
Photoelectrochemical Conversion of Methane into Value-Added Products


Abstract
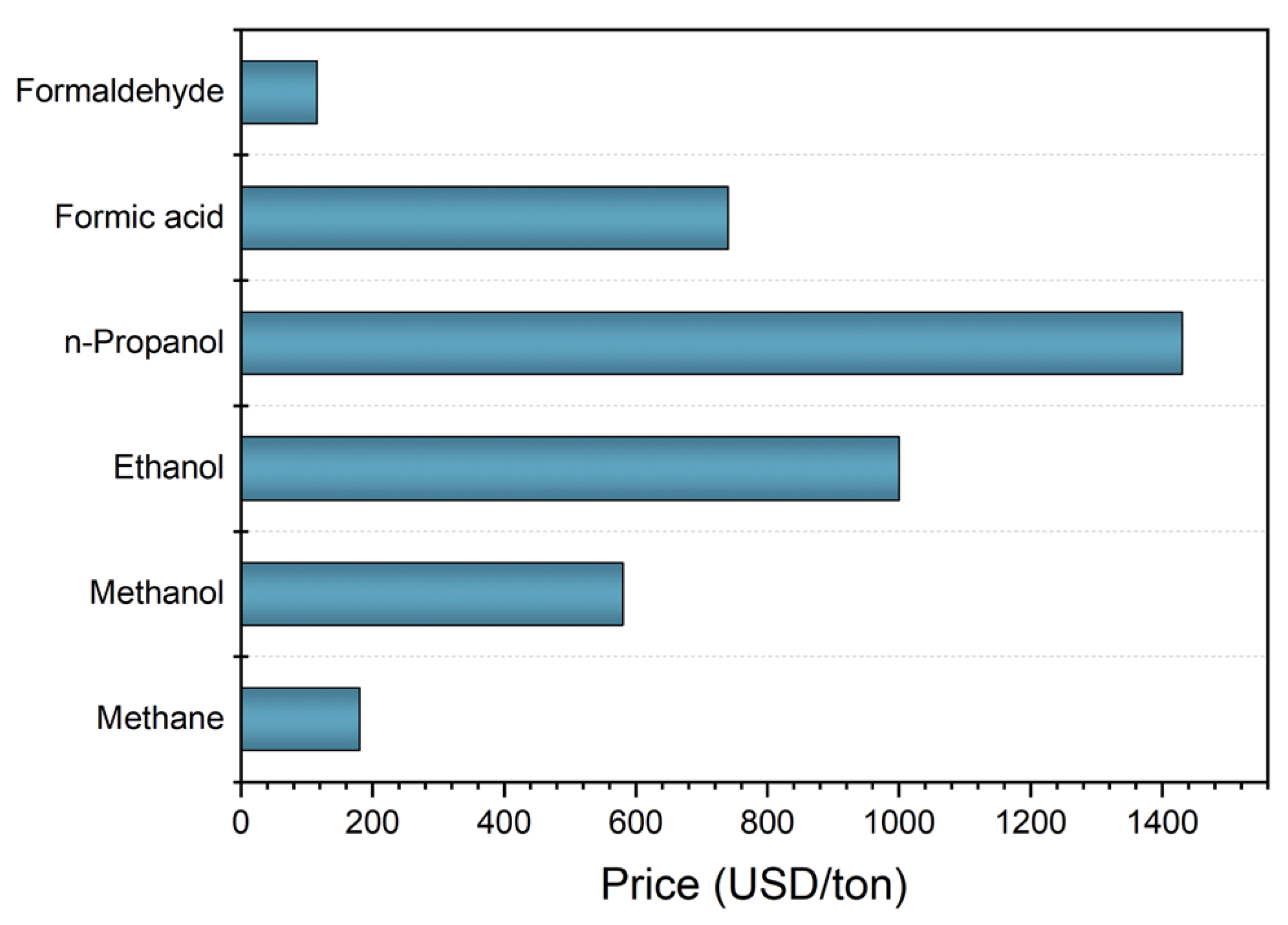
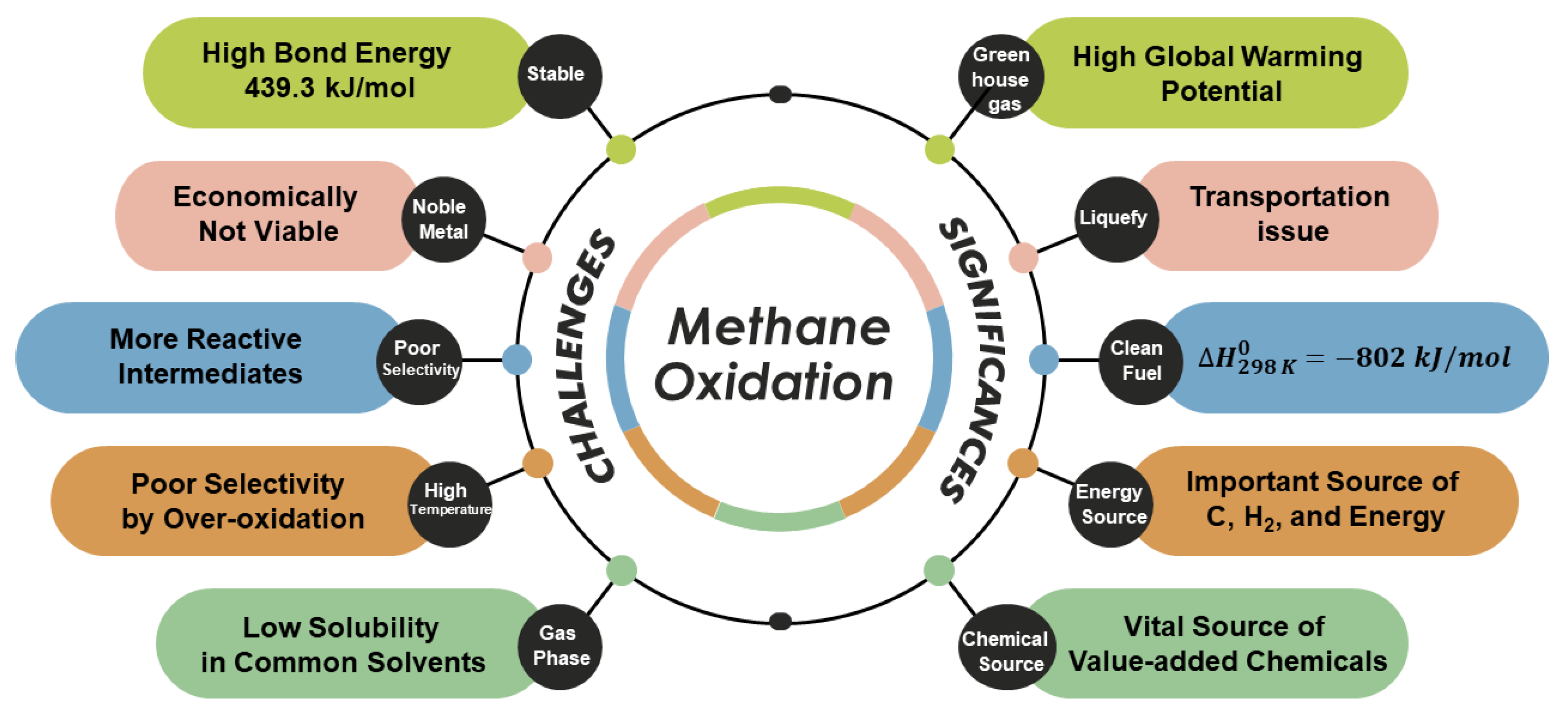
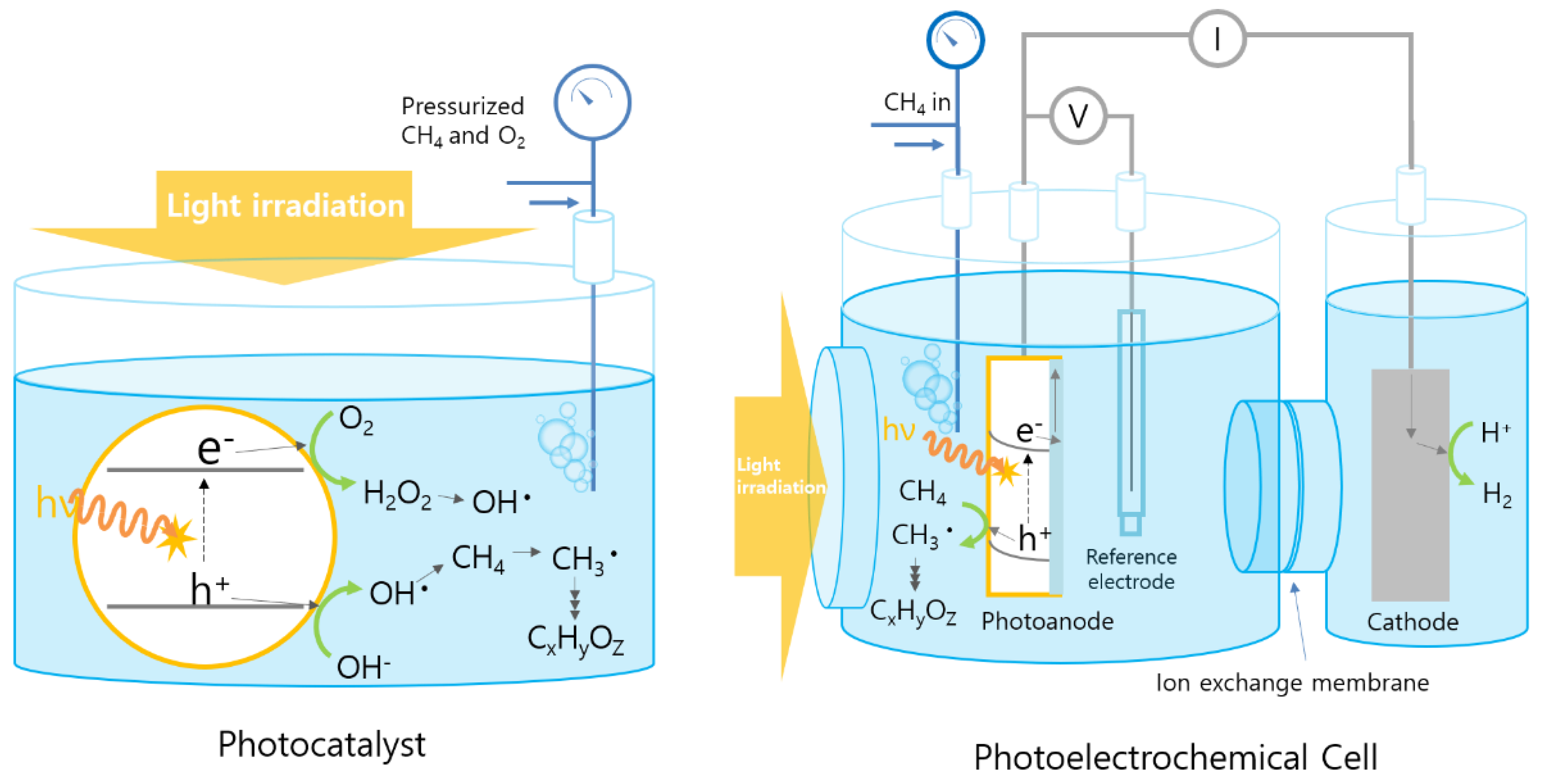
:1. Introduction
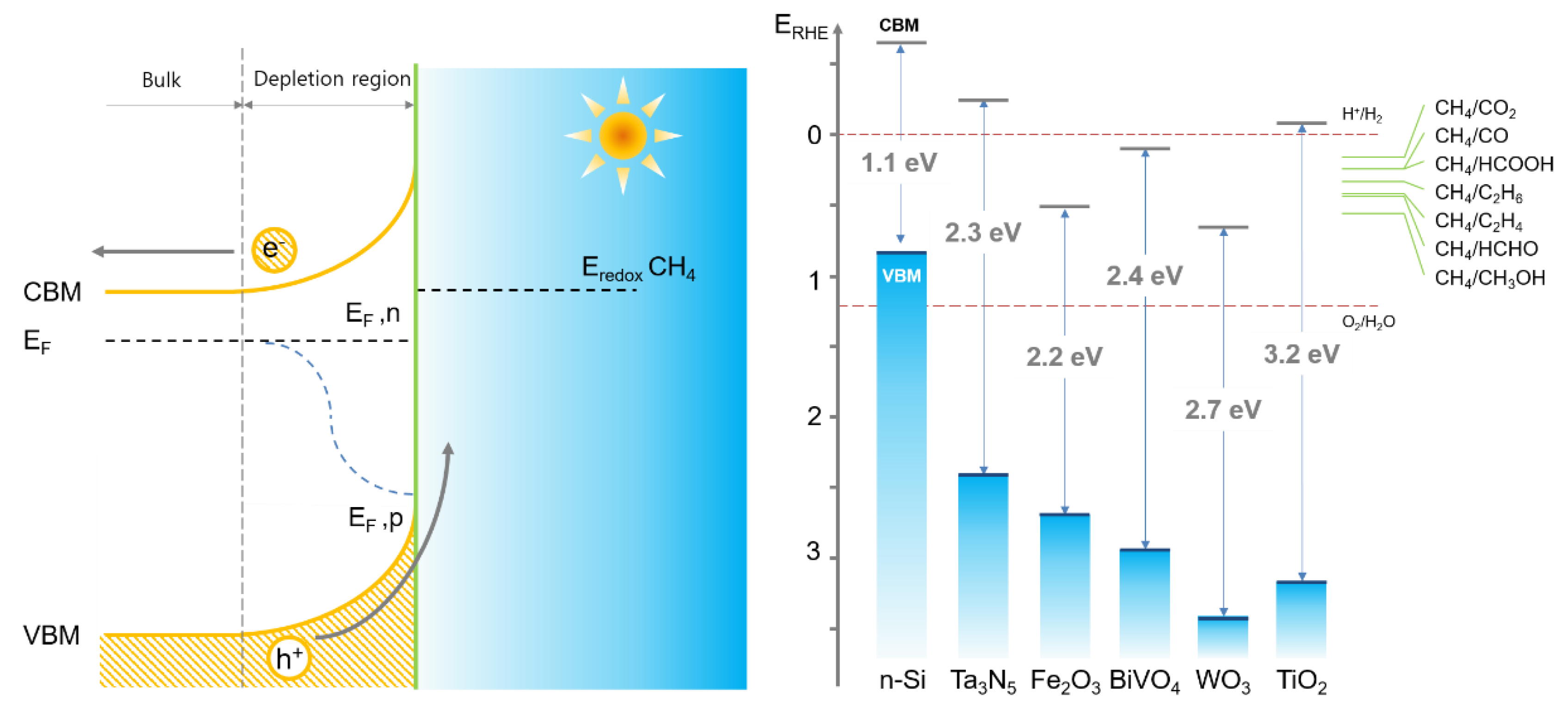
2. Photoelectrochemical (PEC) Methane Oxidation
2.1. TiO2
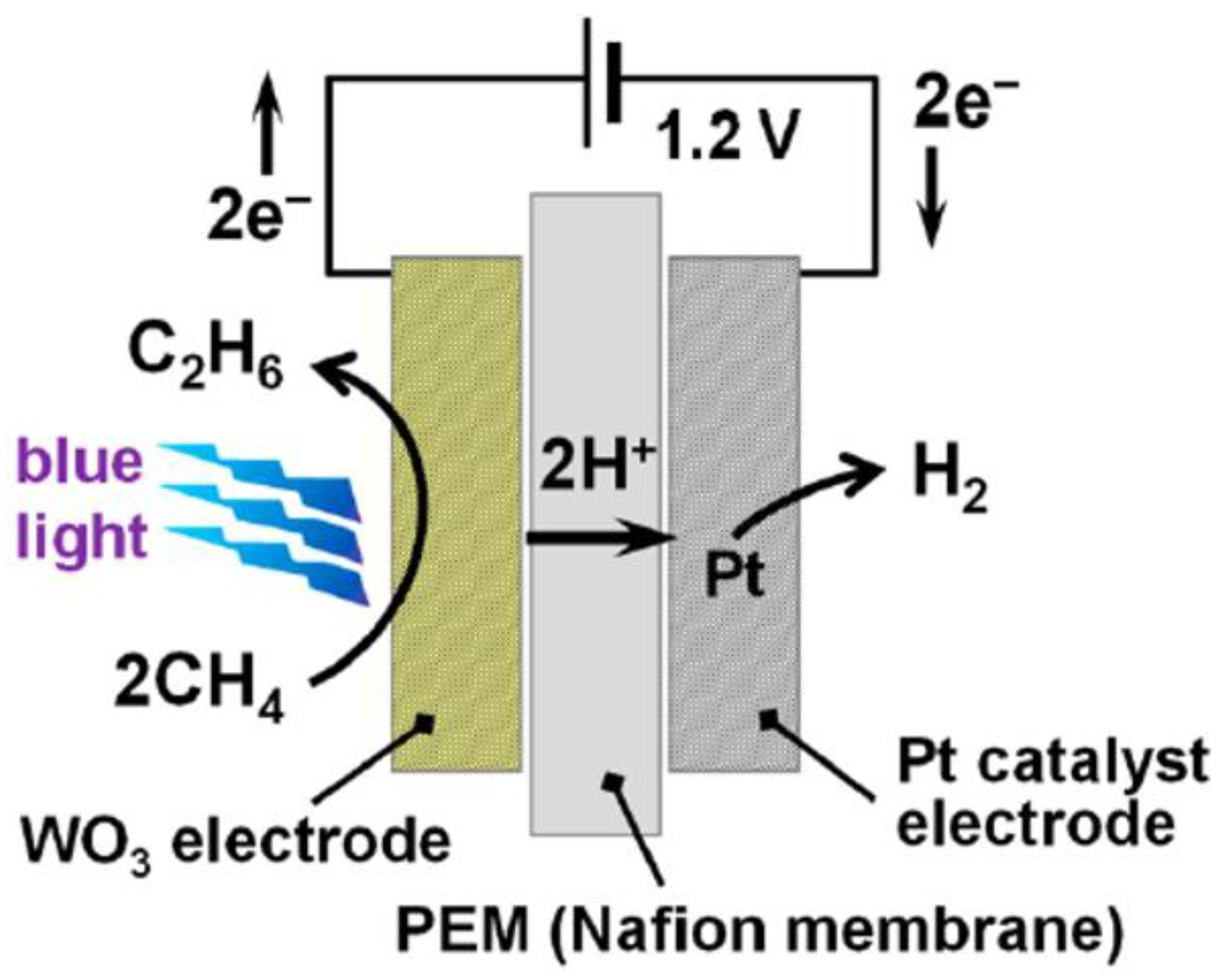
2.2. WO3
2.3. ZnO
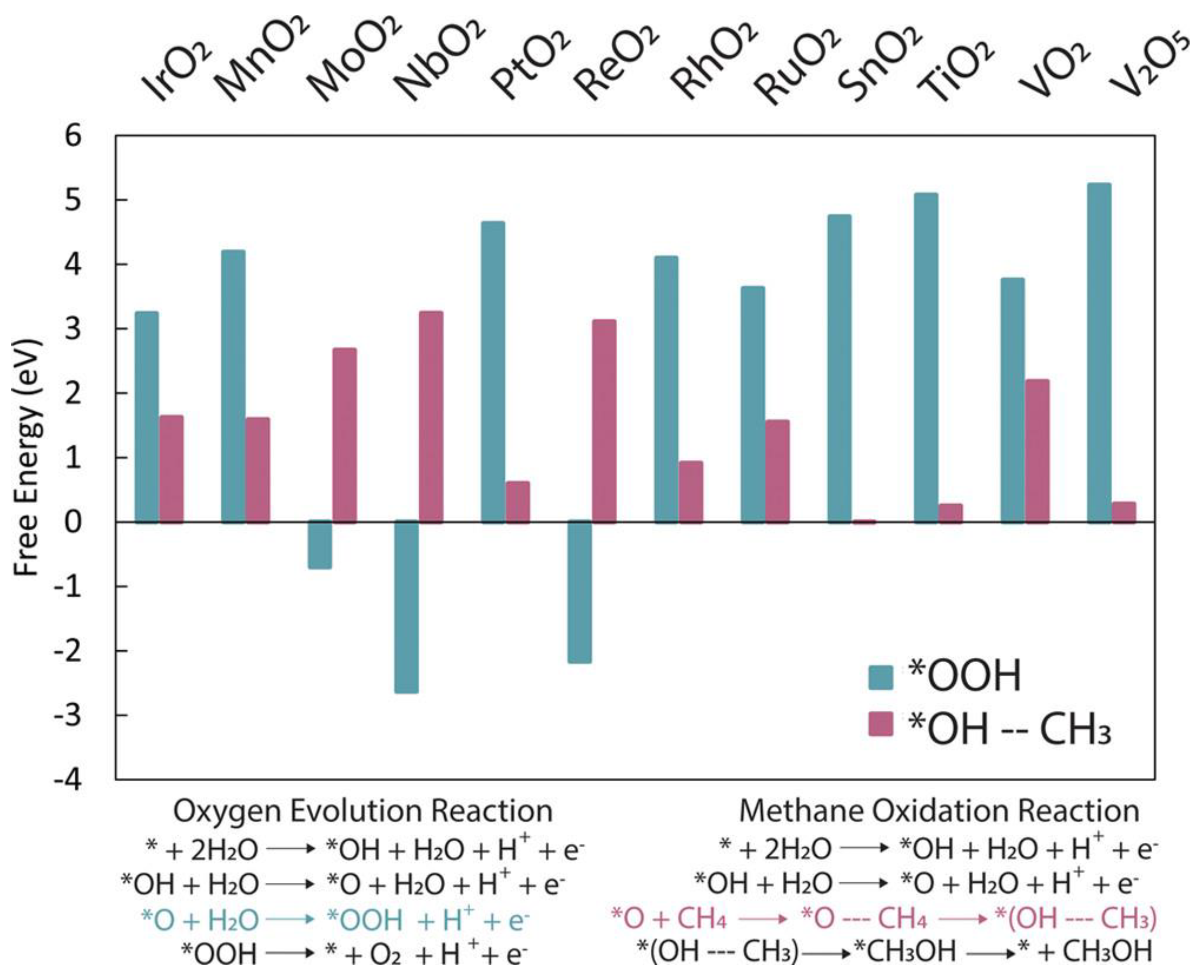
2.4. Potential Semiconductor Materials
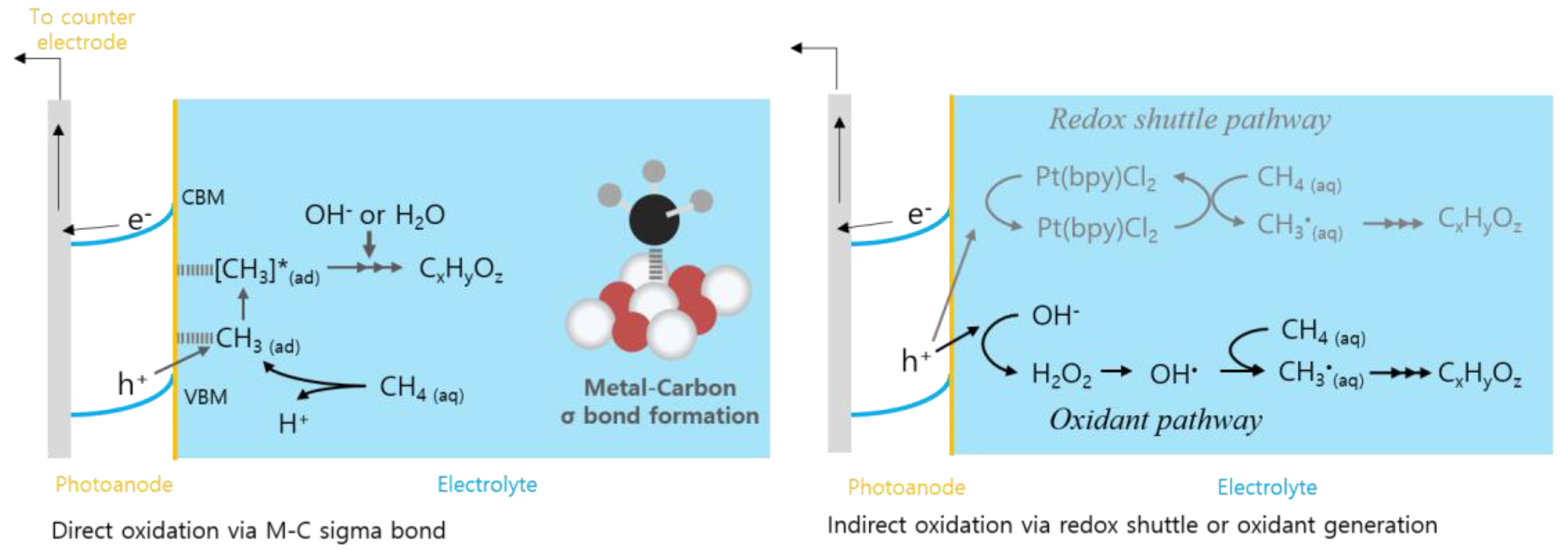
3. Mechanistic Insights for High Selectivity during Methane Oxidation
3.1. Direct Oxidation: Co-Catalyst Approach
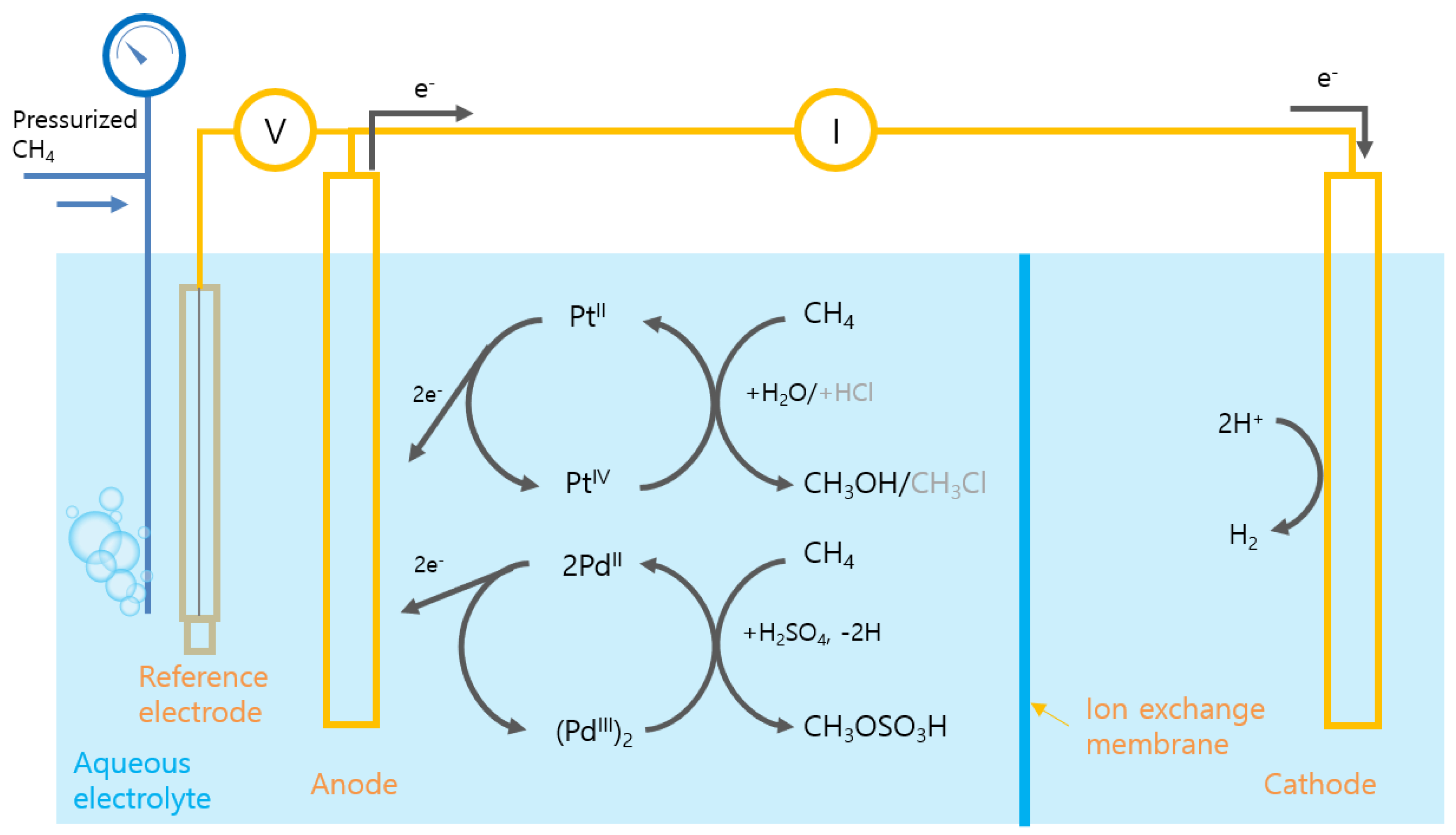
3.2. Indirect Oxidation: PEC Production of Oxidant
4. Other Factors That Affect Methane Oxidation
5. Effect of Catalyst Structure on Reaction Selectivity
6. Activity Evaluation for PEC Methane Oxidation
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- McFarland, E. Unconventional chemistry for unconventional natural gas. Science 2012, 338, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ouyang, Y.; Li, H.; Wang, L.; Zeng, J. Photocatalytic Conversion of Methane: Recent Advancements and Prospects. Angew. Chem. Int. Ed. 2021. [Google Scholar] [CrossRef]
- Hu, D.; Ordomsky, V.V.; Khodakov, A.Y. Major routes in the photocatalytic methane conversion into chemicals and fuels under mild conditions. Appl. Catal. B 2021, 286, 119913. [Google Scholar] [CrossRef]
- Sher Shah, M.S.A.; Oh, C.; Park, H.; Hwang, Y.J.; Ma, M.; Park, J.H. Catalytic Oxidation of Methane to Oxygenated Products: Recent Advancements and Prospects for Electrocatalytic and Photocatalytic Conversion at Low Temperatures. Adv. Sci. 2020, 7, 2001946. [Google Scholar] [CrossRef]
