Abstract
The removal of mercury from flue gases in scrubbers is greatly facilitated if the mercury is present as water-soluble oxidized species. Therefore, increased mercury oxidation upstream of scrubber devices will improve overall mercury removal. For this purpose heterogeneous catalysts have recently attracted a great deal of interest. Selective catalytic reduction (SCR), noble metal and transition metal oxide based catalysts have been investigated at both the laboratory and plant scale with this objective. A review article published in 2006 covers the progress in the elemental mercury (Hgel) catalytic oxidation area. This paper brings the review in this area up to date. To this end, 110 papers including several reports and patents are reviewed. For each type of catalyst the possible mechanisms as well as the effect of flue gas components on activity and stability are examined. Advantages and main problems are analyzed. The possible future directions of catalyst development in this environmental research area are outlined.
1. Introduction
Mercury emissions are of global concern due to their persistence, long-range mobility in the atmosphere, bio-accumulation in aquatic ecosystems and their neurotoxic impact on human health [1,2,3]. Therefore, the control and reduction of mercury emissions have become a major concern at national, regional and international level. For instance, within the United Nations Environmental Programme (UNEP), negotiations have started with a view to achieving an international mercury treaty to reduce the risks from global mercury emissions [1]. In December 2011, U.S. EPA issued the Mercury and Air Toxics Standards (MATS), with the aim of reducing emission levels of mercury and other toxic pollutants from power plants [4]. In Europe, the 2010/75/EU Industrial Emissions Directive [5] sets the average mercury emission limit value at 50 μg/m3 for waste incinerators. Concerns over the environmental effects of mercury emissions have triggered discussions on more stringent regulations in many countries. Consequently, mercury emissions abatement has become a new challenge for environmental engineering.
According to UNEP, estimates for 2005, flue gases from waste incinerators contribute 40 t and cement plants 190 t. Coal fired power and industrial plants are the major source, with between 500 t and 1920 t of global anthropogenic mercury emissions to air [1]. During the combustion process all forms of mercury in fuel decompose into gaseous elemental mercury (Hgel) [6]. As the combustion gas cools down to 400 °C, this elemental mercury is partially oxidized via gas phase reactions involving oxygen and halogen species [6]. HgCl2 and HgO are the volatile oxidized mercury (Hgox) species most likely to occur in flue gases. Oxidized mercury in flue gases is both reactive and water soluble, and therefore is easily captured by scrubbing processes. In wet flue gas desulfurization systems (WFGD) oxidized mercury is removed as a co-benefit. The effectiveness of the WFGD in the removal of oxidized mercury from flue gases depends on the operation conditions [7]. Generally, small concentrations of particle-bound mercury in flue gases are effectively removed in electrostatic precipitators or fabric filters.
Elemental mercury is fairly insoluble in water (nearly 50 µg/L) and not effectively removed in scrubbers such as WFGD units. Therefore, processes that oxidize Hgel in flue gases improve the effectiveness of mercury removal by wet scrubbers. In this context, one of the main challenges of mercury control strategies is the efficient conversion of elemental mercury into the oxidized form. Thermodynamic equilibrium calculation shows that for temperatures below 450 °C, mercury in flue gases should exist mostly in the oxidized form. However, the cooling of flue gases in boilers under technical conditions is fast so that the mercury equilibrium at low temperatures is not reached. Here, the gas phase oxidation is kinetically limited. A selective heterogeneous catalyst, upstream of the scrubber, is needed to increase the speed of mercury oxidation reaction in the cooled flue gases [8].
The status of the Hgel oxidation catalysts up to 2005 is covered in a review paper by Presto and Granite [9]. Since then this subject has gained increasing interest. Figure 1 presents a yearly frequency of the published literature over the past two decades. This figure is based on the compiled literature cited in the Presto and Granite paper [9] and the literature covered in this review. Starting in 2000, an increase in the number of papers covering the topic of catalytic oxidation of gaseous mercury is observed. To date, more than 200 papers are available on the topic.
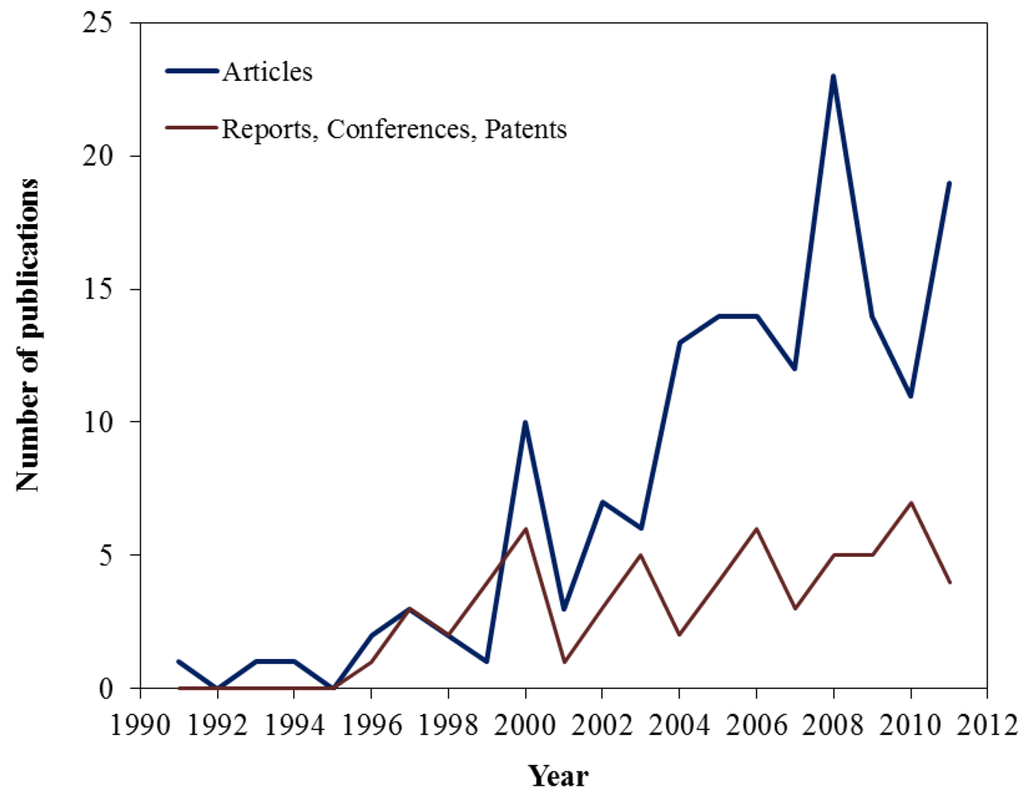
Figure 1.
Yearly Frequency of Published Literature on Mercury Oxidation.
The present review article will discuss recent developments as well as some older papers which are excluded in the previous review paper, but whose importance is nowadays seen differently. For the sake of clarity this review will follow a similar structure as the previous review. Our understanding of the mechanism underlying the catalytic mercury oxidation process will be discussed first. Chapters concentrating on different mercury catalysts follow. Laboratory and plant scale investigations of noble metal and transition metal oxide based catalysts are discussed. Selective catalytic reduction (SCR)-DeNOx catalysts still attract the largest interest. The chapter on this group of catalysts is therefore very extensive.
Another aspect discussed in this review article involves the novel methods in the area of catalytic mercury oxidation. In recent years, a number of patent applications for mercury oxidation catalysts have appeared. A brief summary of their main features will be presented as well.
2. Operation Condition and Constraints of Hg Oxidation Catalysts in Flue Gases
All fuels, including renewables such as wood or gaseous, like natural gas, contain mercury. This mercury is almost completely volatized during the combustion process. Generally, the mercury content of different fuels varies between 0.01 and 2 µg/g, resulting in flue gases with a mercury content of 1 to 200 µg/m3 [2]. The mercury content of natural gas is removed at the well, whereas the mercury from crude oil is removed at the refineries [10]. Therefore, the use of these fuels will not result in any appreciable mercury emissions. Consequently, the mobile combustion sources do not present a mercury emission issue. Mercury in coal is often associated with the pyrite fraction, and pyrite cleaning of coal does reduce Hg levels. However, fine grained pyrite (<70 μm) is not removed by cleaning. Consequently, just one third of mercury in coal is removed by standard coal cleaning procedures [11]. Mercury from solid fuels is not completely removed by cleaning the fuel, and has to be captured from the flue gas.
The stable mercury species at combustion temperatures of around 1000 °C are exclusively in the elemental form. At temperatures below 450 °C, equilibrium in flue gases is dominated by oxidized species such as the oxide and the halogenides (X = Cl, Br and I). This low temperature equilibrium is generally not established in power plants and industrial incinerators. The halogen content of the fuel and of the flue gas determines the extent to which the Hgel is transformed into Hgox. The higher the halogen content the larger will be the fraction of oxidized species of the total mercury [12]. Below500 °C, the kinetics of the oxidation reaction of Hgel in flue gases are low in comparison to the flue gas residence times in incineration plants.
The halogen content of coal-like solid fuels is dominated by chlorine species. Chlorine appears in flue gases predominantly as HCl. At low temperatures, Cl2 becomes the most stable species; however low kinetics do not allow for Cl2 formation in flue gases. The HCl content of flue gases from coal-combustion varies between 1 and 500 mg/m3. The bromine content in coal fuels is generally between 1 and 4% of the chlorine content [13]. Bromine species in the flue gas are more effective in oxidizing Hgel. The iodine content of coal has not been often analyzed but is below the level of bromine.
Mercury oxidation catalysts do not decrease the mercury content of flue gases from industrial and power plant incinerators as such, but rather in combination with a scrubber or, less frequently, an absorber. For this purpose, the mercury oxidation catalyst has to be placed upstream of the flue gas scrubber. Wet scrubbing removes the acid components of flue gas, mainly SO2, SO3, HCl and HBr, as well as the oxidized mercury.
The mercury catalyst may be placed before (high dust) and after (low dust) the dust removal device, which are often operated at temperatures around 280–400 °C. Low dust placement is less prone to clogging by large dust particles. Here, all the volatile acid constituents of the flue gas are still present except for the nitrogen oxides NOx. These are partially removed by ammonia-based SCR—or selective non-catalytic reduction (SNCR)—DeNOx technologies thereby introducing a slip concentration of 5 mg/m³ and higher of NH3.
In the high dust version, the most likely position to place the mercury catalyst is within the SCR-DeNOx reactor. This reactor is operated in SO2-containing flue gases at temperatures between 280 and 400 °C. At this temperature range, the concentration of volatile arsenic (As) and selenium (Se) will still be high and needs special attention in the context of catalyst deactivation.
Another important constraint for mercury oxidation catalyst operation in a SO2-containing gas is its effect on the conversion reaction of SO2 to SO3. SO3 is poorly captured in most scrubbers. It forms sulfuric acid mist in flue gases that leave the plant. Another unwanted side reaction to be considered is the oxidation of NO to NO2. Although some NO2 is removed in scrubbers, as little as 10 to 15 ppm of NO2 in the flue gas may cause a brownish plume from the chimney.
The oxygen content of flue gases resulting from combustion processes varies between 2 and 8 vol.-%. This might also influence the performance of mercury oxidation catalysts.
It should be mentioned that because of the large volumes to be treated, mercury oxidation catalysts, like any other flue gas catalysts, has to be applied in a honeycomb or plate-like structure. This arrangement is preferred in order to reduce both pressure loss and energy requirements.
3. Proposed Mechanisms for the Catalytic Oxidation of Elemental Mercury
Schematically, the oxidation of Hgel in the presence of a catalytic material can be described as follows:
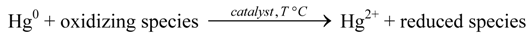
The reaction rate of the gas phase oxidation of Hgel in flue gases is small at temperatures below 500 °C. In order to achieve reasonable oxidation rate in this temperature region, the catalyst has to provide a very active oxidizing species or has to activate the mercury on its surface. For the adsorption process, the Lewis base and electron pair donor character of gaseous Hgel are important. Cation vacancies or Lewis acid sites on the surface of the catalytic active material can provide respectively active sites for the primary step or physical adsorption of elemental mercury. Other flue gas components compete with Hgel for the active Lewis acid sites on the prospective catalytic material.
In recent years, papers on mercury oxidation catalyst increasingly discuss the underlying oxidation mechanisms. In the following sections, the mechanisms proposed to date are briefly discussed. It should be noted that, for almost all the mercury oxidation catalysts studied so far, the HCl and HBr constituents of flue gases to be treated are of vital importance. In the absence of these constituents, the oxidation rate is considerably reduced.
3.1. Deacon Reaction
Because of the importance of the halogen species, it is proposed that the Hgel oxidation in flue gases could be enhanced by chlorine atoms (Cl•) or molecules (Cl2) produced by the Deacon reaction [14]. Vosteen et al. [15,16] pointed out that the bromine species might be even more important, and that a high Hgel oxidation could be achieved in a tail-end SCR-DeNOx system even at low bromine concentrations by the bromine-Deacon reaction.
The Deacon reaction produces chlorine by catalytic oxidation of HCl with air or oxygen according to the overall reaction 2. The reaction is reversible and exothermic.
