1. Introduction
Energy is the key element in the interaction between nature and society and is considered as an essential requirement for the economic development [
1]. Nowadays, 87% of the primary energy demand is obtained from non-renewable resources with limited availability as fossil and nuclear sources [
2]. In addition, the energy production from traditional sources is considered to be polluting and unsustainable. Carbon dioxide emissions during the energy generation from the fossil fuels have become one of the principal environmental preoccupations in the last few years, since it is considered as a major participant to global warming. In the near future, it is necessary to produce energy using alternative sources, considered as renewable, sustainable, efficient, safe and economically attractive [
3]. For this purpose, the use of hydrogen as an efficient and clean fuel is an interesting alternative to the future way to produce energy, but only if obtained from a sustainable source. Hydrogen produced from biofuels such as bioethanol, obtained by biomass fermentation, has a great potential to produce sustainable energy [
4,
5].
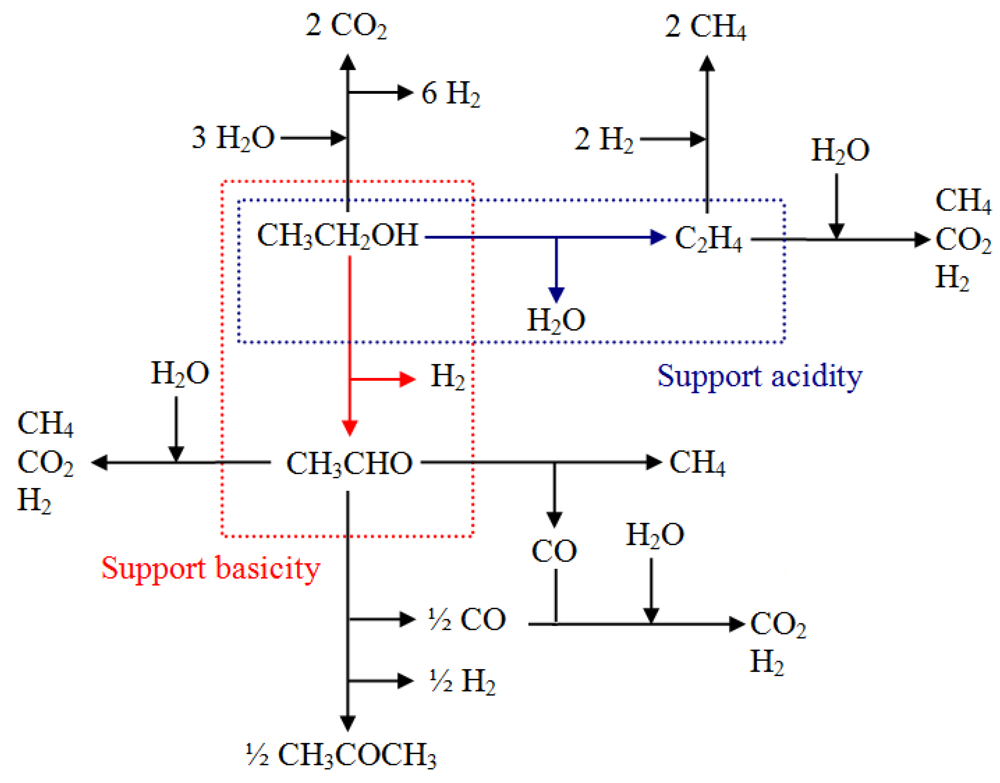
The global reaction of steam reforming of ethanol can be represented by the following general Equation 1:
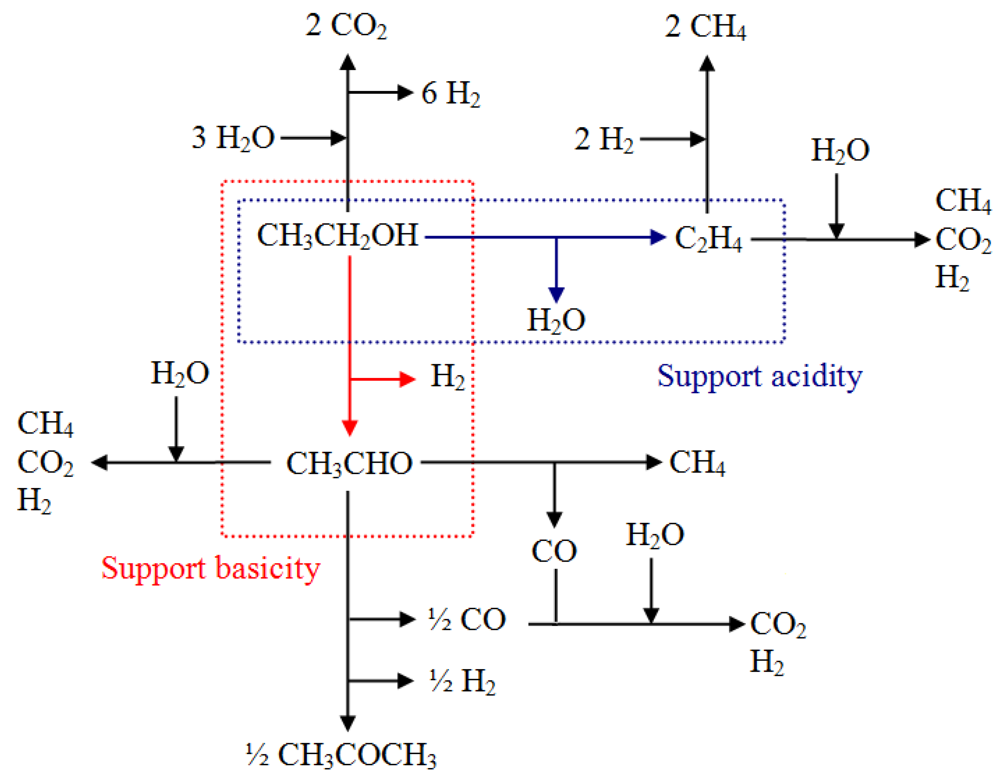
However, different parallel reaction paths exist, such as dehydration into ethylene, dehydrogenation into acetaldehyde, condensation to acetone and cracking reactions [
6,
7,
8]. The relative contribution of the different routes depends on the nature of the support, the active phase and the conditions of the process, as can be seen in
Scheme 1.
Scheme 1.
Possible path route in steam reforming of ethanol.
Scheme 1.
Possible path route in steam reforming of ethanol.
Many active species have been evaluated for the steam reforming of ethanol: noble (Rh, Ru, Re, Pd, Ir, Pt, Rh-Pt, Rh-Pd, Ru-Pt,) and transition (Ni, Cu, Co) metals supported on metal oxides, with a wide range of acid-base and redox properties, which have been the subject of several reviews in the last years [
4,
5,
9,
10]. Some catalysts based on copper and nickel revealed interesting activities [
11,
12,
13,
14] but among the systems reported in the literature, the use of cobalt or rhodium-based catalysts as active phase seems to give the most promising results [
15,
16]. Although bimetallic noble/transition-based catalyst (Rh-Co, Ru-Co, Rh-Ni) studied for this reaction showed interesting results [
17,
18], supports with oxygen mobility, such as CeO
2 and CeO
2-ZrO
2, have received special attention since they are expected to act as promoters in improving the catalytic stability and preventing carbonaceous formations [
19,
20].