- Baek, J.; Rungtaweevoranit, B.; Pei, X.; Park, M.; Fakra, S.C.; Liu, Y.-S.; Matheu, R.; Alshmimri, S.A.; Alshehri, S.; Trickett, C.A.; et al. Bioinspired Metal–Organic Framework Catalysts for Selective Methane Oxidation to Methanol. J. Am. Chem. Soc. 2018, 140, 18208–18216. [Google Scholar] [CrossRef]
- Mostaghimi, A.H.B.; Al-Attas, T.A.; Kibria, M.G.; Siahrostami, S. A review on electrocatalytic oxidation of methane to oxygenates. J. Mater. Chem. A 2020, 8, 15575–15590. [Google Scholar] [CrossRef]
- Tomkins, P.; Ranocchiari, M.; van Bokhoven, J.A. Direct Conversion of Methane to Methanol under Mild Conditions over Cu-Zeolites and beyond. Acc. Chem. Res. 2017, 50, 418–425. [Google Scholar] [CrossRef]
- Meng, X.; Cui, X.; Rajan, N.P.; Yu, L.; Deng, D.; Bao, X. Direct Methane Conversion under Mild Condition by Thermo-, Electro-, or Photocatalysis. Chem 2019, 5, 2296–2325. [Google Scholar] [CrossRef]
- Zakaria, Z.; Kamarudin, S.K. Direct conversion technologies of methane to methanol: An overview. Renew. Sustain. Energy Rev. 2016, 65, 250–261. [Google Scholar] [CrossRef]
- Sivula, K.; van de Krol, R. Semiconducting materials for photoelectrochemical energy conversion. Nat. Rev. Mater. 2016, 1, 15010. [Google Scholar] [CrossRef]
- Kadosh, Y.; Korin, E.; Bettelheim, A. Room-temperature conversion of the photoelectrochemical oxidation of methane into electricity at nanostructured TiO2. Sustain. Energy Fuels 2021, 5, 127–134. [Google Scholar] [CrossRef]
- Li, W.; He, D.; Hu, G.; Li, X.; Banerjee, G.; Li, J.; Lee, S.H.; Dong, Q.; Gao, T.; Brudvig, G.W.; et al. Selective CO Production by Photoelectrochemical Methane Oxidation on TiO2. ACS Cent. Sci. 2018, 4, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Mao, K.; Low, J.; Wang, Z.; Xi, D.; Zhang, W.; Ju, H.; Qi, Z.; Long, R.; Wu, X.; et al. Efficient Photoelectrochemical Conversion of Methane into Ethylene Glycol by WO3 Nanobar Arrays. Angew. Chem. Int. Ed. 2021, 60, 9357–9361. [Google Scholar] [CrossRef] [PubMed]