4 HCl(g) + O2(g) ↔ 2 Cl2(g) + 2 H2O(g) (ΔH° = −28.4 kJ/mol HCl) (2)
The reaction takes place at about 350 to 450 °C in the presence of copper, chromium, vanadium and RuO2 catalysts. This reaction is described by a Mars-van Krevelen type of reaction mechanism involving hydrogen abstraction from adsorbed HCl, recombination and desorption of atomic chlorine, water desorption and oxygen adsorption [17]. HBr reacts in a similar manner, whereby the bromine-Deacon reaction produces more free bromine [18]. However, Griffin [19] suggested that SO2 depletes the Deacon Cl2 in the gas phase through the following gas phase reaction:
Cl2 + SO2 + H2O ↔ 2 HCl + SO3 (3)
In this way the positive effect of the Deacon-generated chlorine species might be inhibited. Bromine reacts to a smaller extent with SO2.
3.2. Eley-Rideal Mechanism
This mechanism assumes that the elemental mercury present in the flue gas could react from the gas-phase (or as a weakly adsorbed) state with an adsorbed oxidant species, most likely HCl. By invoking the Eley-Rideal mechanism, some authors [20] have tried to model the Hgel oxidation under SCR conditions. According to this mechanism, HCl is dissociatively adsorbed on the V2O5-active sites found on the SCR catalyst surface. NH3 and HCl species compete for the active sites. Hgel reacts with the V2O5-chlorinated sites from the gas phase or as a weakly adsorbed species [21,22] to form the final oxidation product, for example, according to the equations (4–6):
2 HCl(g) + 2 V–O–V(s) ↔ 2 V–OH–V–Cl(s) (4)
2 V–OH–V–Cl(s) + Hg0(g) ↔ 2 V–OH–V(s) + HgCl2(g) (5)
2 V–OH–V(s) + 0.5 O2(g) ↔ 2 V–O–V(s) + H2O(g) (6)
In the light of most recent experimental findings, an Eley-Rideal mechanism with adsorbed HCl reacting with gas phase or weakly adsorbed Hgel does not seem plausible. Evidence of HCl adsorption on the catalyst surface was obtained by employing different surface analysis methods [23,24,25]. Following the reaction between HCl and the V2O5 active sites on the catalyst surface, several vanadyl oxychloride complexes such as VOCl2, V2O3(OH)2Cl2, VO2Cl2 are formed [23,24]. However, it has been shown by surface analysis means that Hgel adsorbs on various surfaces as well.
3.3. Langmuir-Hinshelwood Mechanism
Based on the Langmuir-Hinshelwood mechanism, the catalytic oxidation of Hgel takes place between elemental mercury and the oxidant species co-adsorbed on the catalyst surface. This oxidation mechanism is likely to occur in the presence of substrates which can adsorb both HCl and Hgel. A number of materials can adsorb Hgel from the flue gas [26,27,28,29,30]. The adsorption of halogens is mentioned in the previous subchapter.
When investigating the catalytic oxidation of Hgel in the presence of MnOx–CeO2/TiO2, Li et al. [26] proposed the Langmuir-Hinshelwood mechanism as a plausible reaction pathway. They explained the decrease of Hgel oxidation efficiency at temperatures above 250 °C with the decreasing adsorption of Hgel: therefore availability of one of the reaction partners. Eom et al. [25] formulated the rate limiting reaction of the adsorbed mercury and the absorbed chlorine species on a commercial SCR-DeNOx catalyst as:
SCR-cat(V4+–O)Hg+(ads)(s) + 2 SCR-cat(V4+–Cl) ↔ SCR-cat (V4+–O)···HgCl(ads)···(V4+)(s) (7)
3.4. Mars-Maessen Mechanism
Initially, Granite et al. [28] proposed that the catalytic oxidation of Hgel could occur via a Mars-Maessen mechanism. More recent research widely uses the Mars-Maessen mechanism as a most likely pathway for Hgel oxidation in the presence of a metal oxide-based catalyst [31,32,33,34]. According to this mechanism, the Hgel oxidation takes place between the elemental mercury adsorbed on a metal oxide (MxOy) surface and the lattice oxygen, forming a binary mercury oxide. The oxidation of Hgel could be described by the following equations for the CoO/TiO2 system [33]:
Hg(g) + catalyst surface → Hg(ad)(s) (8)
Hg(ad)(s) + CoxOy(s) → HgO–CoxOy–1(s) (9)
HgO–CoxOy–1(s) + ½ O2(g) → HgO(ad)(s) + CoxOy(s) (10)
HgO(ad)(s) + 2 HCl(g) → HgCl2(g) + H2 (11)
Firstly, gaseous Hgel is physically adsorbed on the catalyst surface to form Hgel(ad). Then the Hgel(ad) reacts with the lattice oxygen from the catalyst to form a weakly adsorbed mercuric oxide. Some authors suggest that a part of the Hgel(ad) is directly oxidized by gaseous O2 to form HgO [14,34,35]. The physical adsorption of Hgel on the catalyst surface takes place even at low reaction temperatures, although Hgel(ad) conversion to HgO(ad) is accelerated by increasing the temperature [14]. A further step of the Mars-Maessen mechanism involves the re-oxidation of catalytic metal oxide by gaseous oxygen. In the last step of this mechanism, the HgO(ad) reacts with HCl or HBr to form the volatile mercury halogenides which are released from the catalyst surface.
The formation of mercury oxides and binary mercury oxides was supported by the surface analysis of spent catalysts [33,35]. Liu et al. [33] proposed that the Hgel oxidation product on Co/TiO2 in the absence of O2 is an Hg2O–CoOx oxide. According to their experimental observations this oxidation product reacts with gaseous HCl, forming Hgel and volatile HgCl2 which are released from the catalyst surface.
4. Noble Metal-Based Catalysts for Hgel Oxidation
Platinum group metals and gold are well-known oxidation catalysts in many different areas. For example, platinum group metals are widely used for the treatment of automobile exhaust emissions. Gold was considered to be less active in many oxidation applications in comparison with other noble metals. However, the topic of oxidation catalysis by gold is a fast growing field.
Noble metals catalysts are also promising for mercury oxidation applications due to their ability to adsorb Hgel on their surfaces and to form solid solutions in a process known as amalgamation. This property is well known for gold and has been exploited for decades in gold mining and analytical chemistry. The amalgamation type of adsorption can serve as a first step followed by oxidation and desorption of mercury as a volatile halide.
In the mercury-in-flue-gas oxidation field so far, gold has been the most widely applied noble metal, and will therefore be discussed in a separate chapter. The scarcity and high cost of noble metal-based catalysts limit their use. In Table 1 below, an overview of the investigations on mercury oxidation by noble metals catalysts is given.

Table 1.
Mercury oxidation over noble metal based catalysts.
| Catalyst type | Gas composition | T, °C | Space velocity, h−1 | Hgel oxidation, % | Reference | |||||||
| O2 | H2O | HCl | NO | NH3 | SO2 | Hgel | ||||||
| vol.% | vol.% | ppm | ppm | ppm | ppm | µg/Nm3 | ||||||
| Lab scale | Ru/TiO2●● | - | 4 | 2–12 | 30–300 | 30–260 | 500 | 50 | 150–350 | 79000 | 30–90 | [36] |
| Bench scale | Au/Al2O3 ° | - | - | 0–1000 | 6–18 | 138–160 | 8–10 ■ | ■■ 2.2 × 10−10 | [37] | |||
| Pd/Al2O3 ° | 0–5.25 | - | 0–100 | 500 | - | ■■ 1.6 × 10−10 | ||||||
| Pt/Al2O3 ° | - | - | ■■ 4.1 × 10−10 | |||||||||
| Au/Teflon ° | 6 | 0–8 | 50 | 600 | - | 2000 | 55 | 175–225 | 5–60 | [30] * | ||
| Ir/Al2O3 ° | 8 | 8 | - | <500 | - | <2000 | 12 | 138 | 7.5 ■ | 75 | [38] | |
| Au/TiO2/FF ° | 1200 | 9–65 | ||||||||||
| 4 | 10 | 50 | 100 | - | 1000 | 20–30 | 150 | [29] ● | ||||
| Pd/Al2O3/FF ° | 4800 | 4–84 | ||||||||||
| Pilot scale | Au/γ-Al2O3 ° | ~8 | 9–12 | 1–20 | - | - | 200–1200 | 10–31 | 139–149 | 3200–3600 *** | 40–99 | [39] ** |
| Pd/γ-Al2O3 ° | - | - | 41–87 | |||||||||
| Full scale | Au/γ-Al2O3 ° | 7–9 | 12 | 1.67 | - | - | 501 | 11–14 | ~150 | 21300 | 52–86 | [40] |
●● 0.2–2 wt.-% metal loading; ° 1 wt.-% metal loading; ■ L/min; ■■ reaction rate in the presence of HCl and O2 in (mol Hg2+) × (g catalyst)−1 × s−1; * 3 and 6 ppm Cl2; ● 10 ppm Cl2; FF: fabric filters; *** gas flow rates (m3/h); ** 6–10 ppmv HF, 0–0.6 ppmv Cl2, 0–0.04 ppmv HBr, 0.15 ppmv HI.
4.1. Activity of Platinum Group Based Catalysts for the Oxidation of Hgel
In the absence of halogen species and oxygen, noble metals adsorb elemental mercury even at temperatures as high as 400 °C. This was confirmed in a recent lab scale study conducted by Poulston et al. [41] in fuel gasification gases containing Hgel. Mercury adsorption capacity of platinum Pt and palladium Pd supported on Al2O3 were measured at temperatures between 204 and 388 °C in simulated fuel gases. Up to 14 wt.-% Hg was detected in the Pd-based material [41,42]. In the presence of halogens the Pt group based catalysts oxidizes mercury.
Platinum Pt [8,37], palladium Pd [29,37], ruthenium oxide RuO2 [36], and iridium Ir [38] have been tested on laboratory and pilot scale in order to determine their potential as mercury oxidation catalysts.
The tests involved the use of noble metal catalysts as powders, foil and coated on different supports, such as: alumina beads [37], alumina beads imbedded in fabric filters [29] and titania [36]. The role of the supports is to ensure a high dispersion of the noble metals, maximizing the contact area between flue gas components and the catalytic active centers responsible for the mercury oxidation reaction. In some investigation a marked structure and size effect of the support were observed as well.
In the investigations reported on the mercury oxidation by Pt group metals, the presence of hydrogen halides was needed. The oxygen in flue gas seems to play also a certain part.
Presto and Granite [37] investigated the mercury oxidation at 149 °C under simulated flue gas conditions in the presence of Pd and Pt (1 wt.-%) supported on alumina beads. The HCl and O2 contents were varied. An interesting result was the gradual loss of catalytic activity for the Pd and Pt based catalysts over time. This deactivation behavior was explained by the formation of Pt and Pd oxides during exposure to O2 [37]. Surface bound chlorine seemed to be necessary for the oxidation of Hgel. A marked decrease in mercury oxidation in gases with no HCl was observed. HCl higher than 50 ppm caused no further increase on the oxidation rate. Schofield [8] explained the oxidation of Hgel on Pt as a two-step process. In the first step the non-volatile HgO was formed. When HCl was present, the HgO was converted into the volatile HgCl2 and desorbed, thereby the reaction could proceed. The history of the catalysts investigated had a significant effect on their activity. It should also be noted that the mercury oxidation on Pd and Pt increased with temperature. Pt catalysts for diesel exhaust application are known to effectively oxidize SO2 to SO3 [43].
Results on this undesirable side effect reaction have not been reported so far for the Hgel oxidation catalysts. It is speculated that at the fairly low reaction temperature of 150 °C employed by Presto and Granite [37], this reaction might have not been fast enough. Pt based catalysts have also the potential to actively oxidize the NO in flue gases, as it is well known from the automobile exhaust field [44].
Hrdlicka et al. [29] found that at 150 °C and in the presence of HCl/Cl2 and oxygen the mercury oxidation rate was within the range of 50–80% on 1 wt.-% Pd/Al2O3 coated fabric filters. SO2 and NO had an influence on the mercury oxidation activity.
The published research on RuO2/TiO2 catalysts pointed out that this system might be promising for mercury oxidation in coal-fired flue gases [36]. The lab scale investigation was conducted between 150 and 350 °C with most of the flue gas components present. No appreciable decrease of the mercury oxidation activity over 10 hours was reported. The poisoning effect of SO2 was small. The conversion of SO2 to SO3 by the catalyst was imperceptible. The authors observed that the oxidation activity increased with the HCl content up to 5 ppm and leveled off thereafter. The catalyst was shown to generate some Cl2 from HCl according to the Deacon reaction. In the presence of SO2, no Cl2 could be detected in the gas phase, although Hg oxidation showed only a small decrease. The authors presumed that under these conditions the formation of atomic Cl species still took place and that the mercury reacted with these species [36]. Temperature increased the oxidation rate.
4.2. Activity of Gold Based Catalysts for the Oxidation of Hgel
Gold-based catalysts were found to be effective in a large number of catalytic reactions [45]. Recently, the potential use of gold-based catalysts in mercury pollution control applications has attracted much interest from academia as well as industry [29,30,37,40,46,47]. The studies have concentrated on the temperature range between 140 and 225 °C with, in most cases, 1 wt.-% gold on alumina and in one case with Au on a Teflon-coated quartz filter.