The ethanol steam reforming products (catalytic selectivity) are the result of a complicated network of reactions strongly influenced by the active phase oxidation state [
21,
22], the nature of the support, the activation process, the water/ethanol ratio, the reaction temperature and the contact time between the reactant and the catalyst [
5,
6]. Furthermore, the reaction conditions induce changes in the catalyst physico-chemical properties (
i.e., additional reduction of active phase, solid state reactions, sintering,
etc.) and therefore in the catalytic selectivity and stability.
In some earlier publications, it was reported that the insertion of Co, as active phase [
23,
24,
25] into the Ce-Zr mixed oxides and the subsequent doping with Rh [
26,
27], improve the catalytic stability and selectivity for ethanol steam reforming.
In this work, the influence of gold on Ce-Zr-Co mixed oxides, with different Ce/Zr ratios, is evaluated in the reaction of steam reforming of ethanol. The phase Ce-Zr-Co on its own is already identified as active for this reaction, but the use of gold could promote the activity and affects the route of production of hydrogen going through the acetaldehyde intermediate [
28]. The catalysts were also tested in the reaction of CO oxidation for which gold is known to be the most active metal when present as nanosized particles [
29]. The choice of studying this reaction was made because gold supported on the type of supports as prepared for this work has not yet been explored for this reaction, but also because CO oxidation serves as a probe reaction for both electronic and geometric factors. The oxidation of carbon monoxide by oxygen is a reaction which is described in the literature as being sensitive to the particle size, the support reducibility and the interaction of gold with the support [
30].
3. Results and Discussion
Two samples of Ce-Zr-Co mixed oxides were prepared, Ce1.85Zr1.25Co0.9O7.4 and Ce1.23Zr1.87Co0.9O7.4, and used as supports for gold containing catalysts. The sample with the highest cerium content will be referred to as CeZrCo-A whereas the catalyst with the highest Zr content will be referred to as CeZrCo-B. The corresponding gold samples will then be designed as Au/CeZrCo-A and Au/CeZrCo-B.
The metallic elemental composition for the samples, obtained from ICPS and global EDXS analysis (200 nm), was found to be very close to the expected theoretical value and showed a high degree of homogeneity. The gold loading (ICPS) was close to the theoretical value of 1 wt% and almost reproduced in the system with higher cerium content (0.96 wt%). This has been already observed for the bare oxide systems [
32]. As an example, for catalyst B, the theoretical molar distribution is Ce 30.8%, Zr 46.7%, Co 22.5%. ICP analysis gave Ce 33.2%, Zr 44.8%, Co 22.0% and the composition from EDX analysis was Ce 33.0%, Zr 44.7%, Co 22.3%. The Cl content is under the detection limit (150 ppm) which allows claiming that the catalyst is almost chlorine free. However, from the local composition measurements (14.4 nm), EDXS analysis, some non-homogeneous composition zones are observed. Nevertheless, the sample with higher Ce/Zr ratio in the mixed oxides exhibits the best global and local chemical homogeneity.
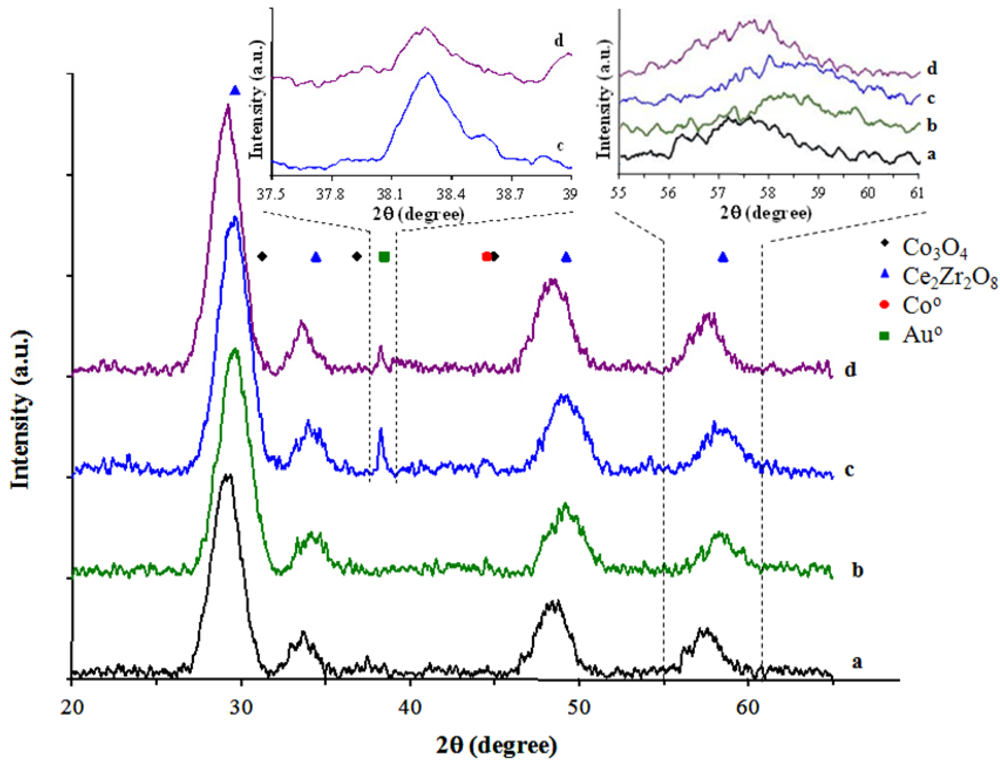
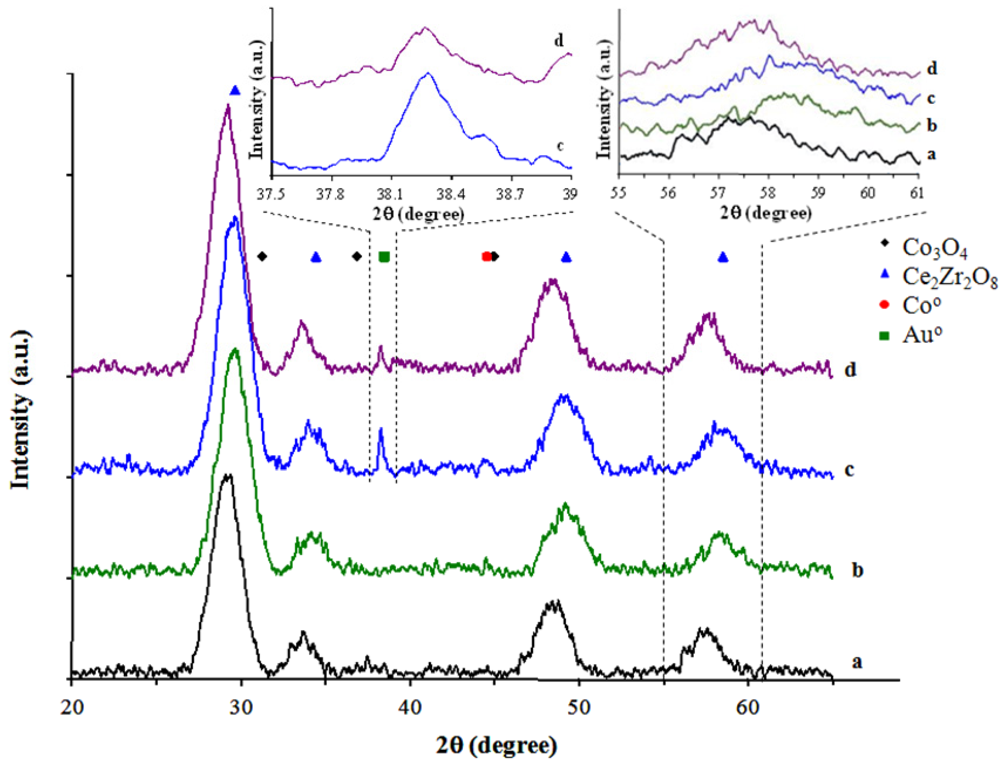
The diffractograms of the CeZrCo mixed oxides, before and after the gold deposition are presented in
Figure 1.