- Amano, F.; Shintani, A.; Tsurui, K.; Mukohara, H.; Ohno, T.; Takenaka, S. Photoelectrochemical homocoupling of methane under blue light irradiation. ACS Energy Lett. 2019, 4, 502–507. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Huang, Z.; Bai, Z.; Gao, Y. Photoelectrocatalytic Oxidation of Methane into Methanol over ZnO Nanowire Arrays Decorated with Plasmonic Au Nanoparticles. Nano 2018, 14, 1950017. [Google Scholar] [CrossRef]
- Alexander, B.D.; Kulesza, P.J.; Rutkowska, I.; Solarska, R.; Augustynski, J. Metal oxide photoanodes for solar hydrogen production. J. Mater. Chem. 2008, 18, 2298–2303. [Google Scholar] [CrossRef]
- Seabold, J.A.; Choi, K.-S. Effect of a Cobalt-Based Oxygen Evolution Catalyst on the Stability and the Selectivity of Photo-Oxidation Reactions of a WO3 Photoanode. Chem. Mater. 2011, 23, 1105–1112. [Google Scholar] [CrossRef]
- Phuan, Y.W.; Ong, W.-J.; Chong, M.N.; Ocon, J.D. Prospects of electrochemically synthesized hematite photoanodes for photoelectrochemical water splitting: A review. J. Photochem. Photobiol. C 2017, 33, 54–82. [Google Scholar] [CrossRef]
- Wu, Y.-C.; Song, R.-J.; Li, J.-H. Recent advances in photoelectrochemical cells (PECs) for organic synthesis. Org. Chem. Front. 2020, 7, 1895–1902. [Google Scholar] [CrossRef]
- Labinger, J.A.; Bercaw, J.E. Understanding and exploiting C–H bond activation. Nature 2002, 417, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.S.; Reis, R.M.; Lanza, M.R.; Bertazzoli, R. Electrosynthesis of methanol from methane: The role of V2O5 in the reaction selectivity for methanol of a TiO2/RuO2/V2O5 gas diffusion electrode. Electrochim. Acta 2013, 87, 606–610. [Google Scholar] [CrossRef]
