Conversion of CO2 via Visible Light Promoted Homogeneous Redox Catalysis

Abstract
:1. Introduction
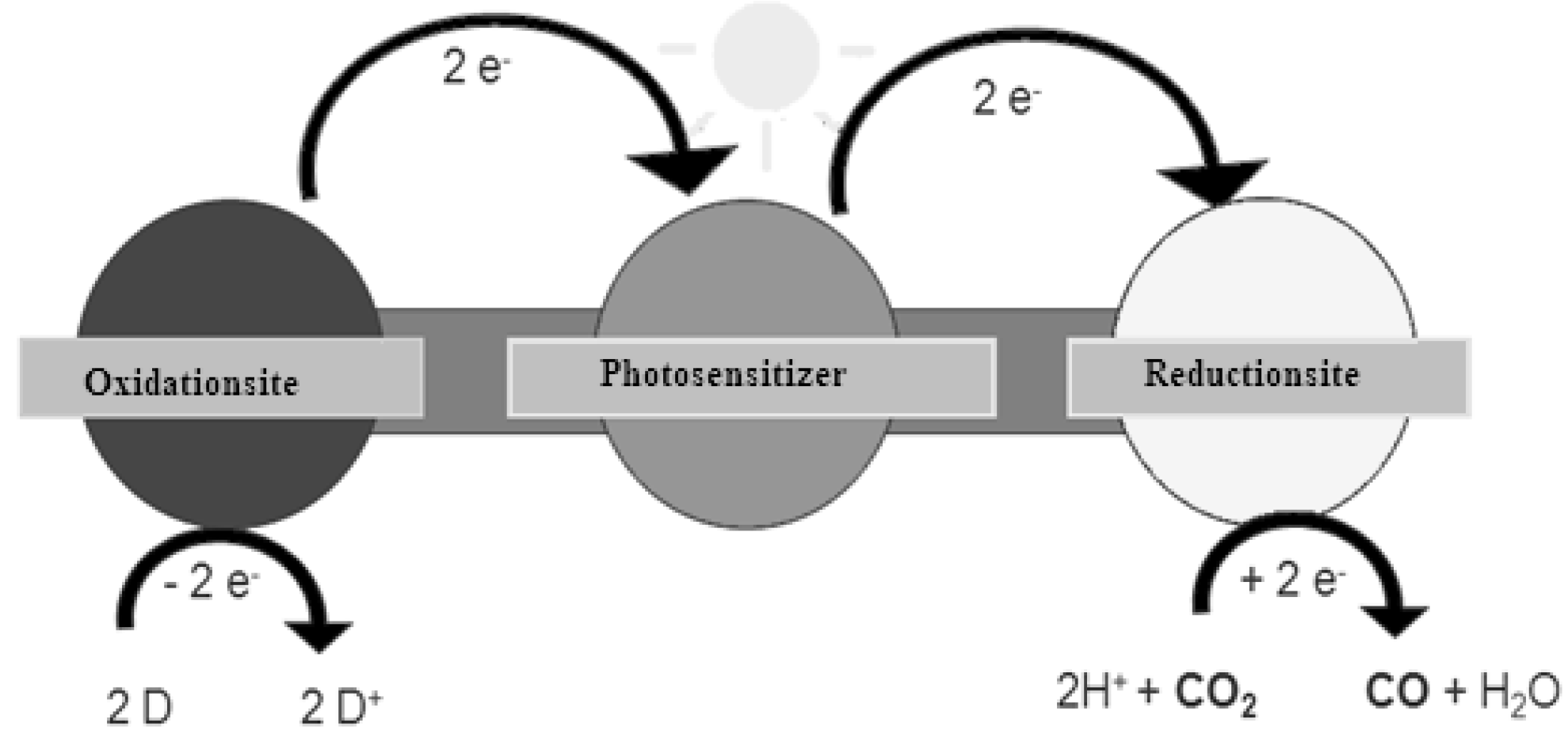
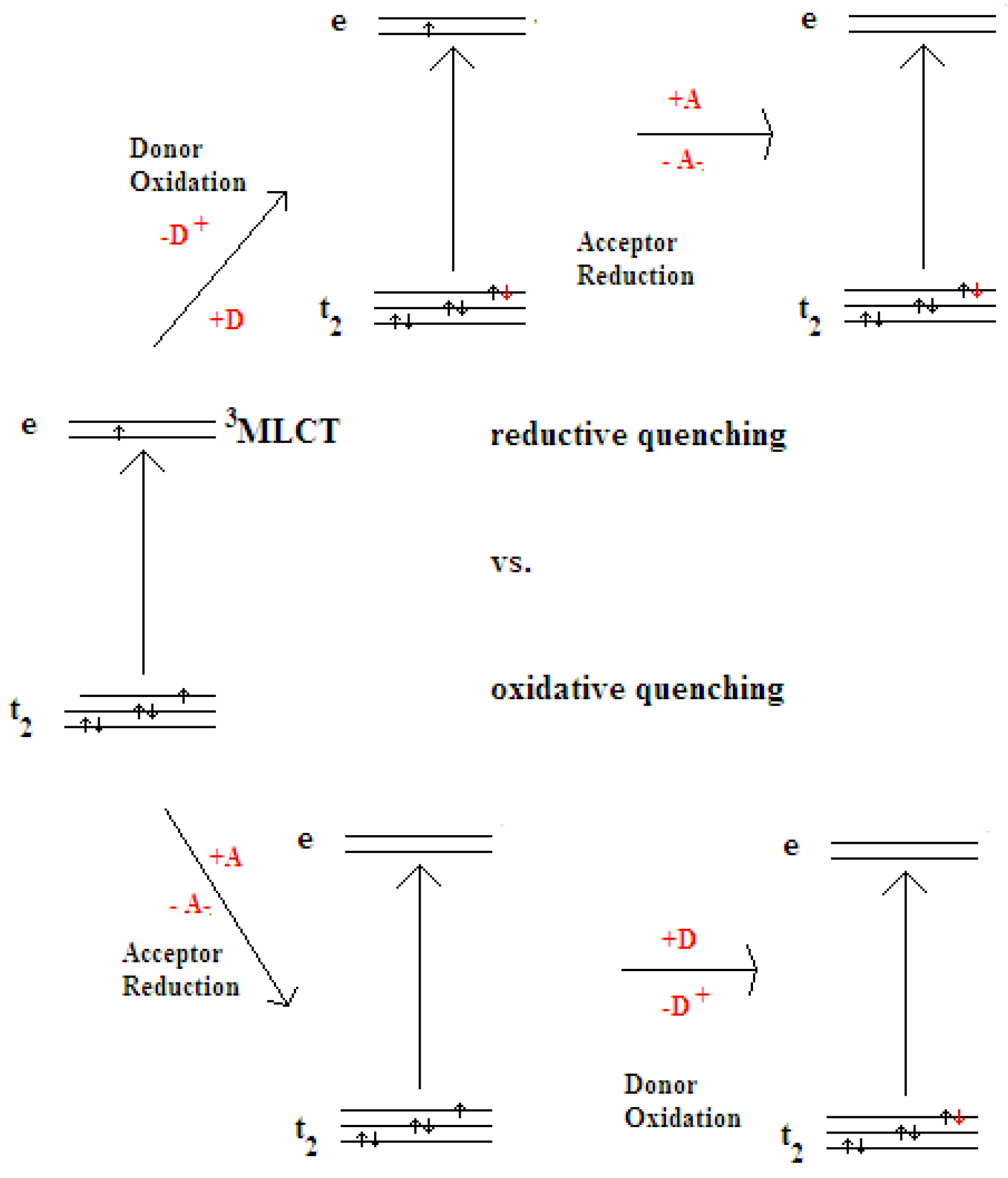
2. Principle of Homogeneous Redox Photocatalysts and Their Building Units
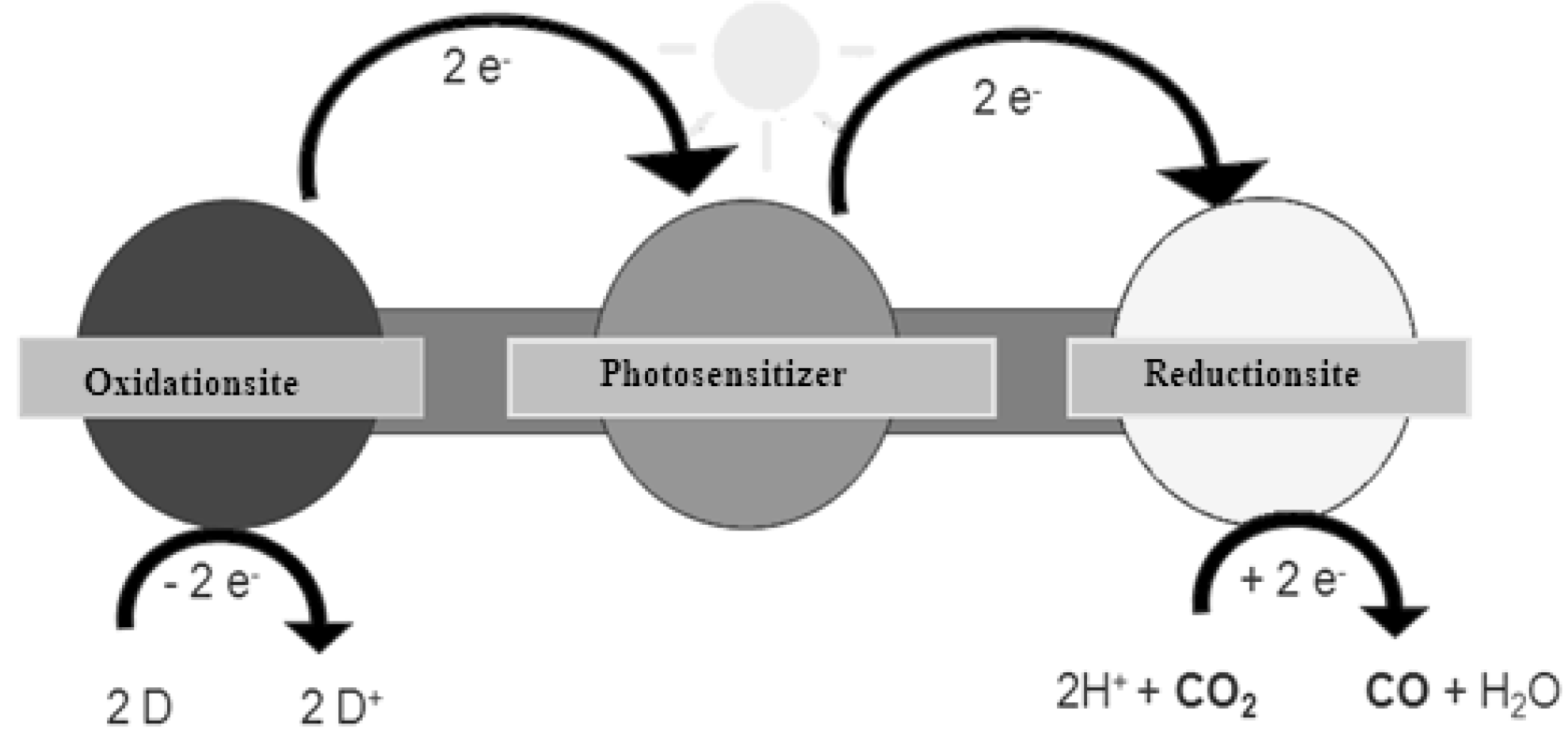
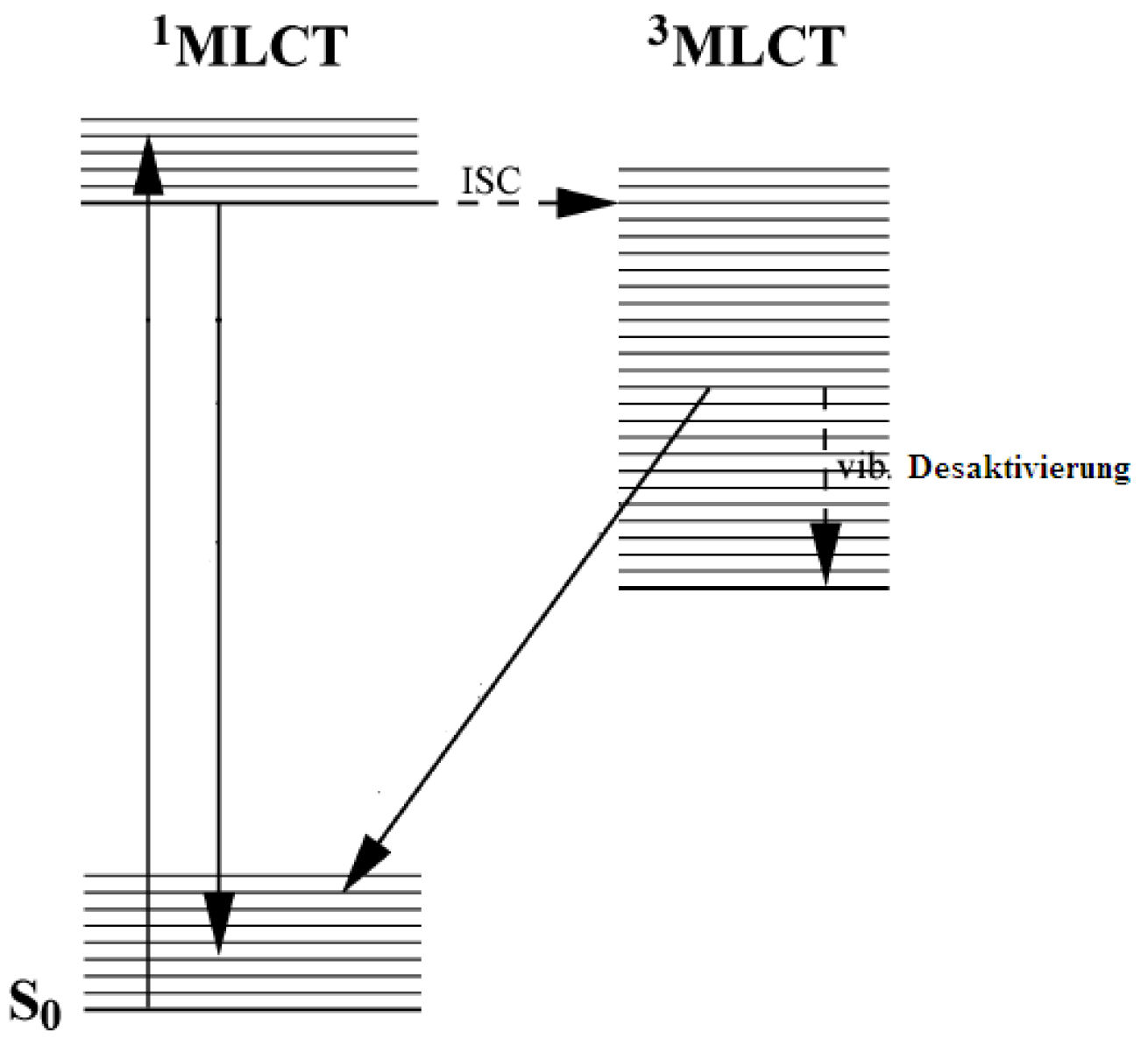
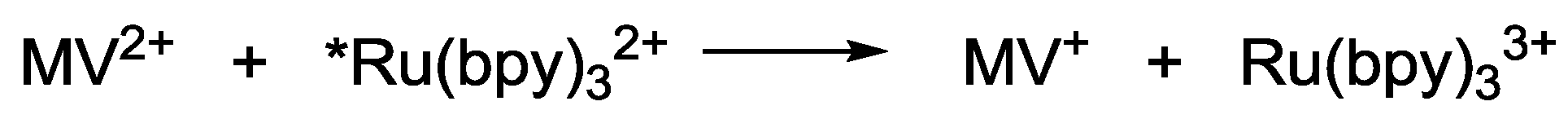
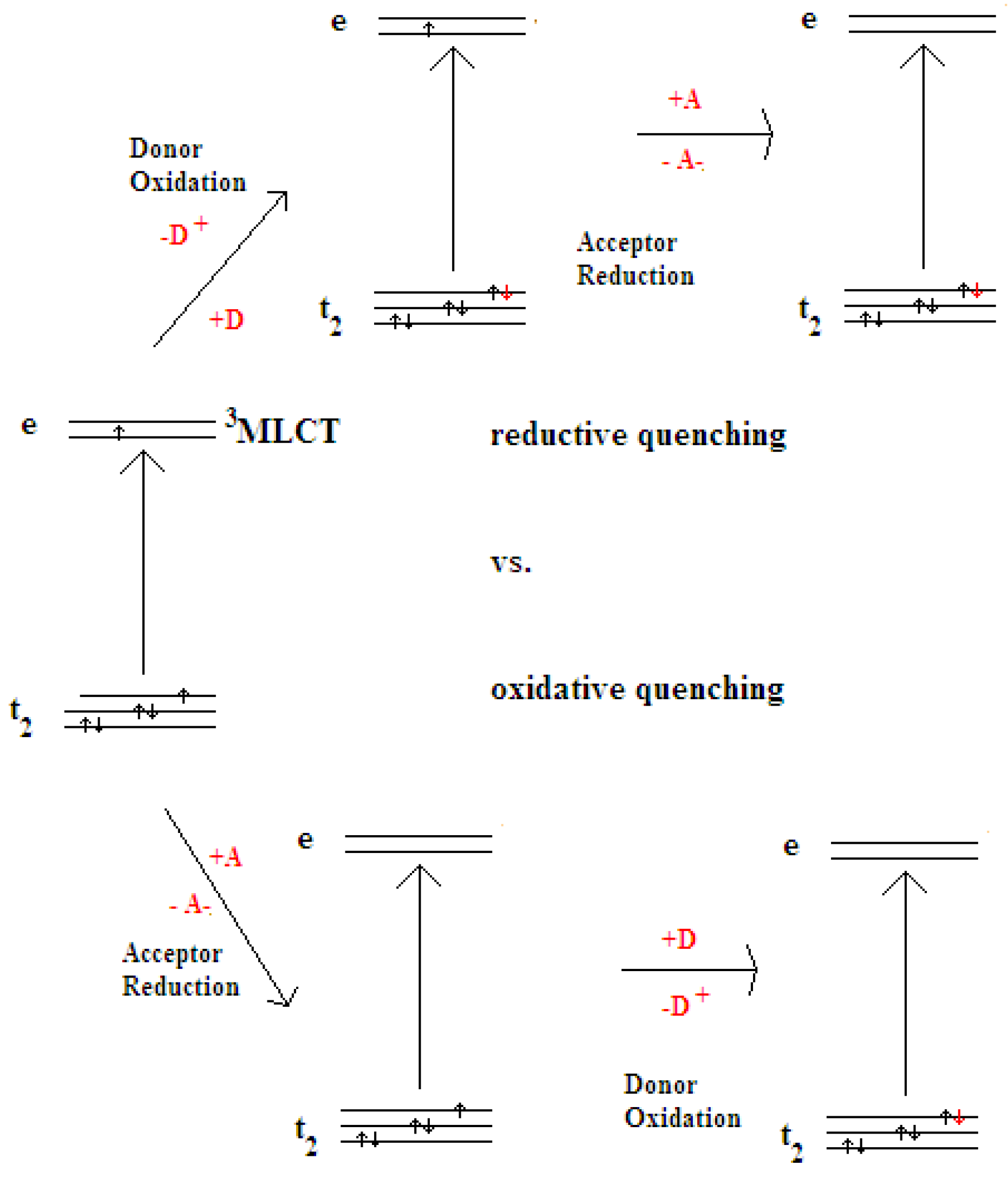
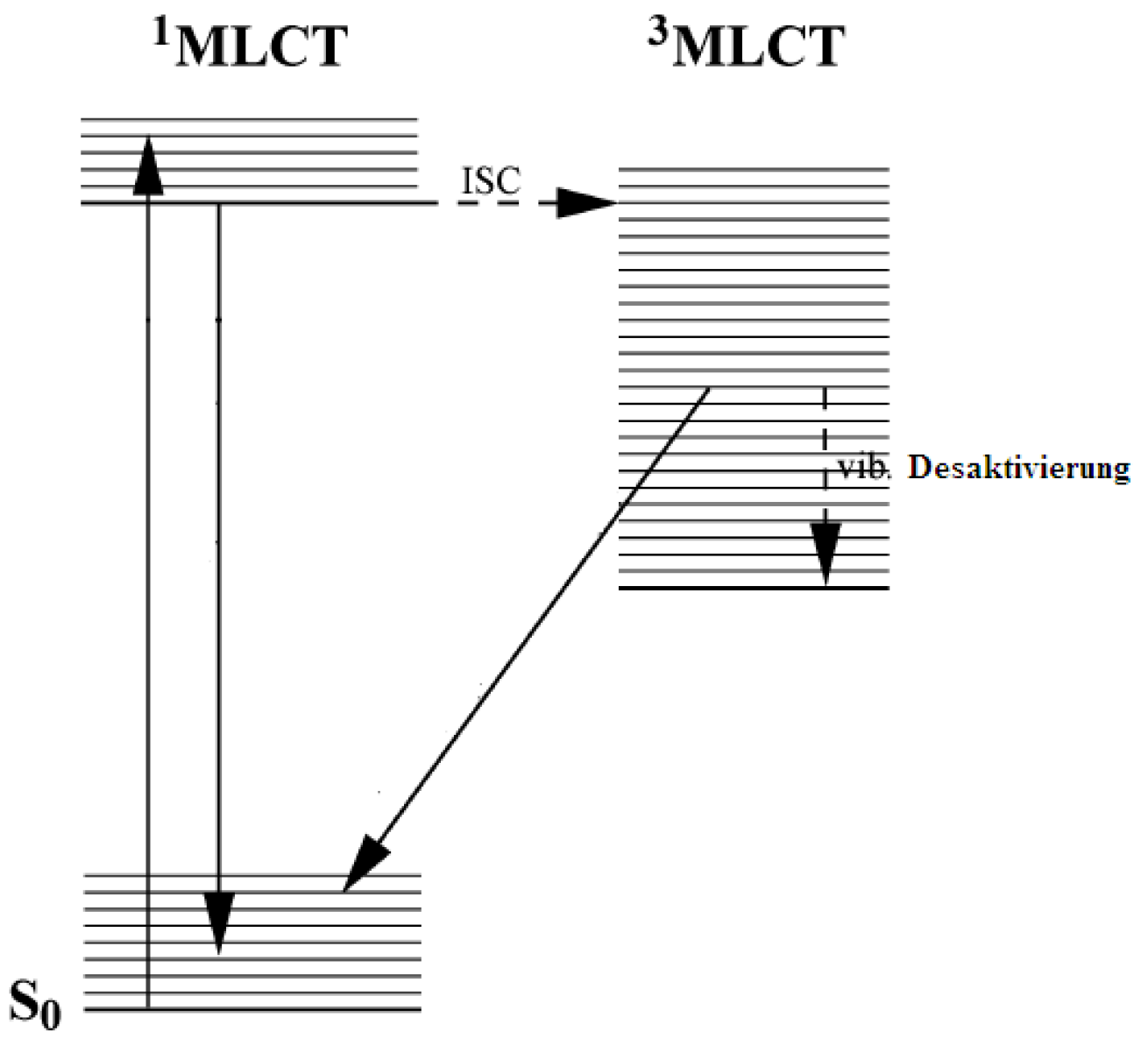
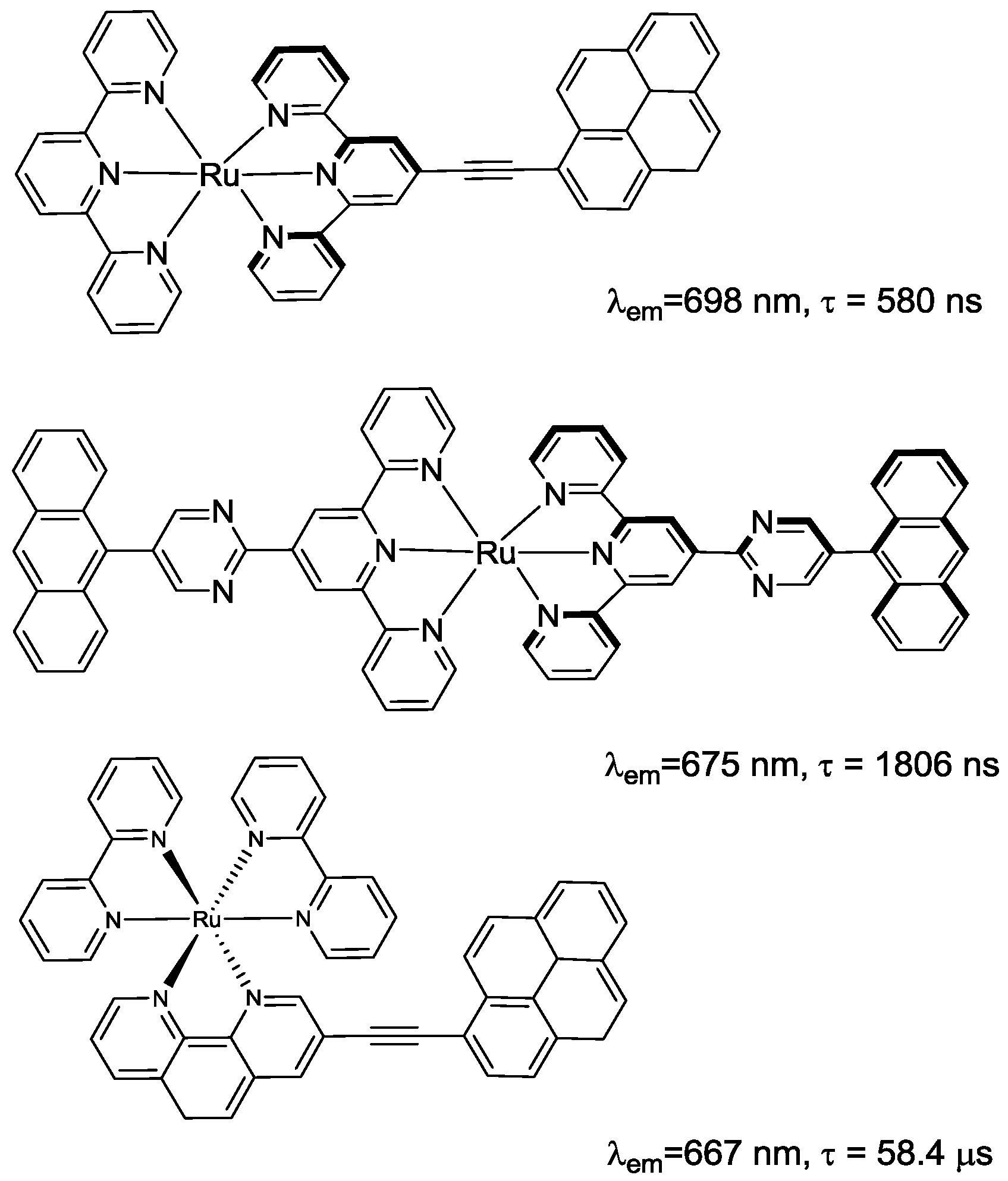
2.1. Chromophores
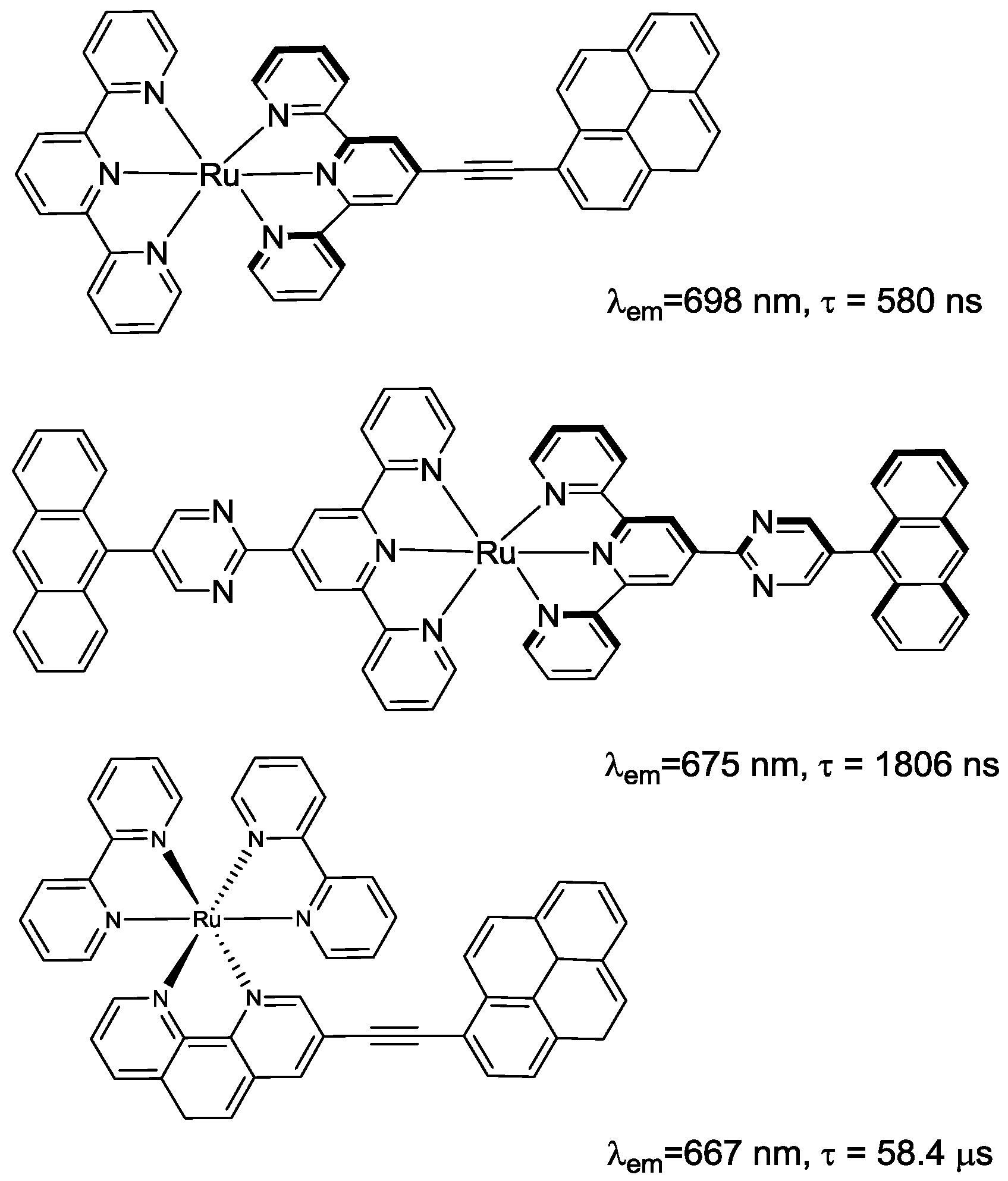

| Emission | Electrochemistry E [V] | ||||||
|---|---|---|---|---|---|---|---|
| Complex | λmax absorption [nm] (103M−1cm−1]) | [nm] | [ns] | ox | red | ref | |
| Ru(bpy)32+ | 452 * (4.16) | 607 * | 0.073 * | 800 * | 1.29 | −1.33, −0.81 a | [48,49] |
| Ru(dmb)32+ | 459 (14.9) | 630 | 0.089 | 840 | +0.80 | −2.22, −1.96, −1.77 b | [17,50,51,52,53,54] |
| Ru(tpy)22+ | 476 (17.7) | - | - | 0.250 | +1.30 | −1.24 a | [28,55] |
| Os(tpy)22+ | 657 (3.65), 477 (13.75) | 718 | 0.014 | 296 | +0.97 | −1.23 a | [56,57,58] |
| Cr(tpy)23+ | 473 | 775 | 50 | +1.43 | −0.17 c | [59,60] | |
| Fe(tpy)22+ | 522 | - | - | 2.5 | +1.05 | −1.17 c | [61] |
| Ir(tpy)23+ | 355 (13.8) | 455 | 70 | [62] | |||
| (bpy)Re(CO)3Cl | 370 (3.42) | 637 | 30 | −1.61 b | [63,64] | ||
| (bpy)Re(CO)3NCS | 375 | 635 | 25 | −1.67 b | [63,65] | ||
| (dmb)Re(CO)3Cl | 366 (3.7) | 601 | 0.0016 | +1.36, 1.85 | −1.43, −1.95 a | [66] | |
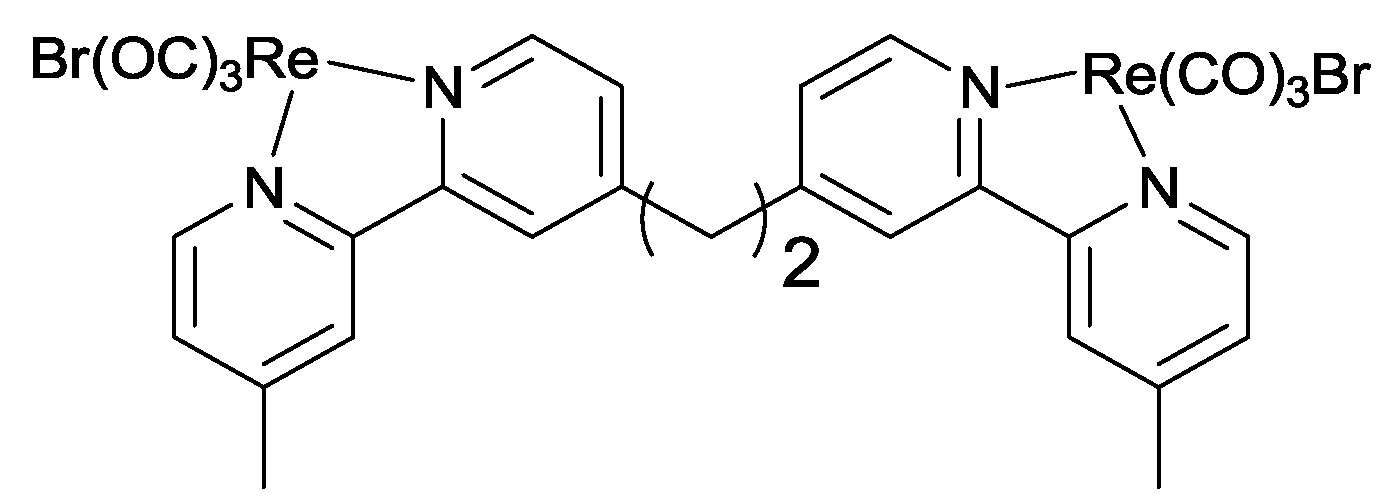
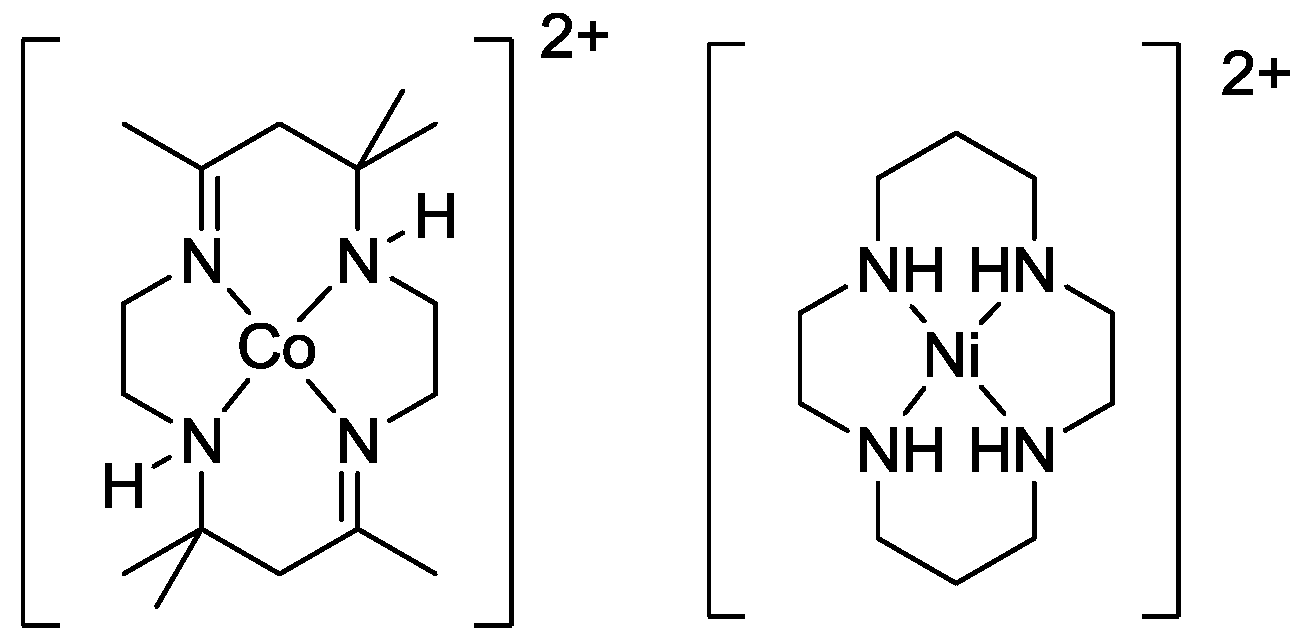
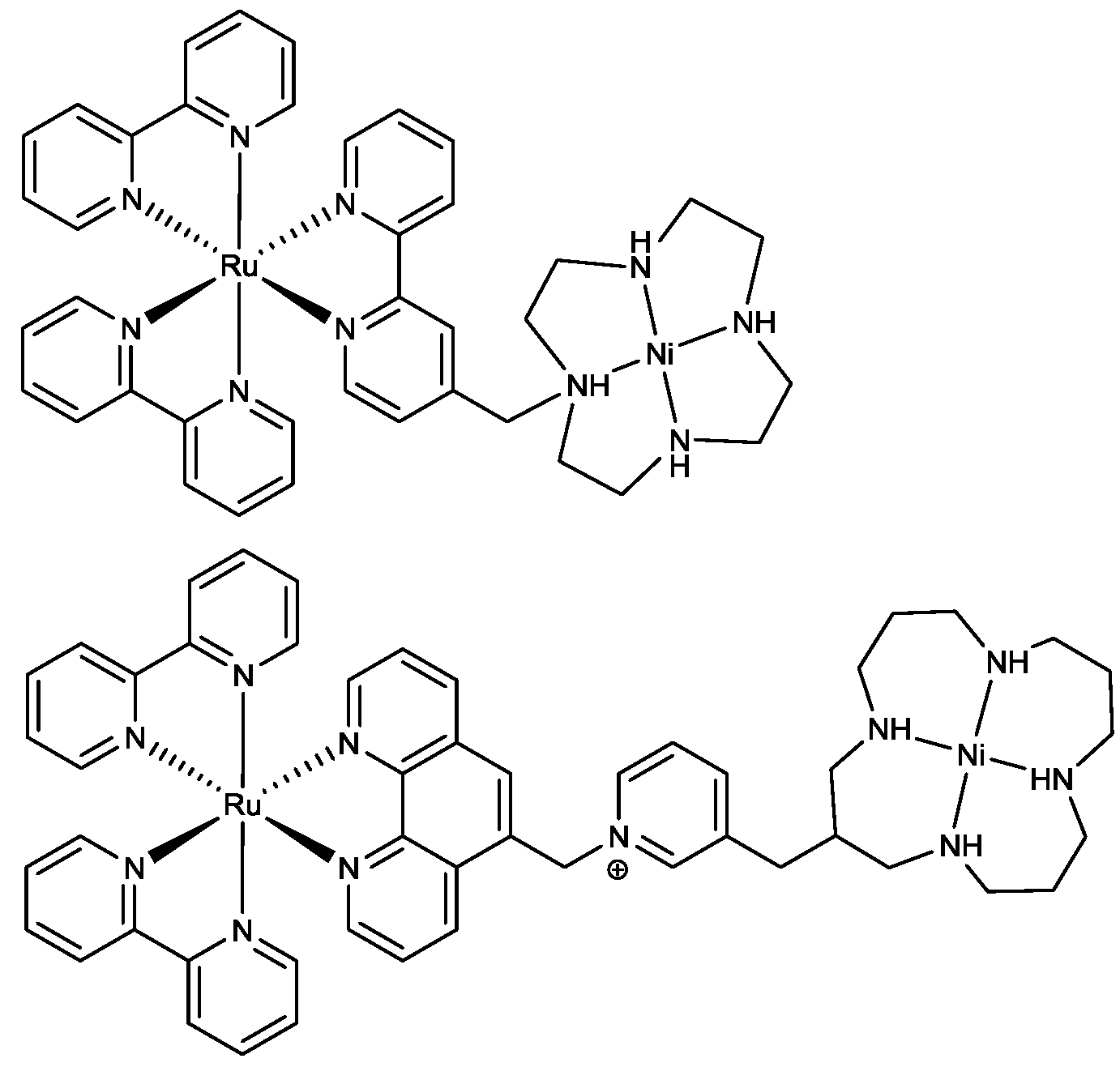
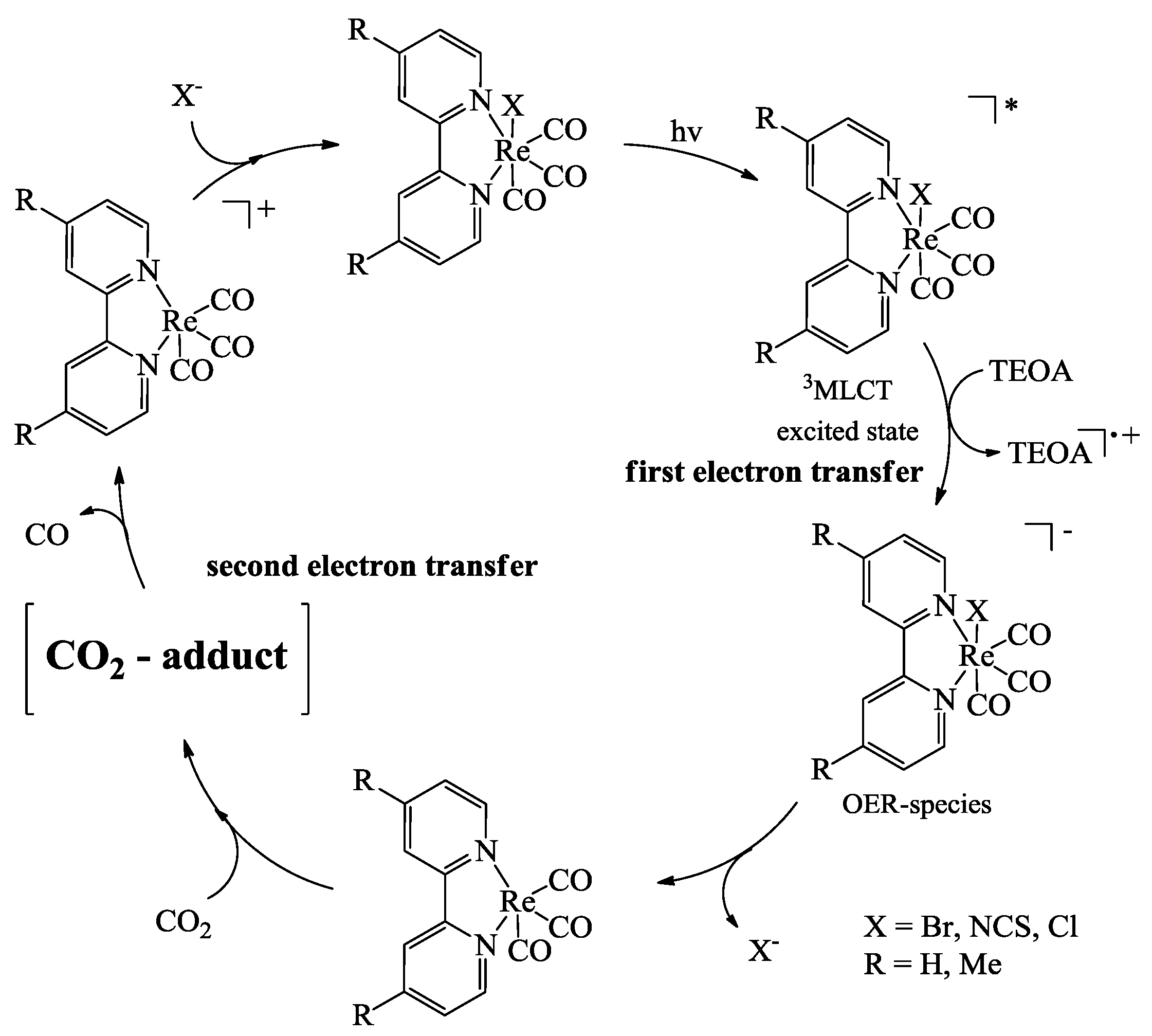
2.2. Design of the Reduction Site
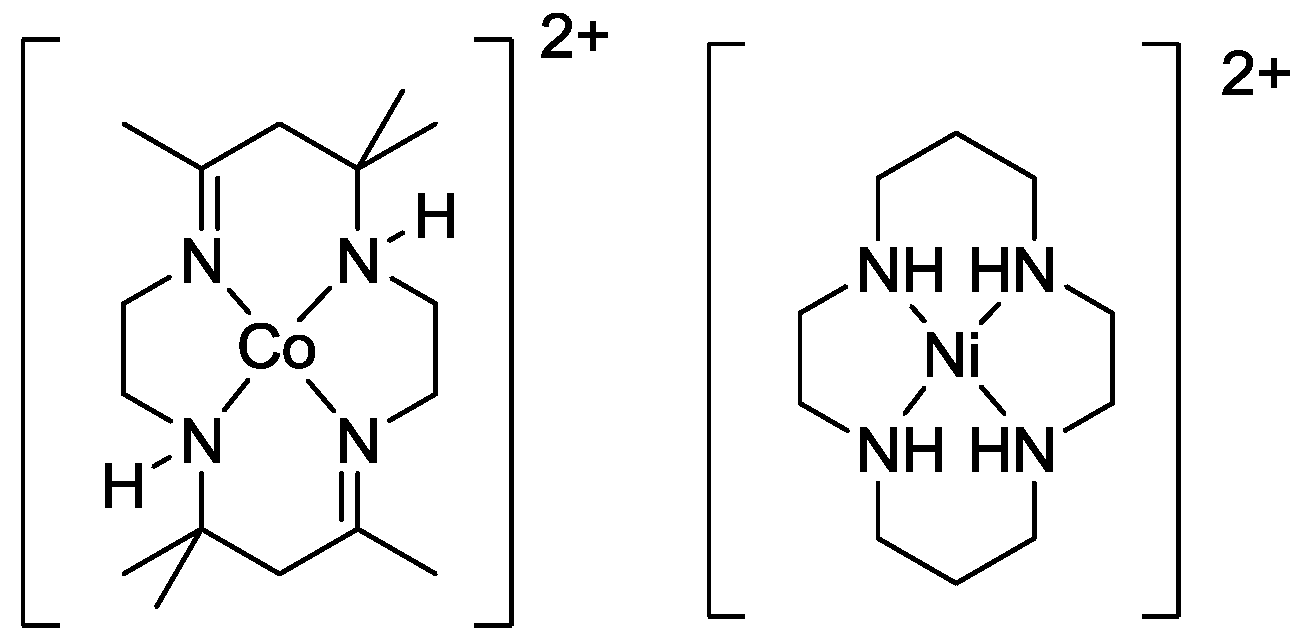
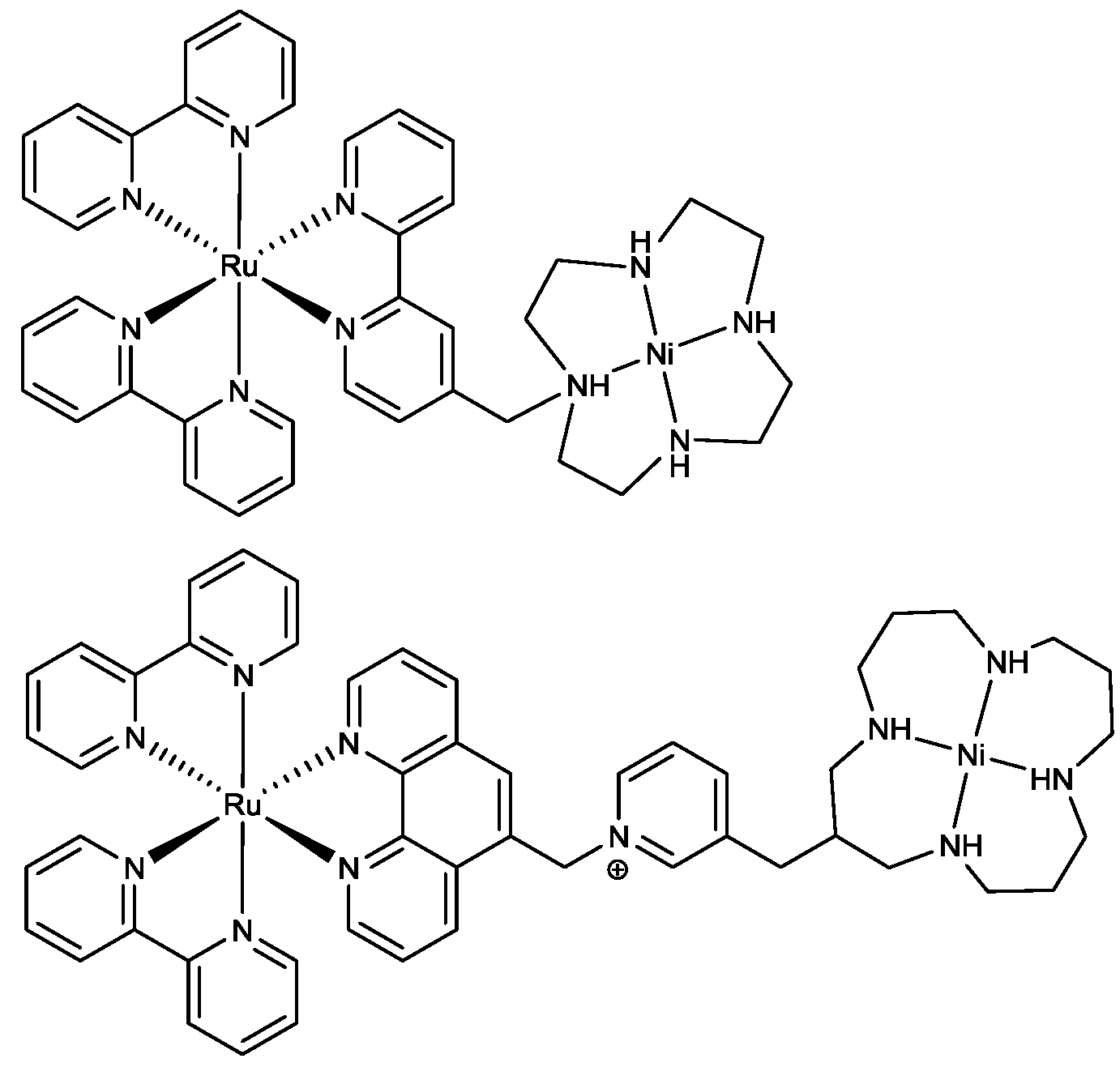
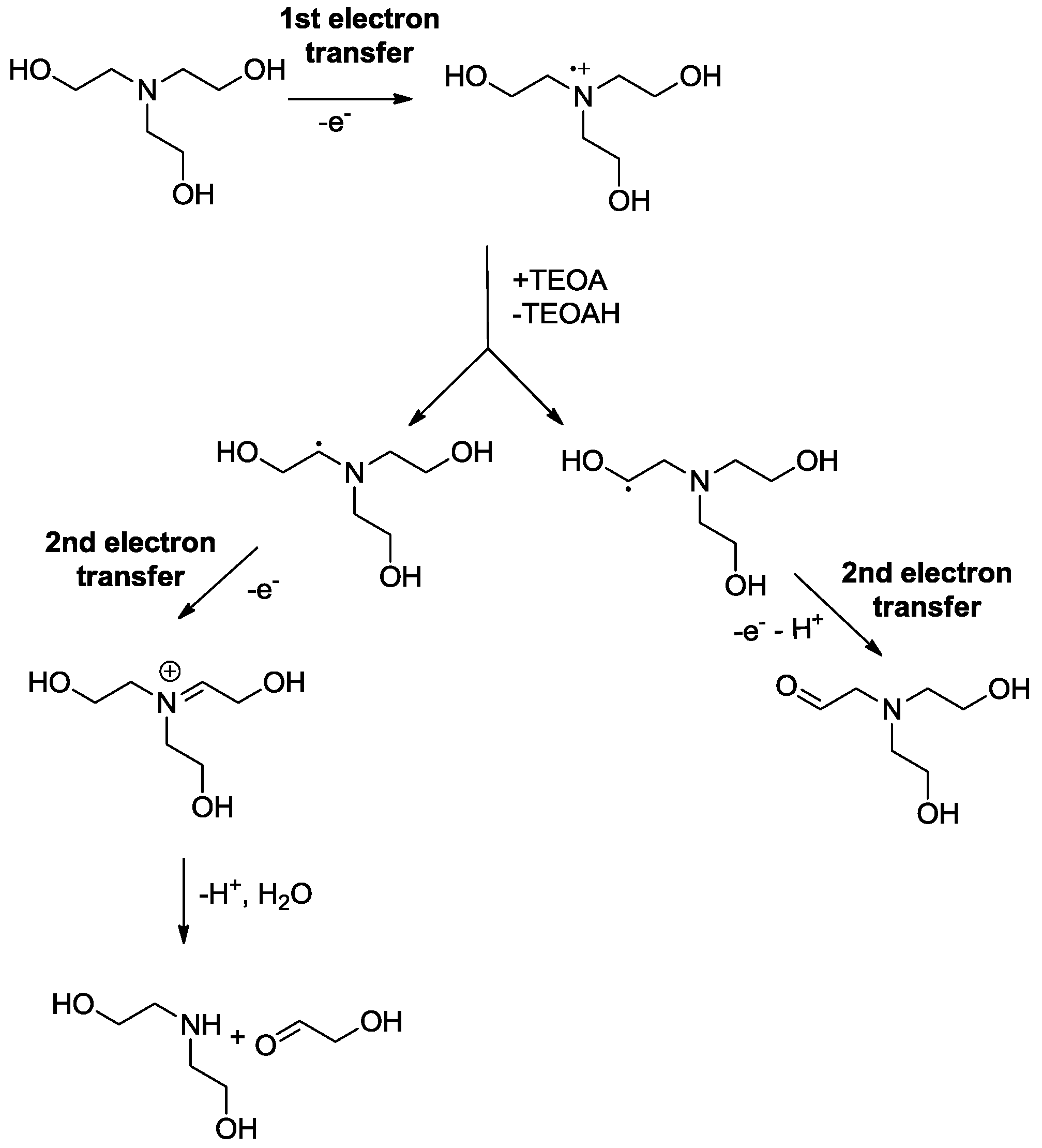
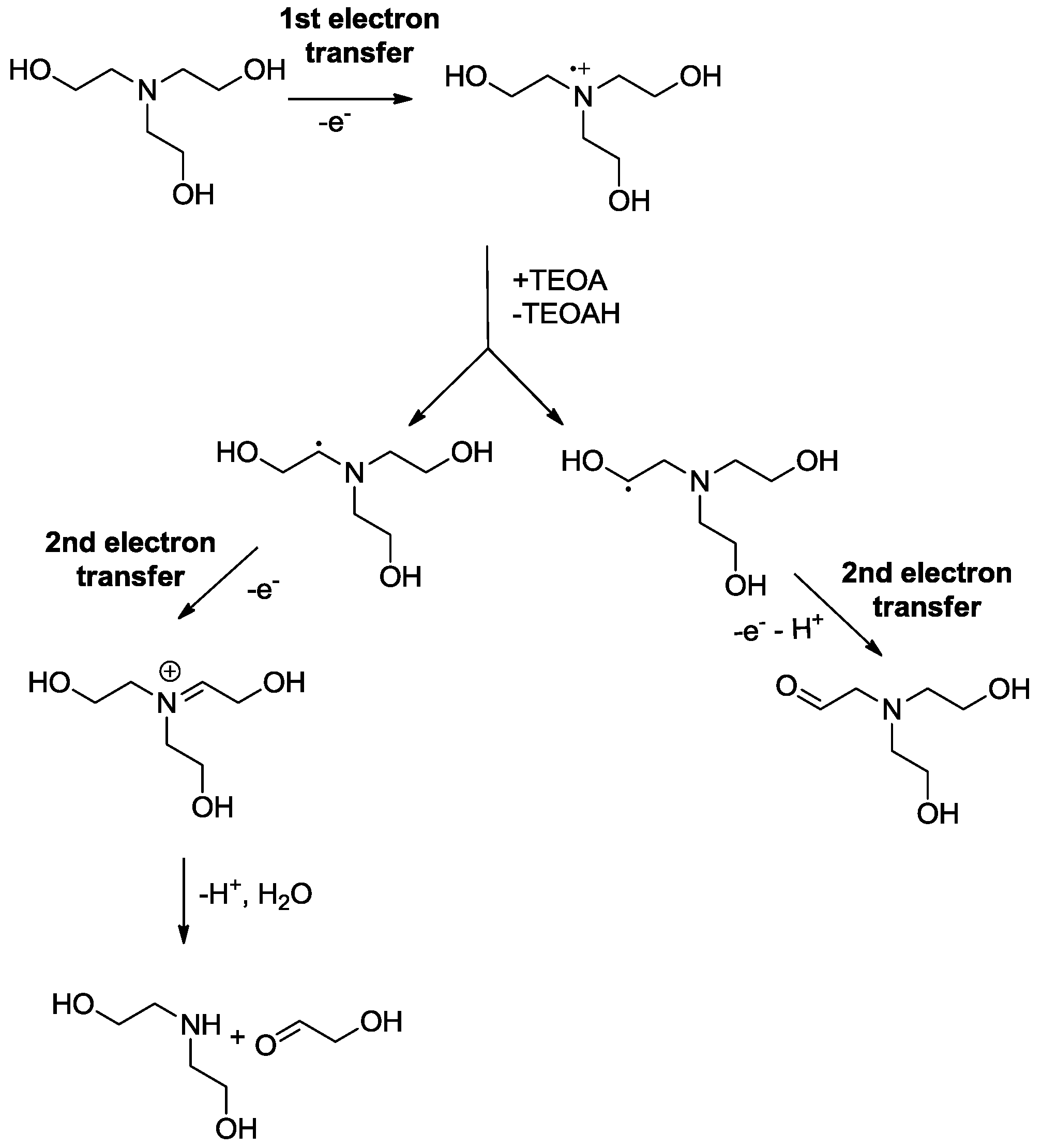
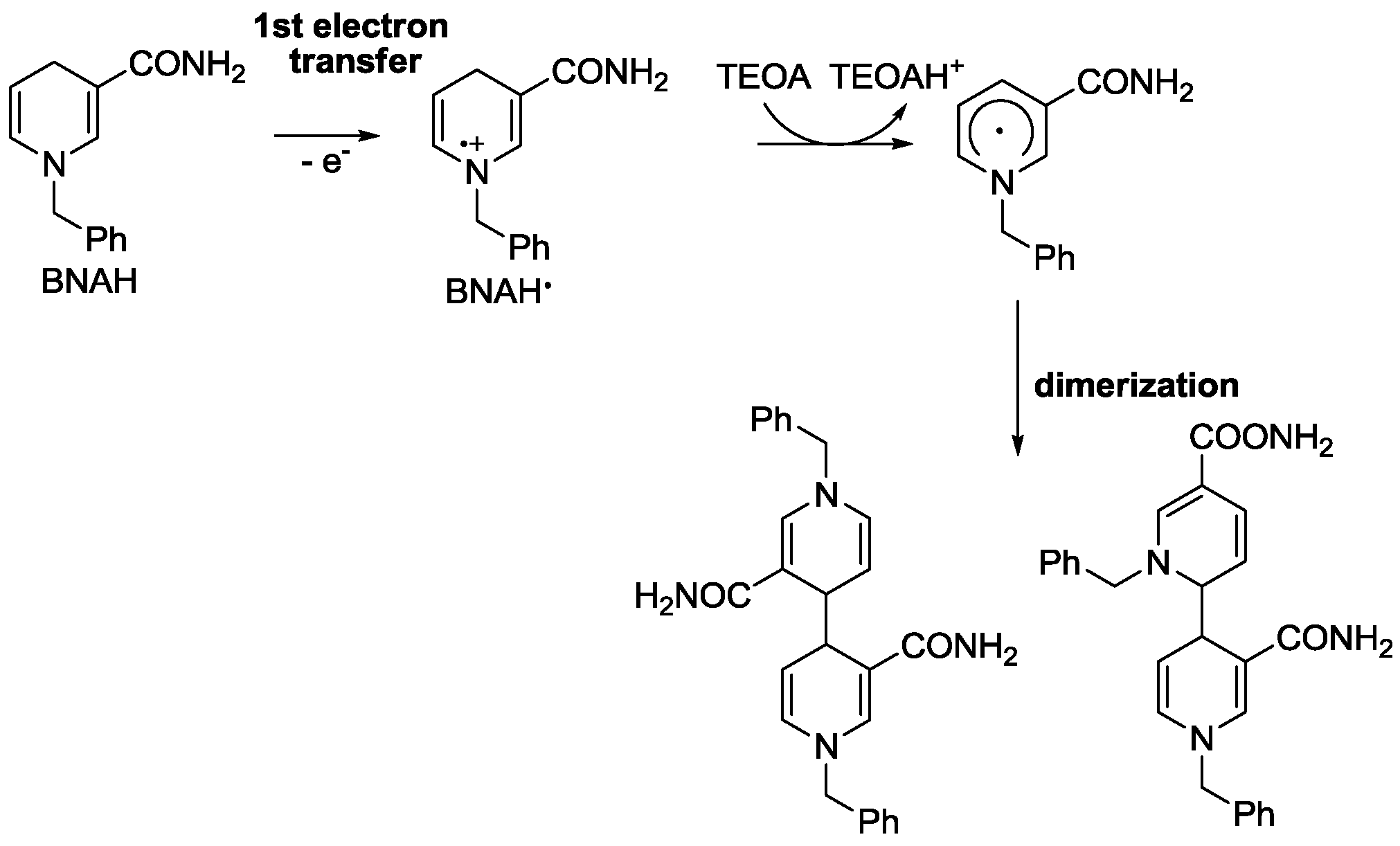
2.3. Oxidation Site

| Donor | kq [107 M−1 s−1] | E0[V] | Lit |
|---|---|---|---|
| N,N,N',N'-Tetramethylbenzidine | 740 | 0.69 | [98] |
| Phenothiazine | 560 | 0.73 | [98] |
| Dimethyl-Dibenzothiofulvalene | 400 | 0.78 | [98] |
| N-Methylpheno-thiazine | 130 | [98] | |
| 1-Benzyl-1,4-dihydronicotinamide | 15 | 0.52 a | [105,106] |
| Diphenylamine | 2.50 | 0.80 | [ 98] |
| Triethanolamine | 0.65 | 0.82 | [12] |
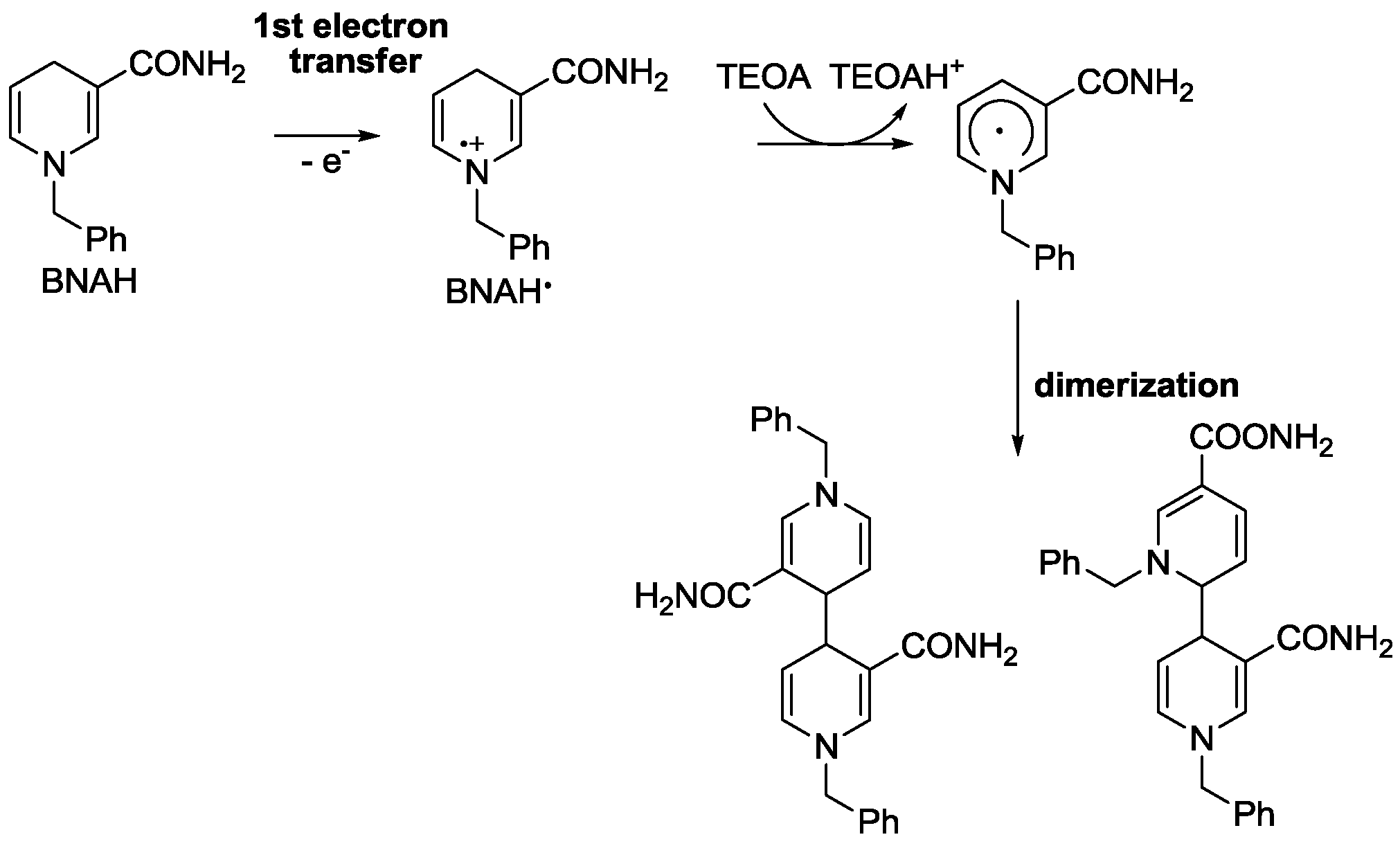
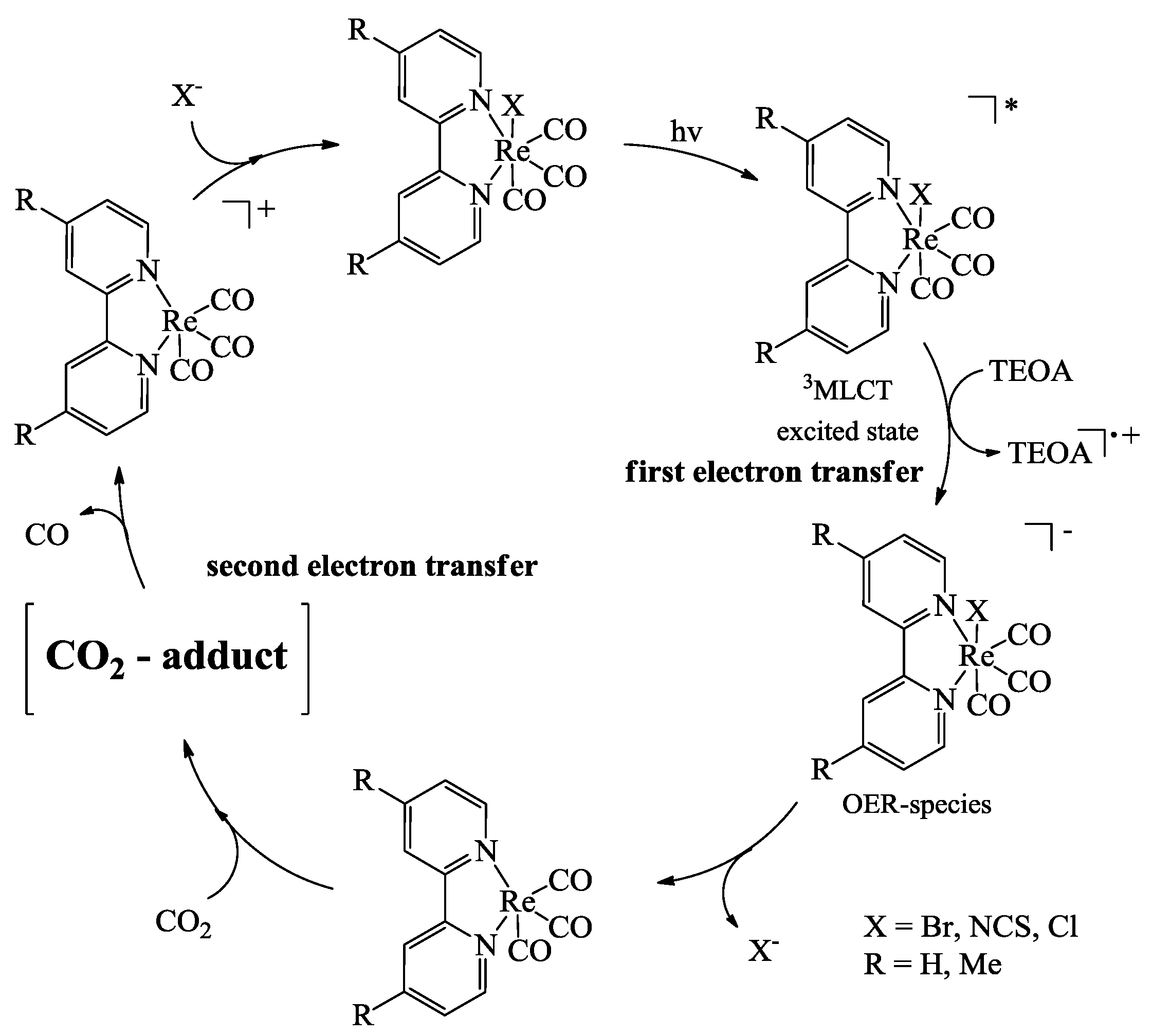
3. Reduction Products of CO2
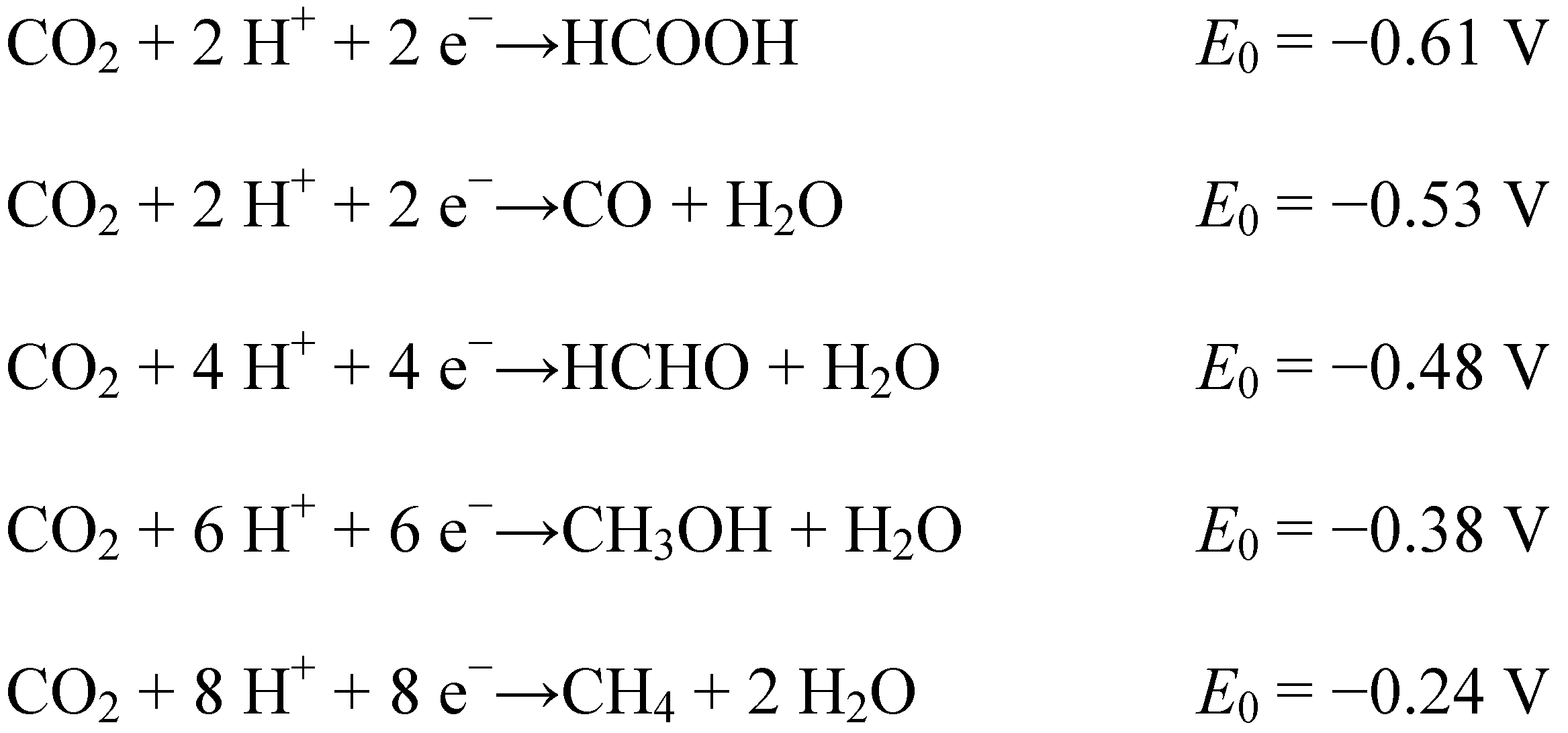
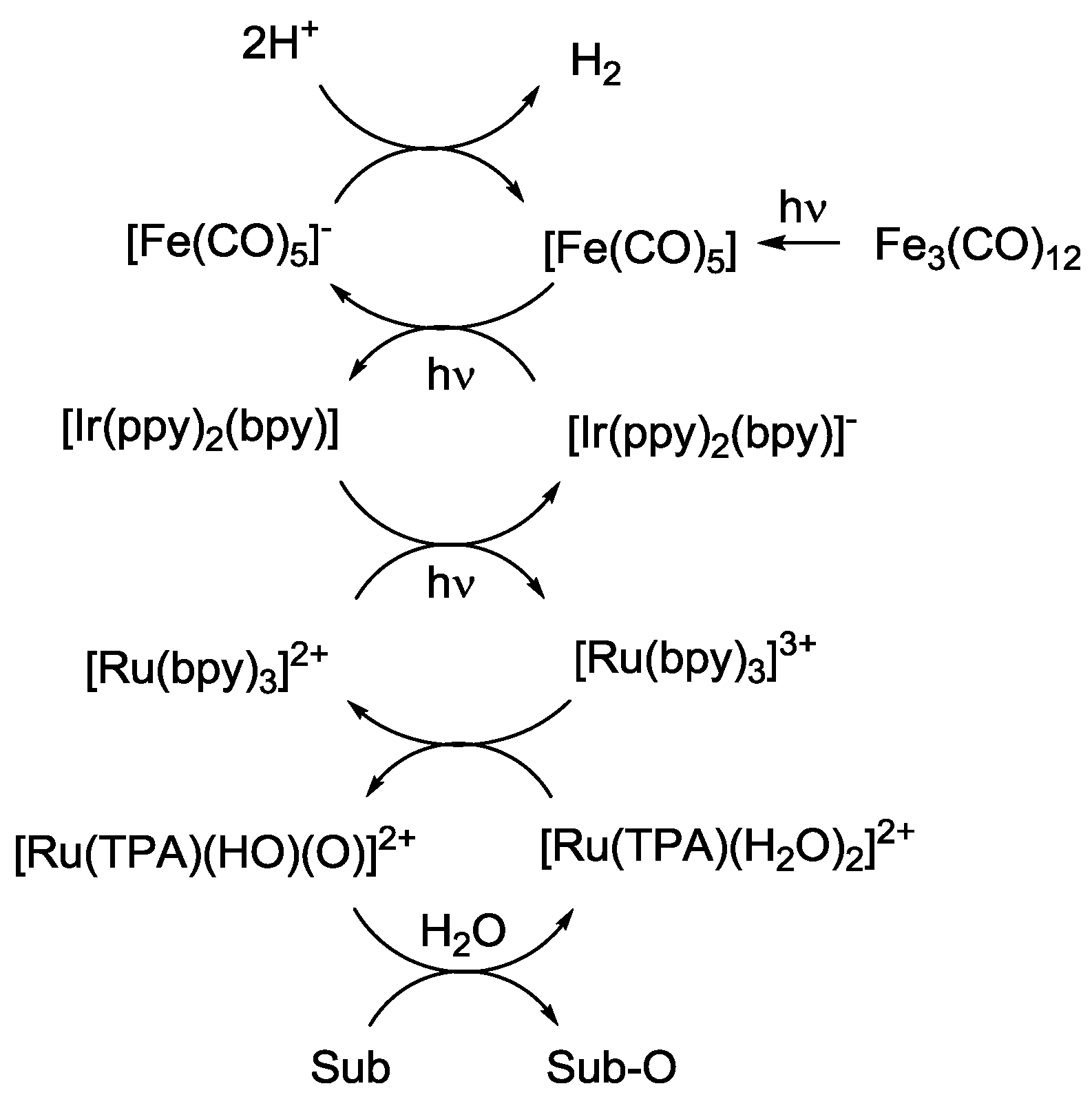
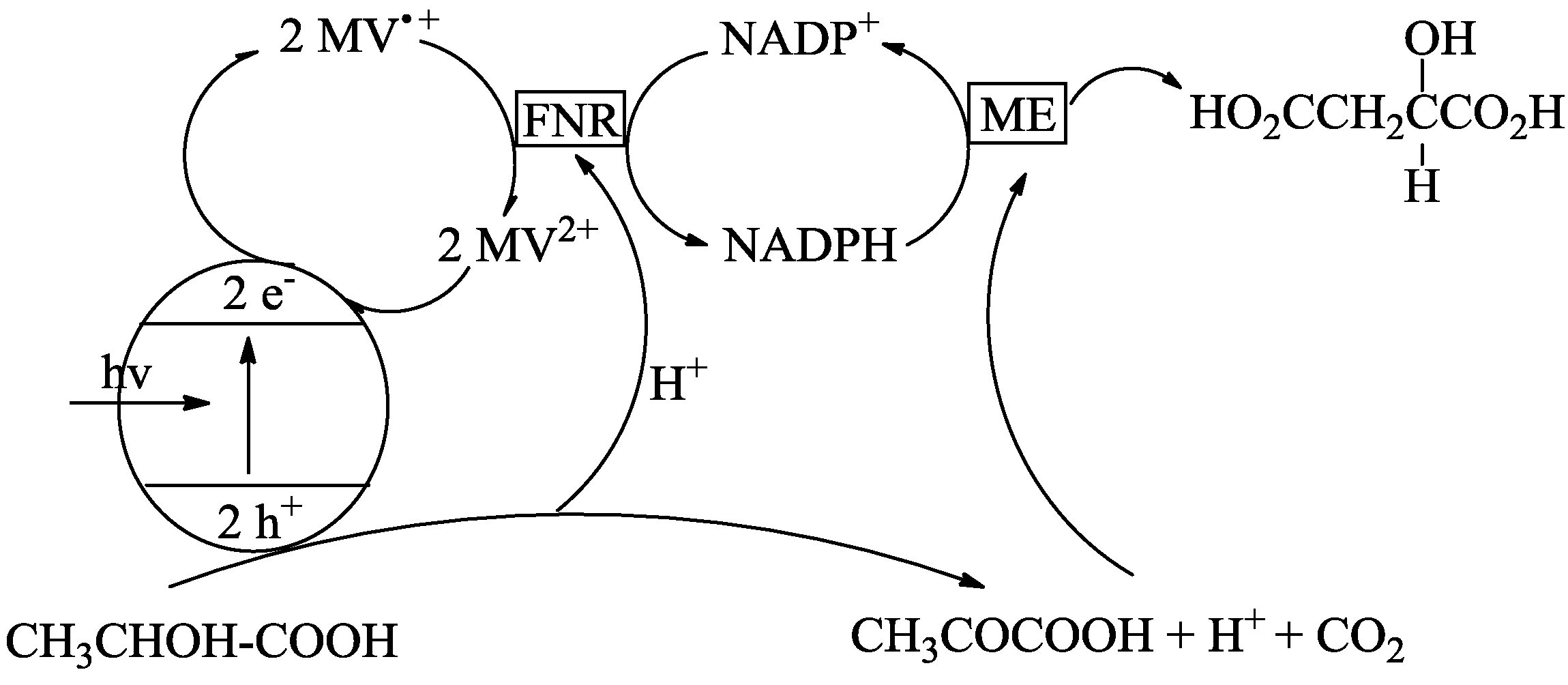
4. Alternatives to Sacrificial Amines
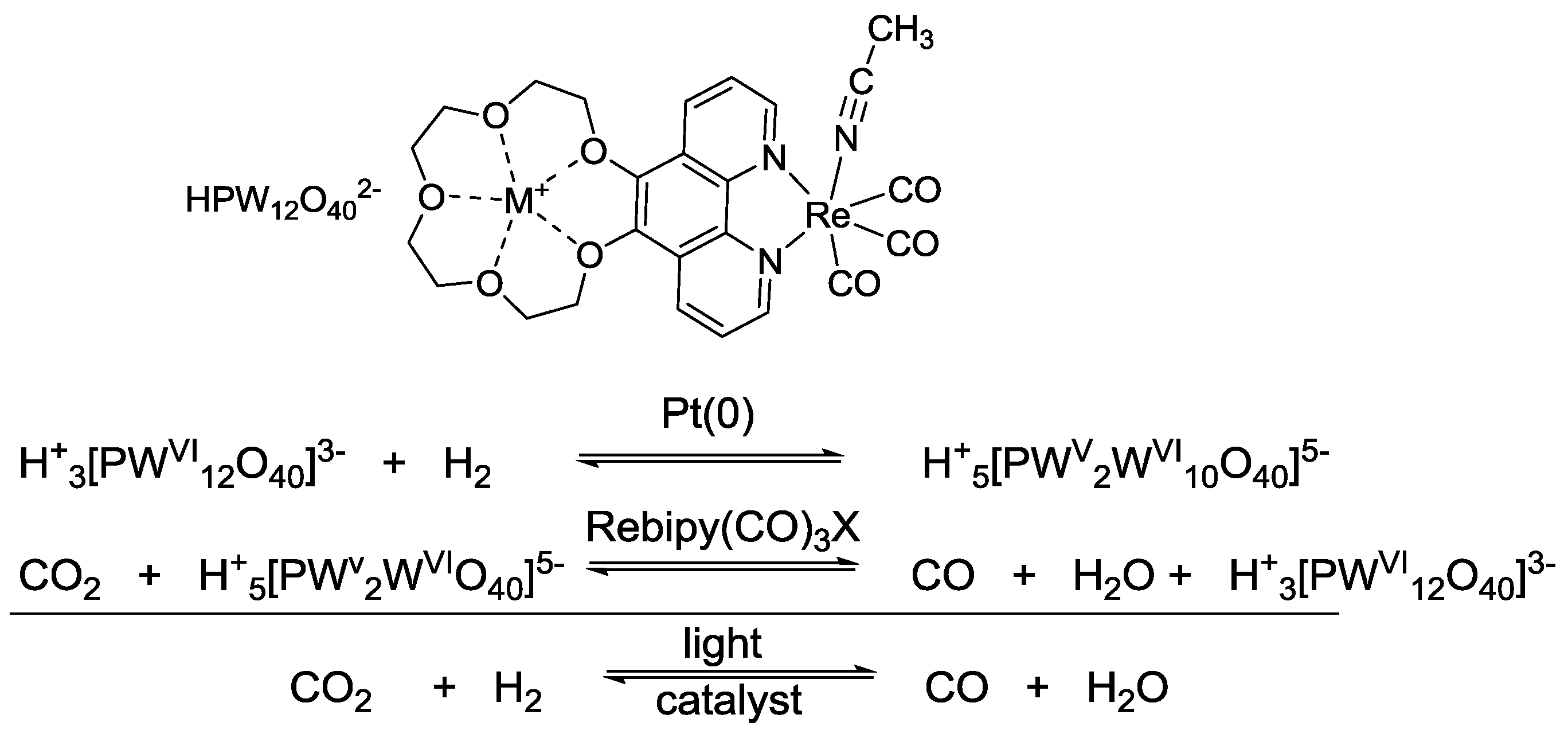
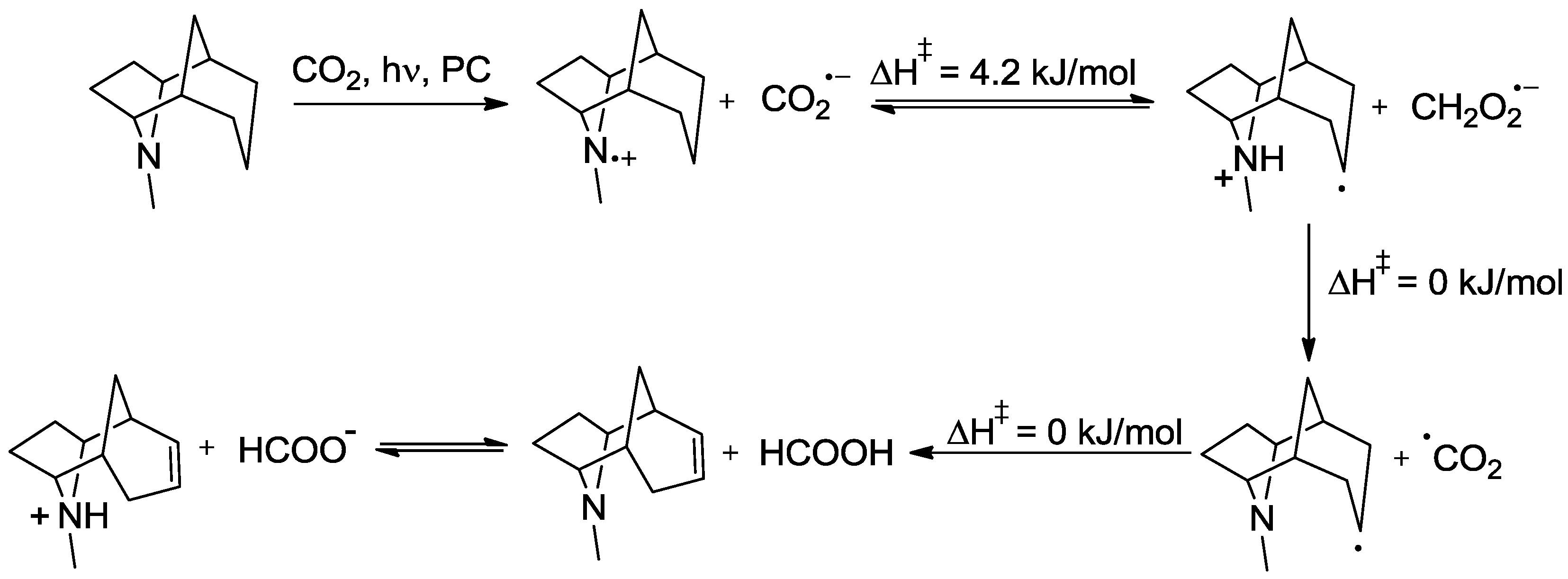
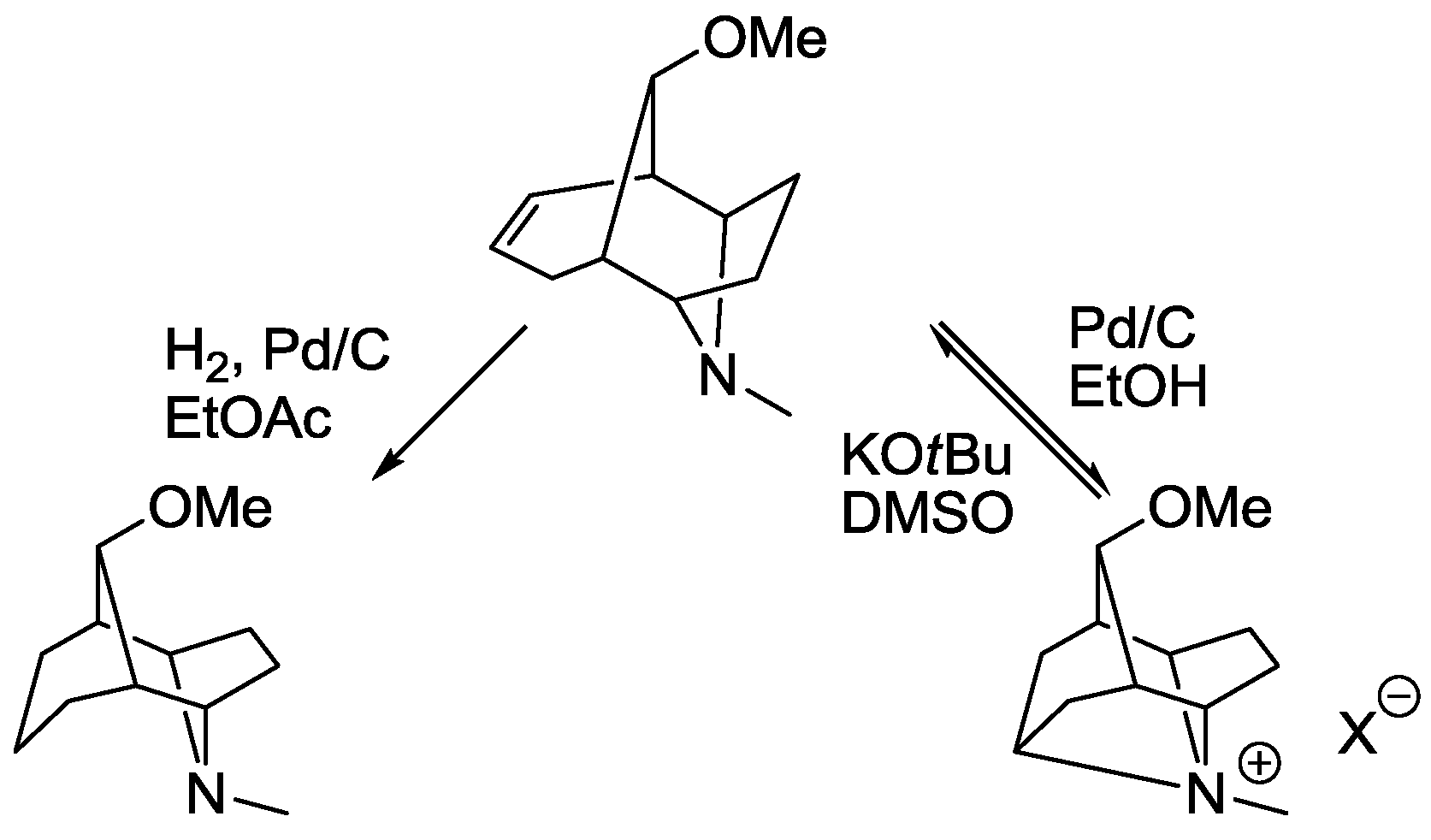
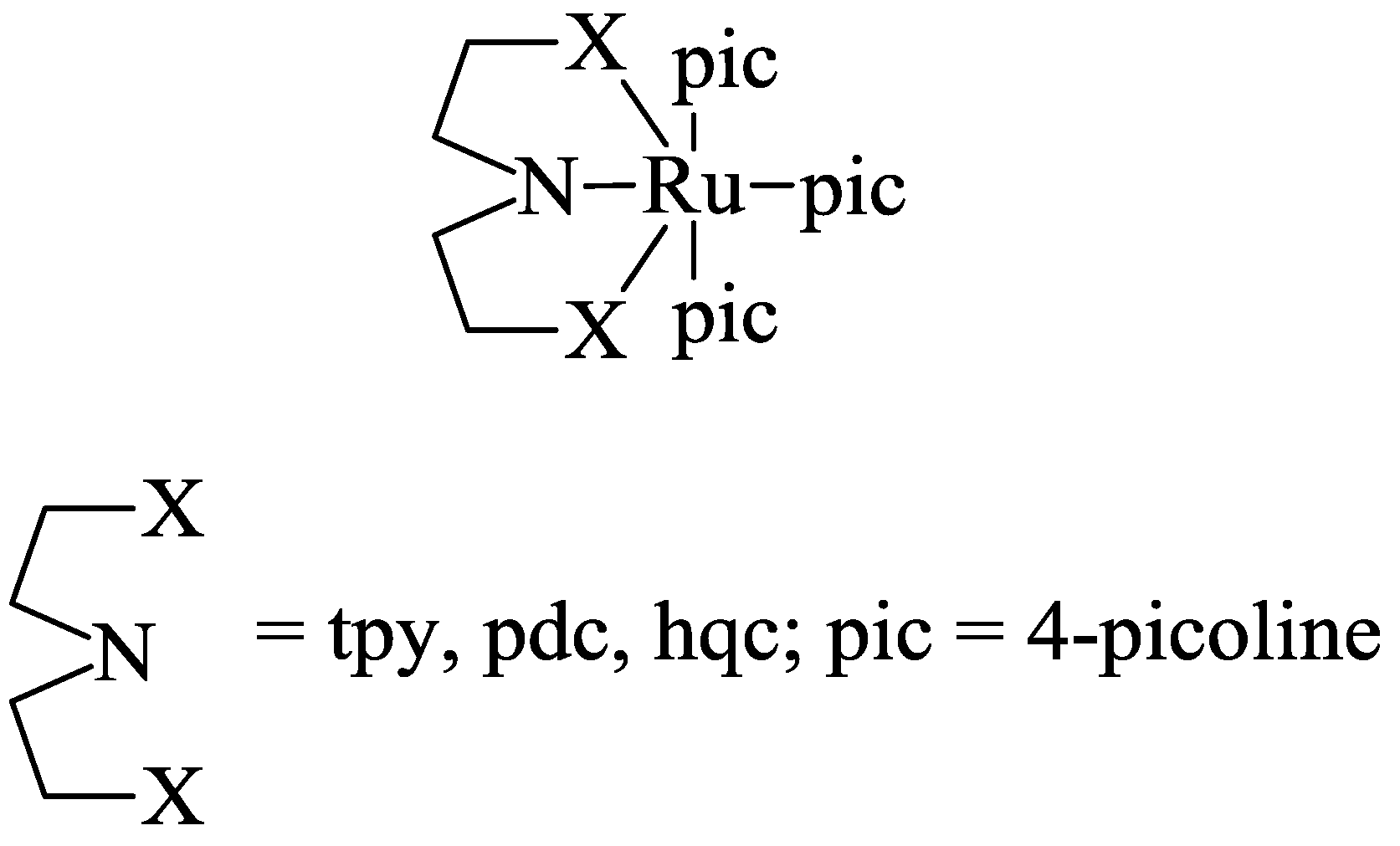
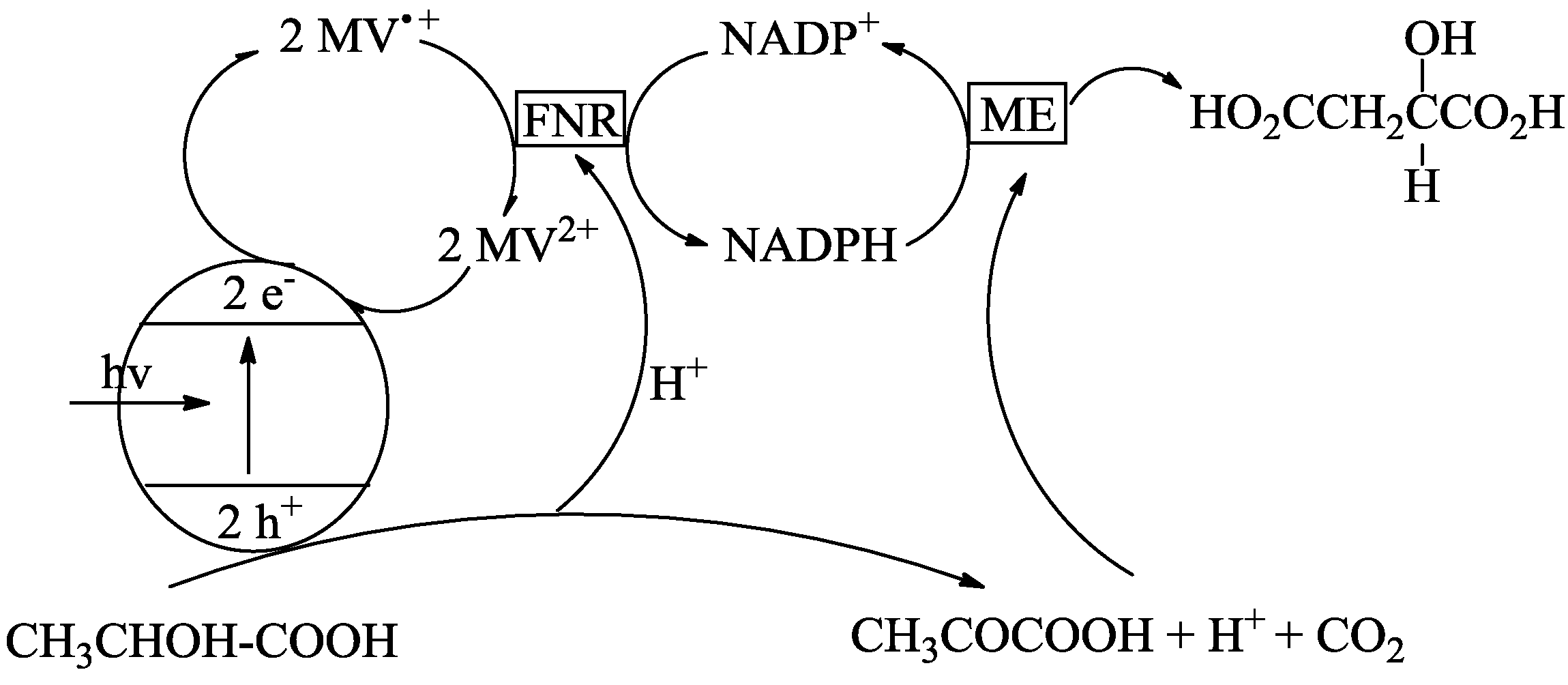
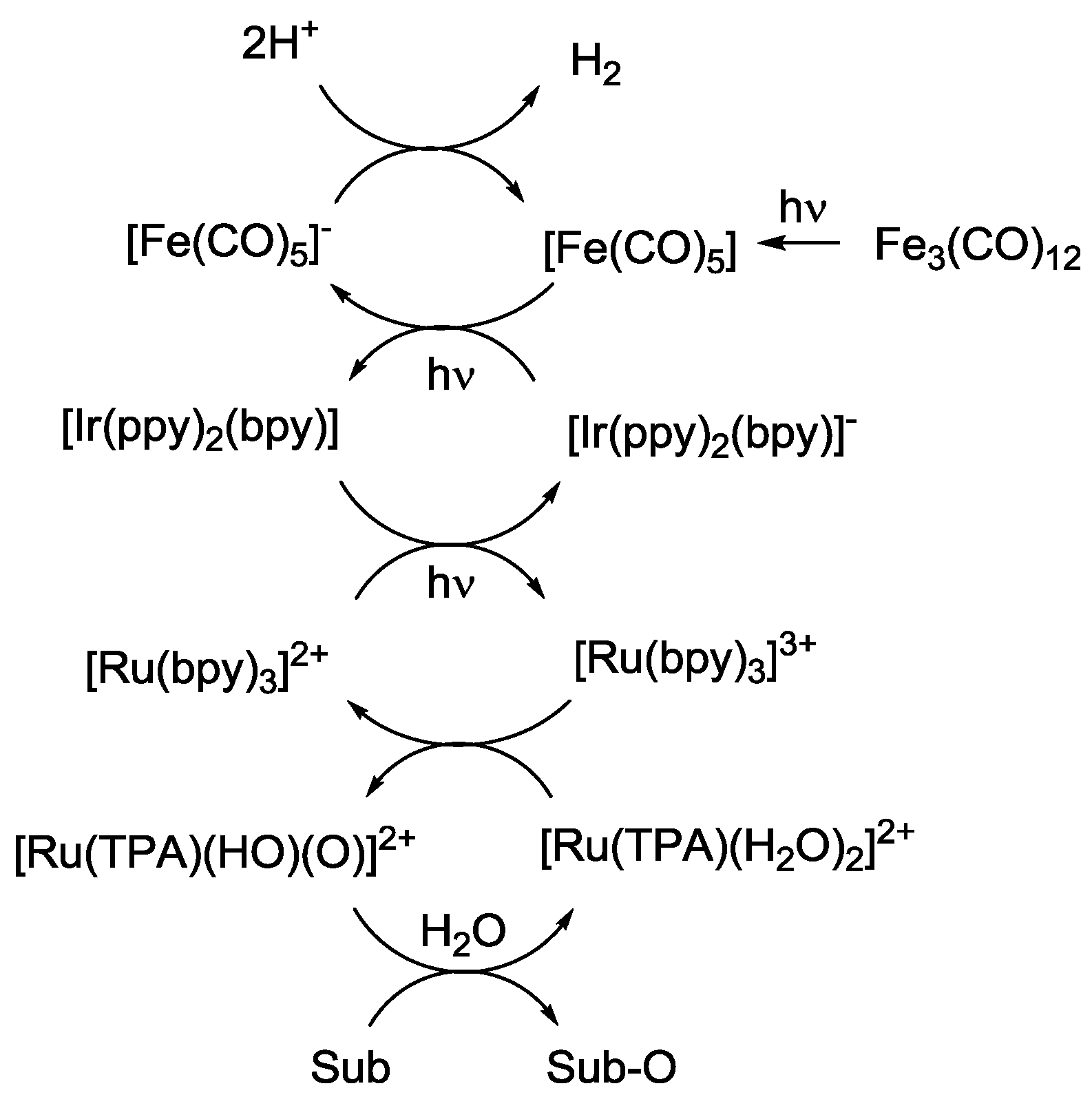
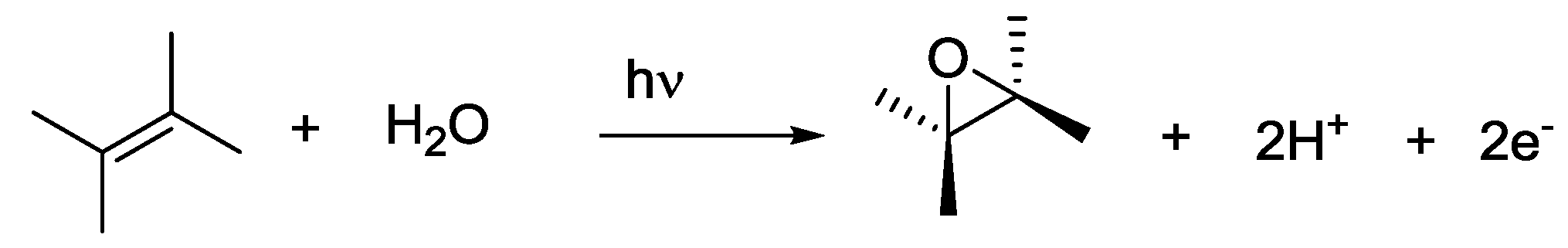
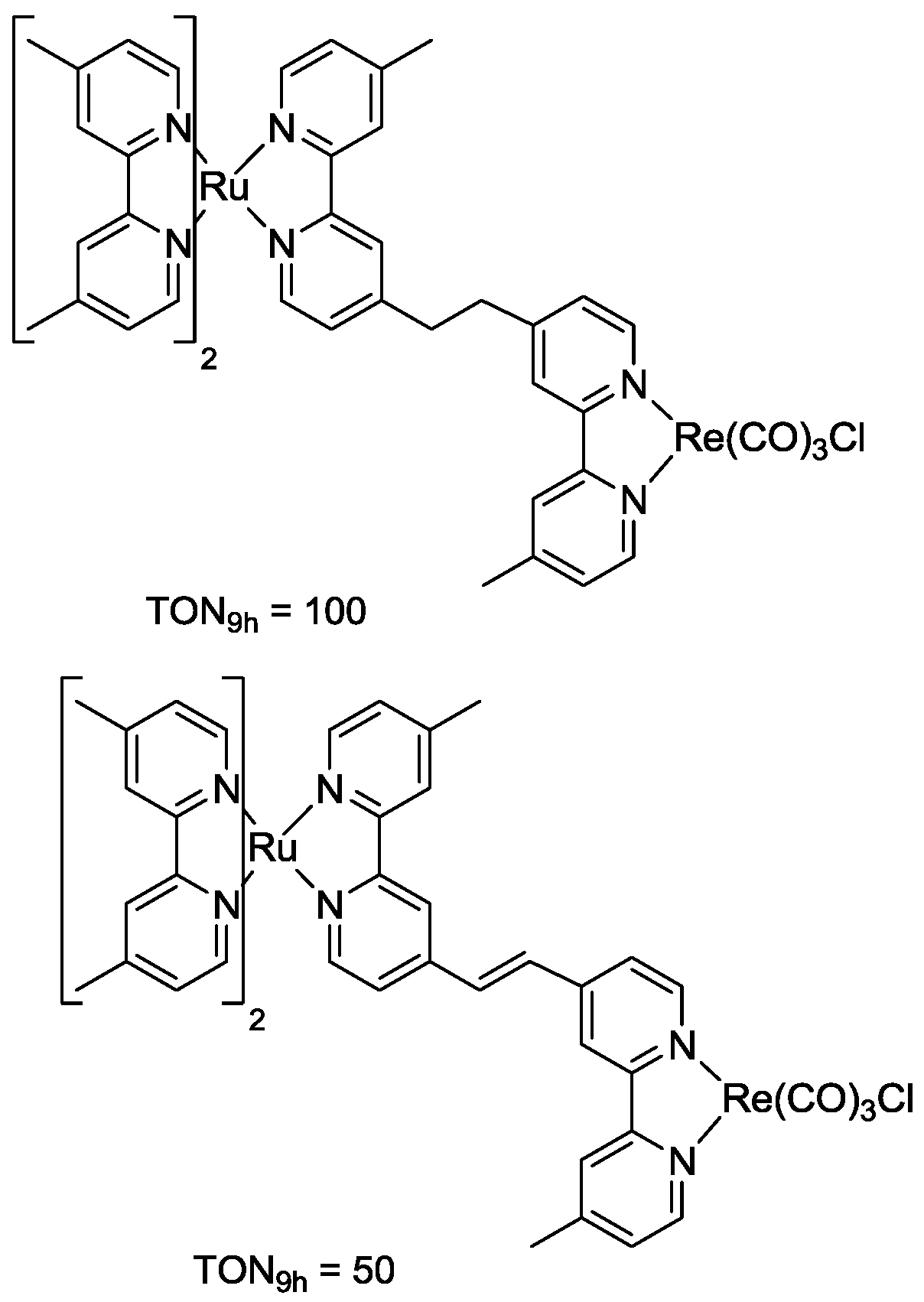
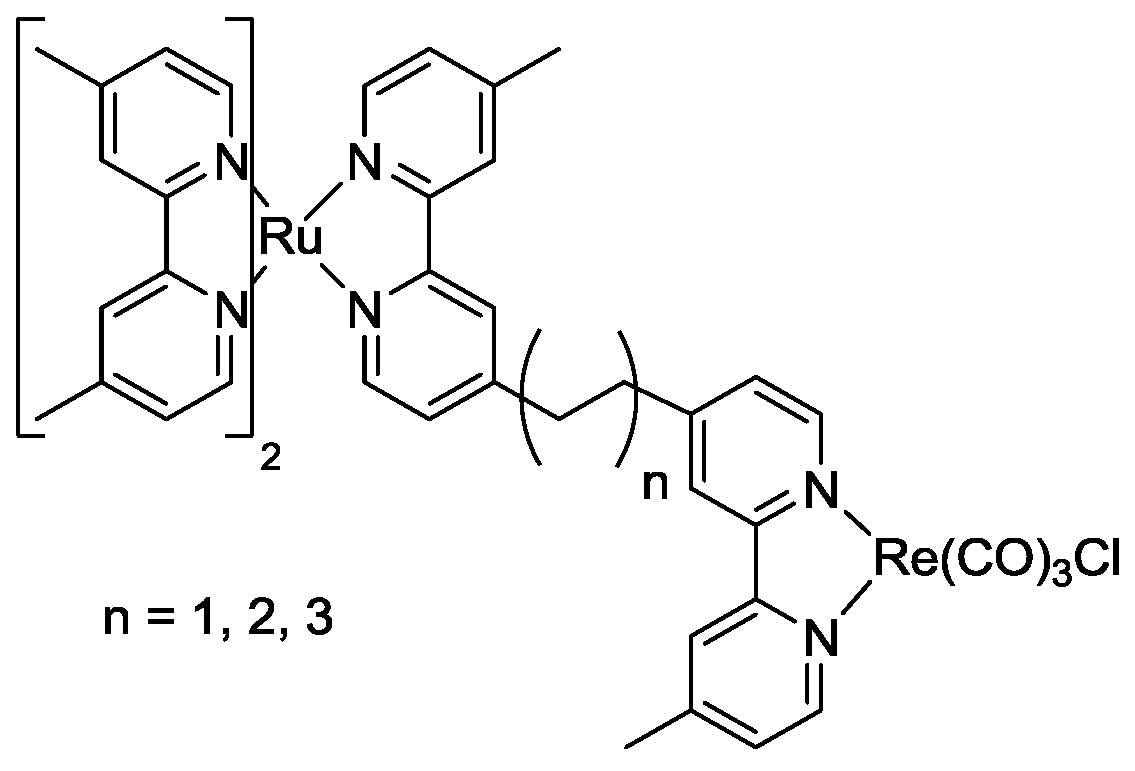
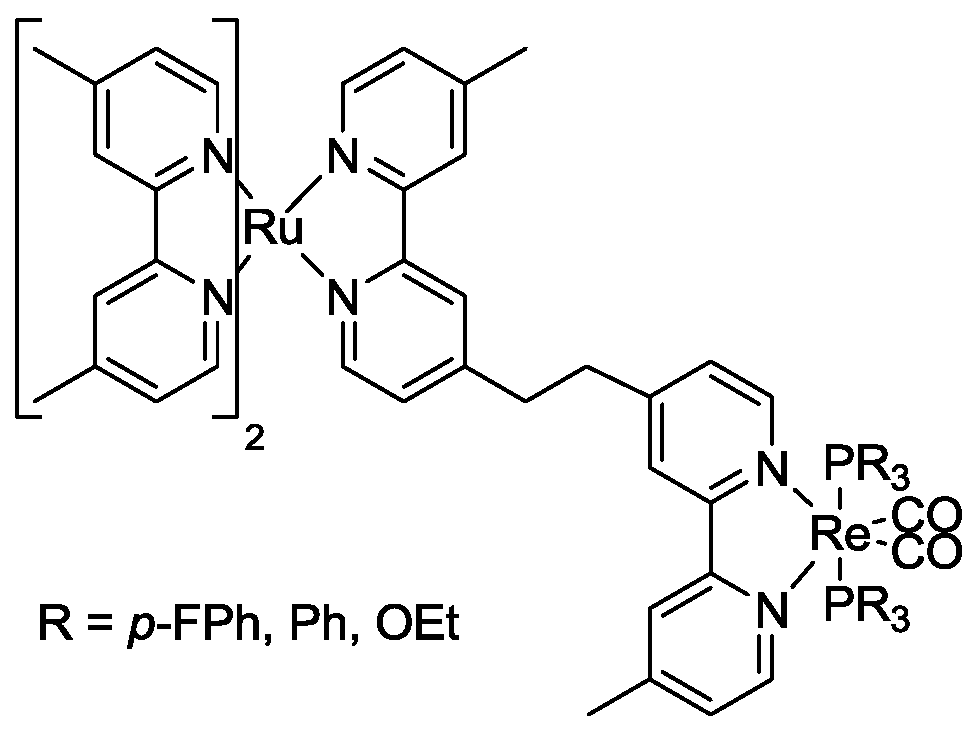
5. Structural Parameters for Supramolecular Catalyst Design
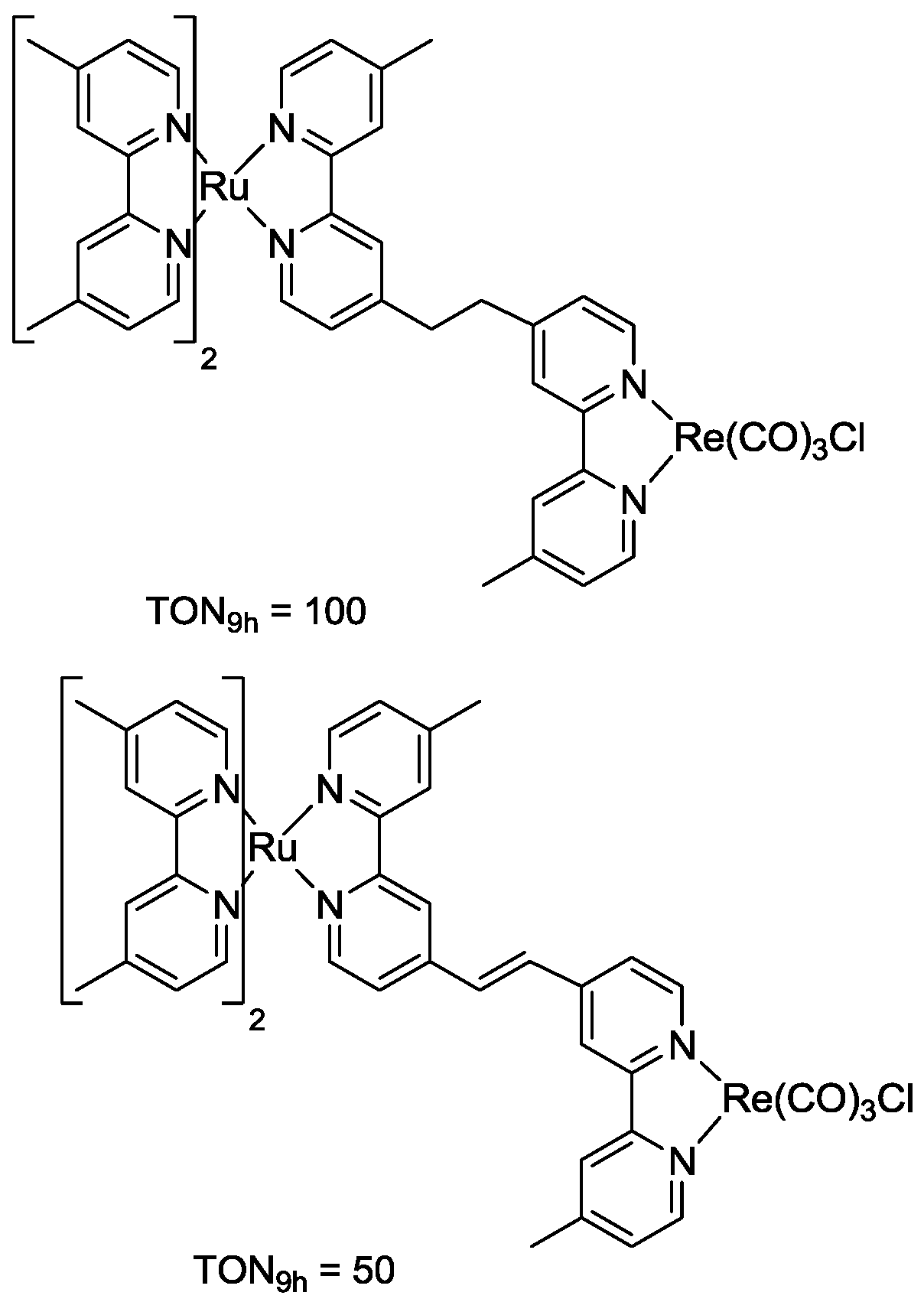
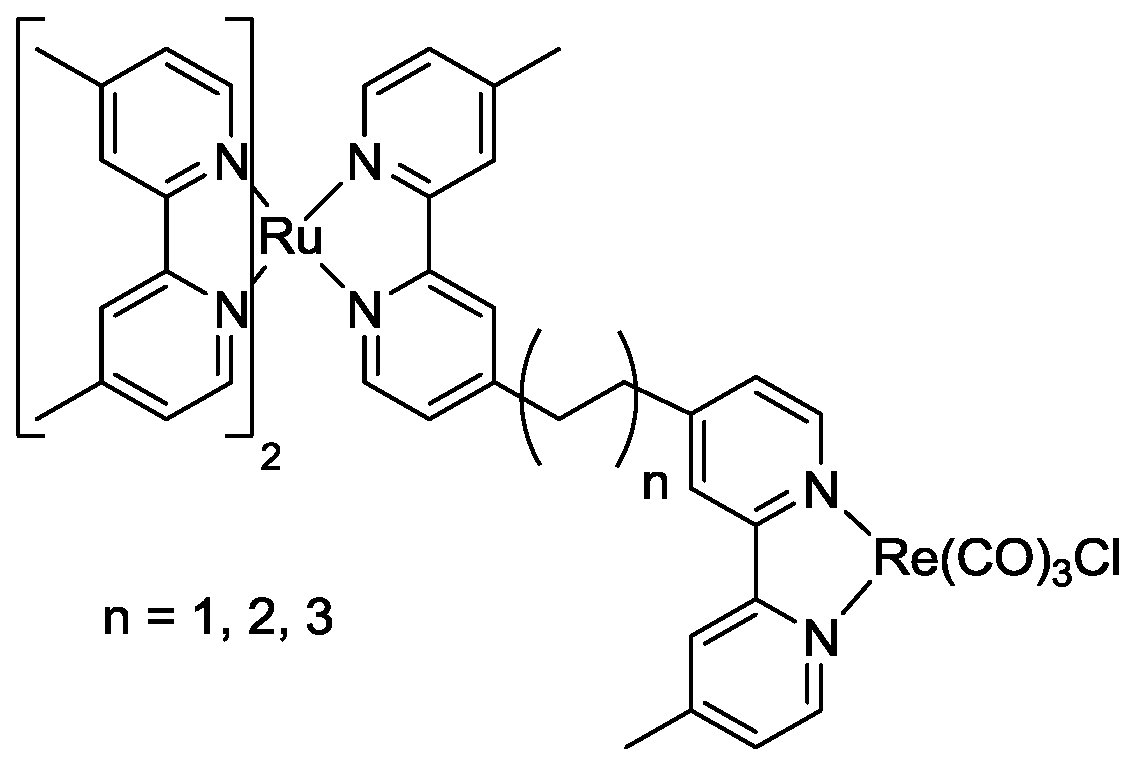
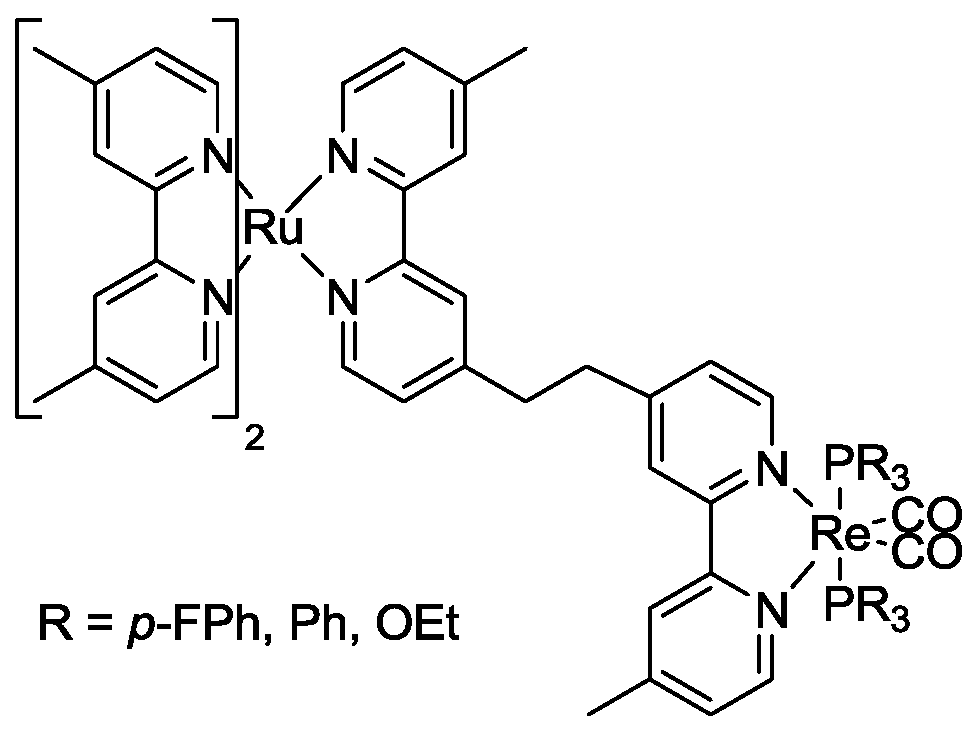
6. Conclusions
Acknowledgments
References
- Aresta, M.; Dibenedetto, D. Utilisation of CO2 as chemical feedstock: opportunities and challenges. Dalton Trans. 2007, 2975–2992. [Google Scholar]
- 2. Solomon, S.; Quin, D.; Manning, M.; Marquis, M.; Averyt, K.; Tignor, M.M.B.; Miller, H.L., Jr.; Chen, Z. The Physical Science Basis, Climate Change, 4th Assessment Report of the IPCC; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Lindzen, R.S.; Quart, J.R. Meteorol. Soc. 1991, 117, 651. [CrossRef]
- Lacis, A.A.; Schmidt, G.A.; Rind, D.; Ruedy, R.A. Atmospheric CO2: Principal Control Knob Governing Earth’s Temperature. Science 2010, 330, 356–359. [Google Scholar]
- Sabine, L.C. The Oceanic Sink for Anthropogenic CO2. Science 2004, 305, 367–371. [Google Scholar] [CrossRef]
- Feely, R.A. Impact of Anthropogenic CO2 on the CaCO3 System in the Oceans. Science 2004, 305, 362–366. [Google Scholar] [CrossRef]
- Langdon, C. Effect of elevated CO2 on the community metabolism of an experimental coral reef. Glob. Biogeochem. Cycles 2003, 17, 1011–1025. [Google Scholar] [CrossRef]
- Gattuso, J.-P.; Frankignoulle, M.; Bourge, I.; Romaine, S.; Buddemeier, R.W. Effect of calcium carbonate saturation of seawater on coral calcification. Glob. Planet Change 1998, 18, 37–46. [Google Scholar] [CrossRef]
- Riebesell, U.; Zondervan, I.; Rost, B.; Tortell, P.D.; Zeebe, R.E.; Morell, F.M. Reduced calcification of marine plankton in response to increased atmospheric CO2. Nature 2000, 407, 364–367. [Google Scholar]
- Rau, S.; Walther, D.; Vos, G.G. Inspired by nature: Light driven organometallic catalysis by heterooligonuclear Ru(II) complexes. Dalton Trans. 2007. [Google Scholar] [CrossRef]
- Young, R.C.; Meyer, T.J.; Whitten, D.G. Electron transfer quenching of excited states of metal complexes. J. Am. Chem. Soc. 1976, 98, 286–287. [Google Scholar] [CrossRef]
- Kalyanasundaram, K. Hydrogen Evolution from Water by Visible Light, a Homogeneous Three Component Test System for Redox Catalysis. Helv. Chim. Acta 1978, 61, 2720–2730. [Google Scholar] [CrossRef]