The mixed oxides preparation method of bare Ce-Zr-Co mixed oxides allows the crystallization without any noticeable phase rejection, showing the main cubic structure of Ce-Zr fluorite type oxide. No single cerium or zirconium oxides are detected (CeO
2, ZrO
2) and only a small weak peak corresponding to cobalt oxide spinel phase is present in the samples at 36° (
Figure 1a,b). Depending upon Ce/Zr ratio, it can be noted a shift of the diffraction peak to lower angles (2θ) for the sample with high Ce/Zr ratio, which could be attributed to the zirconium and cobalt insertion, both having smaller ionic radii compared to cerium ionic radius. The formation of mixed propionates during the polymerization step of the preparation allows the formation of the Co-O-Zr and Co-O-Ce bonds as well as Zr-O-Ce bonds which ensures the insertion of cobalt in the mixed oxide lattice.
After the gold deposition on the mixed Ce-Zr-Co oxide surface, an additional diffraction line is visible at 2θ = 38.4°, corresponding to the Au(111) (
Figure 1c,d), without obvious changes in the main peaks corresponding to the fluorite structure. However, it is possible to observe a shift in the main diffraction lines of the fluorite structure, indicating a modification in the degree of cobalt insertion in the host structure, and resulting in different intensities in the cobalt oxide and gold peaks diffraction. For the lower cerium content samples (
Figure 1b,d), lower diffraction angles are observed after the gold deposition. This behavior is a consequence of the different degree of vacancies initially present in the CeZrCo mixed oxide fluorite structure as compared with those generated in the presence of gold. A similar behavior was noted in gold/ceria catalysts [
33,
34].
Figure 1.
X-Ray Diffractograms of: (a) CeZrCo-A; (b) CeZrCo-B; (c) Au/CeZrCo-A; (d) Au/CeZrCo-B.
Figure 1.
X-Ray Diffractograms of: (a) CeZrCo-A; (b) CeZrCo-B; (c) Au/CeZrCo-A; (d) Au/CeZrCo-B.
From the X-ray diffractograms, it is possible to calculate the particle size of gold by means of Debye-Scherrer equation [
35]. With the increase of zirconium content, the particles size decreases from 28 nm for Au/CeZrCo-A to 24 nm for Au/CeZrCo-B, which could be attributed to a different grafting mode of the gold precursor on the bare supports [
30,
32,
36]. In any case, estimation of the size by means of Debye-Scherrer equation tends to lead to an overestimation of the size as only large particles undergo diffraction. Unfortunately, it was not possible to achieve a more accurate particle size distribution because of the poor contrast obtained by transmission electron microscopy.
As the CeZrCo mixed oxide system was prepared to serve as a precursor of small cobalt particles, which is the active phase in the reforming reaction [
23,
26], a special care of the reducibility of the samples has to be taken. From the reduction profile TPR analysis (
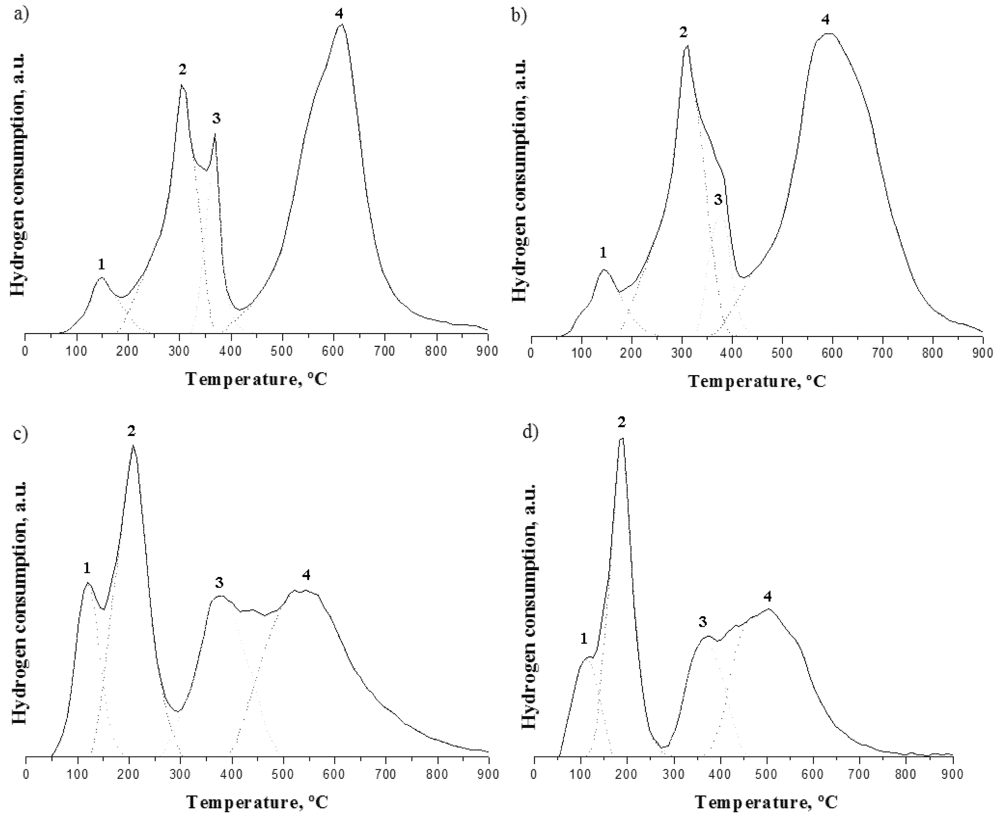
Figure 2), it is possible to describe and discuss the effect of gold. The influence of gold on the TPR profiles is clear, as both the required temperatures to obtain the reduction of the samples and the way that reduction occurs are markedly affected.
