- Misawa, M. Effective Diameter of Molecules and Liquid-gas Critical Point. J. Chem. Phys. 1990, 93, 8401–8402. [Google Scholar] [CrossRef]
- El Haskouri, J.; Cabrera, S.; Roca, M.; Alamo, J.; Beltran-Porter, A.; Beltran-Porter, D.; Marcos, M.D.; Amoros, P. Synthesis of a new mesostructured lamellar oxovanadium phosphate assembled through an (S+X-10) mechanism. Inorg. Chem. 1999, 38, 4243–4248. [Google Scholar]
- Poling, B.E.; Prausnitz, J.M.; O’Connell, J.P. The Properties of Gases and Liquids; McGraw-Hill: New York, NY, USA, 2001; pp. A–50. [Google Scholar]
- Ennaciri, S.A.; Malka, K.; Louis, C.; Barboux, P.; R'kha, C.; Livage, J. Sol-gel synthesis and catalytic properties of vanadium phosphates Bulk materials. Phosphorus Res. Bull. 2000, 11, 87–93. [Google Scholar]
- Kuehl, R.O. Design of Experiments: Statistical Principles of Research Design and Analysis; Brooks/Cole: Pacific Grove, CA, USA, 2000; p. 94. [Google Scholar]
- Amoros, P.; Ibanez, R.; Martinez-Tamayo, E.; Beltran-Porter, A.; Beltran-Porter, D.; Villeneuve, G. New Vandayl Hydrogenphosphate Hydrates—Electronic-Spectra of the VO2+ Ion in the VO(HxPO4)x·yH2O Sysetem. Mater. Res. Bull. 1989, 24, 1347–1360. [Google Scholar] [CrossRef]
- Busca, G.; Cavani, F.; Centi, G.; Trifiro, F. Natura and mechanism of formation of vanadyl pyrophosphate–active phase in normal-butane selective oxidation. J. Catal. 1986, 99, 400–414. [Google Scholar] [CrossRef]
- Albonetti, S.; Cavani, F.; Ligi, S.; Pierelli, F.; Trifiro, F.; Ghelfi, F.; Mazzoni, G. The effect of glycols in the organic preparation of V/P mixed oxide, catalyst for the oxidation of n-butane to maleic anhydride. Studies Surface Sci. Catal. 2002, 143, 963–973. [Google Scholar]
- Utamapanya, S.; Klabunde, K.J.; Schlup, J.R. Nanoscale metal-oxide particle clusters as chemical reagents—synthesis and properties of ultrahigh surface-area magnesium-hydroxide and magnesium-oxide. Chem. Mater. 1991, 3, 175–181. [Google Scholar] [CrossRef]
- Nakamura, M.; Kawai, K.; Fujiwara, Y. Structure and Activity of Vanadyl Phosphate Catalysts. J. Catal. 1974, 34, 345–355. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Salazar, J.M.; Hohn, K.L. Partial Oxidation of n-Butane over a Sol-Gel Prepared Vanadium Phosphorous Oxide. Catalysts 2013, 3, 11-26. https://doi.org/10.3390/catal3010011
Salazar JM, Hohn KL. Partial Oxidation of n-Butane over a Sol-Gel Prepared Vanadium Phosphorous Oxide. Catalysts. 2013; 3(1):11-26. https://doi.org/10.3390/catal3010011
Chicago/Turabian StyleSalazar, Juan M., and Keith L. Hohn. 2013. "Partial Oxidation of n-Butane over a Sol-Gel Prepared Vanadium Phosphorous Oxide" Catalysts 3, no. 1: 11-26. https://doi.org/10.3390/catal3010011
APA StyleSalazar, J. M., & Hohn, K. L. (2013). Partial Oxidation of n-Butane over a Sol-Gel Prepared Vanadium Phosphorous Oxide. Catalysts, 3(1), 11-26. https://doi.org/10.3390/catal3010011