- Takeda, H.; Ishitani, O. Development of efficient photocatalytic systems for CO2 reduction using mononuclear and multinuclear metal complexes based on mechanistic studies. Coord. Chem. Rev. 2010, 254, 346–354. [Google Scholar] [CrossRef]
- Cokoja, M.; Bruckmeier, C.; Rieger, B.; Kühn, F.; Hermann, W.A. Transformation of Carbon Dioxide with Homogeneous Transition-Metal Catalysts: A Molecular Solution to a Global Challenge? Angew. Chem. Int. Ed. 2011, 50, 8510–8537. [Google Scholar]
- Morris, A.J.; Meyer, G.J.; Fujita, E. Molecular Approaches to the Photocatalytic Reduction of Carbon Dioxide for Solar Fuels. Acc. Chem. Res. 2009, 42, 1983–1994. [Google Scholar] [CrossRef]
- Kalyanasundaram, K. Photophysics, photochemistry and solar energy conversion with tris(bipyridyl)ruthenium(II) and its analogues. Coord. Chem. Rev. 1982, 46, 159–244. [Google Scholar] [CrossRef]
- Juris, A.; Balzani, V. Ru(II) polypyridine complexes: photophysics, photochemistry, eletrochemistry, and chemiluminescence. Coord. Chem. Rev. 1988, 84, 85–277. [Google Scholar]
- Meyer, T.J. Photochemistry of metal coordination complexes: metal to ligand charge transfer excited states. Pure Appl. Chem. 1986, 58, 1193–1206. [Google Scholar] [CrossRef]
- Gafney, H.D.; Adainson, A.W. Excited state Ru(bipyr)32+ as an electron-transfer reductant. J. Am. Chem. Soc. 1972, 94, 8238–8239. [Google Scholar]
- Danas, T.N.; Adenson, A.W. Tris (2,2'-bipyridine)ruthenium(II) sensitized reactions of some oxalato complexes. J. Am. Chem. Soc. 1973, 95, 5159–5168. [Google Scholar] [CrossRef]
- Bock, C.R.; Meyer, T.J.; Whitten, D.G. Electron transfer quenching of the luminescent excited state of tris(2,2'-bipyridine)ruthenium(II). Flash photolysis relaxation technique for measuring the rates of very rapid electron transfer reactions. J. Am. Chem. Soc. 1974, 96, 4710–4712. [Google Scholar] [CrossRef]
- Young, R.C.; Meyer, T.J.; Whitten, D.G. Kinetic relaxation measurement of rapid electron transfer reactions by flash photolysis. Conversion of light energy into chemical energy using the tris(2,2'-bipyridine)ruthenium(3+)-tris(2,2'-bipyridine)ruthenium(2+*) couple. J. Am. Chem. Soc. 1975, 97, 4781–4782. [Google Scholar]
- Navon, G.; Sutin, N. Mechanism of the quenching of the phosphorescence of tris(2,2'-bipyridine)ruthenium(II) by some cobalt(III) and ruthenium(III) complexes. Inorg. Chem. 1974, 13, 2159–2164. [Google Scholar] [CrossRef]
- Anderson, C.P.; Salnon, D.J.; Meyer, T.J.; Young, R.C. Photochemical Generation of Ru(bpy)3+ and O2−. J. Am. Chem. Soc. 1977, 99, 1980–1982. [Google Scholar] [CrossRef]
- Bock, C.R.; Meyer, T.J.; Whitten, D.G. Photochemistry of transition metal complexes. Mechanism and efficiency of energy conversion by electron-transfer quenching. J. Am. Chem. Soc. 1975, 97, 2909–2911. [Google Scholar]
- Bock, C.R.; Conner, J.A.; Gutierrez, A.R.; Meyer, T.J.; Whitten, D.G.; Sullivan, B.P.; Nagle, J.K. Estimation of excited-state redox potentials by electron-transfer quenching. Application of electron-transfer theory to excited-state redox processes. J. Am. Chem. Soc. 1979, 101, 4815–4824. [Google Scholar]
- Sauvage, J.-P.; Collin, J.-P.; Chambron, J.-C.; Guillerez, S.; Coudret, C. Ruthenium(II) and Osmium(II) Bis(terpyridine) Complexes in Covalently-Linked Multicomponent Systems: Synthesis, Electrochemical Behavior, Absorption Spectra, and Photochemical and Photophysical Properties. Chem. Rev. 1994, 94, 993–1019. [Google Scholar] [CrossRef]
- Winkler, J.R.; Netzel, T.L.; Creutz, C.; Sutin, N. Direct observation of metal-to-ligand charge-transfer (MLCT) excited states of pentaammineruthenium(II) complexes. J. Am. Chem. Soc. 1987, 109, 2381–2392. [Google Scholar]
- Medlycott, E.A.; Hanan, G.S. Synthesis and properties of mono- and oligo-nuclear Ru(II) complexes of tridentate ligands: The quest for long-lived excited states at room temperature. Coord. Chem. Rev. 2006, 250, 1763–1782. [Google Scholar] [CrossRef]
- Treadway, J.A.; Loeb, B.; Lopez, R.; Anderson, P.A.; Keene, F.R.; Meyer, T.J. Effect of Delocalization and Rigidity in the Acceptor Ligand on MLCT Excited-State Decay. Inorg Chem. 1996, 35, 2242–2246. [Google Scholar] [CrossRef]
- Baranoff, E.; Collin, J.-P.; Flamigni, L.; Sauvage, J.-P. From ruthenium(II) to iridium(III): 15 years of triads based on bis-terpyridine complexes. Chem. Soc. Rev. 2004. [Google Scholar] [CrossRef]
- Xian, X.-Y.; Del Guerzo, A.; Schmehl, R.H. Photophysical behavior of transition metal complexes having interacting ligand localized and metal-to-ligand charge transfer states. J. Photochem. Photobiol.C 2004, 5, 55–77. [Google Scholar] [CrossRef]
- Medlycott, E.A.; Hanan, G.S. Designing tridentate ligands for ruthenium(II) complexes with prolonged room temperature luminescence lifetimes. Chem. Soc. Rev. 2005, 34, 133–142. [Google Scholar] [CrossRef]
- Maestri, M.; Armaroli, N.; Balzani, V.; Constable, E.C.; Thompson, A.M.W.C. Complexes of the Ruthenium(II)-2,2':6',2''-terpyridine Family. Effect of Electron-Accepting and -Donating Substituents on the Photophysical and Electrochemical Properties. Inorg. Chem. 1995, 34, 2759–2767. [Google Scholar]
- Harriman, A.; Mayeux, A.; De Nicola, A.; Ziessel, R. Synthesis and photophysical properties of ruthenium(II) bis(2,2′:6′,2″-terpyridine) complexes constructed from a diethynylated-thiophene residue. Phys. Chem. Phys. 2002, 2229–2235. [Google Scholar]