- Boyd, M.J.; Latimer, A.A.; Dickens, C.F.; Nielander, A.C.; Hahn, C.; Nørskov, J.K.; Higgins, D.C.; Jaramillo, T.F. Electro-Oxidation of Methane on Platinum under Ambient Conditions. ACS Catal. 2019, 9, 7578–7587. [Google Scholar] [CrossRef]
- Arnarson, L.; Schmidt, P.S.; Pandey, M.; Bagger, A.; Thygesen, K.S.; Stephens, I.E.L.; Rossmeisl, J. Fundamental limitation of electrocatalytic methane conversion to methanol. Phys. Chem. Chem. Phys. 2018, 20, 11152–11159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.-H.; Hong, Z.-S.; Chao, T.-H.; Cheng, M.-J. Quantum Mechanical Screening of Metal-N4-Functionalized Graphenes for Electrochemical Anodic Oxidation of Light Alkanes to Oxygenates. J. Phys. Chem. C 2019, 123, 19033–19044. [Google Scholar] [CrossRef]
- Fornaciari, J.C.; Primc, D.; Kawashima, K.; Wygant, B.R.; Verma, S.; Spanu, L.; Mullins, C.B.; Bell, A.T.; Weber, A.Z. A Perspective on the Electrochemical Oxidation of Methane to Methanol in Membrane Electrode Assemblies. ACS Energy Lett. 2020, 5, 2954–2963. [Google Scholar] [CrossRef]
- Lee, B.; Hibino, T. Efficient and selective formation of methanol from methane in a fuel cell-type reactor. J. Catal. 2011, 279, 233–240. [Google Scholar] [CrossRef]
- Heo, P.; Ito, K.; Tomita, A.; Hibino, T. A Proton-Conducting Fuel Cell Operating with Hydrocarbon Fuels. Angew. Chem. Int. Ed. 2008, 47, 7841–7844. [Google Scholar] [CrossRef]
- Yamanaka, I.; Hasegawa, S.; Otsuka, K. Partial oxidation of light alkanes by reductive activated oxygen over the (Pd-black + VO(acac)2/VGCF) cathode of H2–O2 cell system at 298 K. Appl. Catal. A 2002, 226, 305–315. [Google Scholar] [CrossRef]
