
Aerobic Oxidative Desulfurization of Liquid Fuel Catalyzed by P–Mo–V Heteropoly Acids in the Presence of Aldehyde



Abstract
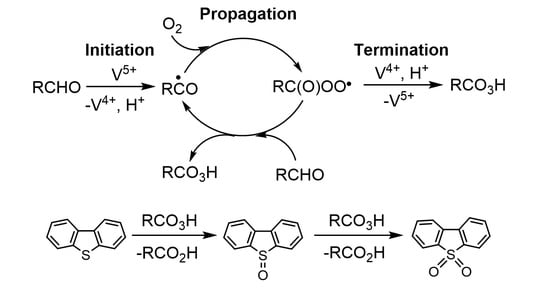
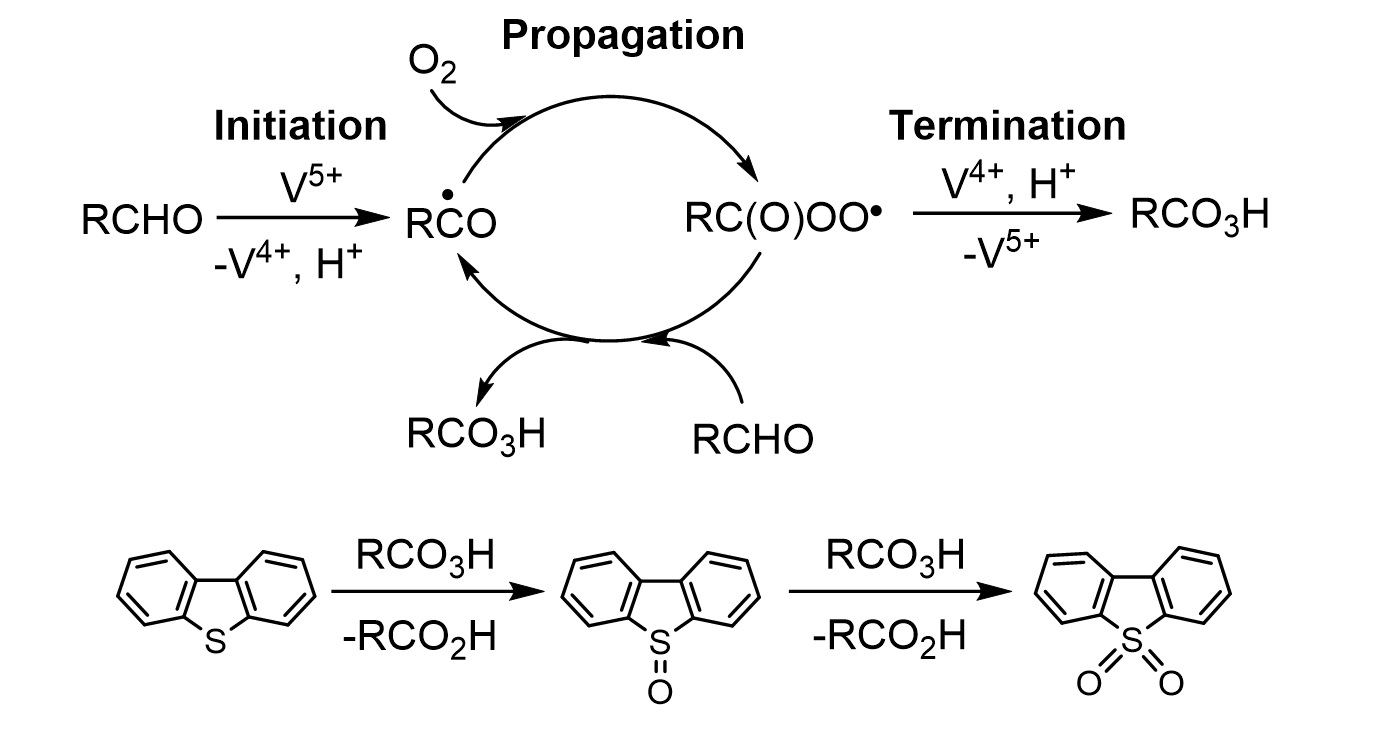
:
1. Introduction
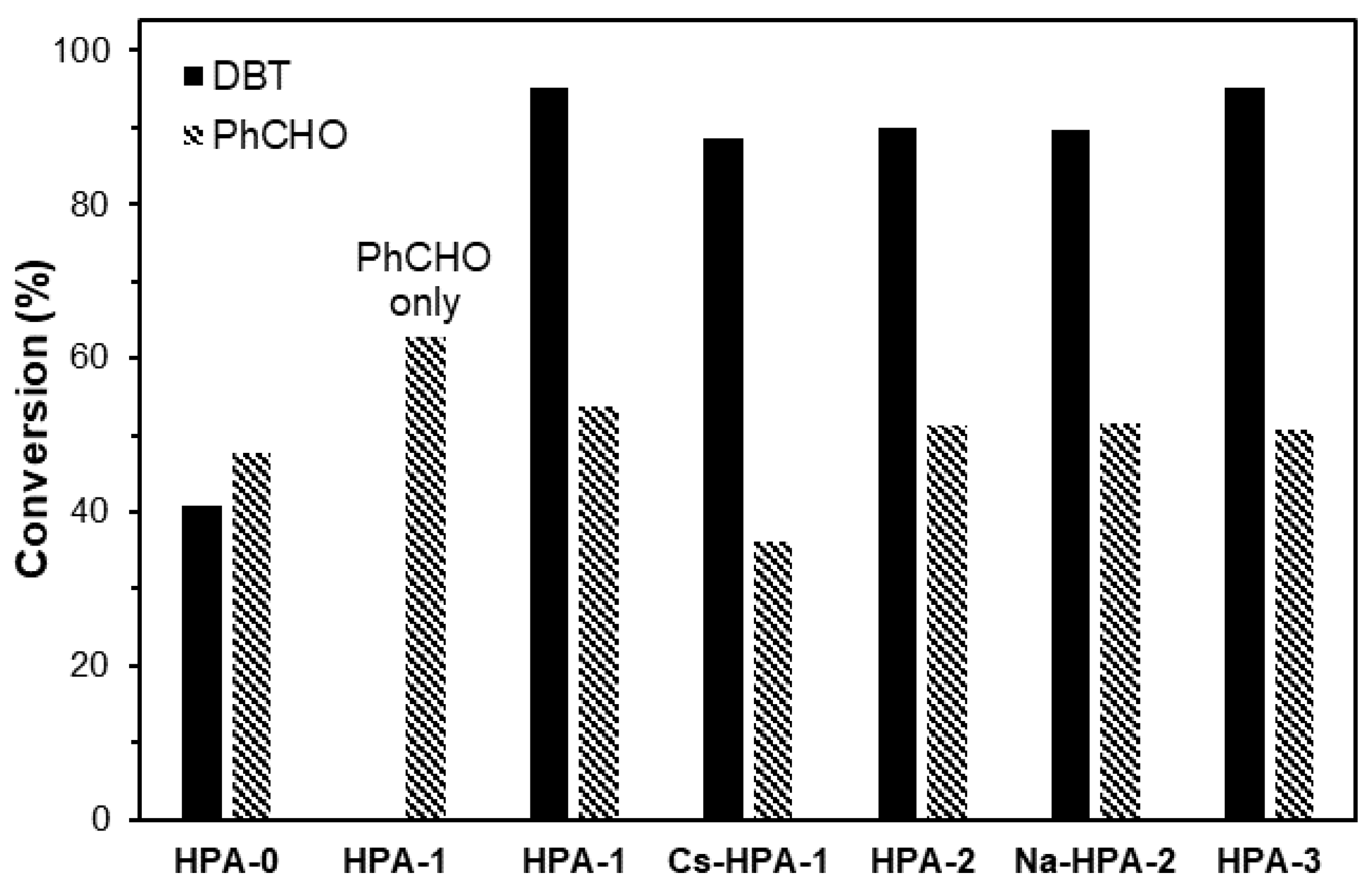
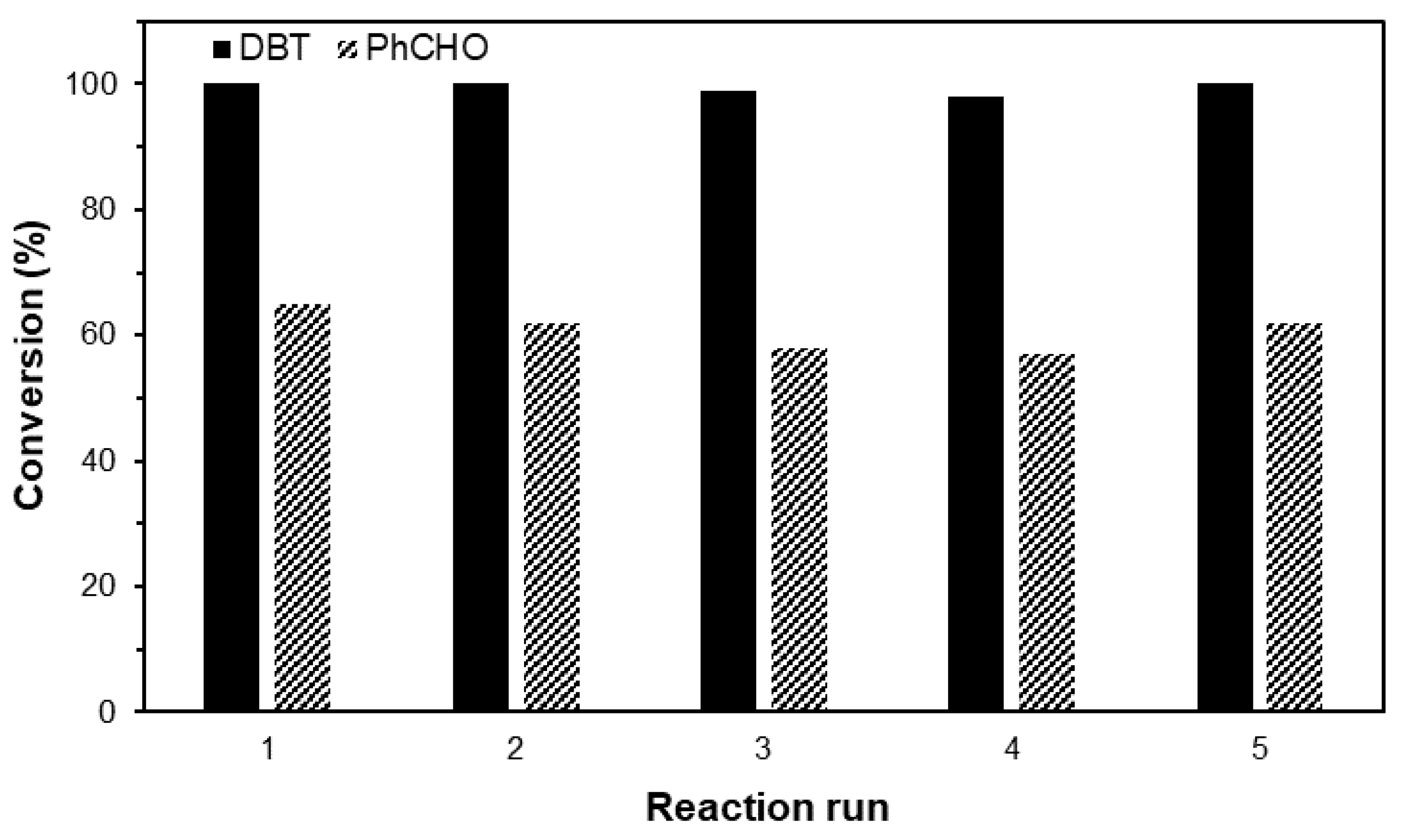
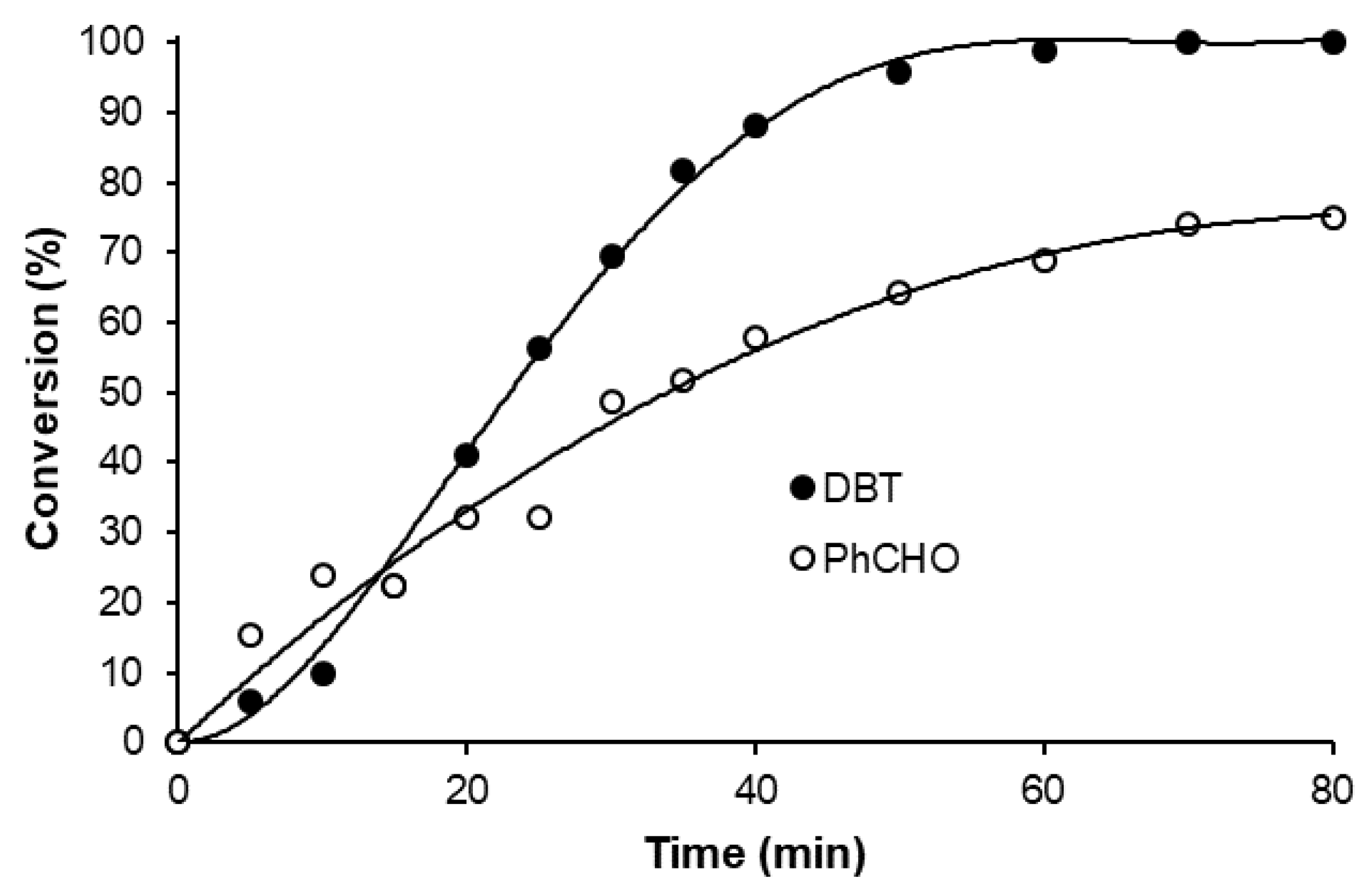
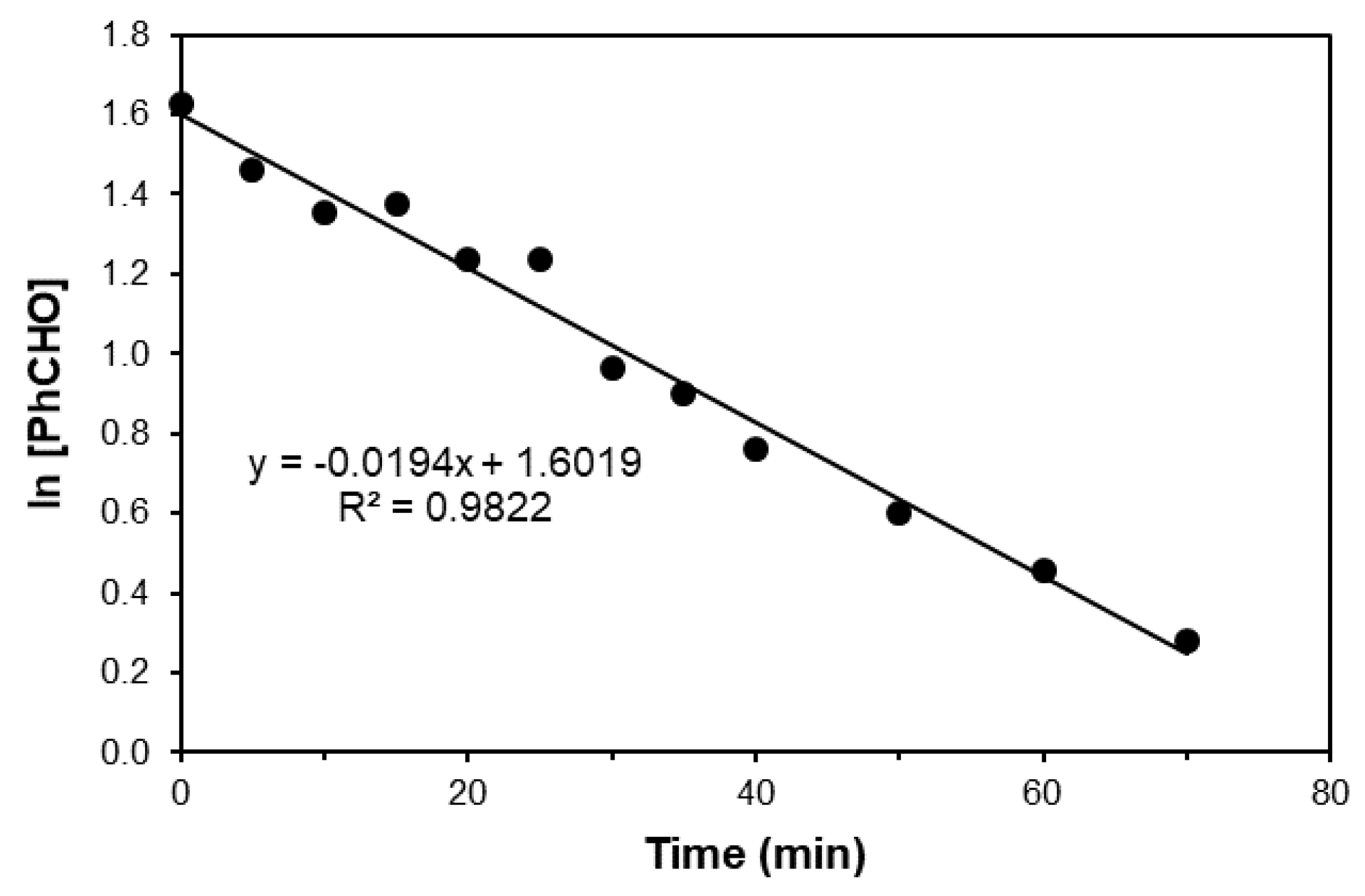
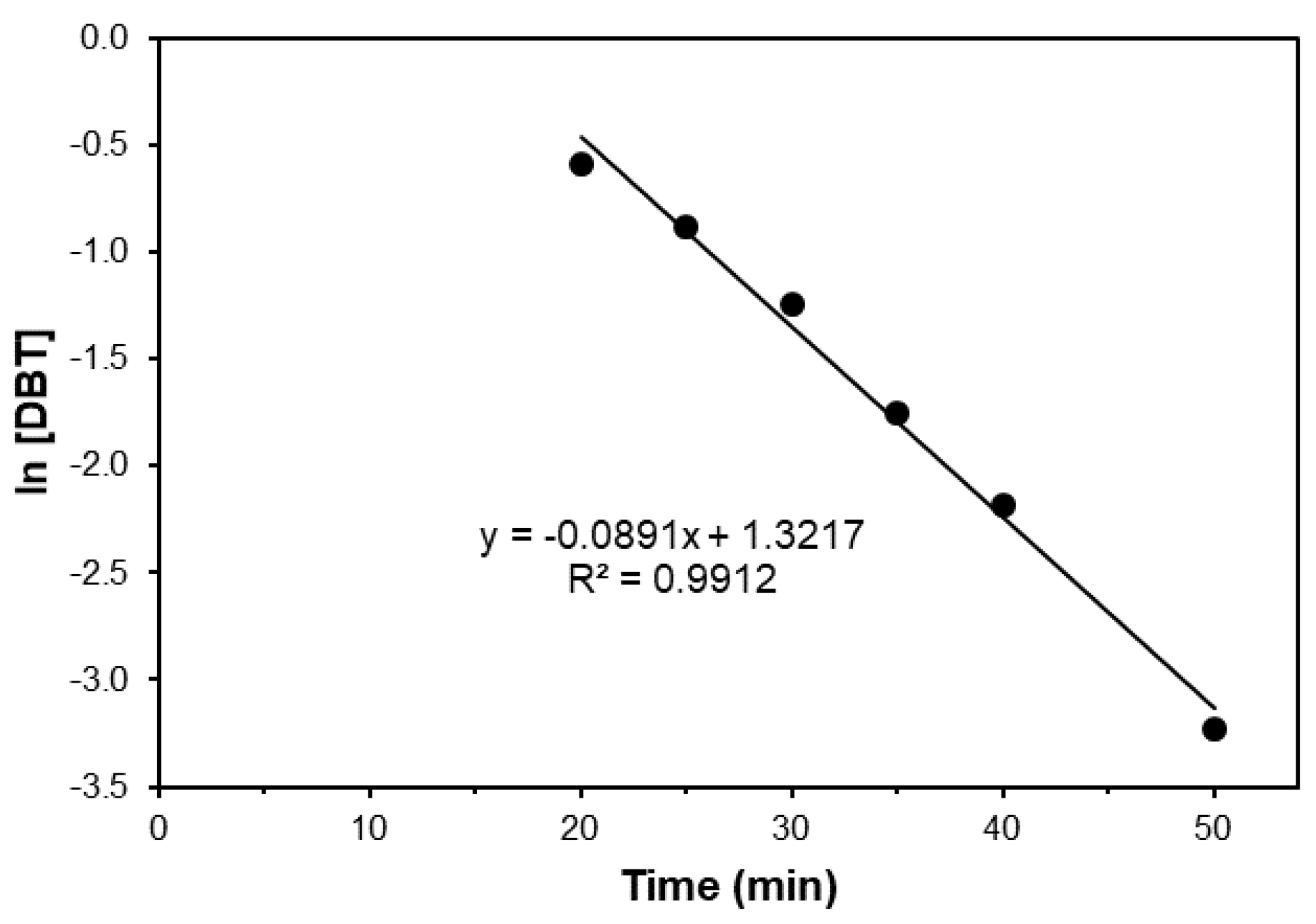
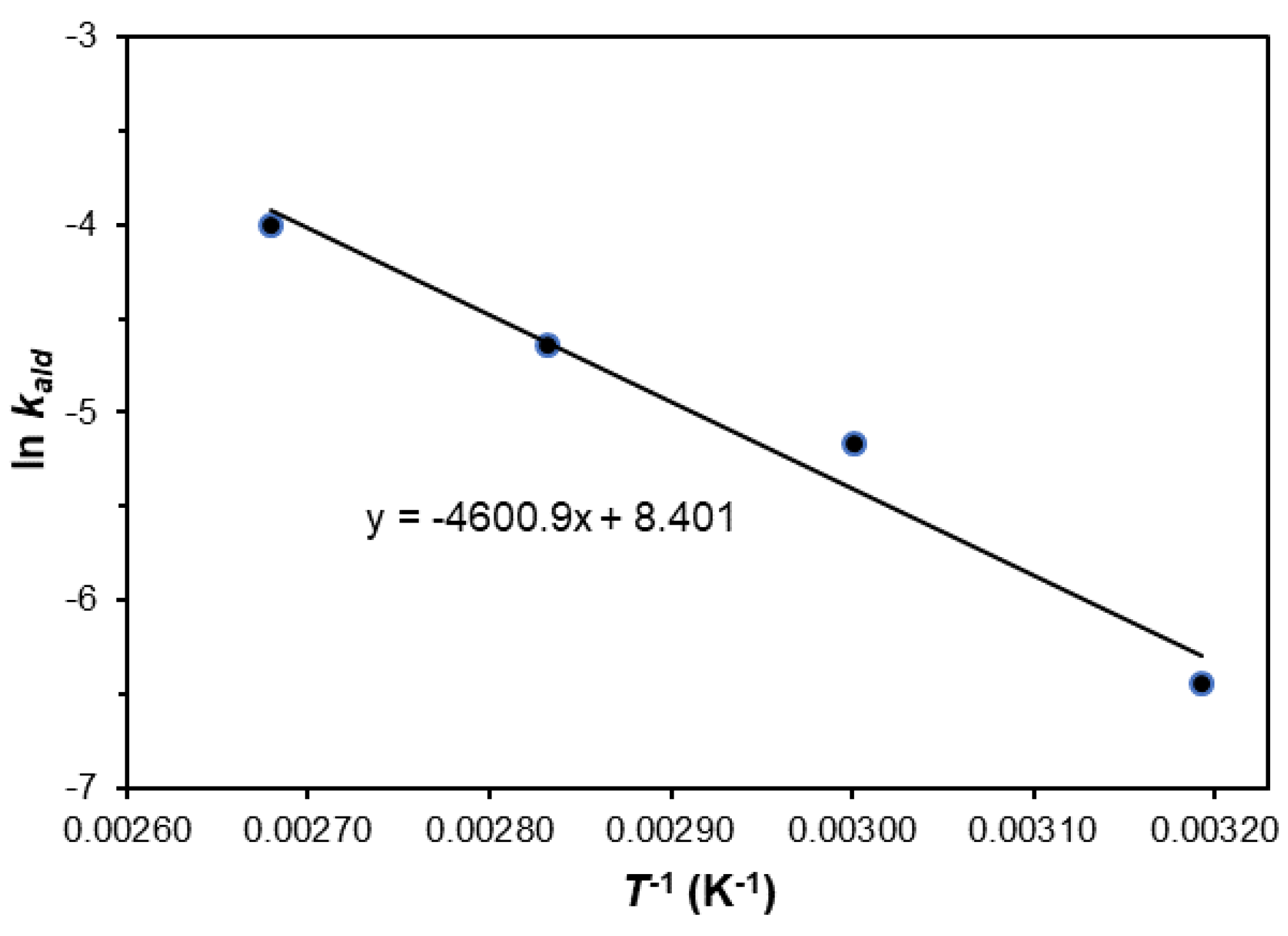
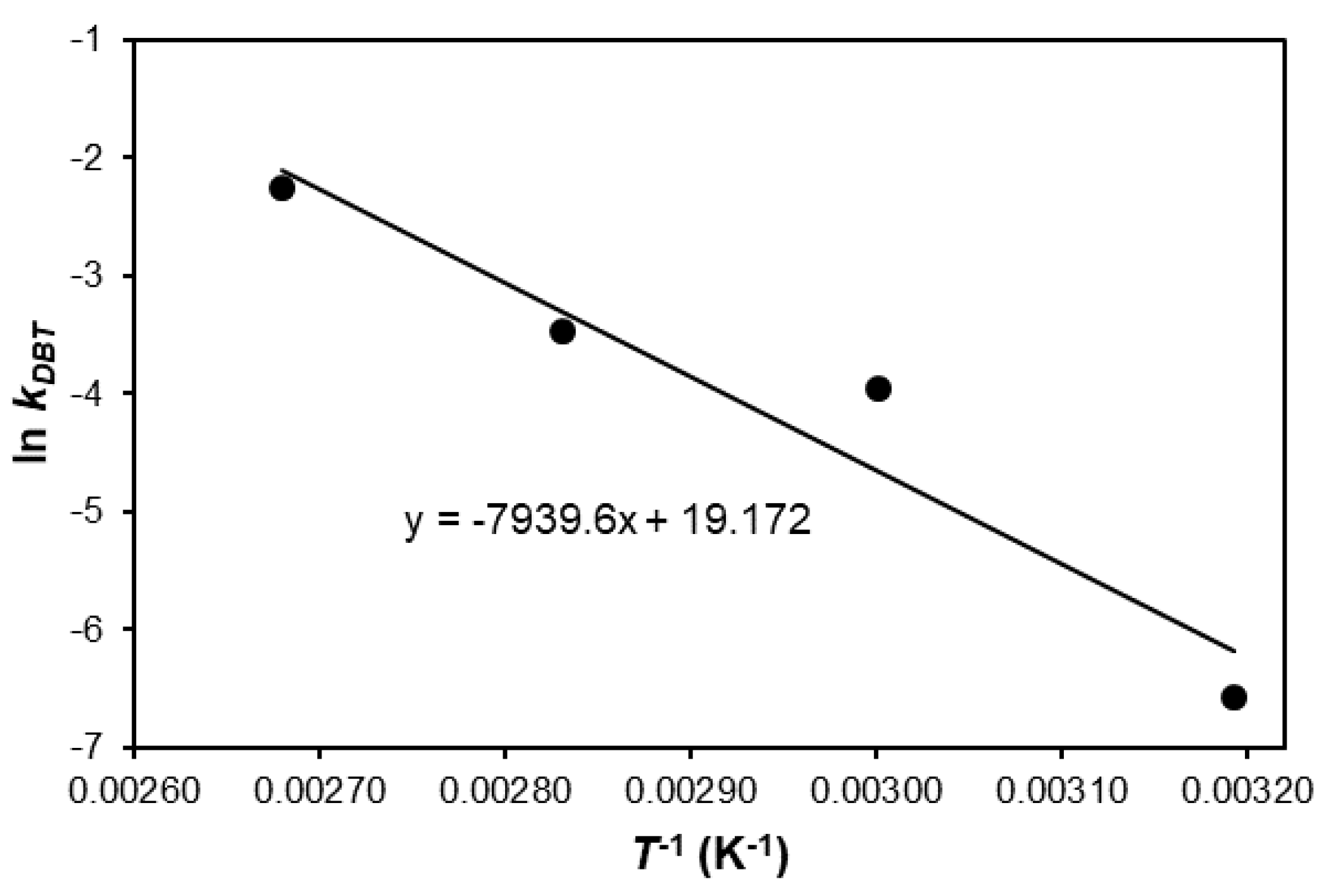
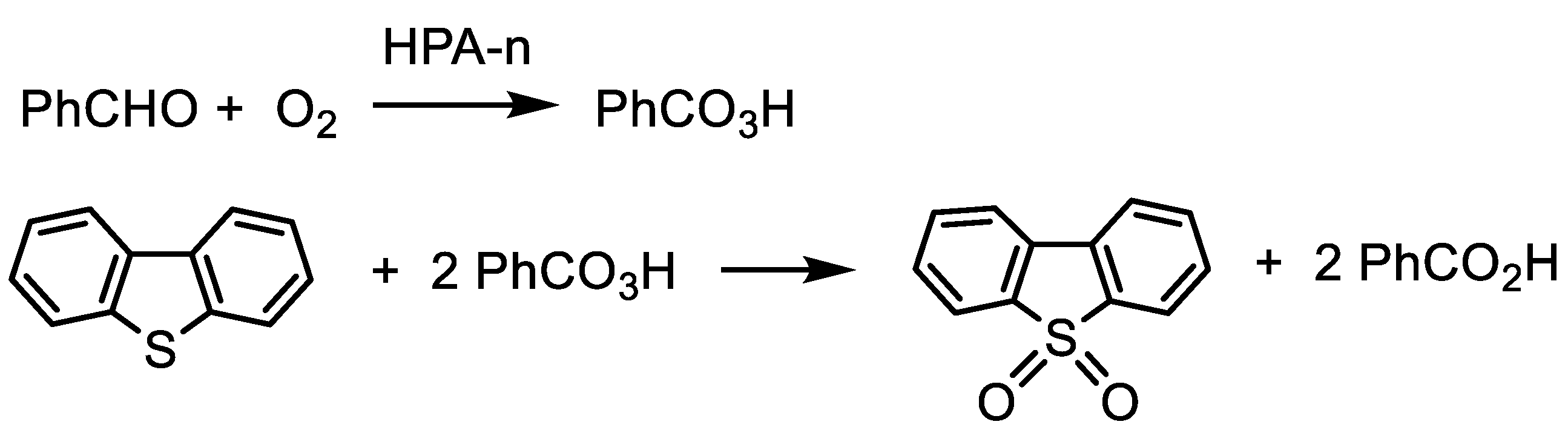
2. Results and Discussion
3. Materials and Methods
3.1. Chemicals and Catalysts
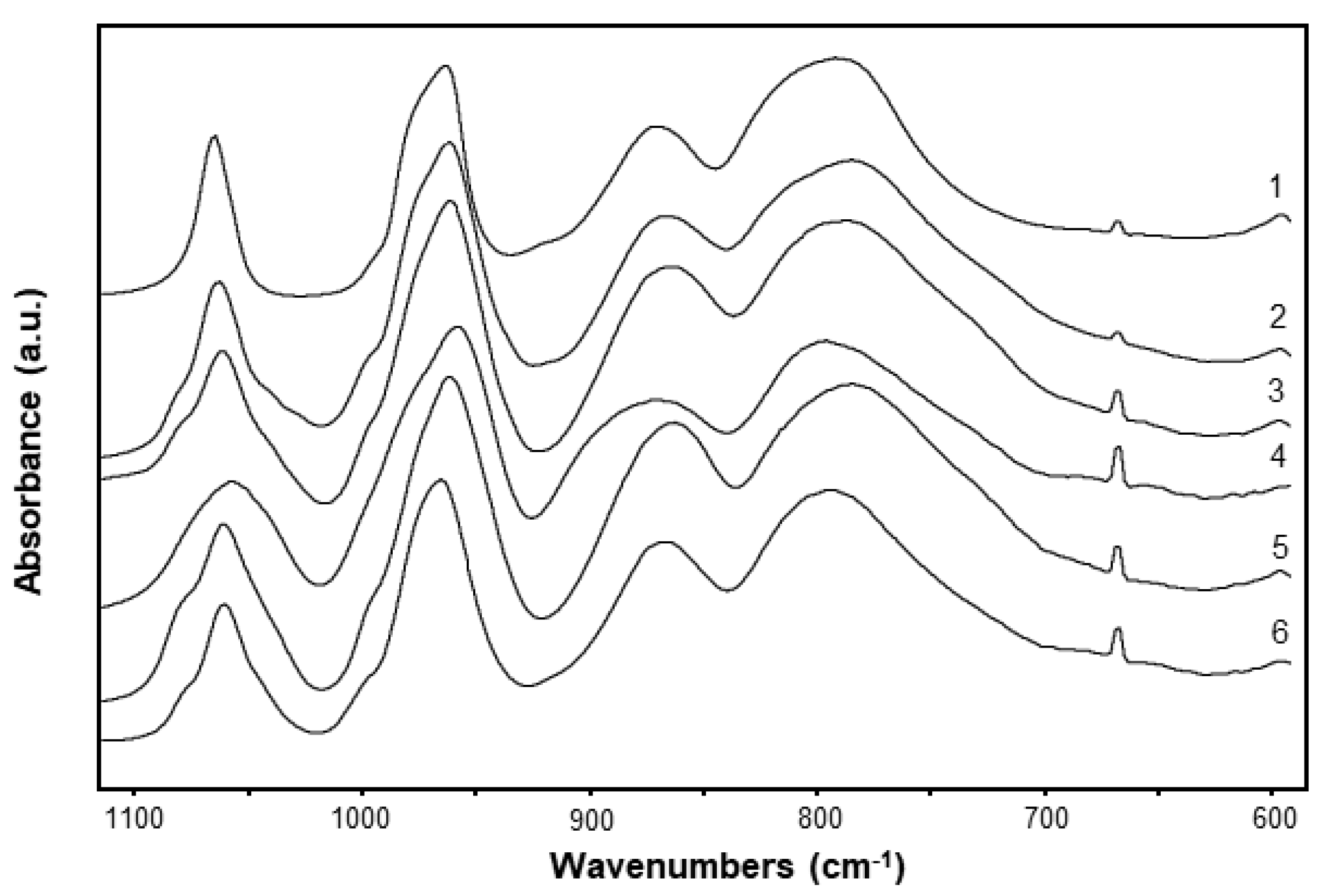
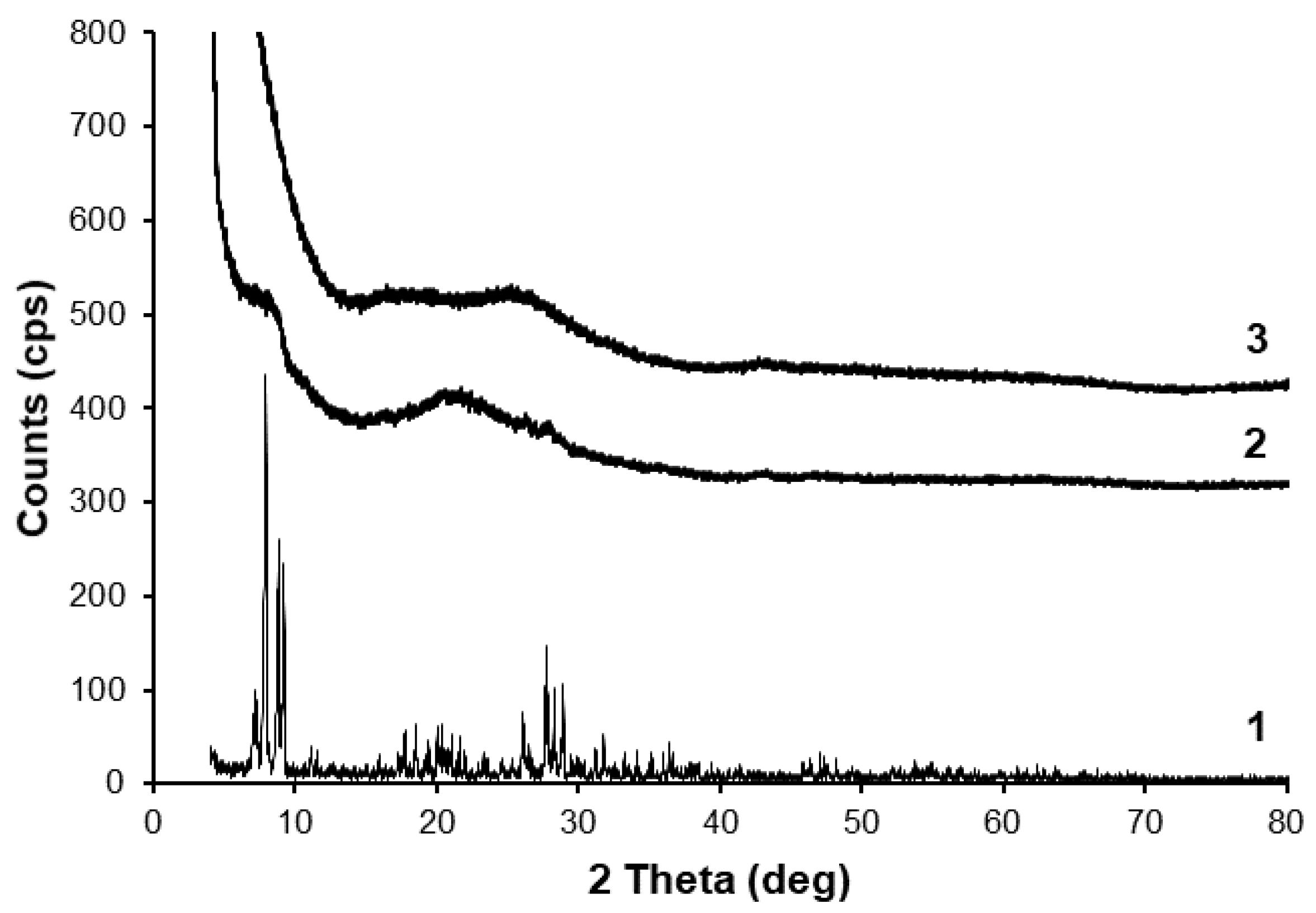
3.2. Catalyst Characterization
3.3. Reaction Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Babich, I.V.; Moulijn, J.A. Science and technology of novel processes for deep desulfurization of oil refinery streams. Fuel 2003, 82, 607–631. [Google Scholar] [CrossRef]
- Prins, R. Hydrotreating. In Handbook of Heterogeneous Catalysis; Ertl, G., Knözinger, H., Schüth, F., Weitkamp, J., Eds.; Wiley-VCH: Hoboken, NJ, USA, 2008; Volume 6, pp. 2695–2718. [Google Scholar]
- Saha, B.; Vedachalam, S.; Dalai, A.K. Review on recent advances in adsorptive desulfurization. Fuel Process. Technol. 2021, 214, 106685. [Google Scholar] [CrossRef]
- Bhutto, A.W.; Abro, R.; Gao, S.; Abbas, T.; Chen, X.; Yu, G. Oxidative desulfurization of fuel oils using ionic liquids: A review. J. Taiwan Inst. Chem. Eng. 2016, 62, 84–97. [Google Scholar] [CrossRef]