Figure 2.
Thermo-programmed reduction (TPR) profiles of samples: (a) CeZrCo-A; (b) CeZrCo-B; (c) Au/CeZrCo-A; (d) Au/CeZrCo-B.
Figure 2.
Thermo-programmed reduction (TPR) profiles of samples: (a) CeZrCo-A; (b) CeZrCo-B; (c) Au/CeZrCo-A; (d) Au/CeZrCo-B.
All the samples, independently of the Ce/Zr ratio or the presence of Au, led to two zones of reduction (
Figure 2). For Au free samples (
Figure 2a,b), the first reduction zone is located below 425 °C, whereas it occurs below 300 °C for the Au-loaded samples (
Figure 2c,d). The first reduction zone is ascribed to the partial reduction of the surface cerium from Ce
4+ to Ce
3+ along with the simultaneous reduction of the cobalt species from Co
3+ and Co
2+ to Co
0. The second reduction zone at high temperature corresponds to the reduction of the Co
2+ incorporated into the fluorite structure and to the reduction of bulk ceria [
25,
37].
The TPR thermograms were decomposed into four peaks. For Ce-Zr-Co mixed oxides there are three peaks in the reduction zone I, at 150, 300, and 380 °C and a broad peak in the reduction zone II, between 400 and 900 °C. The complete description of the thermograms has been published elsewhere for slightly different samples. Briefly, for the gold containing Ce-Zr-Co samples, two peaks at 125 and 200 °C are located in the first reduction zone and the second zone of temperature could also be decomposed into two peaks located at 380 °C and a broad peak between 400 and 900 °C, respectively.
The small consumption of hydrogen centered at around 150 °C (
Figure 2a,b) is attributed to the reduction of highly dispersed cobalt oxide which is not incorporated into the fluorite structure. For the gold-loaded samples, there are both changes in the amount of H
2 which is slightly higher and a shift of reduction temperature towards a lower value. These two phenomena confirm the fact that there are indeed small modifications in the crystalline structure, as proven by XRD. The amount of cobalt oxide not incorporated in the fluorite structure increases,
i.e., small particles of cobalt oxides are present on the support and their reduction is activated by the gold metallic particle.
By numerical integration, it is possible to calculate the contribution of each species to the overall consumption of hydrogen and to estimate the respective proportions of hydrogen allocated to zone 1 and 2. By assuming that, in the fresh samples, gold is in a metallic state—gold usually undergoes autoreduction upon calcination and the presence of metallic gold is proven by XRD—and that cobalt oxide, which is initially present as a stable oxide in the form of (CoO.Co
2O
3) is completely reduced upon hydrogen exposure, it is possible to determine the percentage of Ce
4+ reduced to Ce
3+ during the TPR. The overall hydrogen consumption per gram of catalyst, the percentage of partial reduction (in zone 1) and the percentage of hydrogen consumed by each species are presented in
Table 1.
Table 1.
Total hydrogen consumptions, peak area distribution of samples and percentage of cerium reduced after TPR.
Table 1.
Total hydrogen consumptions, peak area distribution of samples and percentage of cerium reduced after TPR.
| Samples | Peak 1 (%) | Peak 2 (%) | Peak 3 (%) | Peak 4 (%) | Zone I (%) | H2 (mLSTP g−1) | CeIV→CeIII (%) |
|---|
| CeZrCo-A | 5 | 24 | 9 | 61 | 39 | 68 | 47 |
| Au/CeZrCo-A | 11 | 27 | 21 | 41 | 38 | 76 | 70 |
| CeZrCo-B | 5 | 25 | 6 | 63 | 37 | 68 | 56 |
| Au/CeZrCo-B | 10 | 30 | 17 | 43 | 40 | 58 | 26 |
For the Au-free samples, the hydrogen consumption from the first three peaks of reduction is quasi identical (1.2 × 10−3 mol g−1 H2 for CeZrCo-A and 1.1 × 10−3 for CeZrCo-B). The difference between these values and the calculated hydrogen consumption needed for complete cobalt species reduction could directly provide the percentage of the surface ceria reduction. For both samples, 39 and 37% of ceria surface reduction is obtained, respectively. In addition, the unaltered H2 consumption for the peak 4 indicates a constant quantity of reduced bulk Ce+4, no matter the Zr composition. The presence of Zr does not seem to alter the reduction of the bulk cerium content.
However, the reduction of cobalt and cerium changes in the presence of gold being promoted by its presence; the first three peaks of cobalt reduction are completely repositioned. For the first peak, the temperature for reduction is lower, but the volume of consumed hydrogen is doubled, indicating the higher content of cobalt species rejected from the fluorite structure after gold deposition. Moreover, the presence of gold facilitates the Co3+ to Co2+ reduction (peak 2) for which a decrease of the temperature of reduction by 100 °C is observed. The amount of hydrogen consumed in this region is similar for all samples, which confirms that only cobalt is involved in this process. The presence of gold not only induces small structural changes as proven by XRD but modifies the reducibility of the two species corresponding to the low temperature peaks. There is a higher reduction degrees at low temperature observed for both catalysts in the presence of gold. This confirms lower bonding energies of the oxygen on the surface oxidized species and therefore a higher mobility and higher reducibility, which generate vacancies on the surface or species with lower oxidation state which act as active sites (in this case reducing cobalt).
The third peak of reduction seems a little bit more complicated to explain. If the consumption is caused only by Co(II) to Co(0) reduction, the consumption of hydrogen should not change. But an increase in the hydrogen consumption is observed and the temperature of reduction remains unchanged, indicating a higher contribution of the surface ceria reduction. This is confirmed by the fact that for the CeZrCo-B system, where the quantity of Ce is lower, the hydrogen consumption is also lower. However, the calculations of the surface ceria contribution to the reduction process show very similar values, 38% for Au-CeZrCo-A and 40% for Au-CeZrCo-B. The later indicates that the gold presence provokes cobalt rejection of the fluorite structure resulting in a higher H2 consumption at low temperature compensated by a higher surface ceria reduction contribution.
The high temperature peak of reduction (>500 °C) corresponding to the bulk cerium reduction shows a decrease in terms of hydrogen consumption when gold is present which can be directly related to the increase of the surface ceria reduction (
Table 1).
Regardless of the composition of the catalyst, there exists a temperature range free of reduction for all the samples, which allows the possibility to clearly distinguish the zone of partial reduction. This allows the use of catalytic systems in which cobalt is partially reduced and is chemically stable at the temperature at which the reaction of ethanol reforming occurs.