- Kim, R.S.; Surendranath, Y. Electrochemical Reoxidation Enables Continuous Methane-to-Methanol Catalysis with Aqueous Pt Salts. ACS Cent. Sci. 2019, 5, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- O’Reilly, M.E.; Kim, R.S.; Oh, S.; Surendranath, Y. Catalytic Methane Monofunctionalization by an Electrogenerated High-Valent Pd Intermediate. ACS Cent. Sci. 2017, 3, 1174–1179. [Google Scholar] [CrossRef] [Green Version]
- Cha, H.G.; Choi, K.-S. Combined biomass valorization and hydrogen production in a photoelectrochemical cell. Nat. Chem. 2015, 7, 328–333. [Google Scholar] [CrossRef]
- Song, H.; Meng, X.; Wang, Z.-j.; Liu, H.; Ye, J. Solar-Energy-Mediated Methane Conversion. Joule 2019, 3, 1606–1636. [Google Scholar] [CrossRef]
- Lee, J.; Yang, J.; Moon, J.H. Solar Cell-Powered Electrochemical Methane-to-Methanol Conversion with CuO/CeO2 Catalysts. ACS Energy Lett. 2021, 6, 893–899. [Google Scholar] [CrossRef]
- Fuku, K.; Sayama, K. Efficient oxidative hydrogen peroxide production and accumulation in photoelectrochemical water splitting using a tungsten trioxide/bismuth vanadate photoanode. Chem. Commun. 2016, 52, 5406–5409. [Google Scholar] [CrossRef] [PubMed]
- Fuku, K.; Miyase, Y.; Miseki, Y.; Gunji, T.; Sayama, K. Enhanced Oxidative Hydrogen Peroxide Production on Conducting Glass Anodes Modified with Metal Oxides. ChemistrySelect 2016, 1, 5721–5726. [Google Scholar] [CrossRef]
- Murcia-López, S.; Villa, K.; Andreu, T.; Morante, J.R. Improved selectivity for partial oxidation of methane to methanol in the presence of nitrite ions and BiVO4 photocatalyst. Chem. Commun. 2015, 51, 7249–7252. [Google Scholar] [CrossRef]
- Cheng, J.; Li, Z.; Haught, M.; Tang, Y. Direct methane conversion to methanol by ionic liquid-dissolved platinum catalysts. Chem. Commun. 2006, 4617–4619. [Google Scholar] [CrossRef] [Green Version]