- Baba, A.I.; Shaw, J.R.; Simon, J.A.; Thummel, R.P.; Schmehl, R.H. The photophysical behavior of d6 complexes having nearly isoenergetic MLCT and ligand localized excited states. Coord. Chem. Rev. 1998, 171, 43–59. [Google Scholar]
- Passalacqua, R.; Loiseau, F.; Campagna, S.; Fang, Y.-Q.; Hanan, G.S. In Search of Ruthenium(II) Complexes Based on Tridentate Polypyridine ligands that Feature Long-lived Room-Temperature Luminescence: The Multichromophore Approach. Angew. Chem. Int. Ed. 2003, 42, 1608–1611. [Google Scholar] [CrossRef]
- Ji, S.; Wu, W.; Wu, W.; Guo, H.; Zhao, J. Ruthenium(II) Polyimine Complexes with a Long-Lived 3IL Excited State or a 3MLCT/3IL Equilibrium: Efficient Triplet Sensitizers for Low-Power Upconversion. Angew. Chem. Int. Ed. 2011, 50, 1626–1629. [Google Scholar] [CrossRef]
- Fihri, A.; Artero, V.; Pereirab, A.; Fontecavea, M. Efficient H2-producing photocatalytic systems based on cyclometalated iridium- and tricarbonylrhenium-diimine photosensitizers and cobaloxime catalysts. Dalton Trans. 2008. [Google Scholar] [CrossRef]
- Tinker, L.L.; McDaniel, N.D.; Curtin, P.N.; Smith, C.K.; Ireland, M.J.; Bernhard, S. Visible Light Induced Catalytic Water Reduction without an Electron Relay. Chem.–Eur. J. 2007, 13, 8726–8732. [Google Scholar]
- Luong, J.C.; Nadjo, L.; Wrighton, M.S. Ground and excited state electron transfer processes involving fac-tricarbonylchloro(1,10-phenanthroline)rhenium(I). Electrogenerated chemiluminescence and electron transfer quenching of the lowest excited state. J. Am. Chem. Soc. 1978, 100, 5790–5795. [Google Scholar] [CrossRef]
- Du, P.; Schneider, J.; Jarosz, P.; Eisenberg, R. Photocatalytic Generation of Hydrogen from Water Using a Platinum(II) Terpyridyl Acetylide Chromophore. J. Am. Chem. Soc. 2006, 128, 7726–7727. [Google Scholar] [CrossRef]
- Du, P.; Schneider, J.; Li, F.; Zhao, W.; Patel, U.; Castellano, F.N.; Eisenberg, R. Bi- and Terpyridyl Platinum(II) Chloro Complexes: Molecular Catalysts for the Photogeneration of Hydrogen from Water or Simply Precursors for Colloidal Platinum? J. Am. Chem. Soc. 2008, 130, 5056–5058. [Google Scholar] [CrossRef]
- Goldsmith, J.I.; Hudson, W.R.; Lowry, M.S.; Anderson, T.; Bernhard, S. Discovery and High-Throughput Screening of Heteroleptic Iridium Complexes for Photoinduced Hydrogen Production. J. Am. Chem. Soc. 2005, 127, 7502–7510. [Google Scholar]
- Cline, E.D.; Adamson, S.E.; Bernhard, S. Homogeneous Catalytic System for Photoinduced Hydrogen Production Utilizing Iridium and Rhodium Complexes. Inorg. Chem. 2008, 47, 10378–10388. [Google Scholar] [CrossRef]
- Holmann, D.; Gärtner, F.; Ludwig, R.; Barsch, E.; Lunge, H.; Blug, M.; Hoch, S.; Beller, M.; Brückner, A. Insights into the Mechanism of Photocatalytic Water Reduction by DFT-Supported In Situ EPR/Raman Spectroscopy. Angew. Chem. Int. Ed. 2011, 50, 10246–10250. [Google Scholar]
- Miyake, Y.; Nakajima, N.; Sasaki, K.; Saito, R.; Nakanishi, H.; Nishibayashi, Y. Design and Synthesis of Diphosphine Ligands Bearing an Osmium(II) Bis(terpyridyl) Moiety as a Light-Harvesting Unit: Application to Photocatalytic Production of Dihydrogen. Organometallics 2009, 28, 5240–5243. [Google Scholar] [CrossRef]
- Crosby, G.A. Spectroscopic investigations of excited states of transition-metal complexes. Acc. Chem. Res. 1975, 8, 231–238. [Google Scholar] [CrossRef]
- Balzani, V.; Boletta, F.; Gandolfi, M.T.; Maestri, M. Bimolecular Electron Transfer Reactions of Excited States of Transition Metal Complexes. Topics Curr. Chem. 1978, 75, 1–64. [Google Scholar]
- Islangulov, R.R.; Kozlov, D.V.; Castellano, F.N. Low power upconversion using MLCT sensitizers. Chem. Commun. 2005, 30, 3776–3778. [Google Scholar]
- Foxon, S.P.; Alamiry, M.A.H.; Walker, M.G.; Meijer, A.J.H.M.; Sazanovich, I.V.; Weinstein, J.A.; Thomas, J.A. Photophysical Properties and Singlet Oxygen Production by Ruthenium(II) Complexes of Benzo[i]dipyrido[3,2-a:2′,3′-c]phenazine: Spectroscopic and TD-DFT Study. J. Phys. Chem. A 2009, 113, 12754–12762. [Google Scholar]
- Zhou, G.; Wong, W.-Y.; Yao, B.; Xie, Z.; Wang, L. Triphenylamine-Dendronized Pure Red Iridium Phosphors with Superior OLED Efficiency/Color Purity Trade-Offs. Angew. Chem. Int. Ed. 2007, 46, 1149–1151. [Google Scholar] [CrossRef]
- Gholamkhass, B.; Mametsuka, H.; Koike, K.; Tanabe, T.; Furue, M.; Ishitani, O. Architecture of Supramolecular Metal Complexes for Photocatalytic CO2 Reduction: Ruthenium−Rhenium Bi- and Tetranuclear Complexes. Inorg. Chem. 2005, 44, 2326–2336. [Google Scholar] [CrossRef]
- Tamaki, Y.; Watanabe, K.; Koike, K.; Inoue, H.; Morimoto, T.; Ishitani, O. Development of highly efficient supramolecular CO2 reduction photocatalysts with high turnover frequency and durability. Faraday Discuss. 2012, 155, 115. [Google Scholar] [CrossRef]
- Stone, M.L.; Crosby, G.A. Charge-transfer luminescence from ruthenium(II) complexes containing tridentate ligands. Chem. Phys. Lett. 1981, 79, 169–173. [Google Scholar] [CrossRef]
- Beley, M.; Collin, J.; Sauvage, J.-P.; Sugihara, H.; Heisel, F.; Miehe, A. Photophysical and photochemical properties of ruthenium and osmium complexes with substituted terpyridines. Dalton. Trans. 1991. [Google Scholar] [CrossRef]