Due to the strong ability to adsorb Hg on its surface and to form an amalgam with it, gold is considered a very promising candidate for mercury oxidation. An interesting application of this Au-Hg amalgamation property is the MerCAP process [48], which involves Hgel adsorption from flue gas on fixed Au-coated structures followed by thermal regeneration of the gold sorbent and Hg recovery.
For the adsorbed mercury to be oxidized, a reactant is necessary. Chlorine atoms are identified as one likely species for this purpose. Gold readily adsorbs Cl2 molecules, thereby dissociating them into chlorine atoms [29,30]. The chlorine atoms react with mercury. The dissociative adsorption of HCl on Au is presumably weaker, since the dissociation energy of Cl2 is smaller and the bond length higher by comparison to HCl [49]. The importance of HCl adsorption is supported by the declining but continuing oxidation of Hgel when HCl from the gas stream is removed [37]. Presto and Granite [37] showed that increasing the HCl concentration above 50 ppm had no further impact on the reaction rate with Hgel. Zhao et al. [30] observed, for the elemental mercury oxidation across gold on Teflon-coated quartz filters, that in the presence of chlorine Cl2 (10 ppm) the oxidation of Hgel proceeded much faster than with HCl. Apparently HCl decreased the effect of Cl2.
The addition of other flue gas constituents (NO, SO2, H2O) did not seem to influence the elemental mercury oxidation in the presence of Cl2. Compared to the chlorine species, NO, SO2 and H2O seemed to interact to a far lesser degree with Au surfaces [29,30]. The Au/TiO2 coated fabric filters showed an increased mercury oxidation in the presence of HCl (50 ppm) and NO (100 ppm), suggesting that there might be a synergistic effect between HCl and NO in the presence of gold [29].
Removing the oxygen from an HCl containing simulated flue gas caused the Hgel oxidation on Au catalyst to decrease [37]. Mercury oxidation on these catalysts increases with temperature [30,37].
Pilot [39,46] and full scale [40,47] investigations were conducted with the main objective to demonstrate the effectiveness and deactivation properties of a commercial gold catalyst wash coated on γ-alumina honeycomb substrate in promoting Hgel oxidation in flue gases from coal combustion. The catalyst modules were located downstream of a particulate control device [47] and in the flue gas scrubber inlet duct [40]. The flue gas contained only about 2 ppm HCl and 500 pm SO2. The investigation was conducted over a 17 months period.
Even after 6 months of operation in the demonstration plant, the percentage of oxidation of elemental mercury across the catalyst was only 3 percentage points below what was measured immediately after the catalyst was placed in service. However, after 13 months of operation a substantial loss of activity was measured. It is believed that some of this activity loss was real, but that a substantial portion was “artificial” in that it resulted from build-up of fly ash in the catalyst, which blocked the otherwise active surface area of the catalyst. Gas flow to parts of the second and third layers was blocked by fly ash buildup in the first layer [40]. An uneven distribution of the gas flow and the small channel width of 3.2 mm of the honeycomb catalyst contributed to the blocking. In summary it can be said that lab-scale, pilot-scale and full-scale mercury oxidation investigations show that gold-based catalysts are not subjected to fast deactivation under flue gas conditions [37,40,46]. One measurement campaign with respect to the SO2 conversion did not result in an appreciable increase of the SO3 content over the Au catalysts layer. It has to be mentioned that data are scarce on the influence of Au catalysts on the SO2 to SO3 conversion in flue gases. Boreskov et al. [50] measured a much lower SO2 oxidation rate for Au catalysts in comparison to Pt. Therefore the assumption is that this side reaction might be small. The effect of bromine species like HBr has also not been reported yet. Neither has the conversion of NO to NO2 over gold catalysts been studied.
4.3. Summary
Because of their undesirable side reactions, Pt based catalysts do not appear promising for mercury oxidation in flue gases. The recent laboratory results on Ru/TiO2 catalysts for this application do not show appreciable SO2 oxidation but a high mercury oxidation rate, at temperatures around 300 °C and at HCl concentrations in the flue gas as low as 5 ppm. This avenue warrants further investigations.
Despite the plugging issues experienced, the recent demonstration plant results with gold-based catalyst in the low temperature range around 150 °C should encourage further efforts along this line. For this system, basic parameter studies on the effect of the different parameter importance in a flue gas environment and the gold distribution on the support are lacking.
5. Mercury Oxidation by Transition Metal Oxide Catalysts
The class of catalysts discussed in this chapter refers to the oxides of transition metals. A significant number of publications have discussed the activity of transition metal oxides-based catalysts for Hgel oxidation in the 80–500 °C temperature range, see Table 2. Their cost-effectiveness has made this group of catalysts attractive candidates for studies. The catalytic active oxides are deposited most commonly on alumina and titania. The role of the support is to not only stabilize and ensure a high metal dispersion degree but also in certain cases to participate in the Hgel oxidation reaction.
The activity for Hgel oxidation reaction of this catalysts group has so far been investigated mainly at laboratory scale under simulated flue gas conditions. For this purpose a number of parameters have been varied, such as:
- Loading and composition of the metal oxide material;
- Temperature and;
- HCl, Cl2, O2, H2O, NO and SO2 concentrations in the gaseous phase.
Table 2.
Mercury oxidation activity of different supported metals and metal oxides.
| Catalysts | Catalysts characteristics | Reaction temperature, | Mercury oxidation/removal, | Reference | |
| Synthesis method | Metal loading, wt.-% | °C | % | ||
| nano-CuO | commercial | 100 | 90–300 | 20–96 | [51] |
| nano-CuO | commercial | 100 | 150 | 75 | [52] |
| CuCl2/TiO2 | impregnation | 1.5–6 | 350 | 60–100 | [53] |
| CuCl2/TiO2–Al2O3 | wetness impregnation | 0.25–9 | 125–175 | 28–62 | [54] |
| CuCoO4/γ-Al2O3 | thermal decomposition | 1 *** | 100–450 | 10–92 | [55] |
| Co–oxide/TiO2 | sol-gel | 0.5–15 | 90–360 | 10->90 | [33] |
| nano-Fe2O3 | hydrothermal | 100 | 80–400 | <40 | [34] |
| Fe2O3/TiO2 | impregnation | 0.6–5 | 80 | 60–80 | [56] |
| MnOx/Al2O3 | wet impregnation | 1–8 | 100–500 | 45–90 | [14] |
| Mn/α-Al2O3 | wet impregnation | 1 | 100–250 | 30–95 | [57] |
| MnOx/TiO2 | wet impregnation | 10–20 | 175–200 | ~90 | [31] |
| MnOx–CeO2/TiO2 | impregnation | 0.18:0.82:1 ** | 120–400 | 40–>90 | [26] |
| CeO2/TiO2 | impregnation | 0.5–2 * | 120–400 | 40–95 | [58] |
| CeO2/γ-Al2O3 | thermal decomposition | 3–15 | 150–450 | 33–90 | [35] |
| V2O5/TiO2 | sol-gel | 1–10 | 100–500 | 69–100 | [59] |
| SiO2–TiO2 | sol-gel | 12 ● | 135 | 10–90 | [60] |
| SiO2–TiO2–V2O5 | sol-gel | 6–18 ■; 5 ■■ | 135–400 | 40–100 | [61] |
* CeO2:TiO2 mass ratio; ** MnO2:CeO2:TiO2 mass ratio; *** Co:Cu atomic ratio; ● % of TiO2; ■ TiO2; ■■ V2O5.
Ghorishi et al. [62] investigated the influence of different fly ash components (Al2O3, SiO2, Fe2O3, CuO and CaO) on the extent of mercury oxidation. They observed that copper oxide (CuO) and iron (III) oxide (Fe2O3) present in the fly ash composition exhibited good catalytic activity towards the Hgel oxidation reaction. These findings were followed up by Kamata, et al. [63] with an investigation on mercury oxidation by HCl over TiO2 supported metal oxide catalysts (with 1 wt.-% MOx/TiO2, with M = V, Cr, Mn, Fe, Ni, Cu and Mo). TiO2 was essentially inactive. At 350 °C high Hgel oxidation was observed for MoO3, V2O3 and Cr2O3 and the lowest for NiO. In the presence of a NH3/NO molar ratio of 0.33, the unreacted NH3 considerably depressed the Hg oxidation activity of all catalysts. A correlation was found between NO reduction and Hg oxidation activity.
5.1. Copper/Cobalt Based Catalysts
Copper oxide particles and the titania-supported-CuO are potential Hgel oxidation catalysts in the presence of HCl [51,52]. Kamata et al. [52] compared the oxidation of gaseous Hgel at 150 °C in the presence of HCl over different metal oxides. Among the catalysts tested, surface coated CuO nano-particles showed the highest activity. Conversion activity increased with HCl concentration. Formation of Cu2Cl(OH)2 was observed. Yamaguchi et al. [51] investigated further details of the Hgel oxidation activity of CuO nano-particles under simulated flue gas conditions. The CuO nano-particles were dispersed on a quartz filter. An increase in Hgel oxidation (up to 96%) was observed as the reaction temperature decreased from 300 to 90 °C. A decrease in activity was observed when CuO nano-particle size increased from 50 nm to 620 nm. Nano-particle size increased at temperatures above 150 °C because of sintering effects.
The oxidation was HCl dependent and decreased considerably at concentrations of 5 mg/m3 HCl. When employing 1 wt.-% CuO supported on heavy oil-fire fly ash (HOFA) a maximum of 98% Hg oxidation was achieved in the presence of 20 ppm HCl and 350 °C [3].
At temperatures of 300 °C, copper chloride (CuCl2)-impregnated titania exhibited high oxidation activity even in the absence of HCl [53]. The efficiency of CuCl2/TiO2 catalysts increased considerably when the Cu loading was higher than 3 wt.-%. In this case, it was assumed that oxidation of weakly bound Hgel might occur as a result of Cl radicals released following thermal decomposition of CuCl2 on the catalyst surface. The CuCl2 was restored by gas phase HCl. A similar system, copper chloride (CuCl2) on titania/alumina or alumina, was investigated by Miksche et al. [54]. The study focused on a low temperature regime of 125 to 175 °C. The TiO2–Al2O3 substrate provided a more active catalyst with respect to Al2O3.The HCl in the flue gas increased the Hg oxidation whereas SO2 had an adverse effect. In the studied temperature range with 2000 ppm SO2 and 40 ppm HCl in the flue gas, the CuCl2 catalyst provided considerably higher Hg oxidation rates than a conventional SCR-DeNOx catalyst with 1 wt.-% V2O5. However, no long-term stability data were provided.
Mei et al. [55] investigated the Hgel oxidation activity of Al2O3 loaded with 20 wt.-% CuCoO4 and CuCoO4 + NH4Cl or NH4Br. The catalyst was calcined at 400 °C, by which point most of the ammonium salts evaporated; however a small amount of nitrogen doping in the CuCoO4 remained. At 350 °C and in SO2-containing simulated flue gas, mercury oxidations between 72 and 92% were achieved. The NH4Cl and NH4Br doped catalysts showed much higher Hgel oxidation activity than CuCoO4/Al2O3, especially at temperatures between 100 and 350 °C. The SO2 poisoning resistance of CuCoO4 was higher than those of the pure Cu and Co oxide. However, the SO2 tests were extended only over a 2 h period [64].
Cobalt oxide-based catalysts showed Hgel oxidation activity as well [33]. The activity of the Co–oxide/TiO2 catalysts was affected by the Co loading and the oxidation temperature. Hgel oxidation efficiencies higher that 80% were achieved when the Co content was in the 2.5 and 7.5 wt.-% range and at temperatures between 120–330 °C. Liu et al. [33] attributed the good catalytic performance of Co-based catalysts to well-dispersed Co3O4 species in the catalysts. In the absence of HCl, mercury accumulated on the surface of the catalyst. A modified Mars-Maessen mechanism was considered; Hgel was adsorbed on the surface and then reacted with the lattice oxygen to form Hg–OCox bonds. The HCl then reacted with surface bound HgO, releasing volatile HgCl2 and H2O. It should be noted that between 240 and 360 °C, NO was also oxidized by the catalyst to NO2. The influence of SO2 was not investigated. A thermal stability test over a 72 h period showed only a small decrease of activity.
5.2. Iron/Manganese Based Catalysts
Kong et al. [34] studied Hgel oxidation activity of rod-shaped nano-Fe2O3 in a fixed bed reactor arrangement. The Hgel oxidation activity increased considerably with decreasing particle size. Fe2O3 powders with typical size of around 7 μm showed no Hg oxidation activity where the nano-sized samples oxidized 30% of the Hgel. Hgel oxidation increased significantly at temperatures between 75 and 300 °C, and decreased at higher temperatures. The decrease of Hgel-oxidation activity at 400 °C was attributed to the sintering of the Fe2O3 nano-particles. Water vapor as well as oxygen in the flue gases influenced, to a certain extent, the Hgel oxidation by the Fe2O3 nano-particles.