For CeZrCo samples, the zone I is the count of peaks 1, 2, and 3. Otherwise, the zone I in Au/CeZrCo samples is the count of peaks 1 and 2. By comparing the percentage of reduction of zone I and II of gold containing and not containing samples, it is possible to observe a similar area distribution, in which the hydrogen consumption is near to 40% of the total one.
3.1. CO Oxidation
The complete oxidation of carbon monoxide is one of the most studied reactions for gold-containing catalysts since the astonishing discovery of Haruta
et al. [
29] that very small gold nanoparticles exhibit catalytic activity at sub-ambient temperature. The method of preparation has a great influence on the catalytic behavior since it affects the gold particles’ size, morphology and oxidation state. Furthermore, gold catalysts activity is influenced by several parameters such as acidity or basicity of the support, the thermal treatment or the nature of activation process [
30]. Therefore the CO reaction serves as a probe reaction not only for the particle size but also to reveal subtle changes in the electronic state of gold or in the morphology of the particles, modifications which other techniques are at pains to reveal. The conversion of carbon monoxide as a function of the temperature is presented in
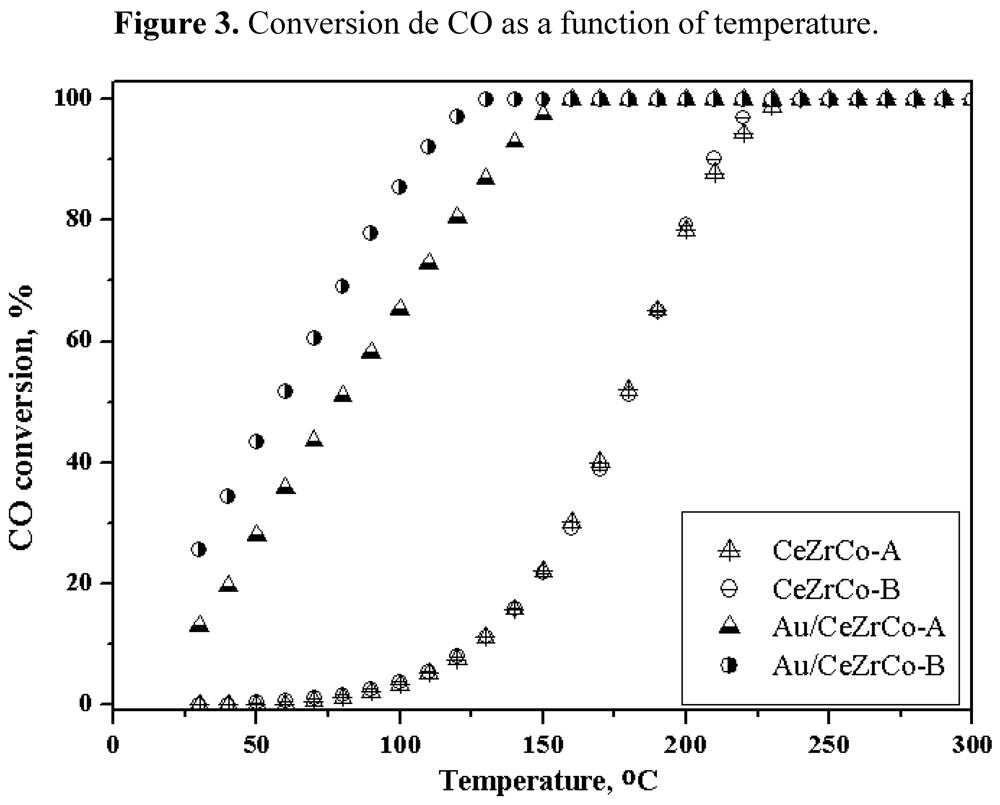
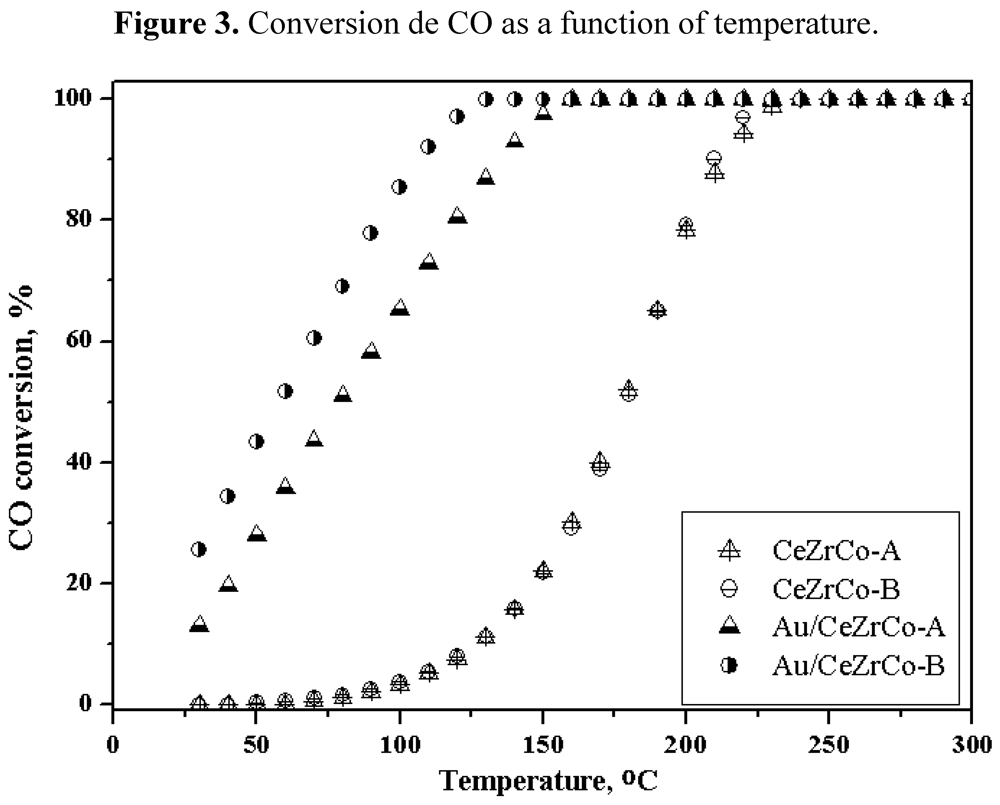
Figure 3 for all the samples with or without gold.
Figure 3.
Conversion de CO as a function of temperature.
Figure 3.
Conversion de CO as a function of temperature.
The Ce/Zr ratio does not influence the activity of the gold free samples. The complete CO oxidation is obtained at 225 °C. When gold is deposited on the mixed oxides phase the behavior of the catalysts changes significantly. They become very active, showing even an activity at room temperature and with a total conversion at ca. 125 °C. The Au/CeZrCo-B sample is slightly more active than Au/CeZrCo-A sample, which could be attributed to the difference in the gold particle size observed for these samples.