- Tomita, A.; Nakajima, J.; Hibino, T. Direct Oxidation of Methane to Methanol at Low Temperature and Pressure in an Electrochemical Fuel Cell. Angew. Chem. Int. Ed. 2008, 47, 1462–1464. [Google Scholar] [CrossRef]
- Santos, M.C.L.; Nunes, L.C.; Silva, L.M.G.; Ramos, A.S.; Fonseca, F.C.; de Souza, R.F.B.; Neto, A.O. Direct Alkaline Anion Exchange Membrane Fuel Cell to Converting Methane into Methanol. ChemistrySelect 2019, 4, 11430–11434. [Google Scholar] [CrossRef]
- Amenomiya, Y.; Birss, V.I.; Goledzinowski, M.; Galuszka, J.; Sanger, A.R. Conversion of Methane by Oxidative Coupling. Catal. Rev. 1990, 32, 163–227. [Google Scholar] [CrossRef] [Green Version]
- Spinner, N.; Mustain, W.E. Electrochemical Methane Activation and Conversion to Oxygenates at Room Temperature. ECS Trans. 2013, 53, 1–20. [Google Scholar] [CrossRef]
- Ma, M.; Jin, B.J.; Li, P.; Jung, M.S.; Kim, J.I.; Cho, Y.; Kim, S.; Moon, J.H.; Park, J.H. Ultrahigh electrocatalytic conversion of methane at room temperature. Adv. Sci. 2017, 4, 1700379. [Google Scholar] [CrossRef]
- Ma, M.; Oh, C.; Kim, J.; Moon, J.H.; Park, J.H. Electrochemical CH4 oxidation into acids and ketones on ZrO2:NiCo2O4 quasi-solid solution nanowire catalyst. Appl. Catal. B 2019, 259, 118095. [Google Scholar] [CrossRef]
- Lu, J.; Zhu, C.; Pan, C.; Lin, W.; Lemmon John, P.; Chen, F.; Li, C.; Xie, K. Highly efficient electrochemical reforming of CH4/CO2 in a solid oxide electrolyser. Sci. Adv. 2018, 4, eaar5100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Hou, S.; Hu, X.; Lu, J.; Chen, F.; Xie, K. Electrochemical conversion of methane to ethylene in a solid oxide electrolyzer. Nat. Commun. 2019, 10, 1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinev, M.Y.; Tulenin, Y.P.; Kalashnikova, O.V.; Bychkov, V.Y.; Korchak, V.N. Oxidation of methane in a wide range of pressures and effect of inert gases. Catal. Today 1996, 32, 157–162. [Google Scholar] [CrossRef]