Mn–Fe spinels [32,65] and nanosized cation-deficient Fe–Ti spinels [66,67] were presented as effective materials for Hgel capture in the absence of HCl. The Hgel sorption capacity of Mn–Fe spinels was promoted by higher Mn contents. The sorption capacity was reduced in the presence of SO2. Sulfates were formed on the surface in this case. The formation of gas phase SO3 by these materials has not been assessed so far. 5 ppm HCl in the simulated gas phase reduced the adsorption and caused the release of oxidized mercury in effluent gas. Results on the Hgel oxidation in the presence of higher HCl concentrations in the gaseous phase are not available yet. It appears that the Hgel, which is a Lewis base, is firstly physically adsorbed by Lewis acid sites on the Fe–Ti spinel and subsequently oxidized by Fe3+ cations present on the spinel surface [66]. When employing Mn–Fe spinels, Hgel was captured and oxidized by Mn4+ cations from the surface [32,65]. The assumption, based on the available publication, is that the Mn–Fe and Fe–Ti spinels are more promising as Hg capture agents at temperatures around 150 °C, and not as mercury oxidation catalysts in flue gases resulting from combustion processes.
In another investigation conducted at temperatures between 175 and 200 °C, a 10 wt.-% MnO2 on TiO2 strongly chemisorbed Hgel in an HCl free gas [31]. SO2 (200 ppm) had a negative effect on Hgel capture. Mercury was also captured in the absence of gas phase oxygen. This observation suggests a Mars-Maessen type of adsorption.
Several other studies suggested that manganese oxide-based catalysts may display good Hgel oxidation efficiency, at least under simulated flue gas conditions [14,57]. Qiao et al. [14] investigated the MnOx/Al2O3 catalyst activity in the wide temperature range of 100 to 500 °C. In the absence of HCl from the flue gas, the MnOx effectively adsorbed the gaseous Hgel with an optimum at 230 °C. Adsorption was suppressed and high mercury oxidation efficiencies achieved in HCl or Cl2-containing flue gases. It appears that Cl2 (2 ppm) is as effective as HCl (20 ppm) in promoting the oxidation of Hgel. HCl and Cl2 obviously desorb oxidized mercury, thereby regenerating the surface of the catalysts. The flue gas components NO, H2O and CO2 had no significant impact on oxidation activity. SO2 exhibited a relatively low inhibitory effect, especially in the presence of Cl2. In a subsequent paper [57], mercury adsorption and oxidation in the presence of Mn oxide (1 wt.-% Mn), supported on α-Al2O3, was studied within the 100–250 °C range. According to the XRD results, the Mn species on the support consisted of a mixture of MnO2 and Mn2O3. It appears that the catalyst was less active at temperatures below 250 °C. Doping the Mn–oxide/α-Al2O3 catalysts with molybdenum (Mo) resulted in high Hgel oxidation in gases with 5 ppm HCl, even in the presence of SO2. Most likely the Mo doping improved MnOx particle dispersion, thereby increasing the Hgel oxidative potential at low temperatures [57]. The presence of Mo–Mn complexes was also supposed to increase the SO2 tolerance. It is speculated that the Deacon reaction, with its reactive chlorine generating capabilities, might be important for the overall reaction. The mercury oxidation rate was pseudo first order in the mercury concentration and dependent to the power 0.36 with respect to the HCl concentration in the flue gas.
5.3. Cerium Based Catalysts
Hg-oxidation activity of cerium oxide-based catalysts has also been reported in the literature [35,58]. The significant Hgel to Hgox conversion activity of CeO2-based catalysts may be due to the high oxygen storage capacity of CeO2. Wen et al. [35] studied the chemisorption of Hgel by CeO2/γ-Al2O3 in the absence of HCl from the gaseous phase. The sorption capacity increased from 150 to 350 °C and then decreased at higher temperatures. The presence of SO2 and H2O in the gas inhibited the adsorption of mercury. At higher temperatures, Ce(SO4)2 is formed on the surface, preventing the contact between gaseous Hgel and CeO2 active sites. Li et al. [58] investigated the catalytic properties of the CeO2/TiO2 system in HCl, NO, and SO2 containing simulated flue gases. A fairly high CeO2/TiO2 mass ratio of 1.5 considerably improved oxidation efficiency. Oxidation activity increased with rising temperatures from 120 to 250 °C, and then decreased at higher temperatures up to 400 °C. At this temperature almost no oxidation activity remained. HCl in the gas phase considerably increased the activity in the presence of oxygen. NO and SO2 in the flue gas were found to have positive effects on Hgel oxidation [58]. In a subsequent paper, Li et al. [26] investigated the Hgel oxidation activity of TiO2 supported Mn–Ce mixed oxides under simulated low-rank coal combustion flue gas. Oxidation activity increased with temperature from 120 to 250 °C, and then dramatically decreased when the temperature reached 300 °C. Significant Hg desorption was observed in the 250–300 °C range. NH3 reduced the Hg oxidation activity by competing with Hgel for the active sites or/and by consuming the surface oxygen which was responsible for Hg oxidation. Once the NH3 was cut off, Hg oxidation activity completely recovered in the presence of O2.
It was noted that the Ce3+ containing catalyst promoted the oxidation of NO to NO2 and also of SO2 to SO3. These side reactions are of importance in combustion systems and should be considered during further development of this catalyst system. Hgel oxidation performance was explained with a Langmuir-Hinshelwood mechanism. In this mechanism, an active surface species reacts with adsorbed mercury. HCl reacts to a certain degree with the active component CeO2. However, mercury oxidation proceeded also in the absence of HCl from the gaseous phase [58].
5.4. Various Metal Based Catalysts
Several other metal oxide and mixed metal oxide-based catalysts were developed as potential Hg capture and oxidation materials [22,59,60,61]. SiO2–TiO2 nano-composite was found to oxidize and capture Hgel under UV light at 135 °C. HCl and SO2 positively affected Hg oxidation and capture activity [60]. A more recent study by Li et al. [61] investigated the Hg oxidation and capture potential of a SiO2–TiO2–V2O5 catalyst under simulated low-rank coal combustion flue gas. The TiO2 content was varied between 6–18 wt.-%, while the V2O5 was kept at 5 wt.-%. Hg oxidation activity decreased as the temperature increased from 135 to 300 °C. However, an increase in Hg oxidation under SCR conditions was observed as the TiO2 content increased up to 18 wt.-%. In the presence of O2, the flue gas components HCl, SO2 and NO exhibited a positive effect on Hg oxidation.
In an experiment conducted by Lee and Bae [59], Hgel was completely adsorbed and oxidized by a high surface area 10 wt.-% V2O5/TiO2 aerogel at 100–200 °C temperature range. With an increase in temperature to 300 °C, mercury started to desorb from the aerogel surface. The amount of Hgel desorbed was higher as temperatures reached 500 °C.
5.5. Summary
The halogens content of flue gases strongly influences Hg oxidation activity in the transition metal systems which otherwise tend to chemisorb mercury at temperatures below 200 °C. SO2 reacts with transition metal active sites forming metal sulfates, thereby potentially deactivating the catalyst. In this way SO2/SO3 conversion is promoted by some transition metal oxides. Not much is known on the effect of Hg-oxidation-active transition metal oxides on the NO/NO2 conversion. Both conversion reactions are undesirable under combustion flue gas conditions. From the research conducted so far, it is obvious that pure transition metal oxides will not be suitable mercury oxidation catalysts for a flue gas environment. These catalysts must be modified in order to be suitable for flue gas environment applications.
6. Mercury Oxidation on SCR Catalysts
SCR catalysts are employed for the reduction of nitrogen oxides (NOx) in the presence of ammonia (NH3) at temperatures higher than 300 °C if SO2 is present. The catalyst consists of a porous titanium dioxide monolithic substrate on which vanadium pentoxide (V2O5), tungsten trioxide (WO3) or molybdenum trioxide (MoO3) is dispersed [68]. Besides NOx reduction, SCR catalysts are also active in the oxidation of volatile organic compounds [69,70]. In an additional side reaction, SCR catalysts also convert gaseous Hgel to Hgox, particularly in the presence of HCl [21,23,25,71]. Currently, SCR catalysts are being developed as multi-pollutant control devices.
In this section, several aspects regarding Hgel oxidation on SCR catalysts will be evaluated. The influence of flue gas components (halogens/hydrogen halides, NH3, SO2/SO3, NOx), composition of the SCR catalysts and temperature will be considered first. The role of flue gas species on deactivation of SCR catalysts for mercury oxidation reaction will be covered as well. A brief discussion on mechanistic pathways and modeling of Hgel oxidation over SCR catalysts will follow. Modifications of SCR catalysts for achieving high Hgel oxidation performance are of interest as well.
In order to determine their efficiency in oxidizing Hgel from flue gases, the SCR catalysts were tested at in the laboratory and in pilot [71,72,73,74] and full scale conditions(e.g., [75]). Some of these Hgel oxidation results are presented in Table 3. Operating conditions and flue gas composition are also given in the table.
6.1. Mercury Adsorption on SCR-Catalysts
Hg adsorption on catalyst surfaces has been mentioned in a number of studies [25,72,73]. Hgel adsorption on the SCR catalyst surface was only observed in the absence of HCl [76,77]. When HCl or NH3 was added to the simulated flue gas, a rapid mercury desorption was observed [23,76,77]. Eswaran and Stenger [76] also reported strong Hg adsorption in the presence of H2SO4. Hgel adsorption slightly increased when SO2 was present [73]. Straube et al. [73] investigated the effect of different parameters on Hg adsorption on SCR catalysts under tail-end conditions (low HCl and SO2 concentration). They suggested that mercury adsorption involved chemisorption and the formation of Hg–O bonding on the SCR catalyst surface. Another interesting finding was the observed link between the V2O5 content and the Hg adsorption extent on the SCR catalysts. In this case, Hg adsorption increased with increasing V2O5 content from 2.5 to 4.5 wt% [73].
Table 3.
Elemental mercury oxidation on SCR-DeNOx catalysts.
| Gas composition | T, °C | Space velocity,h −1 | Hgel oxidation, % | Reference | ||||||||
| O2 vol.% | H2 Ovol.% | HCl ppm | NO ppm | NH3 ppm | SO2 ppm | SO3 ppm | Hgel µg/Nm3 | |||||
| Lab scale (simulated flue gases) | ||||||||||||
| 6 | - | 50 | 400 | 400 | - | - | 36–39 | 350 | 4000 | 3–91 | [25] | |
| 6 | 8 | 0–35 | 400 | 360 | 1000 | - | 10–20 | 371 | 4000 | 12–70 | [76] ■ | |
| 5 | 1.8 | 0–20 | 150 | - | 500 | - | 30 | 350 | 72 ● | 70–90 | [3] | |
| 3 | - | 10–50 | 500 | 500 | - | - | 50 | 250–350 | 120 ● | 85–98 | [78] | |
| 3 | 8 | 5–35 | 400 | 360 | - | - | ~20 | 390 | 3600 | 40–86 | [79] ■,** | |
| Bench scale (simulated flue gases) | ||||||||||||
| - | 15 | 0.3–3 | 400 | 300 | 70 | - | 160 | 260–320 | 170 ● | 50–90 | [73] | |
| 6 | 8 | 0–50 | 600 | 550 | 0–2000 | 0–50 | 13 | 343 | 20–71 | [72] | ||
| 3.5 | 5.3 | 0–204 | 350 | 315 | 280–2891 | - | 19 | 350 | 2609 | 0–>90 | [77] | |
| 7.1 | 6.8 | 0–20 | 200 | 180 | 500 | - | 20–25 | 350–400 | 20004000 | 30–88 | [80] | |
| Pilot scale (flue gases) | ||||||||||||
| 2.7–4.4 | - | 246 | 960 | 765 | 222–2921 | - | 5–10 | 300–400 | 2943 | 9–20 | [74] | |
| 3 | - | 500 | 250 | 275 | 2000 | 50 | 120 | 300–350 | 1800 | <80 | [71] * | |
* 15 ppm HBr and 25 ppm Cl2; ■ 10 ppm H2SO4; ** 2 ppm HI and 2 ppm HBr; ● gas volume flow (L/h).
6.2. Influence of Flue Gas Constituents
Laboratory-scale studies proved that the activity of SCR catalysts for Hgel oxidation under simulated flue gas conditions is strongly linked to the halide species present and their concentrations [3,21,23,53,71,78,80]. Since HCl is the dominant halide species in flue gases, Hgel oxidation is considered to proceed according to the following overall equation:
Hg(g) + 2 HCl(g) + 0.5 O2° HgCl2(g) + H2O(g) (12)
He et al. [23] reported Hgel adsorption and oxidation over an HCl pretreated SCR catalyst. When a Hgel/N2 gas mixture was passed over an untreated SCR catalyst, no obvious Hgel adsorption or oxidation was observed. The authors suggested that in a pure N2 environment at 300 °C, the physical adsorption of Hgel on catalyst surface was weak. Adsorption and a certain degree of oxidation were recorded when employing the HCl pretreated catalyst, suggesting that adsorbed HCl reacted with adsorbed Hgel to form Hgox. Evidence of HCl adsorption on SCR catalyst surface was obtained by employing XPS [23,25] and FT-IR [23,24] surface analysis methods. HCl might also react with the active component V2O5 of the SCR catalyst. Several oxychloride complexes such as VOCl2, V2O3(OH)2Cl2, VO2Cl2 [23,24] might be formed on the SCR-DeNOx catalyst surface.