3.2. Ethanol Steam Reforming
In order to elucidate the influence of gold on the Ce-Zr-Co catalytic behavior, the reaction of ethanol steam reforming was carried out at low temperature (440 °C). This temperature favors the production of acetone, by acetaldehyde condensation, and the deactivation by carbonaceous species is impeded. For the same reasons, the catalysts were not reduced before the catalytic test, and were therefore used as prepared [
25]. The gas phase distribution—
i.e., the selectivity—is strongly influenced by the reaction temperature and the active phase reduction state (metallic phase). At temperatures higher than 550 °C, only the main products (H
2, CO, CO
2, CH
4) are formed, as a consequence of the transformation of the intermediate adsorbed compounds [
8,
25] and of some thermodynamics restrictions. A similar behavior is observed when the active phase is in the metallic state, which is so influential that it hides the role of promoters occurring only in the deactivation route and affects long-term stability. The presence of trace of propylene can be attributed to acetone hydrogenation [
38].
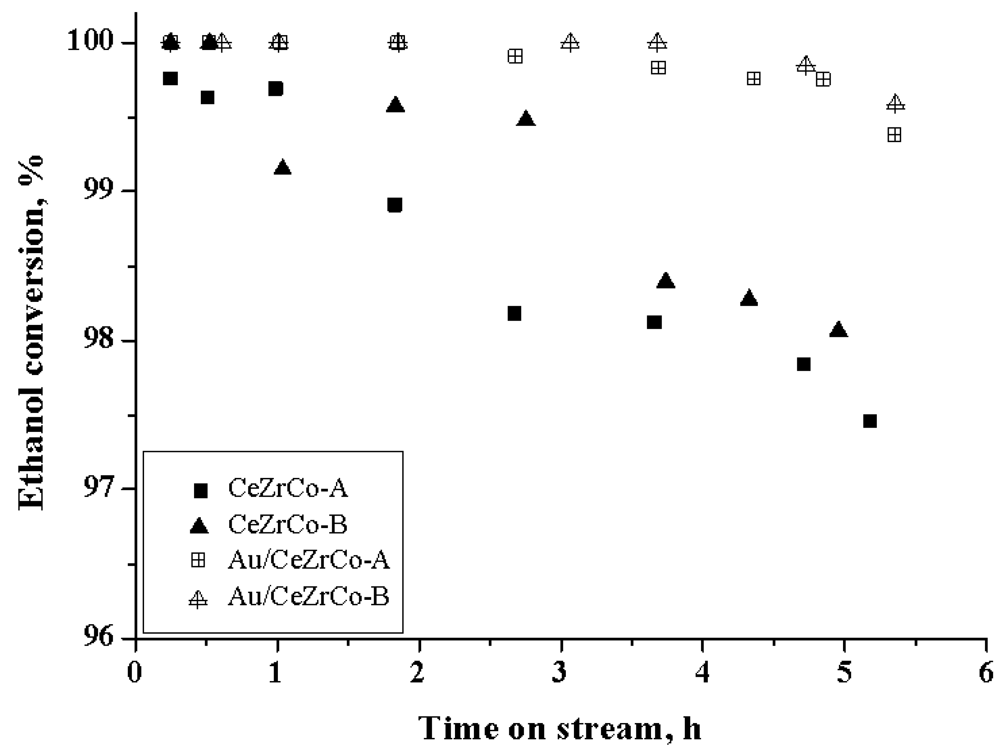
The evolution of the catalytic conversion of the samples with the reaction time is given in
Figure 4.
Figure 4.
Ethanol conversion as a function of time on stream.
Figure 4.
Ethanol conversion as a function of time on stream.
All the catalytic systems are very active for the reaction with an ethanol conversion close to 100% in the first hours of the reaction, despite the fact that no reducing treatment was performed. The change in the Ce/Zr ratio does not seem to have a great influence on the ethanol conversion but has an effect in the catalytic selectivity, modifying the acetone and ethylene production. In the case of Ce-Zr-Co samples, the activity decreases slowly down to 98% after 5 h of reaction whereas for the Au/CeZrCo samples, the activity remains higher than 99.8% for the first 5 h of reaction. The gold addition seems to change the kinetics of deactivation and to stabilize the conversion for a longer time probably due to a higher activity to transform the adsorbed intermediates or by changing the reaction route. The process of carbonaceous deposition and/or active surface change by the adsorption of intermediary products, as principal causes of deactivation [
39] seems to be inhibited or delayed.
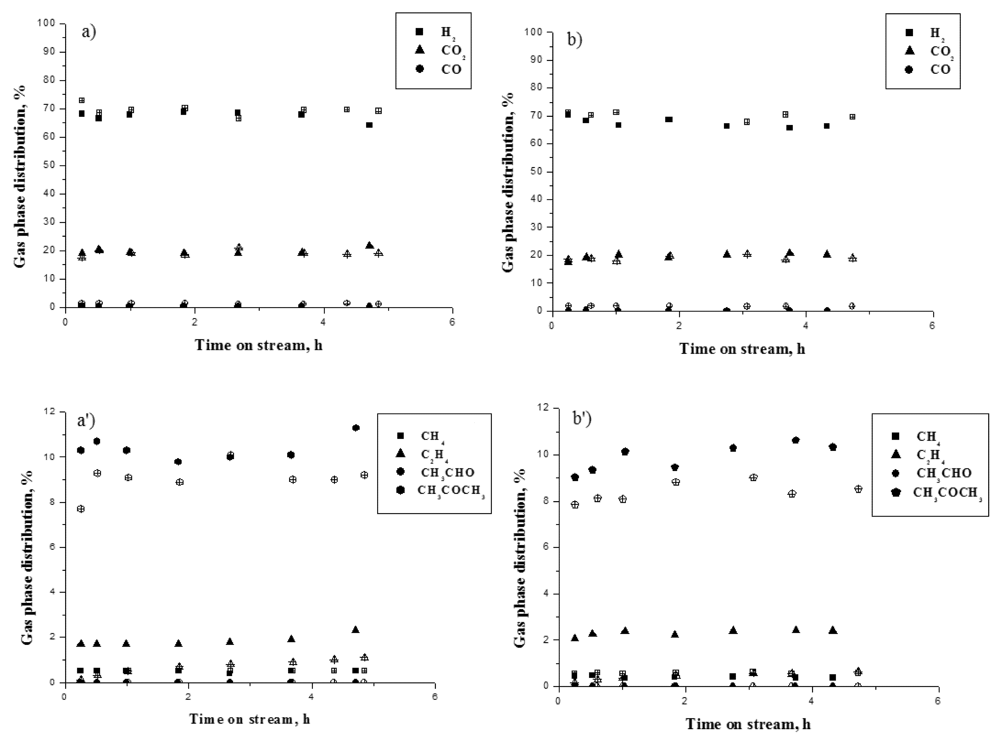
In
Figure 5, the gas phase distribution for both gold containing catalysts compared with the bare Ce-Zr-Co catalysts. In the
Figure 5a,a′ the molar product distribution of the main products (H
2, CO
2 and CO) is presented, whereas in
Figure 5b,b′ the molar distribution of the secondary products (CH
4, C
2H
4, CH
3COCH
3) are given, which gives some insights into the reaction path.