- Yamamoto, S.; Alcauskas, J.B.; Crozier, T.E. Solubility of methane in distilled water and seawater. J. Chem. Eng. Data 1976, 21, 78–80. [Google Scholar] [CrossRef]
- Wei, J.; Yang, J.; Wen, Z.; Dai, J.; Li, Y.; Yao, B. Efficient photocatalytic oxidation of methane over β-Ga2O3/activated carbon composites. RSC Adv. 2017, 7, 37508–37521. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Xu, C.-Q.; Hung, S.-F.; Liu, W.; Cai, W.; Zeng, Z.; Jia, C.; Chen, H.M.; Xiao, H.; Li, J.; et al. Breaking Long-Range Order in Iridium Oxide by Alkali Ion for Efficient Water Oxidation. J. Am. Chem. Soc. 2019, 141, 3014–3023. [Google Scholar] [CrossRef]
- Liang, Z.; Li, T.; Kim, M.; Asthagiri, A.; Weaver Jason, F. Low-temperature activation of methane on the IrO(110) surface. Science 2017, 356, 299–303. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | E (V) vs. RHE |
|---|---|
| 2H2O (l) → O2 (g) + 4H+ (aq) + 4e− | 1.23 |
| CH4 (g) + 2H2O (l) → 2CO2 (g) + 8H+ (aq) + 8e− | 0.17 |
| CH4 (g) + H2O (l) → CO (g) + 6H+ (aq) + 6e− | 0.26 |
| CH4 (g) + H2O (l) → HCOOH (aq) + 6H+ (aq) + 6e− | 0.26 |
| 2CH4 (g) → C2H6 (g) + 2H+ (aq) + 2e− | 0.35 |
| 2CH4 (g) → C2H4 (g) + 4H+ (aq) + 4e− | 0.44 |
| CH4 (g) + H2O (l) → HCHO (aq) + 4H+ (aq) + 4e− | 0.46 |
| CH4 (g) + H2O (l) → CH3OH + 2H+ (aq) + 2e− | 0.58 |
| Materials | Morphology | Preparation Method | Photocurrent Density (mA·cm−2) | Applied Potential | Light Intensity (mW cm−2) | Light Source | Electrode Area (cm2) | Methane Pressure (Atm) | Resction Mechanism | Faradaic Efficiency (%) | Product Rate | Electrolyte | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TiO2 | Nanotubes array | Anodizing | 0.54 | 0.8 VRHE | 100 | Hg (Xe) lamp (100 mW·cm−2) | 0.23 | 1 | Free radical | HCOOH (16%), CO2 (72%), O2 (12%) | - | 0.05 M H2SO4 | [11] |