- Jiang, Z.; Lü, H.; Zhang, Y.; Li, C. Oxidative desulfurization of fuel oils. Chin. J. Catal. 2011, 32, 707–715. [Google Scholar] [CrossRef]
- Eseva, E.A.; Akopyan, A.V.; Anisimov, A.V.; Maksimov, A.L. Oxidative desulfurization of hydrocarbon feedstock using oxygen as oxidizing agent (a review). Petroleum Chem. 2020, 60, 979–990. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, R. Recent advances on catalysts and systems for the oxidation of thiophene derivatives in fuel oil with molecular oxygen. Mini Rev. Org. Chem. 2018, 15, 488–497. [Google Scholar] [CrossRef]
- Collins, F.M.; Lucy, A.R.; Sharp, C. Oxidative desulphurisation of oils via hydrogen peroxide and heteropolyanion catalysis. J. Mol. Catal. A 1997, 117, 397–403. [Google Scholar] [CrossRef]
- Komintarachat, C.; Trakarnpruk, C. Oxidative desulfurization using polyoxometalates. Ind. Eng. Chem. Res. 2006, 45, 1853–1856. [Google Scholar] [CrossRef]
- Shojaei, A.F.; Rezvani, M.A.; Loghmani, M.H. Comparative study on oxidation desulphurization of actual gas oil and model sulfur compounds with hydrogen peroxide promoted by formic acid: Synthesis and characterization of vanadium containing polyoxometalate supported on anatase crushed nanoleaf. Fuel Process. Technol. 2014, 118, 1–6. [Google Scholar] [CrossRef]
- Mirante, F.; Dias, L.; Silva, M.; Ribeiro, S.O.; Corvo, M.C.; de Castro, B.; Granadeiro, C.M.; Balula, S.S. Efficient heterogeneous polyoxometalate-hybrid catalysts for the oxidative desulfurization of fuels. Catal. Commun. 2018, 104, 1–8. [Google Scholar] [CrossRef]