- Demas, J.M.; Crosby, G.A. Quantum efficiencies on transition metal complexes. II, Charge-transfer luminescence. J. Am. Chem. Soc. 1971, 93, 2841–2847. [Google Scholar]
- Kober, E.M.; Marshall, J.L.; Dressick, W.J.; Sullivan, B.P.; Caspar, J.-V.; Meyer, T.J. Synthetic control of excited states. Nonchromophoric ligand variations in polypyridyl complexes of osmium(II). Inorg. Chem. 1985, 24, 2755–2763. [Google Scholar]
- Brunschwig, B.S.; Sutin, N. Reactions of the excited states of substituted polypyridinechromium(III) complexes with oxygen, iron(II) ions, ruthenium(II) and -(III), and osmium(II) and -(III) complexs. J. Am. Chem. Soc. 1978, 100, 7568–7577. [Google Scholar] [CrossRef]
- Ohno, T.; Kato, S.; Kaiazaki, S.; Hanazaki, I. Singlet-triplet transitions of aromatic compounds coordinating to a paramagnetic chromium(III) ion. Inorg. Chem. 1986, 25, 3853–3858. [Google Scholar] [CrossRef]
- Creutz, C.; Chou, M.; Netzel, T.L.; Okumura, M.; Sutin, N. Lifetimes, spectra, and quenching of the excited states of polypyridine complexes of iron(II), ruthenium(II), and osmium(II). J. Am. Chem. Soc. 1980, 102, 1309–1319. [Google Scholar] [CrossRef]
- Ayala, N.P.; Flynn, C.M.; Sacksteder, L.A.; Demas, J.N.; de Graff, B.A. Synthesis, luminescence, and excited-state complexes of the tris(1,10-phenanthroline)- and bis(terpyridine)iridium(III) cations. J. Am. Chem. Soc. 1990, 112, 3837–3844. [Google Scholar] [CrossRef]
- Kurz, P.; Probst, B.; Spingler, B.; Alberto, R. Ligand Variations in [ReX(diimine)(CO)3] Complexes: Effects on Photocatalytic CO2 Reduction. Eur. J. Inorg. Chem. 2006, 2966–2974. [Google Scholar]
- Kalyanasundaram, K.J. Luminescence and redox reactions of the metal-to-ligand charge-transfer excited state of tricarbonylchloro-(polypyridyl)rhenium(I) complexes. Chem. Soc. Faraday Trans. 1986, 82, 2401–2415. [Google Scholar]
- Takeda, H.; Koike, K.; Inoue, H.; Ishitani, O. Development of an Efficient Photocatalytic System for CO2 Reduction Using Rhenium(I) Complexes Based on Mechanistic Studies. J. Am. Chem. Soc. 2008, 130, 2023–2031. [Google Scholar] [CrossRef]
- van Wallendael, S.; Shaver, R.J.; Rillema, D.P.; Yoblinski, B.J.; Stathis, M.; Guarr, T.F. Ground-state and excited-state properties of monometallic and bimetallic complexes based on rhenium(I) tricarbonyl chloride: effect of an insulating vs a conducting bridge. Inorg. Chem. 1990, 29, 1761–1767. [Google Scholar]
- Kirch, M.; Lehn, J.-M.; Sauvage, J.-P. Hydrogen Generation by Visible Light Irradiation of Aqueous Solutions of Metal Complexes. An approach to the photochemical conversion and storage of solar energy. Helv. Chim. Acta 1979, 62, 1345–1384. [Google Scholar]
- Lehn, J.-M.; Ziessel, R. Photochemical generation of carbon monoxide and hydrogen by reduction of carbon dioxide and water under visible light irradiation. Proc. Natl. Acad. Sci. USA 1982, 79, 701–704. [Google Scholar]
- Matsuoka, S.; Yamamoto, K.; Ogata, T.; Kusaba, M.; Nakashima, N.; Fujita, E.; Yanagida, S. Efficient and selective electron mediation of cobalt complexes with cyclam and related macrocycles in the p-terphenyl-catalyzed photoreduction of carbon dioxide. J. Am. Chem. Soc. 1993, 115, 601–609. [Google Scholar]
- Ogata, T.; Yamamoto, Y.; Wada, Y.; Murakoshi, K.; Kusaba, M.; Nakashima, N.; Ishida, A.; Takamuku, S.; Yanagida, S. Phenazine-Photosensitized Reduction of CO2 Mediated by a Cobalt-Cyclam Complex through Electron and Hydrogen Transfer. J. Phys. Chem. 1995, 99, 11916–11922. [Google Scholar]
- Fisher, B.; Eisenberg, R. Electrocatalytic reduction of carbon dioxide by using macrocycles of nickel and cobalt. J. Am. Chem. Soc. 1980, 102, 7361–7363. [Google Scholar] [CrossRef]
- Grant, J.L.; Goswami, K.; Spreer, L.O.; Otvos, J.W.; Calvin, M. Photochemical reduction of carbon dioxide to carbon monoxide in water using a nickel(II) tetra-azamacrocycle complex as catalyst. J. Chem. Soc. Dalton Trans. 1987. [Google Scholar] [CrossRef]
- Craig, C.A.; Spreer, L.O.; Otvos, J.W.; Calvin, M. Photochemical reduction of carbon dioxide using nickel tetraazamacrocycles. J. Phys. Chem. 1990, 94, 7957–7960. [Google Scholar] [CrossRef]
- Kimura, E.; Bu, X.; Shionoya, M.; Wada, S.; Maruyama, S. A new nickel(II) cyclam (cyclam = 1,4,8,11-tetraazacyclotetradecane) complex covalently attached to tris(1,10-phenanthroline)ruthenium(2+). A new candidate for the catalytic photoreduction of carbon dioxide. Inorg. Chem. 1992, 31, 4542–4546. [Google Scholar] [CrossRef]
- Kimura, E.; Wada, S.; Shionoya, M.; Okazaki, Y. New Series of Multifunctionalized Nickel(II)-Cyclam (Cyclam = 1,4,8,11-Tetraazacyclotetradecane) Complexes. Application to the Photoreduction of Carbon Dioxide. Inorg. Chem. 1994, 33, 770–778. [Google Scholar] [CrossRef]
- Kimura, E.; Wada, S.; Shionoya, M.; Takahashi, T.; Litaka, Y. A novel cyclam-nickel(II) complex appended with a tris-(2,2′-bipyridine) ruthenium(II) complex (cyclam = 1,4,8,11-tetra-azacyclotetradecane). Chem. Commun. 1990. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Efficient photochemical reduction of CO2 to CO by visible light irradiation of systems containing Re(bipy)(CO)3X or Ru(bipy)32+–CO2+ combinations as homogeneous catalysts. J. Chem. Soc. Chem. Commun. 1983. [Google Scholar] [CrossRef]