Enhancement of Hgel oxidation activity was observed as HCl concentrations increased from 1 to 50 ppm [72]. An increase in Hg oxidation with increasing HCl concentrations was also mentioned by Kamata et al. [21]. Actually, many reports discuss the HCl influence on SCR catalyst efficiency in oxidizing Hgel. For this aim, HCl concentrations were varied within the 0–500 ppm range (see Table 3).
Yang and Pan [81] investigated Hgel oxidation on SCR full-scale units as a function of coal-chloride content. When coal-chloride content fell below 100 ppm (below 10 ppmv HCl in flue gas), the Hgel oxidation was lower than 10%. For coals containing over 800 ppm chloride (approximately 80 ppmv HCl in flue gas), the Hgel oxidation leveled off at 60–80%. The pilot scale study by Lee et al. [74] also suggests that Hgel oxidation across SCR units is coal-type/composition dependent.
Eswaran and Stenger [79] employed honeycomb and plate-like SCR catalysts to investigate the effect of HCl, HBr and HI on Hgel oxidation in a lab-scale reactor. Compared to HCl, HBr and HI added in small amounts (2 ppm) had a stronger effect on Hg oxidation (over 85%). The authors noted that in the presence of HBr, a large amount of Hgox was retained on the catalyst surface, while HI caused the previously retained mercury to desorb from the catalyst surface.
Pilot-scale results obtained by Cao et al. [82] revealed that, in the presence of different commercial SCR catalysts and under PRB (power river basin coal with low sulfur and chlorine contents) coal-derived flue gases, Hgel oxidation was enhanced by the addition of hydrogen halides in the following order: HBr, HI, and HCl or HF. However, it must be considered that the measurement of Hgel and Hgox by the authors might have been hampered by the presence of HBr and HI in the flue gas.
A large number of studies mentioned the negative effect of NH3 addition on Hgel oxidation by SCR catalysts in the presence of HCl and NOx [21,25,78,83]. Hong et al. [78] observed that increasing the NH3/NO ratio led to a decrease in Hgel oxidation activity of SCR catalysts at 350 °C. Eswaran and Stenger [76] explained the decrease in Hgel oxidation by the fact that NH3 caused Hgel to desorb from the SCR catalyst surface. Eom et al. [25] suggests that the decrease in Hgel oxidation in the presence of NH3 is caused by the adsorption of both Hgel and NH3 on the same active sites, and the reaction rate of NH3 is much faster than the reaction rate of Hgel.
SO2 and SO3 in the flue gases are known to affect Hgel oxidation by SCR catalysts [71,72,73,76]. The results obtained by Zhuang et al. [72] indicated that SO2 and SO3 had a mitigating effect on Hgel oxidation by SCR catalysts in the presence of HCl. This behavior was attributed to the competitive adsorption of SO2, SO3 and HCl on the SCR catalyst surface active sites [72]. Also, Cao et al. [71] observed that Hgel oxidation decreased once the SO2 concentration in the flue gas was increased. On the other hand, they observed that Hgel oxidation increased with increasing the SO3 content to a maximum of 50 ppm [71]. The addition of H2SO4 to a simulated flue gas increased the Hg oxidation activity of a commercial SCR catalyst, especially in the presence of HCl [76]. A strong adsorption of Hgox on the catalyst was determined to occur in the presence of HCl and SO3.
6.3. Influence of Catalyst Composition, Temperature and Space Velocity
The vanadium content of SCR catalysts affects Hg oxidation capacity as well. Higher vanadium content leads to a higher oxidation activity. One study reported Hgel oxidation of 90% when vanadium content was 1.1–1.2 wt.-% and less than 40% when the content was 0.5 wt.-% [3]. Kamata et al. [24] observed an increase in Hgel oxidation almost linearly with VOx loadings up to 10 wt.-%.
Vanadia-based SCR catalysts were tested at temperatures above 300 °C, temperatures which are consistent with the SCR conditions in coal fired power plants. However, it was observed that high temperatures could limit the extent of Hgel oxidation [73,84], most likely due to the desorption of mercury from the catalyst surface [59]. In a pilot-scale study, Sibley et al. [84] observed that, in the presence of hydrochloric acid (100 ppmv), a decline of mercury oxidation occurred with increasing temperature. Other studies described a similar Hg oxidation activity loss with increasing flue gas temperature and space velocity [75,80,83]. A bench scale study by Lee et al. [80] on a honeycomb catalyst under simulated PRB coal combustion flue gas reported significant Hgel oxidation activity loss (from 83 to 30%) at 400 °C and 4000 h−1 space velocity compared to 88% Hg oxidation at 350 °C and 2000 h−1 space velocity. On the other hand, in the presence of 10 ppm H2SO4, 15 ppm HCl and 1000 ppm SO2 a higher Hgel oxidation was observed once the temperature increased from 340 to 370 °C [76].
6.4. Loss of Hgel Oxidation Activity
Among other factors, long term Hgel oxidation activity of SCR catalysts is dependent on catalyst age. Kamata et al. [21] conducted a series of experiments aiming to clarify the aging process of SCR catalysts. Samples of catalysts which had been in service for different time periods as well fresh catalysts were employed. They observed a loss of Hgel oxidation activity as the operation time increased. This behavior was more pronounced as NH3 concentration increased. Also, the laboratory studies of Eswaran and Stenger [79] showed that Hg oxidation in the presence of HCl was affected by catalyst age, suggesting that catalyst activity diminishes with catalyst history (age and flue gas conditions encountered).
A major problem of SCR catalyst operations under high dust configuration is the loss of catalytic activity caused by fly ash clogging/fouling and/or chemical poisoning. At high-dust SCR reactor operating temperatures (280–400 °C) the concentration of volatile metals and metalloids in flue gas is higher. The most common catalyst poisons include arsenic (As), selenium (Se), potassium (K), sodium (Na), calcium (Ca), phosphorus (P) and sulfur trioxide (SO3). Crocker et al. [85] studied the influence of fly ash components (Ca, Na) and gas phase species (NH3, SO2 and P) on SCR catalyst deactivation by the formation of sulfates and phosphates. Thermogravimetric analysis (TGA) results showed that higher temperatures led to higher sulfation rates. NH3 and P from flue gas enhanced sulfate and phosphate formation on SCR catalysts. Wan et al. [86] investigated the influence of alkali (earth) metal doping on Hgel oxidation by a V2O5-WO3/TiO2 catalyst. Over 90% Hgel oxidation was achieved when using fresh catalysts in the 200–400 °C range. Alkali doping reduced the Hgel oxidation activity down to 70%. H2O vapor had a strong inhibitory effect on Hgel oxidation by alkali-doped catalysts. The deactivation potential of alkali (earth) metals was associated with their basicity value and ordered as follows: K, Na ~ Ca, Mg [86,87].
The DeNOx activity of SCR catalyst decays as gas phase arsenic oxide As2O3 reacts with V2O5 active sites [88,89,90]. A loss in Hg oxidation activity is most likely to occur in this case as well, since As poisoning reduces the number of V2O5 active sites. MoO3-containing catalysts are more resistant to poisoning by As2O3. As2O3 reacts preferentially with MoO3, thus mitigating the deactivation rate of V2O5 active sites [88].
6.5. Mechanism
One of the aims of recent studies is to elucidate the mechanism through which Hgel is oxidized on the SCR catalyst surface. The mercury oxidation mechanism has to consider the critical promotional impact of HCl and the inhibitory effect of ammonia (or the DeNOx reaction). Because of the impact of ammonia, mercury oxidation takes place mostly at the outlet of the SCR reactor after NH3 is consumed (see Figure 2).
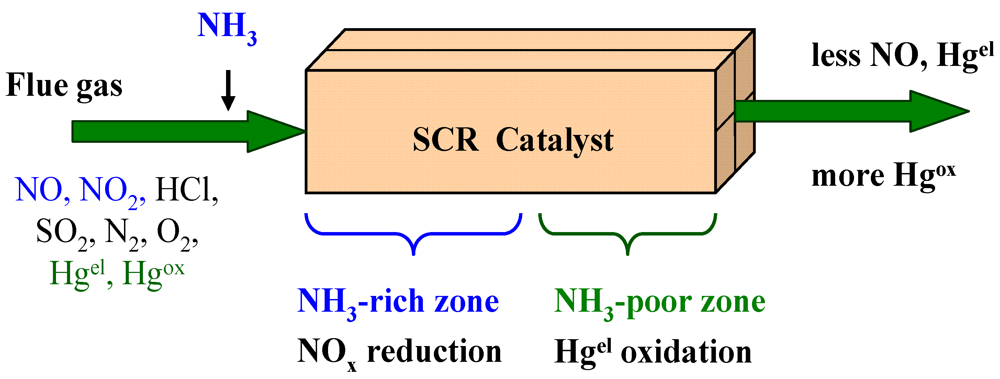
Figure 2.
Schematicof Nitrogen Oxides Reduction and Mercury Oxidation over a SCR Catalyst (adapted from [16]).
An early paper by Niksa and Fujiwara [20] proposed a model that is based on an Hg-oxidation mechanism in which NH3 and HCl species compete for the V2O5-active sites from the catalyst surface. The gaseous Hgel reacts with adsorbed HCl, either from the gas-phase or as a weakly adsorbed species in an Eley-Rideal type of mechanism. This mechanism does not seem to be the most likely in the light of more recent findings. Senior [91] proposed a model based on an Eley-Rideal mechanism in which the adsorbed Hgel reacts with gaseous HCl. According to the Senior model, Hgel competes with NH3 for V2O5 active sites.
The more recently proposed Hg-oxidation mechanisms are based on the surface analysis conducted on SCR catalysts. Evidence of HCl and Hgel adsorption onto the SCR catalyst surface active sites were recently given [23,24,72,73]. He et al. [23] proposed that Hgel oxidation over SCR catalysts occurs via a Langmuir-Hinshelwood mechanism. A similar mercury oxidation mechanism was proposed by Eom et al. [25]. According to the Langmuir-Hinshelwood mechanism, both HCl and Hgel species are adsorbed onto the SCR catalysts active sites, followed by the formation of HgCl2 and its desorption from the catalyst surface.
Eom et al. [25] considered that Hgel oxidation proceeds with the reaction of gaseous Hg with vanadium oxychloride complexes from the catalyst surface, therefore a first layer of Hg(ads) is formed. Once the first layer is formed, the rest of the Hg from flue gas is adsorbed and reacts with chloride complexes from the catalyst surface to form multi-layers of HgCl2.
A recent theoretical study employed a DFT (density functional theory) method to investigate the reactivity of V2O5 surface towards Hgel, HCl, HgCl and HgCl2 [92]. The calculated adsorption energies indicated that adsorption of Hgel on the V2O5 surface was stronger than that of HCl, therefore it is expected that the Hgel(ads) reacts with chlorine species to form an HgCl intermediate. HgCl, strongly bound on the surface, further reacts with chlorine species to form HgCl2(ads) which readily desorbs from the V2O5 surface.
The inhibition of Hg oxidation may be explained by the competitive adsorption of HCl and NH3 [25]. In this case, NH3 adsorption may dominate HCl adsorption on the active sites [21].
Some recent studies have reported reduction of Hgox in the presence of NH3 [93,94]. The experimental results obtained by Madsen et al. [93] suggested that at temperatures higher than 325 °C a part of the Hgox was reduced back to Hgel by NH3. Also, it was observed that VOC removal on SCR catalysts induced some Hgox reduction [94].
HgCl2 + NH3 + 0.25 O2° Hgel + 2 HCl + 0.5 N2 + 0.5 H2O (13)
6.6. Optimization of Hg Oxidation Activity
Improving the mercury oxidation activity of SCR-DeNOx catalysts is of interest for researchers in academia and industry. One of the ways for enhancing Hg-oxidation activity is by applying/impregnating small quantities of metal oxides onto the SCR-DeNOx catalyst surface. Published studies reported results regarding Hg-oxidation activity of RuO2/SCR [36], CuO/SCR, NiO/SCR, ZnO/SCR [95].
It appears that the RuO2-modified SCR catalysts exhibit good Hg-oxidation activity even in the presence of NH3 at 350 °C. Hg-oxidation activity increased up to 90% with increased Ru loading up to 2 wt.-%, and at a 2 to 12 ppm HCl content of the simulated flue gas [36]. Zeng et al. [95] investigated the Hg-oxidation activity of a SCR catalyst impregnated with different metal oxides (Cr2O3, ZnO, CuO, NiO, MnO). Under simulated flue gas conditions (4 % by volume O2, 7 % by volume H2O, 2000 mg/m3 SO2, 100 mg/m3 HCl, NH3/NO = 400 ppm and 390 °C), the Hg-oxidation activity of the non-impregnated reference SCR catalyst was lower than 20%. Under the same conditions, the oxidation efficiency of metal oxide-impregnated SCR catalysts increased up to 45%. Experimental results showed that the CuO impregnated SCR catalyst exhibited the highest Hg-oxidation activity [95]. However, the investigation also showed that the activity improvement by metal oxides might not be stable in SO2-containing flue gases. SO2 present in the flue gas might react with the metal oxide active sites from the SCR surface forming metal sulfates, thereby causing a loss in mercury oxidation activity.