- Craven, M.; Yahya, R.; Kozhevnikova, E.F.; Robertson, C.M.; Steiner, A.; Kozhevnikov, I.V. Alkylaminophosphazenes as efficient and tuneable phase-transfer agents for polyoxometalate-catalyzed biphasic oxidation with hydrogen peroxide. ChemCatChem 2016, 8, 200–208. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V. Catalysts for Fine Chemical Synthesis: Catalysis by Polyoxometalates; Wiley: West Sussex, UK, 2002. [Google Scholar]
- Li, J.; Yang, Z.; Li, S.; Jin, Q.; Zhao, J. Review on oxidative desulfurization of fuel by supported heteropolyacid catalysts. J. Ind. Eng. Chem. 2020, 82, 1–16. [Google Scholar] [CrossRef]
- Taghizadeh, M.; Mehrvarz, E.; Taghipour, A. Polyoxometalate as an effective catalyst for the oxidative desulfurization of liquid fuels: A critical review. Rev. Chem. Eng. 2020, 36, 831–858. [Google Scholar] [CrossRef]
- Xu, J.; Zhu, Z.; Su, T.; Liao, T.; Deng, C.; Hao, D.; Zhao, Y.; Ren, W.; Lü, H. Green aerobic oxidative desulfurization of diesel by constructing an Fe-Anderson type polyoxometalate and benzene sulfonic acid-based deep eutectic solvent biomimetic cycle. Chin. J. Catal. 2020, 41, 868–876. [Google Scholar] [CrossRef]
- Tang, N.; Zhang, Y.; Lin, F.; Lu, H.; Jiang, Z.; Li, C. Oxidation of dibenzothiophene catalyzed by [C8H17N(CH3)3]3H3V10O28 using molecular oxygen as oxidant. Chem. Commun. 2012, 48, 11647–11649. [Google Scholar] [CrossRef] [PubMed]