Figure 5.
Gas phase distribution as a function of time on stream: (a,a′) CeZrCo-A catalysts; (b,b′) CeZrCo-B catalysts. Full symbols—CeZrCo samples, empty symbols—Au/CeZrCo samples.
Figure 5.
Gas phase distribution as a function of time on stream: (a,a′) CeZrCo-A catalysts; (b,b′) CeZrCo-B catalysts. Full symbols—CeZrCo samples, empty symbols—Au/CeZrCo samples.
The
Table 2 is summarizing the average molar distribution of products during the whole time of reaction.
Table 2.
Average distribution (in %mol) in the gas phase on CeZrCo and Au/CeZrCo samples.
Table 2.
Average distribution (in %mol) in the gas phase on CeZrCo and Au/CeZrCo samples.
| Sample | CH4 | CO2 | C2H4 | H2 | CO | C3H6 | CH3COCH3 |
|---|
| CeZrCo-A | 0.5 | 19.6 | 1.8 | 67.4 | 0.3 | 0.1 | 10.4 |
| Au/CeZrCo-A | 0.5 | 19.0 | 0.7 | 69.4 | 1.2 | 0.1 | 9.1 |
| CeZrCo-B | 0.4 | 19.6 | 2.3 | 67.4 | 0.2 | 0.1 | 9.9 |
| Au/CeZrCo-B | 0.6 | 19.0 | 0.5 | 69.7 | 1.7 | 0.1 | 8.4 |
For CeZrCo and Au/CeZrCo samples, independently of Ce/Zr ratio, the molar distribution of the main products in the gas phase remains almost stable with time on stream, independently of the conversion.
However, in the presence of gold, the gas phase distribution of H2 and minor products, with the exception of methane, presents some significant changes resulting from a different reaction pathway, which favors the activation of ethanol through CH3CHO instead of the one through C2H4. The significant decrease of ethylene in the presence of gold tends to show that the dehydration of ethanol (acid way) decreased. Consequently the second activation way by dehydrogenation into acetaldehyde is favored.
In the presence of Au, a high selectivity toward hydrogen is observed, along with a higher CO production. A decrease of both of acetone and ethylene is obtained, but in the case of this latter product, it is more marked. The CH4 and CO2 production remains almost constant despite the addition of gold. This behavior suggests that in the presence of gold, the ethanol-reforming reaction proceeds mainly on CeZrCo actives sites preferentially through the acetaldehyde intermediate. The preferential decomposition of this intermediate by decarboxylation produces CO and CH4, or its corresponding adsorbed species. Therefore, acetone production is affected since it is formed by acetaldehyde condensation reaction.
The adsorbed species are rapidly evolved in the steam reforming reaction increasing the CO and H
2 amounts (
Scheme 1). The major CO interaction in the presence of gold could be confirmed by the catalytic results as shown in the total oxidation of CO. Otherwise, the slight increase in CO
2 amounts in gold containing samples could be explained through the water gas shift reaction (WGSR). It is also interesting to note the absence of acetaldehyde formation during the course of the reaction, despite the slight decrease in the conversion, thus confirming the high rate of conversion of acetaldehyde to other products.
Another interesting result concerns the increase in the amount of ethylene produced at a higher Ce/Zr ratio, which is attributed to the inclusion of Zr in the structure, decreasing the acidity of the support and therefore favoring the reaction pathway to the acetone production. The effect of Ce/Zr molar ratio on redox behavior and of the acid-base properties acid-base is well known. A catalyst with acidic properties favors the dehydration route to produce ethylene and water whereas catalysts with basic properties favor the dehydrogenation route producing acetaldehyde and hydrogen. At low temperatures of reaction, acetaldehyde is converted by ketonization reactions to acetone.
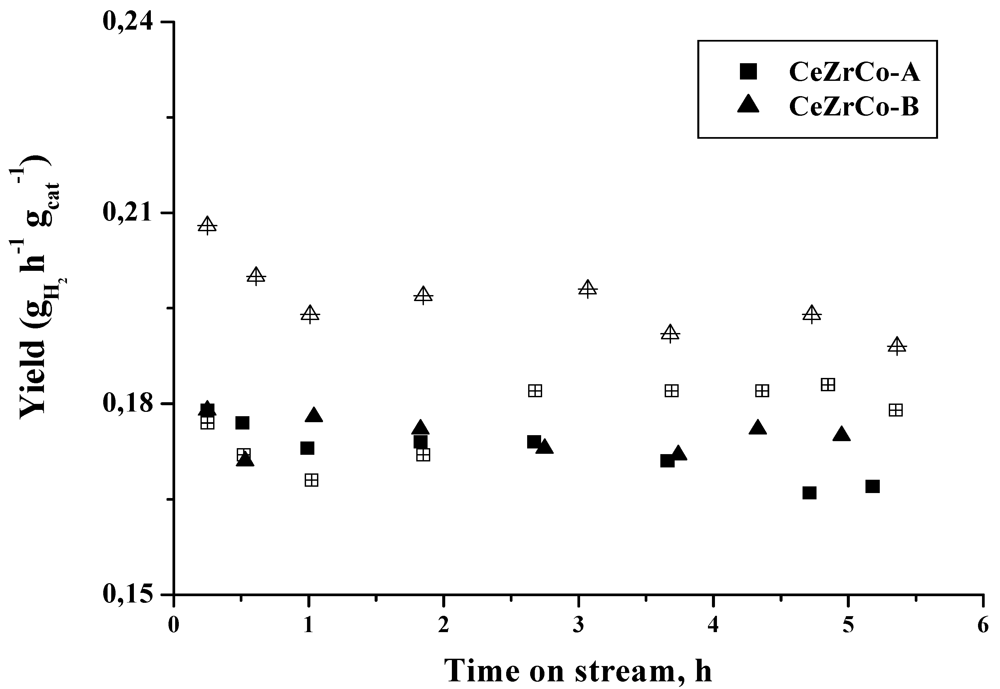
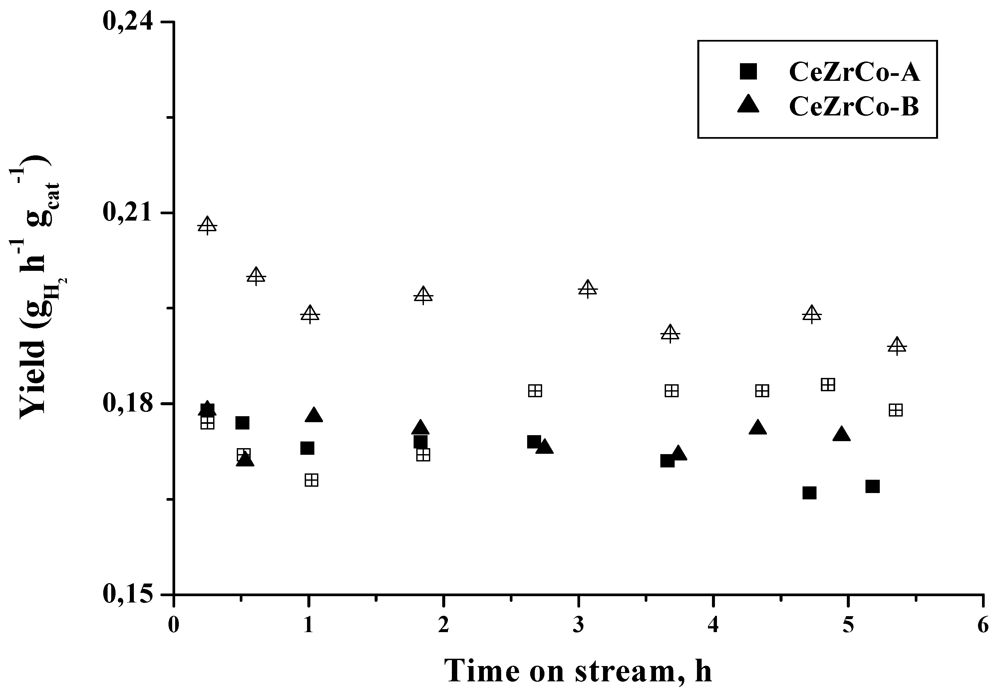
When the hydrogen yield is calculated (
Figure 6), a higher hydrogen production, by gram of catalyst and per hour is obtained for the gold containing catalysts.