- Hawecker, J.; Lehn, J.-M.; Ziessel, R. Photochemical and Electrochemical Reduction of Carbon Dioxide to Carbon Monoxide Mediated by (2,2′-Bipyridine)tricarbonylchlororhenium(I) and Related Complexes as Homogeneous Catalysts. Helv. Chim. Acta 1986, 69, 1990–2012. [Google Scholar] [CrossRef]
- Tam, W.; Lin, G.-Y.; Wong, W.-K.; Kiel, W.A.; Wong, V.-K.; Gladysz, J.A. Synthesis and electrophile-induced disproportionation of the neutral formyl triphenylphosphinenitrosyl-.eta.-cyclopentadienylrhenium formyl ((.eta.-C5H5)Re(NO)(PPh3)(CHO)). J. Am. Chem. Soc. 1982, 104, 141–152. [Google Scholar]
- Barrientos-Penna, C.F.; Gilchrist, A.B.; Klahn-Oliva, A.H.; Hanlan, A.J.L.; Sutton, D. Aryldiazenido (N2Ar) complexes. Carboxylate, formate, hydroxycarbonyl, and hydride derivatives of [(.eta.-C5H5)Re(CO)2(N2Ar)][BF4] and [(.eta.-C5Me5)Re(CO)2(N2Ar)][BF4] (Ar = aryl). Organometallics 1985, 4, 478–485. [Google Scholar]
- Aresta, M. Activation of Small Molecules. In Carbon Dioxide Reduction and Uses as a Chemical Feedstock; Tolman, W.B., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 1–41. [Google Scholar]
- Hori, H.; Johnson, F.P.A.; Koike, K.; Ishitani, O.; Ibusuki, T. Efficient photocatalytic CO2 reduction using [Re(bpy) (CO)3{P(OEt)3}]+. Photochem. J. Photobiol. A 1996, 96, 171–174. [Google Scholar]
- Hayashi, Y.; Kita, S.; Brundschwig, B.S.; Fujita, E. Involvement of a Binuclear Species with the Re−C(O)O−Re Moiety in CO2 Reduction Catalyzed by Tricarbonyl Rhenium(I) Complexes with Diimine Ligands: Strikingly Slow Formation of the Re−Re and Re−C(O)O−Re Species from Re(dmb)(CO)3S (dmb = 4,4‘-Dimethyl-2,2‘-bipyridine, S = Solvent). J. Am. Chem. Soc. 2003, 125, 11976–11987. [Google Scholar] [CrossRef]
- Gibson, D.H.; Xiaolong, Y. Synthesis and Reactions of fac-Re(dmbpy)(CO)3X (dmbpy = 4,4'-dimethyl-2,2'-bipyridyl; X = COOH, COOMe, H, OH, and OCHO). J. Am. Chem. Soc. 1998, 120, 11200–11201. [Google Scholar] [CrossRef]
- Gibson, D.H.; Xiaolong, Y. Transformations of fac-Re(dmbpy)(CO)3(CO2H) in the presence of carbon dioxide (dmbpy=4,4′-dimethyl-2,2′-bipyridine). Chem. Commun. 1999, 1411–1412. [Google Scholar]
- Bruckmeier, C.; Lehenmeier, M.W.; Reithmeier, R.; Rieger, B.; Herranz, J.; Kavakli, C. Binuclear rhenium(I) complexes for the photocatalytic reduction of CO2. Dalton Trans. 2012, 41, 5026–5037. [Google Scholar]
- Ishida, H.; Fujiki, K.; Ohba, T.; Ohkubo, K.; Tanaka, K.; Terada, T.; Tanaka, T. Ligand effects of ruthenium 2,2′-bipyridine and 1,10-phenanthroline complexes on the electrochemical reduction of CO2. J. Chem. Soc. Dalton Trans. 1990. [Google Scholar] [CrossRef]
- Collomb-Dunand-Sauthier, M.-N.; Deronzier, A.; Ziessel, R. Electrocatalytic Reduction of Carbon Dioxide with Mono(bipyridine)carbonylruthenium Complexes in Solution or as Polymeric Thin Films. Inorg. Chem. 1994, 33, 2961–2967. [Google Scholar] [CrossRef]
- Fujita, E. Photochemical carbon dioxide reduction with metal complexes. Coord. Chem. Rev. 1999, 185–186, 373–384. [Google Scholar]
- Collomb-Dunand-Sauthier, M.-N.; Deronzier, A.; Ziessel, R. Electrocatalytic reduction of CO2 in water on a polymeric [{Ru0(bpy)(CO)2}n](bpy = 2,2′-bipyridine) complex immobilized on carbon electrodes. J. Chem. Soc. Chem. Commun. 1994. [Google Scholar] [CrossRef]
- Collomb-Dunand-Sauthier, M.-N.; Deronzier, A.; Ziessel, R. Electrochemical characterization of [RuII(bpy)(CO)2Cl2] (bpy = 2,2'-bipyridine): Isolation of a new complex: [RuII(bpy)(CO)Cl2][Me4N]. J. Electroanal. Chem. Interfacial Electrochem. 1991, 319, 347–353. [Google Scholar] [CrossRef]
- Ishida, H.; Tanaka, K.; Tanaka, T. The electrochemical reduction of CO2 catalyzed by ruthenium carbonyl complexes. Chem. Lett. 1985, 405–406. [Google Scholar]
- Ishida, H.; Tanaka, K.; Tanaka, T. Electrochemical CO2 reduction catalyzed by ruthenium complexes [Ru(bpy)2(CO)2]2+ and [Ru(bpy)2(CO)Cl]+. Effect of pH on the formation of CO and HCOO−. Organometallics 1987, 6, 181–186. [Google Scholar]
- Ishida, H.; Tanaka, H.; Tanaka, K.; Tanaka, T. Selective formation of HCOO– in the electrochemical CO2 reduction catalysed by [Ru(bpy)2(CO)2]2+(bpy = 2,2′-bipyridine). J. Chem. Soc. Chem. Commun. 1987, 131–132. [Google Scholar]
- Ishida, H.; Terada, T.; Tanaka, K.; Tanaka, T. Photochemical carbon dioxide reduction catalyzed by bis(2,2'-bipyridine)dicarbonylruthenium(2+) using triethanolamine and 1-benzyl-1,4-dihydronicotinamide as an electron donor. Inorg. Chem. 1990, 29, 905–911. [Google Scholar] [CrossRef]
- Lehn, J.-M.; Ziessel, R. Photochemical reduction of carbon dioxide to formate catalyzed by 2,2
![]() -bipyridine- or 1,10-phenanthroline-ruthenium(II) complexes. J. Organomet. Chem. 1990, 382, 157–173. [Google Scholar]
-bipyridine- or 1,10-phenanthroline-ruthenium(II) complexes. J. Organomet. Chem. 1990, 382, 157–173. [Google Scholar] - Chardon-Noblat, S.; Collomb-Dunand-Sauthier, M.-N.; Deronzier, A.; Ziessel, R.; Zsoldos, D. Formation of Polymeric [{Ru0(bpy)(CO)2}n] Films by Electrochemical Reduction of [Ru(bpy)2(CO)2](PF6)2: Its Implication in CO2 Electrocatalytic Reduction. Inorg. Chem. 1994, 33, 4410–4412. [Google Scholar] [CrossRef]