6.7. Commercial Development
The Babcock-Hitachi Company is currently offering a SCR catalyst with improved Hgel oxidation efficiency and a low SO2/SO3 conversion rate (below 0.5%) under typical DeNOx operating conditions [96]. Kai and Kato [97] developed a plate-like honeycomb SCR catalyst in which a molybdenum-vanadium complex oxide MoV2O8 is the active compound for Hgel oxidation reaction. The Mo–V complex oxide coated on various inorganic porous carriers exhibits a relatively high Hgel oxidation activity (above 72%), and small SO2/SO3 conversion rate below 1%. The catalyst also exhibits high NOx removal efficiency of approximately 98%.
In their patent application, Nochi et al. [98] provided a method for producing a novel modified SCR-type Hg oxidation catalyst. The catalyst consists of V2O5 and MoO3 as active compounds supported on a TiO2 carrier. In catalyst formulation, one of the following elements is added: W, Cu, Co, Ni, Zn. More than 82% Hg oxidation can be achieved under typical high dust DeNOx plant conditions.
Several patents and patent applications discuss the improvement of vanadium‑based catalysts for high Hg oxidation activity. For example, the addition of iridium Ir [99], manganese compound [100], silver halide [101] to the vanadium based-catalyst composition is expected to improve their reactivity towards Hg oxidation reaction.
6.8. Summary
Mercury oxidation activity of SCR catalysts is nowadays regarded as an important co-benefit of SCR systems in coal fired power plants.
The activity of SCR catalysts is correlated with boiler operating conditions and flue gas chemistry. Based on pilot and full scale data, several side approaches to maximize Hgel oxidations have been suggested [83]. Increasing halide/(chlorine) content of the flue gases is one of them. When low chlorine coals are burned, the resulting HCl content in the flue gas is low; therefore low Hgel oxidations are achieved. By blending low chlorine coal with high chlorine coal or by injecting different halogen containing additives (e.g., NH4Cl, CaCl2, Br2, etc) into the flue gas [102,103,104] or by adding bromide salts to the coal [15], a higher Hgel oxidation could be obtained.
The conversion of SO2 to SO3 over SCR catalysts is a major problem. Therefore, the vanadium content of a SCR catalyst has to be limited in order to mitigate the SO2/SO3 reaction rate. However, the Hgel oxidation rate will also decrease if the vanadium content decreases [3].
Operating the SCR catalyst in the tail-end configuration, where the SO2 concentrations of the flue gas are low, would allow the use of higher vanadium contents and would benefit mercury oxidation. However, the HCl contents are in most cases low, thereby severely inhibiting the Hg oxidation rate. Operating the SCR catalysts at lower temperatures could improve oxidation activity, since it has been reported that higher temperatures inhibit Hgel oxidation [83,84].
Even though the amount of research regarding Hg oxidation on SCR catalysts has considerably increased over the past five years, further investigations are necessary for a better understanding of the role of flue gas components and the mechanism of Hgel oxidation. Also, the development and industrial implementation of SCR catalysts with high Hg oxidation activity are of great importance and intensively investigated.
7. Novel Catalytic Methods for Mercury Oxidation in Flue Gases
Besides the development of conventional catalysts for Hgel oxidation in flue gases, researchers are also investigating and implementing novel methods to achieve the same goal. Over recent years, various processes, such as photo-catalytic oxidation [105,106,107] and membrane delivery with catalytic oxidation [108], have been presented as possible alternatives to traditional catalytic oxidation methods.
Among the recently developed methods, the photo-catalytic oxidation of Hgel by UV (λ = 320–400 nm)-irradiated TiO2 surfaces has received a great deal of attention, since it can be performed even at room temperature. The aim of the experimental research was not only to study the photo-catalytic oxidation of Hgel but also to achieve new modified TiO2 structures [22,60,105,107] which might exhibit greater effectiveness in oxidizing Hgel in flue gases. For instance, Jeon et al. [107] investigated the photo-catalytic ability of nanotitanosilicate fibers for oxidizing gas-phase Hgel under UV black light, fluorescent light and sunlight. The highest Hgel oxidation activity (88%) was achieved under fluorescent light. Wang et al. [105] investigated the Hgel oxidation by titania nanotubes with high surface area and porosity. More than 90% Hgel oxidation efficiency was achieved during a period of 100 h. This mercury oxidation efficiency was due to the photo-catalyst structure as well the synergistic effect between the Hgel oxidation and adsorption.
The proposed mechanistic pathway of UV-assisted photo-catalytic oxidation involves HO• radical formation by adsorption of O2 or H2O on the TiO2 surface. The HO• radicals thus created readily react with adsorbed Hgel to form HgO [105,106]. In order to gain a better understanding of the Hgel photo-catalytic oxidation reaction, more experimental data is required. Also, the role of other flue gas constituents is of great interest.
Another innovative method, proposed by Guo et al. [108], for Hgel removal from flue gases is the membrane delivery catalytic oxidation system (MDCOs). The concept of MDCOs involves the use of an Al2O3 porous tubular membrane which serves as a carrier for the MnOx/Al2O3 catalyst. The system combines the controlled delivery of oxidants (e.g., HCl) with the catalytic oxidation of Hgel. By employing this system, more than 90% Hgel oxidation was achieved in the 150–300 °C temperature range. The oxidation mechanism on MDCOs involves the reaction of adsorbed Hgel with adsorbed atomic chlorine (Cl•) to form HgCl2.
Sorbent injection technology is promising for elemental and oxidized mercury removal from flue gases. Due to their ability to adsorb not only Hgel [27,109,110], but also nitrogen oxide [111], sulfur dioxide [112,113] and/or hydrochloric acid [112], carbon-based materials promote elemental mercury oxidation. However, the aspects of carbon based materials used for Hgel oxidation will not be covered in the present review.
The novel catalytic methods discussed above present a number of advantages as well as disadvantages compared with the traditional oxidation methods employed so far. For instance, when employing the MDCO system, high Hgel oxidation efficiency is achieved with lower HCl consumption. Also, the inhibitory effect of SO2 seems to be less significant. However, the implementation of MCDO system at industrial scale requires an elaborate design.
The photo-catalytic oxidation of Hgel is an attractive method since it can be performed even at room temperature and with high Hgel concentrations in the flue gas. The cost of TiO2 material is relatively low; however the cost of providing continuous UV light in a power plant configuration remains an issue. In power plants, large flue gas volumes have to be treated. The design of large photo-catalytic reactors is unexplored territory.
8. Conclusions and Future Research
Over the past six years a significant amount of research has been devoted to developing and implementing new catalysts for elemental mercury Hgel oxidation in flue gases. Academia and industry have focused their efforts towards this purpose. Research has been carried out at laboratory, pilot and full scale.
It is now known that the activity of almost all mercury oxidation catalysts studied so far depends on a certain concentration of HCl and HBr in the flue gases to be treated. The role of the hydrogen halides could be restricted to transforming the primary formed non-volatile HgO into the volatile HgX2. The halides could also be the source of chlorine and bromine atoms, the reactive intermediates which oxidize the elemental mercury. Most catalysts are not effective in flue gases from coals containing low levels of halogens.
SCR-DeNOx catalysts were intensively studied due to their potential as co-oxidation agents for Hgel. Ways for improving their Hg oxidation activity were proposed. Coating/impregnating the SCR monolith with different metals and metal oxides was reported to cause an increase in Hg oxidation activity. The latest reports on RuO2 addition to SCR catalyst seems to be promising in this respect.
Recently it was established by different research groups that SCR-catalysts can also reduce oxidized mercury back to the elementary valence state. This aspect has been overlooked so far and should be an integral part of future research. The prevention of reduction of oxidized mercury by catalysts will result in an increase of the overall oxidation activity of SCR-DeNOx catalysts.
The rate constant of mercury oxidation reaction on a standard commercial high-dust SCR-DeNOx catalyst is of the order of the DeNOx reaction. Therefore the mercury oxidation will take place in the upper layer of the porous catalyst. Consequently, the pore structure of catalysts will be of importance in the effort to optimize the oxidation activity for mercury.
Recently, some publications have discussed the likely pathways for mercury oxidation over SCR catalysts. Hgel oxidation involves V2O5 active sites and HCl and HBr species. At this point, further research is necessary in order to gain a better understanding of the mechanistic pathways for both Hg oxidation and the role of other flue gas components. This will aid future catalyst development.
The laboratory-based research conducted so far showed that transition metal oxides are able to adsorb and oxidize Hgel in the presence of gaseous HCl or HBr. The activity of these catalysts is affected by temperature, other flue gas components and metal loading. Langmuir-Hinshelwood and Mars-Maessen mechanisms were proposed for Hg oxidation over metal oxide catalysts. The SO2/SO3 and NO/NO2 conversion ability has to be considered when implementing metal oxide based catalysts in full scale applications. Investigations so far have shown that transition metal oxides are prone to poisoning by SO2 in the flue gas.
Laboratory and pilot scale studies have centered on noble metal catalysts for mercury oxidation applications in flue gases. Noble metals have the ability to adsorb mercury on their surface. They might affect the undesired SO2/SO3 and NO/NO2 conversion reaction. From this group, gold-based catalysts seem to be promising candidates. These catalysts are highly active even at low metal loadings (1 wt.-%) and low temperatures (<200 °C). Pilot scale studies showed that the gold based catalysts are not easily subjected to deactivation under flue gas conditions. Further investigation into the mechanism, the role of other flue gas components and temperature on mercury oxidation by gold catalysts are necessary.
Several new methods for catalytic oxidation of mercury from flue gases were proposed. UV-assisted oxidation by TiO2 nano-particles or fibers, catalytic membranes and dielectric barrier discharge are the proposed novel methods. However, at this stage the number of publications on these topics is relatively limited and these methods have a long way to go until they might be ready for industrial application.
Acknowledgments
The authors would like to thank Ion Balasanian from Gheorghe Asachi Technical University of Iasi, Romania, and Katharina Zeng from Martin-Luther University Halle-Wittenberg, Germany for their support.
Financial support through the European Union scholarship (Erasmus) funding is acknowledged.
References
- Division of Technology, Industry and Economics (DTIE) Chemicals Branch, Study on Mercury Sources and Emissions and Analysis of the Cost and Effectiveness of Control Measures; UNEP: Geneva, Switzerland, 2010.
- Pirrone, N.; Cinnirella, S.; Feng, X.; Finkelman, B.R.; Friedli, R.H.; Leaner, J.; Mason, R.; Mukherjee, B.A.; Stracher, G.; Streets, G.D.; Telmer, K. Mercury Fate and Transport in the Global Atmosphere—Emissions, Measurements and Models; Springer: New York, NY, USA, 2009. [Google Scholar]
- Lee, B.J.; Lee, M.S.; Lee, Y.I. The characteristics of catalysts for mercury oxidation in thermal power plants. Proc. World Acad. Sci. Eng. Technol. 2008, 44, 256–257. [Google Scholar]
- United States Environmental Protection Agency (U.S. EPA). Mercury News, December 2011. Available online: http://www.epa.gov/hg/ (accessed on 2 January 2012).
- European Commission. Directive 2010/75/EU of the European Parliament and of the Council on industrial emissions (integrated pollution prevention and control). Off. J. Eur. Union 2010, L334, 17–117.
- Pavlish, J.H.; Sondreal, E.A.; Mann, D.M.; Olson, S.E.; Galbreath, C.K.; Laudal, L.; Benson, A.S. Status review of mercury control options for coal-fired power plants. Fuel Process. Technol. 2003, 82, 89–165. [Google Scholar] [CrossRef]
- Pavlish, H.J.; Hamre, L.L.; Zhuang, Y. Mercury control technologies for coal combustion and gasification systems. Fuel 2010, 89, 838–847. [Google Scholar] [CrossRef]
- Schofield, K. Fuel-mercury combustion emissions: An important heterogeneous mechanism and an overall review of its implications. Environ. Sci. Technol. 2008, 42, 9014–9030. [Google Scholar] [CrossRef]
- Presto, A.A.; Granite, E.J. Survey of catalysts for oxidation of mercury in flue gas. Environ. Sci. Technol. 2006, 40, 5601–5609. [Google Scholar] [CrossRef]
- Wilhelm, S.M. Estimate of mercury emissions to the atmosphere from petroleum. Environ. Sci. Technol. 2001, 35, 4704–4710. [Google Scholar] [CrossRef]
- Yudovich, Y.E.; Ketris, M.P. Mercury in coal: A review. Part 2 coal use and environmental problems. Int. J. Coal Geol 2005, 62, 136–165. [Google Scholar]
- Sondreal, E.A.; Benson, S.A.; Pavlish, S.A.; Ralston, N.V.C. An overview of air quality III: Mercury, trace elements, and particulate matter. Fuel Process. Technol. 2004, 85, 425–440. [Google Scholar] [CrossRef]
- Vosteen, B.W. Native Halogens in Coals from US, China and Elsewhere—Low Chlorine Coal Need Bromide Addition for Effective Mercury Capture. In Proceedings of MEC7—Mercury Emissions from Coal—International Experts Workshop, Glasgow, UK, 16–18 June 2010.