| TiO2 | - | Atomic layer deposition | 0.158 | 0.6 VRHE | 0.1 | UV lamp (254 nm) | 1 | 1 | M-C σ bond | CO (52%), CO2 (11%), O2 (37%) | - | 1 M NaOH (pH 3.3) | [12] |
| WO3 | Nanoplate | Hydrothermal | 4 | 0.7 VRHE | 100 | Xenon lamp (simulated solar light, 100 mW·cm−2) | 1 | - | Free radical | HOCH2CH2OH (23.9%), C2H6 (2.1%), CH3OH (2.3%), CO (4.3%), O2 (63.5%) | 0.47 µmol·cm−2·h−1 | 0.1 M Na2SO4 | [13] |
| WO3 | Nanoparticle | Solution-based dip coating | 4.2 | 1.2 V (two-electrode system) | 6.8 | 3-W blue light LED lamps | 16 | 1 | - | C2H6 (12%), CO2 (75.3%), CO (6.3%), O2 (0.8%) | 0.15 µmol·h−1 | - | [14] |
| ZnO | Nanowire array | Hydrothermal | 0.22 | 1 VAg/AgCl | 100 | Xe lamp (simulated solar light, 100 mW·cm−2) | 10 | 1 | Free radical | CH3OH (11.69%) | 0.571 µmol·min−1 | 0.05 M Na2SO4 | [15] |
| ZnO/Au | Nanowire array | Hydrothermal | 0.35 | 1 VAg/AgCl | 100 | Xe lamp (simulated solar light, 100 mW·cm−2) | 10 | 1 | Free radical | CH3OH (32.11%) | 1.407 µmol·min−1 | 0.05 M Na2SO4 | [15] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehmood, A.; Chae, S.Y.; Park, E.D. Photoelectrochemical Conversion of Methane into Value-Added Products. Catalysts 2021, 11, 1387. https://doi.org/10.3390/catal11111387
Mehmood A, Chae SY, Park ED. Photoelectrochemical Conversion of Methane into Value-Added Products. Catalysts. 2021; 11(11):1387. https://doi.org/10.3390/catal11111387
Chicago/Turabian StyleMehmood, Adeel, Sang Youn Chae, and Eun Duck Park. 2021. "Photoelectrochemical Conversion of Methane into Value-Added Products" Catalysts 11, no. 11: 1387. https://doi.org/10.3390/catal11111387