- Probst, B.; Rodenberg, A.; Guttentag, M.; Hamm, P.; Alberto, R. A Highly Stable Rhenium−Cobalt System for Photocatalytic H2 Production: Unraveling the Performance-Limiting Steps. Inorg. Chem. 2010, 49, 6453–6460. [Google Scholar] [CrossRef]
- Chan, S.F.; Chou, M.; Creutz, C.; Matsubara, T.; Sutin, N. Mechanism of the formation of dihydrogen from the photoinduced reactions of poly(pyridine)ruthenium(II) and poly(pyridine)rhodium(III) complexes. J. Am. Chem. Soc. 1981, 103, 369–379. [Google Scholar] [CrossRef]
- Delaive, P.J.; Foreman, T.K.; Giannotti, C.; Whitten, D.G. Photoinduced electron transfer reactions of transition-metal complexes with amines. Mechanistic studies of alternate pathways to back electron transfer. J. Am. Chem. Soc. 1980, 102, 5627–5631. [Google Scholar]
- Neshvad, G.; Hoffman, M.Z. Reductive quenching of the luminescent excited state of tris(2,2'-bipyrazine)ruthenium(2+) ion in aqueous solution. J. Phys. Chem. 1989, 93, 2445–2452. [Google Scholar] [CrossRef]
- Kishore, K.; Dey, G.R.; Mukherjee, T. OH radical reactions with ethanolamine: formation of reducing as well as oxidizing radicals. Res. Chem. Intermed. 2004, 30, 837–845. [Google Scholar] [CrossRef]
- Kutal, C.; Corbin, A.J.; Ferraudi, G. Further studies of the photoinduced reduction of carbon dioxide mediated by tricarbonylbromo(2,2'-bipyridine)rhenium(I). Organometallics 1987, 6, 553–557. [Google Scholar]
- Kutal, C.; Weber, M.A.; Ferraudi, G.; Geiger, D. A mechanistic investigation of the photoinduced reduction of carbon dioxide mediated by tricarbonylbromo(2,2'-bipyridine)rhenium(I). Organometallics 1985, 4, 2161–2166. [Google Scholar]
- Pac, C.; Miyauchi, Y.; Ishitani, O.; Ihama, M.; Yasuda, M.; Sakurai, H. Redox-photosensitized reactions. 11. Ru(bpy)32+-photosensitized reactions of 1-benzyl-1,4-dihydronicotinamide with aryl-substituted enones, derivatives of methyl cinnamate, and substituted cinnamonitriles: electron-transfer mechanism and structure-reactivity relationships. J. Org. Chem. 1984, 49, 26–34. [Google Scholar]
- Kobayashi, A.; Konno, H.; Sakamoto, K.; Sekine, A.; Ohashi, Y.; Iida, M.; Ishitani, O. Transition Metal Complexes Coordinated by an NAD(P)H Model Compound and their Enhanced Hydride-Donating Abilities in the Presence of a Base. Chem. Eur. J. 2005, 11, 4219–4226. [Google Scholar] [CrossRef]
- Maestri, M.; Grätzel, M. 530 nm-Laser Photolysis Studies of the Photo Reduction of Tris (2,2-bipyridine)-ruthenium(II) by Organic Donors. Ber. Bunsenges. Phys. Chem. 1977, 81, 504–507. [Google Scholar] [CrossRef]
- Suga, K.; Mizota, H.; Kanzaki, Y.; Aoyagui, S. ΔG0-dependence of the chemical transfer coefficient and the potential dependence of the electrochemical transfer coefficient. Electroanal. Chem. Interfacial Chem. 1973, 41, 313–321. [Google Scholar] [CrossRef]
- Marcus, R.A. On the theory of oxidation-reduction reactions involving electron transfer. V. Comparison and properties of electrochemical and chemical rate constants. J. Phys. Chem. 1963, 67, 853–857. [Google Scholar] [CrossRef]
- Bian, Z.Y.; Sumi, K.; Furue, M.; Sato, S.; Koikeand, K.; Ishitani, O. A Novel Tripodal Ligand, Tris[(4′-methyl-2,2′-bipyridyl-4-yl)methyl]carbinol and Its Trinuclear RuII/ReI Mixed-Metal Complexes: Synthesis, Emission Properties, and Photocatalytic CO2 Reduction. Inorg. Chem. 2008, 47, 10801–10803. [Google Scholar]
- Tamaki, Y.; Watanabe, K.; Koike, K.; Inoue, H.; Morimto, T.; Ishitani, O. Development of highly efficient supramolecular CO2 reduction photocatalysts with high turnover frequency and durability. Faraday Discuss. 2012, 155, 115–127. [Google Scholar] [CrossRef]
- Woolerton, T.W.; Sheard, S.; Reisner, E.; Pierce, E.; Ragsdale, S.W.; Armstrong, F.A. Efficient and Clean Photoreduction of CO2 to CO by Enzyme-Modified TiO2 Nanoparticles Using Visible Light. J. Am. Chem. Soc. 2010, 132, 2132–2133. [Google Scholar]
- Waugh, K.C. Methanol Synthesis. Catal. Today 1992, 15, 51–75. [Google Scholar]
- Matsuoka, S.; Kohzuki, T.; Pac, C.; Ishida, A.; Takamuku, S.; Kusaba, M.; Nakashima, N.; Yanagida, S. Photocatalysis of oligo(p-phenylenes): photochemical reduction of carbon dioxide with triethylamine. J. Phys. Chem. 1992, 96, 4437–4442. [Google Scholar] [CrossRef]
- Ishida, H.; Tanaka, K.; Tanaka, T. Electrochemical CO2 reduction catalyzed by ruthenium complexes [Ru(bpy)2(CO)2]2+ and [Ru(bpy)2(CO)Cl]+. Effect of pH on the formation of CO and HCOO−. Organometallics 1987, 6, 181. [Google Scholar]
- Ishida, H.; Fujiki, K.; Ohba, T.; Ohkubo, K.; Tanaka, K.; Terada, T.; Tanaka, T. Ligand effects of ruthenium 2,2′-bipyridine and 1,10-phenanthroline complexes on the electrochemical reduction of CO2. J. Chem. Soc. Dalton Trans. 1990, 2155–2160. [Google Scholar]
- Bloomfield, L.A.; Freeman, R.R.; Brown, W.A. Photofragmentation of Mass-Resolved Si2–12+ Clusters. Phys. Rev. Lett. 1985, 54, 2246–2249. [Google Scholar] [CrossRef]
- Geusic, M.E.; McIlrath, T.J.; Jarrold, M.F.; Bloomfield, L.A.; Freeman, R.R.; Brown, W.L. Photofragmentation of mass-resolved carbon cluster ions: Observation of a ‘‘magic’’ neutral fragment. J. Chem. Phys. 1986, 84, 2421–2422. [Google Scholar]
- Mandich, M.L.; Reents, W.D., Jr.; Bondybey, V.E. Reactivity of ionic silicon clusters with methylsilane studied by Fourier transform ion cyclotron resonance mass spectrometry. J. Phys. Chem. 1986, 90, 2315–2319. [Google Scholar] [CrossRef]