- Qiao, S.; Chen, J.; Li, J.; Qu, Z.; Liu, P.; Yan, N.; Jia, J. Adsorption and catalytic oxidation of gaseous elemental mercury in flue gas over MnOx/alumina. Ind. Eng. Chem. Res. 2009, 48, 3317–3322. [Google Scholar]
- Vosteen, W.B.; Kanefke, R.; Köser, H. Bromine-enhanced mercury abatement from combustion flue gases-recent industrial applications and laboratory research. VGB PowerTech 2006, 86, 70–75. [Google Scholar]
- Vosteen, W.B.; Straube, S.; Köser, H. Mercury Sorption and Mercury Oxidation by Chlorine and Bromine at SCR-DeNOx Catalysts—Part A: Oxidation. In Proceedings of 9th Annual EPA, DOE, EPRI, EEI Conference on Clean Air, Global Warming & Renewable Energy, Tucson, AZ, USA, 24–25 January 2006.
- Lopez, N.; Gomez-Segura, J.; Marin, R.P.; Perez-Ramirez, J.P. Mechanism of HCl oxidation (Deacon process) over RuO2. J. Catal. 2008, 255, 29. [Google Scholar]
- Bierögel, S.; Baltin, G.; Koeser, H. Important side reactions in DeNOx facilities—Investigation for halogen formation. Chem. Ing. Tech. 2003, 75, 1066–1067. [Google Scholar]
- Griffin, R.D. A new theory of dioxin formation in municipal solid waste combustion. Chemophere 1986, 15, 1987–1990. [Google Scholar] [CrossRef]
- Niksa, S.; Fujiwara, N. A predictive mechanism for mercury oxidation on selective catalytic reduction catalyst under coal-derived flue gas. J. Air Waste Manag. Assoc. 2005, 55, 1866–1875. [Google Scholar] [CrossRef]
- Kamata, H.; Uueno, S.I.; Naito, T.; Yukimura, A. Mercury oxidation over the V2O5(WO3)/TiO2 commercial SCR catalyst. Ind. Eng. Chem. Res. 2008, 47, 8136–8141. [Google Scholar] [CrossRef]
- Li, Y.; Murphy, D.P.; Wu, C.Y.; Powers, W.K.; Bonzongo, J.C. Development of silica/vanadia/titania catalysts for removal of elemental mercury from coal-combustion flue gas. Environ. Sci. Technol. 2008, 42, 5304–5309. [Google Scholar]
- He, S.; Zhou, J.; Zhu, Y.; Luo, Z.; Ni, M.; Cen, K. Mercury oxidation over a vanadia-based selective catalytic reduction catalyst. Energy Fuels 2009, 23, 253–259. [Google Scholar] [CrossRef]
- Kamata, H.; Uueno, S.I.; Naito, T.; Yamaguchi, A.; Ito, S. Mercury oxidation by hydrochloric acid over a VOx/TiO2 catalyst. Catal. Commun. 2008, 9, 2441–2444. [Google Scholar] [CrossRef]
- Eom, Y.; Jeon, H.S.; Ngo, A.T.; Kim, J.; Lee, G.T. Heterogeneous mercury reaction on a selective catalytic reduction (SCR) catalyst. Catal. Lett. 2008, 121, 219–225. [Google Scholar] [CrossRef]
- Li, H.; Wu, Y.C.; Li, Y.; Zhang, J. Superior activity of MnOx–CeO2/TiO2 catalyst for catalytic oxidation of elemental mercury at low flue gas temperatures. Appl. Catal. B Environ. 2012, 111–112, 381–388. [Google Scholar] [CrossRef]
- Hu, C.; Zhou, J.; Luo, Z.; Cen, K. Oxidative adsorption of elemental mercury by activated carbon in simulated coal-fired flue gas. Energy Fuels 2011, 25, 154–158. [Google Scholar] [CrossRef]
- Granite, J.E.; Pennline, H.W.; Hargis, A.R. Novel sorbents for mercury removal from flue gas. Ind. Eng. Chem. Res. 2000, 39, 1020–1029. [Google Scholar] [CrossRef]
- Hrdlicka, J.A.; Seames, W.S.; Mann, M.D.; Muggli, D.S.; Horabik, C.A. Mercury oxidation in flue gas using gold and palladium catalysts on fabric filters. Environ. Sci. Technol. 2008, 42, 6677–6682. [Google Scholar]
- Zhao, Y.; Mann, M.D.; Pavlish, J.H.; Mibeck, B.A.F.; Dunham, E.G.; Olson, E.S. Application of gold catalyst for mercury oxidation by chlorine. Environ. Sci. Technol. 2006, 40, 1603–1608. [Google Scholar]
- Ji, L.; Sreekanth, M.P.; Smirniotis, G.P.; Thiel, W.S.; Pinto, G.N. Manganese oxide/Titania materials for removal of NOx and elemental mercury from flue gas. Energy Fuels 2008, 22, 2299–2306. [Google Scholar] [CrossRef]
- Yang, S.; Guo, Y.; Yan, N.; Wu, D.; He, H.; Qu, Z.; Jia, J. Elemental mercury capture from flue gas by magnetic Mn-Fe spinel: Effect of chemical heterogeneity. Ind. Eng. Chem. Res. 2011, 50, 9650–9656. [Google Scholar]
- Liu, Y.; Wang, Y.; Wang, H.; Wu, Z. Catalytic oxidation of gas-phase mercury over Co/TiO2 catalysts prepared by sol-gel method. Catal. Commun. 2011, 12, 1291–1294. [Google Scholar] [CrossRef]
- Kong, F.; Qui, J.; Liu, H.; Zhao, R.; Ai, Z. Catalytic oxidation of gas-phase elemental mercury by nano-Fe2O3. J. Environ. Sci. 2011, 24, 699–704. [Google Scholar]
- Wen, X.; Li, C.; Fan, X.; Gao, H.; Zhang, W.; Chen, L.; Zeng, G.; Zhao, G. Experimental study of gaseous elemental mercury removal with CeO2/γ-Al2O3. Energy Fuels 2011, 25, 2939–2944. [Google Scholar] [CrossRef]
- Yan, N.; Chen, W.; Chen, J.; Qu, Z.; Guo, Y.; Yang, S.; Jinping, J. Significance of RuO2 modified SCR catalysts for elemental mercury oxidation in coal-fired flue gas. Environ. Sci. Technol. 2011, 45, 5725–5730. [Google Scholar]
- Presto, A.A.; Granite, E.J. Noble Metal Catalysts for mercury oxidation in utility flue gas. Platin. Met. Rev. 2008, 52, 144–154. [Google Scholar] [CrossRef]
- Granite, J.E.; Pennline, H.W. Catalysts for oxidation of mercury in flue gases. U.S. Patent 7,776,780 B1, 17 August 2010. [Google Scholar]
- Blythe, M.G.; Braman, C.; Dombrowski, K.; Machalek, T. Pilot Testing of Mercury Oxidation Catalysts for Upstream of Wet FGD Systems—Final Technical Report; Cooperative Agreement No. DE-FC26-04NT41992; DOE-NETL: Austin, TX, USA, 2010. [Google Scholar]
- Blythe, M.G.; Paradis, J. Full-Scale Testing of a Mercury Oxidation Catalyst Upstream of a Wet FGD System, Final Technical Report; Cooperative Agreement No. DE-FC26-06NT42778; DOE-NETL: Austin, TX, USA, 2010. [Google Scholar]
- Poulston, S.; Granite, J.E.; Pennline, W.H.; Myers, R.C.; Stanko, P.D.; Hamilton, H.; Rowsell, L.; Smith, J.W.A.; Ilkenhans, T.; Chu, W. Metal sorbents for high temperature mercury capture from fuel gas. Fuel 2007, 86, 2201–2203. [Google Scholar] [CrossRef]
- Granite, J.E.; Pennline, H.W. Method for high temperature mercury capture from gas streams. U.S. Patent 7,033,419 B1, 25 April 2006. [Google Scholar]
- Koutsopoulos, S.; Johannessen, T.; Eriksen, K.M.; Fehrmann, R. Titania-supported Pt and Pt–Pd nanoparticle catalysts for the oxidation of sulfur dioxide. J. Catal. 2006, 238, 206–213. [Google Scholar]
- Ji, Y.; Toops, T.J.; Graham, U.M.; Jacobs, G.; Crocker, M. A kinetic and DRIFTS study of supported Pt catalysts for NO oxidation. Catal. Lett. 2006, 110, 29–37. [Google Scholar] [CrossRef]
- Bond, G.C.; Louis, C.; Thompson, D.T. Catalysis by Gold; Imperial College Press: London, UK, 2006. [Google Scholar]
- Blythe, G.; Dombrowski, K.; Machalek, T.; Richardson, C.; Richardson, M. Pilot Testing of Mercury Oxidation Catalysts for Upstream of Wet FGD Systems, Final Report; Cooperative Agreement No. DE-FC26-01NT41185; DOE-NETL: Austin, TX, USA, 2006. [Google Scholar]
- Blythe, G.; Miller, C.; Freeman, B.; Madrid, J. Full-scale demonstration of oxidation catalyst for enhanced mercury control by wet FGD. In Proceedings of Power Plant Air Pollutant Control“Mega” Symposium, Baltimore, MD, USA, 25–28 August 2008.
- Richardson, C. Evaluation of MerCAP for Power Plant Mercury Control—Final Project Report for US Department of Energy—NETL; Cooperative Agreement No. DE-FC26-03NT41993; DOE-NETL: Austin, TX, USA, 2009. [Google Scholar]
- Tao, F.M. A new approach to the efficient basis set for accurate molecular calculations: Applications to diatomic molecules. J. Chem. Phys. 1994, 100, 3645–3650. [Google Scholar]
- Boreskov, G.K.; Slinko, M.G.; Volkova, E.I. Catalysis of the conversion of sulfur dioxide to sulfur trioxide by metals and platinum-gold alloys. Dokl. Akad. Nauk SSSR 1953, 92, 109–120. [Google Scholar]
- Yamaguchi, A.; Akiho, H.; Ito, S. Mercury oxidation by copper oxides in combustion flue gases. Powder Technol. 2008, 180, 222–226. [Google Scholar] [CrossRef]
- Kamata, H.; Mouri, S.; Uueno, S.I.; Takano, K.; Watanabe, K.; Yamaguchi, A.; Ito, S. Mercury oxidation by hydrogen chloride over the CuO based catalysts. Stud. Surf. Sci. Catal. 2007, 172, 621–622. [Google Scholar]
- Kim, H.M.; Ham, S.W.; Li, J.B. Oxidation of gaseous elemental mercury by hydrochloric acid over CuCl2/TiO2-based catalysts in SCR process. Appl. Catal. B Environ. 2010, 99, 272–278. [Google Scholar] [CrossRef]
- Miksche, J.S.; Ghorishi, B.S. Catalytic Oxidation of Elemental Mercury at Low Temperatures-Technical Paper. In Proceedings of 32nd International Technical Conference on Coal Utilization & Fuel Systems, Clearwater, FL, USA, 10–15 June 2007.