- Li, J.K.; Xu, Y.Q.; Hu, C.W. In situ synthesis of a novel dioxidovanadium-based nickel complex as catalyst for deep oxidative desulfurization with molecular oxygen. Inorg. Chem. Commun. 2015, 60, 12–14. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, J.; Li, H.; Wei, Y.; Fu, Y.; Liao, W.; Zhu, L.; Chen, G.; Zhu, W.; Li, H. Tuning the electrophilicity of vanadium-substituted polyoxometalate based ionic liquids for high-efficiency aerobic oxidative desulfurization. Appl. Catal. B 2020, 271, 118936. [Google Scholar] [CrossRef]
- Claußnitzer, J.; Bertleff, B.; Korth, W.; Albert, J.; Wasserscheid, P.; Jess, A. Kinetics of triphase extractive oxidative desulfurization of benzothiophene with molecular oxygen catalyzed by HPA-5. Chem. Eng. Technol. 2020, 43, 465–475. [Google Scholar] [CrossRef]
- Wang, R.; Zhao, Y.; Kozhevnikov, I.V.; Zhao, J. An ultrasound enhanced catalytic ozonation process for the ultra-deep desulfurization of diesel oil. New J. Chem. 2020, 44, 15467–15474. [Google Scholar] [CrossRef]
- Pope, M.T. Heteropoly and Isopoly Oxometalates; Springer: Berlin/Heidelberg, Germany, 1983. [Google Scholar]
- Murata, S.; Murata, K.; Kidena, K.; Nomura, M. A novel oxidative desulfurization system for diesel fuels with molecular oxygen in the presence of cobalt catalysts and aldehydes. Energy Fuels 2004, 18, 116–121. [Google Scholar] [CrossRef]
- Lu, H.; Gao, J.; Jiang, Z.; Yang, Y.; Song, B.; Li, C. Oxidative desulfurization of dibenzothiophene with molecular oxygen using emulsion catalysis. Chem. Commun. 2007, 150–152. [Google Scholar] [CrossRef] [PubMed]