- Bondybey, V.E.; Schwartz, G.P.; English, J.H. Laser vaporization of alloys: Laser induced fluorescence of heteronuclear metal clusters. J. Chem. Phys. 1983, 78, 11–15. [Google Scholar] [CrossRef]
- Maidan, R.; Willner, I. Neutral gas-phase analogs of condensed-phase post-transition-metal cluster ions: laser vaporization and photoionization of tin-bismuth and lead-antimony alloys. J. Am. Chem. Soc. 1986, 108, 8101–8102. [Google Scholar] [CrossRef]
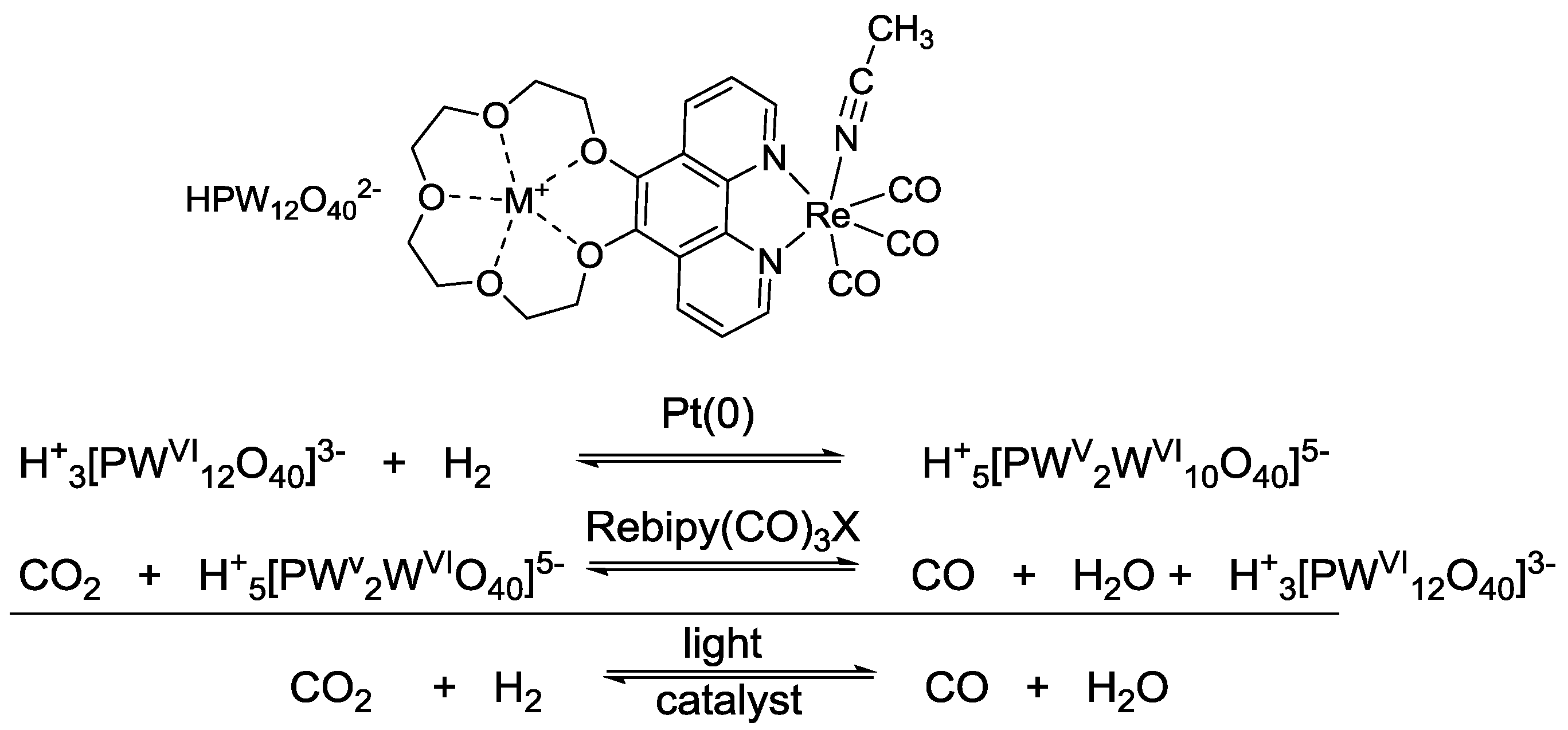
- Ettedgui, J.; Diskin-Posner, Y.; Weiner, L.; Neumann, R. Photoreduction of Carbon Dioxide to Carbon Monoxide with Hydrogen Catalyzed by a Rhenium(I) Phenanthroline−Polyoxometalate Hybrid Complex. J. Am. Chem. Soc. 2011, 133, 188–190. [Google Scholar]
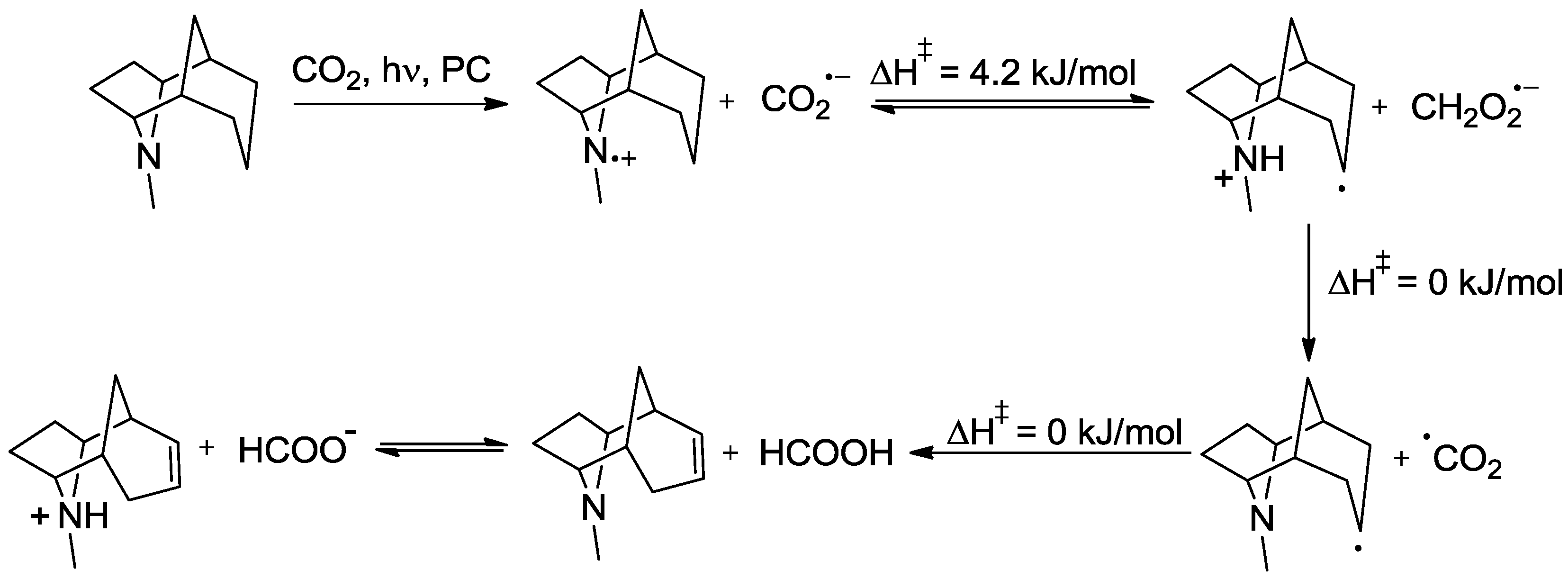
- Richardson, R.D.; Holland, E.J.; Carpenter, B.K. A renewable amine for photochemical reduction of CO2. Nature Chem. 2011, 3, 301–303. [Google Scholar] [CrossRef]
- Cohen, S.G.; Parola, A.; Parson, G.H. Photoreduction by amines. Chem. Rev. 1973, 73, 141–161. [Google Scholar]
- Cape, J.L.; Hurst, J.K. Detection and Mechanistic Relevance of Transient Ligand Radicals Formed during [Ru(bpy)2(OH2)]2O4+-Catalyzed Water Oxidation. J. Am. Chem. Soc. 2008, 130, 827–829. [Google Scholar]
- Geletii, Y.V.; Huang, Z.; Hou, Y.; Musaev, D.G.; Lian, T.; Hill, C.L. Homogeneous Light-Driven Water Oxidation Catalyzed by a Tetraruthenium Complex with All Inorganic Ligands. J. Am. Chem. Soc. 2009, 131, 7522–7523. [Google Scholar]
- Besson, C.; Huang, Z.; Geletii, Y.V.; Lense, S.; Hardcasle, K.L.; Musaev, D.G.; Lian, T.; Proust, A.; Hill, C.L. Cs9[(γ-PW10O36)2Ru4O5(OH)(H2O)4], a new all-inorganic, soluble catalyst for the efficient visible-light-driven oxidation of water. Chem. Commun. 2010, 46, 2784–2786. [Google Scholar]
- Orlandi, M.; Argazzi, R.; Sartorel, A.; Carraro, M.; Scorrano, G.; Bonchio, M.; Scandola, F. Ruthenium polyoxometalate water splitting catalyst: very fast hole scavenging from photogenerated oxidants. Chem. Commun. 2010, 46, 3152–3154. [Google Scholar]
- Puntoriero, F.; Ganga, G.L.; Sartorel, A.; Carraro, M.; Scorrano, G.; Bonchio, M.; Campagna, S. Photo-induced water oxidation with tetra-nuclear ruthenium sensitizer and catalyst: A unique 4 × 4 ruthenium interplay triggering high efficiency with low-energy visible light. Chem. Commun. 2010, 46, 4725–4727. [Google Scholar]
- Roeser, S.; Farras, P.; Bozoglian, F.; Martínez-Belmonte, M.; Benet-Buchholz, J.; Llobet, A. Chemical, Electrochemical, and Photochemical Catalytic Oxidation of Water to Dioxygen with Mononuclear Ruthenium Complexes. ChemSusChem 2011, 4, 197–207. [Google Scholar]
- Duan, L.; Xu, Y.; Zhang, P.; Wang, M.; Sun, L. Chemical, Electrochemical, and Photochemical Catalytic Oxidation of Water to Dioxygen with Mononuclear Ruthenium Complexes. Inorg. Chem. 2010, 49, 209–215. [Google Scholar]
- Duan, L.; Xu, Y.; Gorlov, M.; Tong, L.; Andersson, S.; Sun, L. Chemical and Photochemical Water Oxidation Catalyzed by Mononuclear Ruthenium Complexes with a Negatively Charged Tridentate Ligand. Chem. Eur. J. 2010, 16, 4659–4668. [Google Scholar]
- Xu, Y.; Duan, L.; Tong, L.; Akermark, B.; Sun, L. Visible light-driven water oxidation catalyzed by a highly efficient dinuclear ruthenium complex. Chem. Commun. 2010, 46, 6506–6508. [Google Scholar] [CrossRef]
- Xu, Y.; Fischer, A.; Duan, L.; Tong, L.; Gabrirlsson, E.; Akermark, B.; Sun, L. Chemical and Light-Driven Oxidation of Water Catalyzed by an Efficient Dinuclear Ruthenium Complex. Angew. Chem. Int. Ed. 2010, 49, 8934–8937. [Google Scholar]
- Li, F.; Jiang, Y.; Zhang, B.; Huang, F.; Gao, Y.; Sun, L. Towards A Solar Fuel Device: Light-Driven Water Oxidation Catalyzed by a Supramolecular Assembly. Angew. Chem. Int. Ed. 2012, 51, 2417–2420. [Google Scholar]
- Huang, Z.; Luo, Z.; Geletii, Y.V.; Vickers, J.W.; Yin, Q.; Wu, D.; Hou, Y.; Ding, Y.; Song, J.; Musaev, D.G.; et al. Efficient Light-Driven Carbon-Free Cobalt-Based Molecular Catalyst for Water Oxidation. J. Am. Chem. Soc. 2011, 133, 2068–2071. [Google Scholar]
- McCool, N.S.; Robinson, D.M.; Sheats, J.E.; Dismukes, G.C. A Co4O4 “Cubane” Water Oxidation Catalyst Inspired by Photosynthesis. J. Am. Chem. Soc. 2011, 133, 11446–11449. [Google Scholar]
- la Ganga, G.; Puntoriero, F.; Campagna, S.; Bazzan, I.; Berardi, S.; Bonchio, M.; Sartorel, A.; Natalic, M.; Scandola, F. Light-driven water oxidation with a molecular tetra-cobalt(III) cubane cluster. Faraday Discuss. 2012, 155, 177–190. [Google Scholar] [CrossRef]
- Tong, L.; Wang, Y.; Duan, L.; Xu, Y.; Cheng, X.; Fischer, A.; Ahlquist, M.S.G.; Sun, L. Water Oxidation Catalysis: Influence of Anionic Ligands upon the Redox Properties and Catalytic Performance of Mononuclear Ruthenium Complexes. Inorg. Chem. 2012, 51, 3388–3398. [Google Scholar]
- Iizuka, K.; Wato, T.; Miseki, Y.; Saito, K.; Kudo, A. Photocatalytic Reduction of Carbon Dioxide over Ag Cocatalyst-Loaded ALa4Ti4O15 (A = Ca, Sr, and Ba) Using Water as a Reducing Reagent. J. Am. Chem. Soc. 2011, 133, 20863–20868. [Google Scholar] [CrossRef]
- Sato, S.; Arai, T.; Morikawa, T.; Uemura, K.; Suzuki, T.M.; Tanaka, H.; Kajino, T. Selective CO2 Conversion to Formate Conjugated with H2O Oxidation Utilizing Semiconductor/Complex Hybrid Photocatalysts. J. Am. Chem. Soc. 2011, 133, 15240–15243. [Google Scholar]