- Mei, Z.; Shen, Z.; Mei, Z.; Zhang, Y.; Xiang, F.; Chen, J.; Wang, W. The effect of N-doping and halide-doping on the activity of CuCoO4 for the oxidation of elemental mercury. Appl. Catal. B Environ. 2008, 78, 112–119. [Google Scholar] [CrossRef]
- Wu, S.; Ozaki, M.; Uddin, A.Md.; Sasaoka, E. Development of iron-based sorbents for Hg0 removal from coal derived fuel gas: Effect of hydrogen chloride. Fuel 2008, 87, 467–474. [Google Scholar] [CrossRef]
- Li, J.; Yan, N.; Qu, Z.; Qiao, S.; Yang, S.; Guo, Y.; Liu, P.; Jia, J. Catalytic oxidation of elemental mercury over the modified catalyst Mn/α-Al2O3 at lower temperatures. Environ. Sci. Technol. 2010, 44, 426–431. [Google Scholar]
- Li, H.; Wu, Y.C.; Li, Y.; Zhang, J. CeO2–TiO2 catalysts for catalytic oxidation of elemental mercury in low-rank coal combustion flue gas. Environ. Sci. Technol. 2011, 45, 7394–7400. [Google Scholar]
- Lee, W.; Bae, G.N. Removal of elemental mercury Hg0 by nanosized V2O5/TiO2 catalysts. Environ. Sci. Technol. 2009, 43, 1522–1527. [Google Scholar] [CrossRef]
- Li, Y.; Murphy, P.; Wu, Y.C. Removal of elemental mercury from simulated coal-combustion flue gas using a SiO2–TiO2 nanocomposite. Fuel Process. Technol. 2008, 89, 567–573. [Google Scholar] [CrossRef]
- Li, H.; Li, Y.; Wu, Y.C.; Zhang, J. Oxidation and capture of elemental mercury over SiO2-TiO2-V2O5 catalysts in simulated low-rank coal combustion flue gas. Chem. Eng. J. 2011, 169, 186–193. [Google Scholar] [CrossRef]
- Ghorishi, S.B.; Lee, W.C.; Jozewicz, S.W.; Kilgroe, D.J. Effects of fly ash transition metal content and flue gas HCl/SO2 ration on mercury speciation on waste combustion. Environ. Eng. Sci. 2005, 22, 221–231. [Google Scholar] [CrossRef]
- Kamata, H.; Uueno, S.I.; Sato, N.; Naito, T. Mercury oxidation by hydrochloric acid over TiO2 supported metal oxide catalysts in coal combustion flue gas. Fuel Proc. Technol. 2009, 90, 947–951. [Google Scholar] [CrossRef]
- Mei, Z.; Shen, Z.; Zhao, Q.; Yuan, T.; Zhang, Y.; Xiang, F.; Wang, W. Removing and recovering gas-phase elemental mercury by CuxCo3−xO4 0.75 ≤ x ≤ 2.25, in the presence of sulphur compounds. Chemosphere 2008, 70, 1399–1404. [Google Scholar] [CrossRef]
- Yang, S.; Guo, Y.; Yan, N.; Wu, D.; He, H.; Xie, J.; Qu, Z.; Jia, J. Remarkable effect of the incorporation of titanium on the catalytic activity and SO2 poisoning resistance of magnetic Mn–Fe spinel for elemental mercury capture. Appl. Catal. B Environ. 2011, 101, 698–708. [Google Scholar] [CrossRef]
- Yang, S.; Guo, Y.; Yan, N.; Wu, D.; He, H.; Qu, Z.; Yang, C.; Zhou, Q.; Jia, J. Nanosized cation-deficient Fe–Ti spinel: A novel magnetic sorbent for elemental mercury capture from flue gas. Appl. Mater. Interfaces 2011, 3, 209–217. [Google Scholar]
- Yang, S.; Yan, N.; Guo, Y.; Wu, D.; He, H.; Qu, Z.; Li, J.; Zhou, Q.; Jia, J. Gaseous elemental mercury capture from flue gas using magnetic nanosized (Fe3−xMnx)1-δO4. Environ. Sci. Technol. 2011, 45, 1540–1546. [Google Scholar]
- Granger, P.; Parlvulescu, V.I. Past and Present in DeNOx Catalysis—From Molecular Modeling to Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Finocchio, E.; Baldi, M.; Busca, G.; Pistarino, C.; Romezzano, G.; Bregani, F.; Toledo, G.P. A study of the abatement of VOC over V2O3–WO3–TiO2 and alternative SCR catalysts. Catal. Today 2000, H59, 261–268. [Google Scholar]
- Lazar, L.; Köser, H.; Balasanian, I.; Bandrabur, F. Catalytic destruction of aromatic VOCs on SCR-DeNOx commercial catalyst. Environ. Eng. Manag. J. 2007, 6, 13–20. [Google Scholar]
- Cao, Y.; Chen, B.; Wu, J.; Cui, H.; Smith, J.; Chen, K.C.; Chu, P.; Pan, P.W. Study of mercury oxidation by a selective catalytic reduction catalyst in a pilot-scale slipstream reactor at a utility boiler burning bituminous coal. Energy Fuels 2007, 21, 145–156. [Google Scholar]
- Zhuang, Y.; Laumb, J.; Liggett, R.; Holmes, M.; Pavlish, J. Impacts of acid gases on mercury oxidation across SCR catalyst. Fuel Process. Technol. 2007, 88, 929–934. [Google Scholar] [CrossRef]
- Straube, S.; Hahn, T.; Koeser, H. Adsorption and oxidation of mercury in tail-end SCR-DeNOx plants-Bench scale investigations and speciation experiments. App. Catal. B Environ. 2008, 79, 286–295. [Google Scholar] [CrossRef]
- Lee, W.C.; Srivastava, K.R.; Ghorishi, B.S.; Kaewowski, J.; Hastings, W.T.; Hirschi, C.J. Pilot-scale study of the effect of selective catalytic reduction catalysts on mercury speciation in Illinois and Powder River Basin coal combustion flue gases. J. Air Waste Manag. Assoc. 2006, 56, 643–649. [Google Scholar] [CrossRef]
- Senior, C. Mercury oxidation across SCRs in coal-fired power plants. In Proceedings ofMercury Control Technology R&D Program Review, Pittsburgh, PA, USA, 12–14 July 2005.
- Eswaran, S.; Stenger, H.G. Understanding mercury conversion in selective catalytic reduction (SCR) catalysts. Energy Fuels 2005, 19, 2328–2334. [Google Scholar] [CrossRef]
- Lee, W.C.; Srivastava, K.R.; Ghorishi, B.S.; Hastings, W.T.; Stevens, M.F. Investigation of selective catalytic reduction impact on mercury speciation under simulated NOx emission control conditions. J. Air Waste Manag. Assoc. 2004, 54, 1560–1566. [Google Scholar] [CrossRef]
- Hong, J.H.; Ham, W.S.; Kim, H.M.; Lee, M.S.; Lee, B.J. Characteristics of commercial selective catalytic reduction catalysts for the oxidation of gaseous elemental mercury with respect to reaction conditions. Korean J. Chem. Eng. 2010, 27, 1117–1122. [Google Scholar] [CrossRef]
- Eswaran, S.; Stenger, H.G. Effect of halogens on mercury conversion in SCR catalysts. Fuel Process. Technol. 2008, 89, 1153–1159. [Google Scholar] [CrossRef]
- Lee, W.C.; Serre, D.S.; Zhao, Y.; Lee, J.S.; Hastings, W.T. Mercury oxidation promoted by a selective catalytic reduction catalyst under simulated powder river basin coal combustion conditions. J. Air Waste Manag. Assoc. 2008, 58, 484–493. [Google Scholar] [CrossRef]
- Yang, H.M.; Pan, W.P. Transformation of mercury speciation through the SCR system in power plants. J. Environ. Sci. 2007, 19, 181–184. [Google Scholar] [CrossRef]
- Cao, Y.; Gao, Z.; Zhu, J.; Wang, Q.; Huang, Y.; Chiu, C.; Parker, B.; Chu, P.; Pan, W.P. Impacts of halogen addition in mercury oxidation, in a slipstream selective catalyst reduction (SCR), reactor when burning sub-bituminous coal. Environ. Sci. Technol. 2008, 42, 256–261. [Google Scholar]
- Senior, C. Oxidation of Mercury Across SCR Catalysts in Coal-Fired Power Plants Burning Low Rank Fuels. Final Report to U.S. DOE/NETL; U.S. Department of Energy Agreement No. DE-FC26-03NT41728; Reaction Engineering International: Salt Lake City, UT, USA, 2004. [Google Scholar]
- Sibley, F.A.; Dene, C.; Jimenez, A.; Hinton, W.S. Pilot scale studies on mercury oxidation by SCR catalyst. In Proceedings of Power Plant Air Pollutant Control “Mega” Symposium Symposium, Baltimore, MD, USA, 25–28 August 2008.
- Crocker, R.C.; Benson, A.S.; Laumb, D.J. SCR catalyst blinding due to sodium and calcium sulphate formation. Prepr. Pap. Am. Chem. Soc. Div. Fuel Chem. 2004, 49, 169–172. [Google Scholar]
- Wan, Q.; Duan, L.; Li, J.; Chen, L.; He, K.; Hao, J. Deactivation performance and mechanism of alkali (earth) metals on V2O5–WO3/TiO2 catalyst for oxidation of gaseous elemental mercury in simulated coal-fired flue gas. Catal. Today 2011, 175, 189–195. [Google Scholar]
- Guo, X.; Beutler, J.; Anderson, C.; Nackos, A.; Ashton, J.; Bartholomew, C.; Hecker, W.; Baxter, L. Poisoning/Deactivation Study of V2O5/TiO2 SCR Catalysts. In Proceedings of ACERC Annual Conference, Provo, UT, USA, 26 February 2008.
- Staudt, E.J.; Engelmeyer, T.; Weston, H.W.; Sigling, R. The impact of arsenic on coal fired power plants equipped with SCR. In Presented at ICAC Forum, Houston, TX, USA, 12–13 February 2002.
- Thowarth, H. Trace Element Behavior in Pulverised Fuel Fired Power Plants—Impact of Fuels and Emission Control Technologies; Cuvillier Verlag: Göttingen, Germany, 2007. [Google Scholar]
- Senior, C.; Lignell, O.D.; Sarofim, F.A.; Mehta, A. Modelling arsenic partitioning in coal-fired power plants. Combust. Flame 2006, 1473, 209–221. [Google Scholar]
- Senior, C. Oxidation of mercury across selective catalytic reduction catalysts in coal fired power plants. J. Air Waste Manag. Assoc. 2006, 56, 23–31. [Google Scholar] [CrossRef]
- Liu, J.; He, M.; Zheng, C.; Chang, M. Density functional theory study of mercury adsorption on V2O5(001) surface with implications for oxidation. Proc. Combust. Inst. 2011, 33, 2771–2777. [Google Scholar] [CrossRef]
- Madsen, K.; Jensen, D.A.; Frandsen, J.F.; Thogersen, R.J. A mechanistic Study on the Inhibition of the DeNOx Reaction on the Mercury Oxidation over SCR catalysts. In Proceedings of Air Quality VIII Conference, Arlington, VA, USA, 24–27 October 2011.
- Stolle, R.; Köser, H.; Gutberlet, H. Determination of Hg-oxidation-activity of SCR-DeNOx-catalysts. In Presented at VGB Workshop “Flue Gas Cleaning 2008”, Vilnius, Lithuania, 3–4 June 2008.
- Zeng, K.; Stolle, R.; Köser, H. Quecksilberoxidation an metalleoxiddotierten SCR-DeNOx-Katalysatoren. In Proceedings of ProzessNet Conference, Mannheim, Germany, 8–10 September 2009.
- Kai, K.; Kato, Y.; Kikkawa, H.; Imada, N.; Nagai, Y. New SCR Catalysts with Improved Mercury Oxidation for Bituminous Coal-Fired Boilers. In Proceedings of Air Quality Conference, Arlington, VA, USA, 24–27 September 2007.
- Kai, K.; Kato, Y. Catalysts for oxidation of metal mercury. WIPO Patent Application WO/2008/035773, 27 March 2008. [Google Scholar]
- Nochi, K.; Yonemura, M.; Kiyosawa, M. Mercury oxidation catalyst and method for producing the same. U.S. Patent Application 2011/0082028A1, 7 April 2011. [Google Scholar]
- Kai, K.; Kato, Y. Oxidation treatment of metallic mercury in flue gas using oxidation catalysts. JP Patent 2010/088984A, 22 April 2010. [Google Scholar]
- Kai, K.; Kato, Y.; Imada, N. Method and catalyst for oxidation treatment of mercury metal in flue gas. JP Patent 2010/088983A, 22 April 2010. [Google Scholar]
- Kai, K.; Kato, Y. Method for waste gas treatment with a catalyst. JP Patent 2009226238A, 8 October 2009. [Google Scholar]
- Nagayasu, T.; Nakashoji, H.; Imai, N. Multi-pollutant control capabilities of double contact flow scrubber (DCFS). VGB PowerTech 2011, 91, 68–73. [Google Scholar]
- Zhuang, Y.; Thompson, S.J.; Zxgarlicke, J.C.; Pavlish, H.J. Impact of calcium chloride addition on mercury transformations and control in coal flue gas. Fuel 2007, 86, 2351–2359. [Google Scholar] [CrossRef]
- Niksa, S.; Padak, B.; Krishnakumar, B.; Naik, V.C. Process Chemistry of Br addition to utility flue gas for Hg emissions control. Energy Fuels 2010, 24, 1020–1029. [Google Scholar] [CrossRef]
- Wang, H.; Zhou, S.; Xiao, L.; Wang, Y.; Liu, Y.; Wu, Z. Titania nanotubes—A unique photocatalyst and adsorbent for elemental mercury removal. Catal. Today 2011, 175, 202–208. [Google Scholar]
- Snider, G.; Ariya, P. Photo-catalytic oxidation reaction of gaseous mercury over titanium dioxide nanoparticle surfaces. Chem. Phys. Lett. 2010, 491, 23–28. [Google Scholar] [CrossRef]
- Jeon, H.S.; Eom, Y.; Lee, G.T. Photocatalytic oxidation of gas-phase elemental mercury by nanotitanosilicate fibers. Chemosphere 2008, 71, 969–974. [Google Scholar] [CrossRef]
- Guo, Y.; Yan, N.; Yang, S.; Qu, Z.; Wu, Z.; Liu, Y.; Liu, P.; Jia, J. Conversion of elemental mercury with a novel membrane delivery catalytic oxidation system (MDCOs). Environ. Sci. Technol. 2011, 45, 706–711. [Google Scholar]
- Granite, J.E.; Freeman, C.M.; Hargis, A.R.; O’Dowd, J.W.; Pennline, H.W. The thief process for mercury removal from flue gas. J. Environ. Manag. 2006, 84, 628–634. [Google Scholar]
- Dunham, E.G.; DeWall, A.R.; Senior, L.C. Fixed-bed studies of the interactions between mercury and coal combustion fly ash. Fuel Process. Technol. 2003, 82, 197–213. [Google Scholar] [CrossRef]
- Claudino, A.; Soares, J.L.; Moreira, M.P.F.R.; José, J.H. Adsorption equilibrium and breakthrough analysis for NO adsorption on activated carbons at low temperatures. Carbon 2004, 42, 1483–1490. [Google Scholar] [CrossRef]
- Ochiai, R.; Uddin, A.Md.; Sasaoka, E.; Wu, S. Effects of HCl and SO2 concentration on mercury removal by activated carbon sorbents in coal-derived flue gas. Energy Fuels 2009, 23, 4734–4739. [Google Scholar] [CrossRef]
- Presto, A.A.; Granite, E.J. Impact of sulfur oxides on mercury capture by activated carbon. Environ. Sci. Technol. 2007, 41, 6579–6584. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).