- Dumont, V.; Oliviero, L.; Mauge, F.; Houalla, M. Oxidation of dibenzothiophene by a metal–oxygen–aldehyde system. Catal. Today 2008, 130, 195–198. [Google Scholar] [CrossRef]
- Rao, T.V.; Krishna, P.M.; Paul, D.; Nautiyal, B.R.; Kumar, J.; Sharma, Y.K.; Nanoti, S.M.; Sain, B.; Garg, M.O. The oxidative desulfurization of HDS diesel: Using aldehyde and molecular oxygen in the presence of cobalt catalysts. Petrol. Sci. Technol. 2011, 29, 626–632. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Z.; Zhu, W.; Wu, P.; Jiang, W.; Zhang, M.; Li, H.; Zhu, W.; Li, H. One-pot extraction and oxidative desulfurization of fuels with molecular oxygen in low-cost metal-based ionic liquids. Energy Fuels 2017, 31, 1376–1382. [Google Scholar] [CrossRef]
- Teles, J.H.; Hermans, I.; Franz, G.; Sheldon, R.A. Oxidation, Ullmann’s Encyclopedia of Industrial Chemistry; Wiley–VCH: Weinheim, Germany, 2015. [Google Scholar]
- Weissermel, K.; Arpe, H.J. Industrial Organic Chemistry, 4th ed.; Wiley–VCH: Weinheim, Germany, 2003. [Google Scholar]
- Li, J.; Chu, X.; Tian, S. Research on determination of total acid number of petroleum using mid-infrared attenuated total reflection spectroscopy. Energy Fuels 2012, 26, 5633–5637. [Google Scholar]
- Kozhevnikov, I.V.; Matveev, K.I. Homogeneous catalysts based on heteropoly acids. Appl. Catal. 1983, 5, 135–150. [Google Scholar] [CrossRef]
- Kozhevnikov, I.V. Catalysis by heteropoly acids and multicomponent polyoxometalates in liquid-phase reactions. Chem. Rev. 1998, 98, 171–198. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, I.A.; Schreiber, R.E.; Neumann, R. Dioxygen in polyoxometalate mediated reactions. Chem. Rev. 2018, 118, 2680–2717. [Google Scholar] [CrossRef]