Figure 6.
Hydrogen yield as a function of time on stream. (Full symbols: CeZrCo samples, empty symbols: Au/CeZrCo samples).
Figure 6.
Hydrogen yield as a function of time on stream. (Full symbols: CeZrCo samples, empty symbols: Au/CeZrCo samples).
The differences in the hydrogen yield obtained in CeZrCo and Au/CeZrCo catalyst, are influenced by the decrease of the conversion but not by the hydrogen selectivity changes and as a consequence, the hydrogen yield is higher in the gold containing samples.
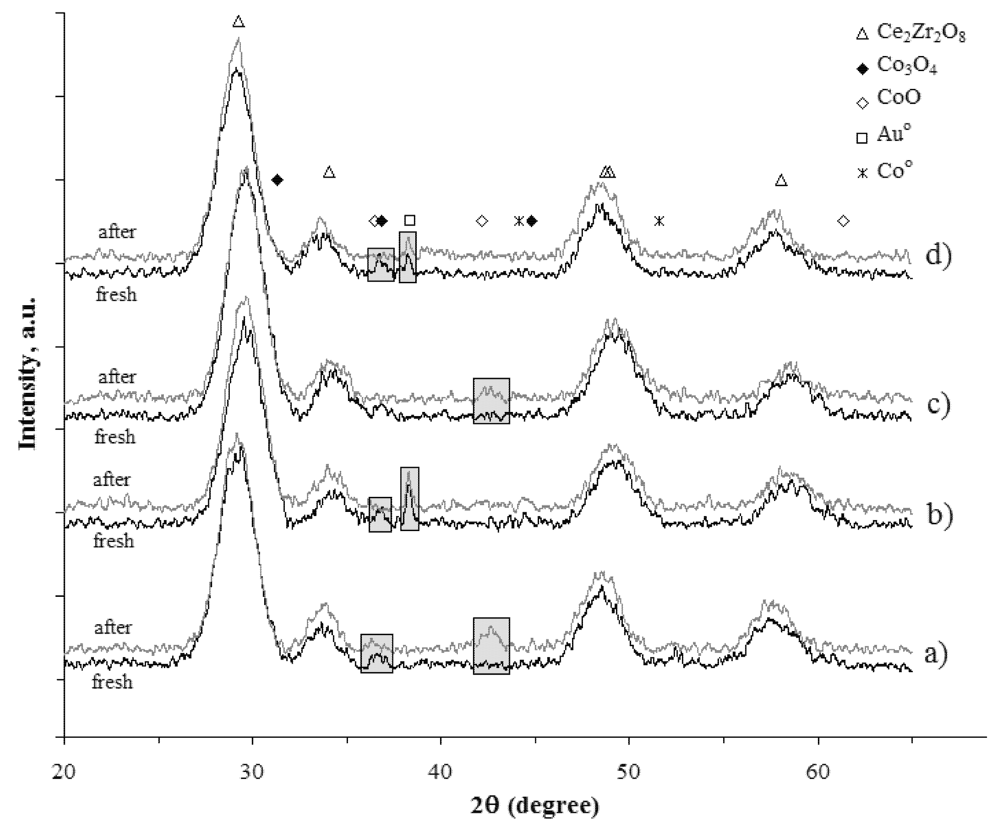
In
Figure 7 the diffractograms of the CeZrCo and Au/CeZrCo catalyst, before and after steam reforming reaction are presented.
The samples were evaluated as catalyst without any reduction process, in order to study the gold influence in the Ce-Zr-Co mixed oxides catalytic activity and stability. By comparing the X-ray diffraction before and after the test, it is possible to observe the reduction under the reactive mixture. In all samples, the diffraction pattern corresponding to Co3O4 in the spinel phase, as observed in the case of the fresh samples, is no longer observed after the catalytic test, proving the reduction of inserted cobalt to a lower oxidation state.
For the gold free CeZrCo catalyst (
Figure 7a,b) a peak related with CoO is observed at
ca. 2θ = 43°, whereas for the gold containing CeZrCo catalyst (
Figure 7b,c) the apparition of this peak is not obtained and no indication of a deeper reduction into Co
0 is demonstrated by XRD. This difficulty could be attributed to a smaller particle size of cobalt in the reduced state, therefore not easily detectable but most probably, it corresponds to a higher degree of dispersion due to the presence of gold. It is possible to state the reduction of the outer cobalt structure that occurred, as based on the observation of the Co
3O
4 peak decreasing. However, the reduction of the cobalt inserted in the structure was not obtained since no changes in the fluorite structure oxide peak diffraction were observed. In respect to the gold stability, it seems that sintering under the reactive mixture during the catalytic test did not occur, since no remarkable changes in gold corresponding structure are observed.
Figure 7.
X-Ray Diffractograms of catalysts, before and after ethanol steam reforming, test of: (a) CeZrCo-A; (b) Au/CeZrCo-A; (c) CeZrCo-B; (d) Au/CeZrCo-B.
Figure 7.
X-Ray Diffractograms of catalysts, before and after ethanol steam reforming, test of: (a) CeZrCo-A; (b) Au/CeZrCo-A; (c) CeZrCo-B; (d) Au/CeZrCo-B.

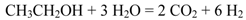

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}