- Inoue, H.; Yamachika, M.; Yoneyama, H.J. Photocatalytic conversion of lactic acid to malic acid through pyruvic acid in the presence of malic enzyme and semiconductor photocatalysts. Chem. SOC. Faraday Trans. 1992, 88, 2215–2219. [Google Scholar] [CrossRef]
- Walther, D.; Ruben, M.; Rau, S. Carbon dioxide and metal centres: from reactions inspired by nature to reactions in compressed carbon dioxide as solvent. Coord. Chem. Rev. 1999, 182, 67–100. [Google Scholar]
- Singh, W.M.; Pegram, D.; Duan, H.; Kalita, D.; Simone, P.; Emmert, G.L.; Zhao, X. Hydrogen Production Coupled to Hydrocarbon Oxygenation from Photocatalytic Water Splitting. Angew. Chem. Int. Ed. 2012, 51, 1653–1656. [Google Scholar]
- Endicott, J.F.; Chen, Y.; Xie, P. Electron-transfer spectroscopy: donor–acceptor electronic coupling, reorganizational energies, reaction pathways and dynamics. Coord. Chem. Rev. 2005, 249, 343–373. [Google Scholar]
- Gabrielsson, A.; Smith, J.R.L.; Perutz, R.N. Remote site photosubstitution in metalloporphyrin-rhenium tricarbonylbipyridine assemblies: photo-reactions of molecules with very short lived excited states. Dalton Trans. 2008. [Google Scholar] [CrossRef]
- Easun, T.L.; Alsindi, W.Z.; Towrie, M.; Ronayne, K.L.; Sun, X.Z.; Ward, M.D.; George, M.W. Photoinduced Energy Transfer in a Conformationally Flexible Re(I)/Ru(II) Dyad Probed by Time-Resolved Infrared Spectroscopy: Effects of Conformation and Spatial Localization of Excited States. Inorg. Chem. 2008, 47, 5071–5078. [Google Scholar]
- Lazarides, T.; Sykes, D.; Faulkner, S.; Barbieri, A.; Ward, M.D. On the Mechanism of d–f Energy Transfer in RuII/LnIII and OsII/LnIII Dyads: Dexter-Type Energy Transfer Over a Distance of 20 Å. Chem.–Eur. J. 2008, 14, 9389–9399. [Google Scholar]
- Welter, S.; Lafolet, F.; Cecchetto, E.; Vergeer, F.; de Cola, L. Energy Transfer by a Hopping Mechanism in Dinuclear IrIII/RuII Complexes: A Molecular Wire? Chem. Phys. Chem. 2005, 6, 2417–2427. [Google Scholar] [CrossRef]
- Patoux, C.; Launay, J.; Beley, M.; Chodorowski-Kimmes, S.; Collin, J.; James, S.; Sauvage, J. Long-Range Electronic Coupling in Bis(cyclometalated) Ruthenium Complexes. J. Am. Chem. Soc. 1998, 120, 3717–3725. [Google Scholar]
- Callahan, R.W.; Brown, G.M.; Meyer, T.J. Effects of weak metal-metal interactions in ligand-bridged complexes of ruthenium. Dimeric complexes containing ruthenium ions in different coordination environments. Inorg. Chem. 1975, 14, 1443–1453. [Google Scholar]
- Kimura, E.; Wada, S.; Shionoya, M.; Takahashi, T.; Litaka, Y. A novel cyclam–nickel(II) complex appended with a tris-(2,2′-bipyridine) ruthenium(II) complex (cyclam = 1,4,8,11-tetra-azacyclotetradecane). J. Chem. Soc. Chem. Commun. 1990, 397–398. [Google Scholar]
- Bian, Z.Y.; Sumi, K.; Furue, M.; Sato, S.; Koike, K.; Ishitani, O. Synthesis and properties of a novel tripodal bipyridyl ligand tb-carbinol and its Ru(II)–Re(I) trimetallic complexes: investigation of multimetallic artificial systems for photocatalytic CO2 reduction. Dalton Trans. 2009, 983–993. [Google Scholar]
- Bian, Z.-Y.; Chi, S.-M.; Lia, L.; Fu, W. Conjugation effect of the bridging ligand on the CO2 reduction properties in difunctional photocatalysts. Dalton Trans. 2010, 39, 7884–7887. [Google Scholar] [CrossRef]
- Koike, K.; Hori, H.; Ishizuka, M.; Westwell, J.R.; Takeuchi, K.; Ibusuki, T.; Enjouji, K.; Konno, H.; Sakamoto, K.; Ishitani, O. Key Process of the Photocatalytic Reduction of CO2 Using [Re(4,4‘-X2-bipyridine)(CO)3PR3]+ (X = CH3, H, CF3; PR3 = Phosphorus Ligands): Dark Reaction of the One-Electron-Reduced Complexes with CO2. Organometallics 1997, 16, 5724–5729. [Google Scholar] [CrossRef]
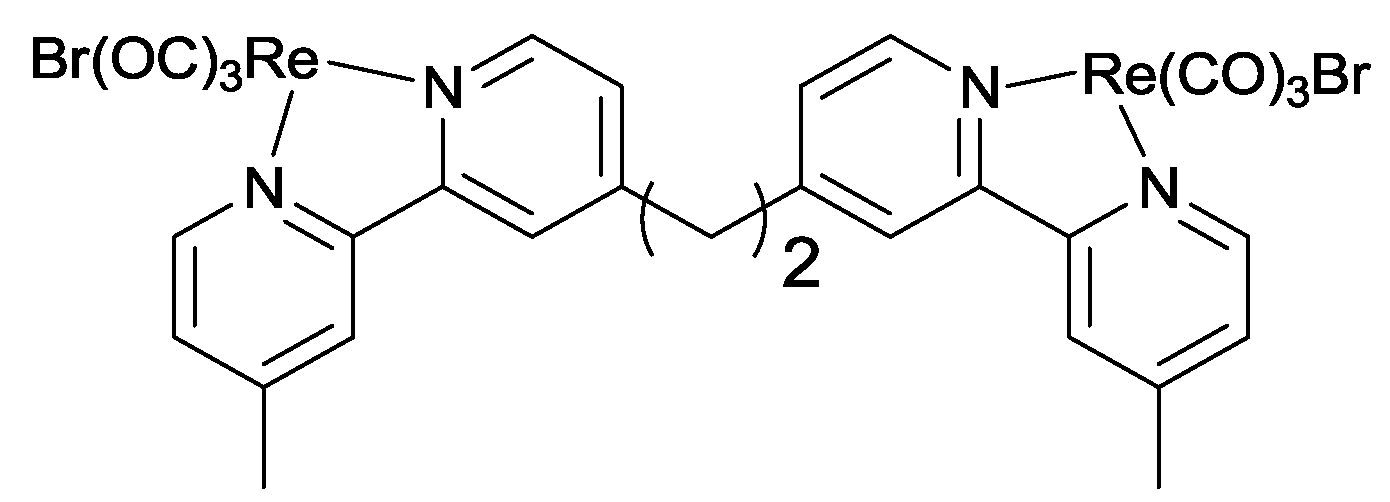
- Koike, K.; Naito, S.; Sato, S.; Tamaki, Y.; Ishitani, O. Architecture of supramolecular metal complexes for photocatalytic CO2 reduction: III: Effects of length of alkyl chain connecting photosensitizer to catalyst. J. Photochem. Photobiol. A 2009, 207, 109–114. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
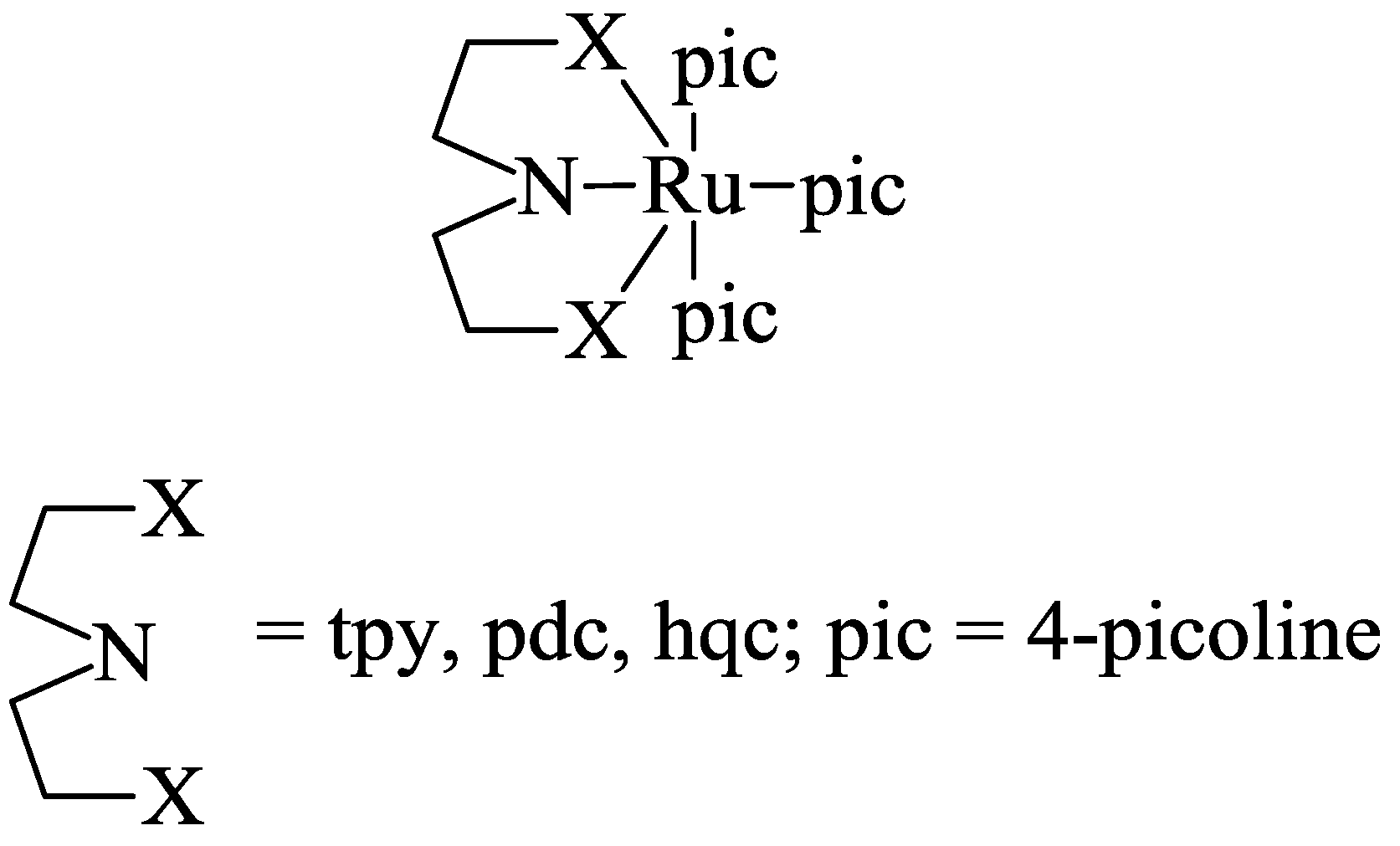{kind=link}
{kind=link}
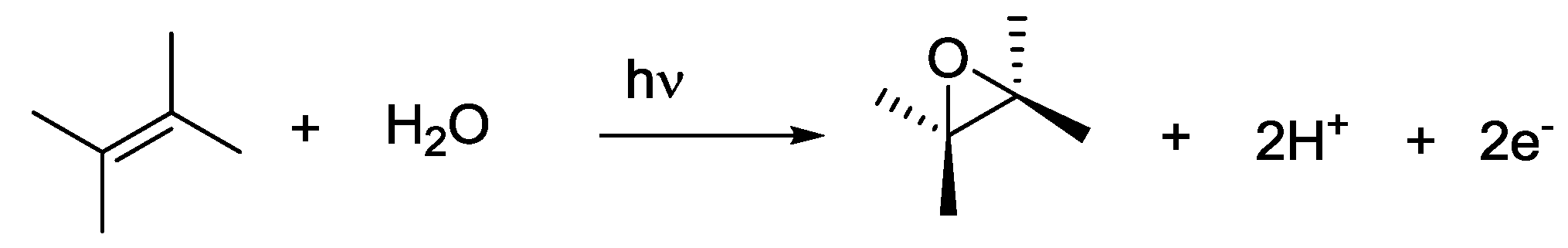{kind=link}
{kind=link}
{kind=link}
{kind=link}
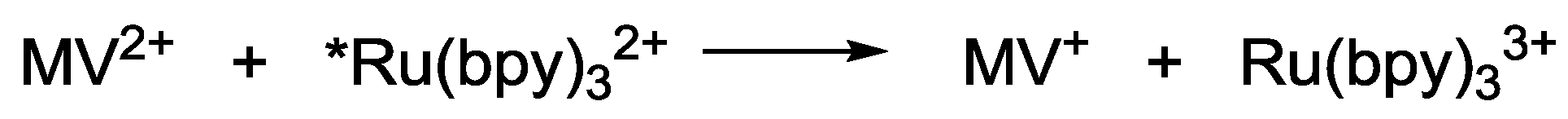{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
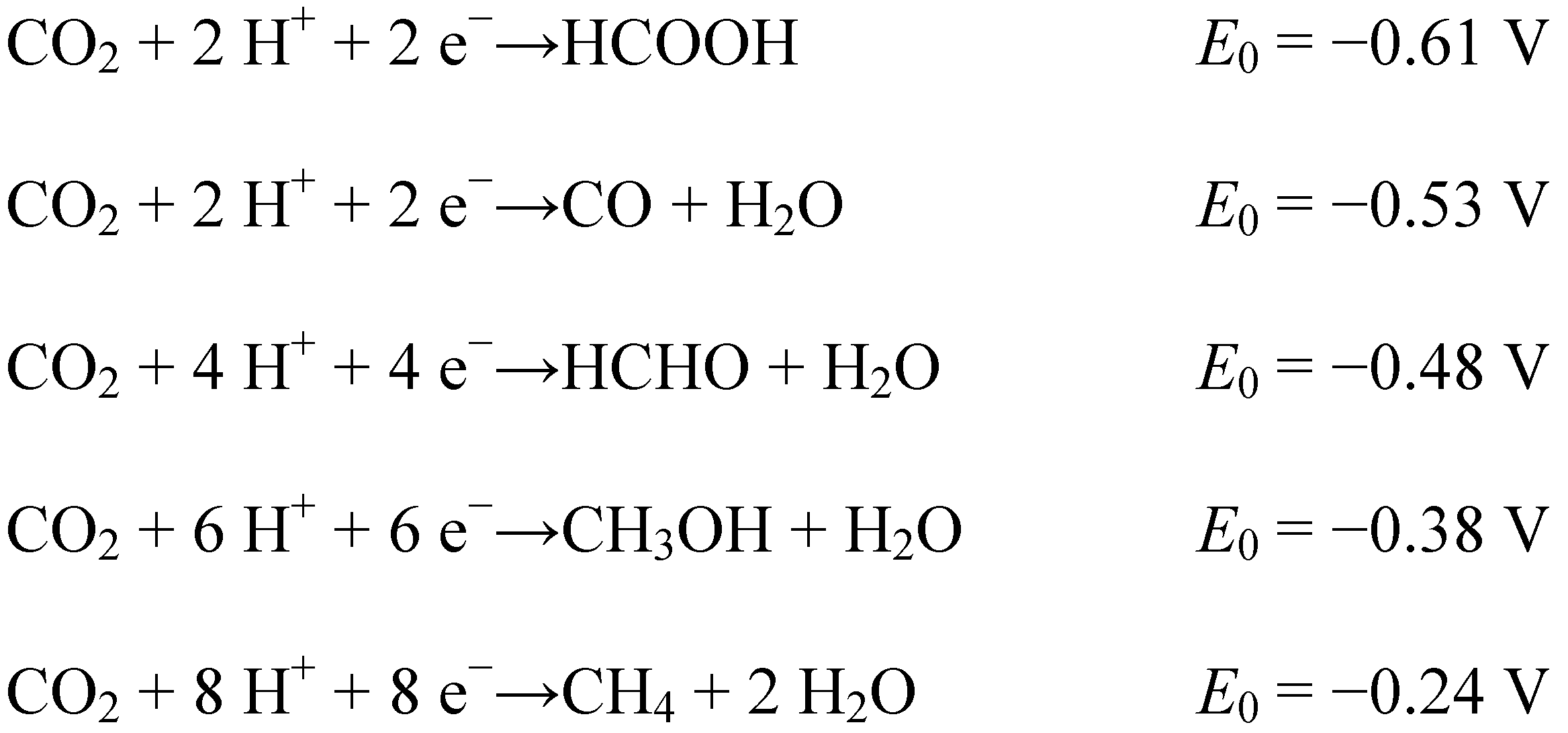{kind=link}
{kind=link}
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Reithmeier, R.; Bruckmeier, C.; Rieger, B. Conversion of CO2 via Visible Light Promoted Homogeneous Redox Catalysis. Catalysts 2012, 2, 544-571. https://doi.org/10.3390/catal2040544
Reithmeier R, Bruckmeier C, Rieger B. Conversion of CO2 via Visible Light Promoted Homogeneous Redox Catalysis. Catalysts. 2012; 2(4):544-571. https://doi.org/10.3390/catal2040544
Chicago/Turabian StyleReithmeier, Richard, Christian Bruckmeier, and Bernhard Rieger. 2012. "Conversion of CO2 via Visible Light Promoted Homogeneous Redox Catalysis" Catalysts 2, no. 4: 544-571. https://doi.org/10.3390/catal2040544
APA StyleReithmeier, R., Bruckmeier, C., & Rieger, B. (2012). Conversion of CO2 via Visible Light Promoted Homogeneous Redox Catalysis. Catalysts, 2(4), 544-571. https://doi.org/10.3390/catal2040544