- Tong, J.; Wang, W.; Su, L.; Li, Q.; Liu, F.; Ma, W.; Lei, Z.; Bo, L. Highly selective oxidation of cyclohexene to 2-cyclohexene-1-one over polyoxometalate/metal–organic framework hybrids with greatly improved performances. Catal. Sci. Technol. 2017, 7, 222–230. [Google Scholar] [CrossRef]
- Tsigdinos, G.A.; Hallada, C.J. Molybdovanadophosphoric acids and their salts. 1. Investigation of methods of preparation and characterization. Inorg. Chem. 1968, 7, 437–441. [Google Scholar] [CrossRef]
- Kanno, M.; Yasukawa, T.; Ninomiya, W.; Ooyachi, K.; Kamiya, Y. Catalytic oxidation of methacrolein to methacrylic acid over silica-supported 11-molybdo-1-vanadophosphoric acid with different heteropolyacid loadings. J. Catal. 2010, 273, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Khenkin, A.M.; Rosenberger, A.; Neumann, R. Reaction of aldehydes with the H5PV2Mo10O40 polyoxometalate and co-oxidation of alkanes with molecular oxygen. J. Catal. 1999, 182, 82–91. [Google Scholar] [CrossRef]
- Sankar, M.M.; Nowicka, E.; Carter, E.; Murphy, D.M.; Knight, D.M.; Bethell, D.; Hutchings, G.J. The benzaldehyde oxidation paradox explained by the interception of peroxy radical by benzyl alcohol. Nat. Commun. 2014, 5, 3332. [Google Scholar] [CrossRef] [PubMed]
- Ingold, K.U.; Pratt, D.A. Advances in radical-trapping antioxidant chemistry in the 21st century: A kinetics and mechanisms perspective. Chem. Rev. 2014, 114, 9022–9046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamamoto, M.; Nakayama, K.; Nishiyama, Y.; Ishii, Y. Oxidation of organic substrates by molecular oxygen/aldehyde/heteropolyoxometalate system. J. Org. Chem. 1993, 58, 6421–6425. [Google Scholar] [CrossRef]
- Ghubayra, R.; Nuttall, C.; Hodgkiss, S.; Craven, M.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Oxidative desulfurization of model diesel fuel catalyzed by carbon-supported heteropoly acids. Appl. Catal. B 2019, 253, 309–316. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst | H2O 1 (wt%) | SBET 2 (m2g−1) | Pore Volume 3 (cm3g−1) | Pore Diameter 4 (Å) |
|---|---|---|---|---|
| HPA-0 | 13.5 | 14.8 | 0.018 | 48 |
| HPA-1 | 12.3 | 15.5 | 0.020 | 52 |
| HPA-2 | 12.8 | 2.4 | 0.0044 | 72 |
| HPA-3 | 12.0 | 2.6 | 0.0060 | 93 |
| Cs-HPA-1 | 5.2 | 8.9 | 0.15 | 67 |
| Na-HPA-2 | 13.0 | 1.7 | 0.0046 | 106 |
| 15%HPA-1/SiO2 | 236 | 1.29 | 219 | |
| 15%HPA-1/C | 928 | 0.81 | 35 |
| Entry | DBT (mmol) | PhCHO (mmol) | Temperature (°C) | Time | Conversion (%) | |
|---|---|---|---|---|---|---|
| (h) | DBT | PhCHO | ||||
| 1 | 0 | 4.9 | 60 | 2.0 | - | 63 |
| 2 | 0.50 | 0 | 60 | 2.0 | 0 | 0 |
| 3 | 0.50 | 1.0 | 60 | 2.0 | 1.6 | 11 |
| 4 | 0.50 | 2.0 | 60 | 2.0 | 23 | 27 |
| 5 | 0.50 | 2.9 | 60 | 2.0 | 31 | 30 |
| 6 | 0.50 | 4.9 | 60 | 2.0 | 86 | 50 |
| 7 | 0.50 | 5.9 | 60 | 2.0 | 100 2 | 63 |
| 8 3 | 0.50 | 5.9 | 60 | 2.0 | 10 | 42 |
| 9 4 | 0.50 | 5.9 | 60 | 2.0 | 14 | 26 |
| 10 | 0.50 | 4.9 | 40 | 2.0 | 18 | 28 |
| 11 | 0.50 | 4.9 | 80 | 2.0 | 93 | 71 |
| 12 | 0.50 | 4.9 | 100 | 1.2 | 100 2 | 75 |
| 13 | 0.50 | 1.8 | 100 | 2.0 | 55 | 64 |
| 14 | 0.50 | 2.8 | 100 | 2.0 | 100 2 | 77 |
| 15 | 0.50 | 3.7 | 100 | 1.3 | 100 2 | 69 |
| 16 5 | 0.50 | 4.9 | 60 | 2.0 | 84 | 53 |
| Catalyst | Temperature (°C) | Conversion (%) | |
|---|---|---|---|
| DBT | PhCHO | ||
| 15%HPA-1/SiO2 | 100 | 61 | 48 |
| 15%HPA-1/SiO2 | 120 | 80 | 53 |
| 15%HPA-1/C | 100 | 11 | 60 |
| Bulk HPA-1 2 | 100 | 100 | 69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghubayra, R.; Hindle, R.; Yahya, R.; Kozhevnikova, E.F.; Kozhevnikov, I.V. Aerobic Oxidative Desulfurization of Liquid Fuel Catalyzed by P–Mo–V Heteropoly Acids in the Presence of Aldehyde. Catalysts 2021, 11, 988. https://doi.org/10.3390/catal11080988
Ghubayra R, Hindle R, Yahya R, Kozhevnikova EF, Kozhevnikov IV. Aerobic Oxidative Desulfurization of Liquid Fuel Catalyzed by P–Mo–V Heteropoly Acids in the Presence of Aldehyde. Catalysts. 2021; 11(8):988. https://doi.org/10.3390/catal11080988
Chicago/Turabian StyleGhubayra, Reem, Rachel Hindle, Rana Yahya, Elena F. Kozhevnikova, and Ivan V. Kozhevnikov. 2021. "Aerobic Oxidative Desulfurization of Liquid Fuel Catalyzed by P–Mo–V Heteropoly Acids in the Presence of Aldehyde" Catalysts 11, no. 8: 988. https://doi.org/10.3390/catal11080988
APA StyleGhubayra, R., Hindle, R., Yahya, R., Kozhevnikova, E. F., & Kozhevnikov, I. V. (2021). Aerobic Oxidative Desulfurization of Liquid Fuel Catalyzed by P–Mo–V Heteropoly Acids in the Presence of Aldehyde. Catalysts, 11(8), 988. https://doi.org/10.3390/catal11080988