CO2 Methanation over Rh/CeO2 Studied with Infrared Modulation Excitation Spectroscopy and Phase Sensitive Detection


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Catalyst Adsorbates during Steady-State Conditions
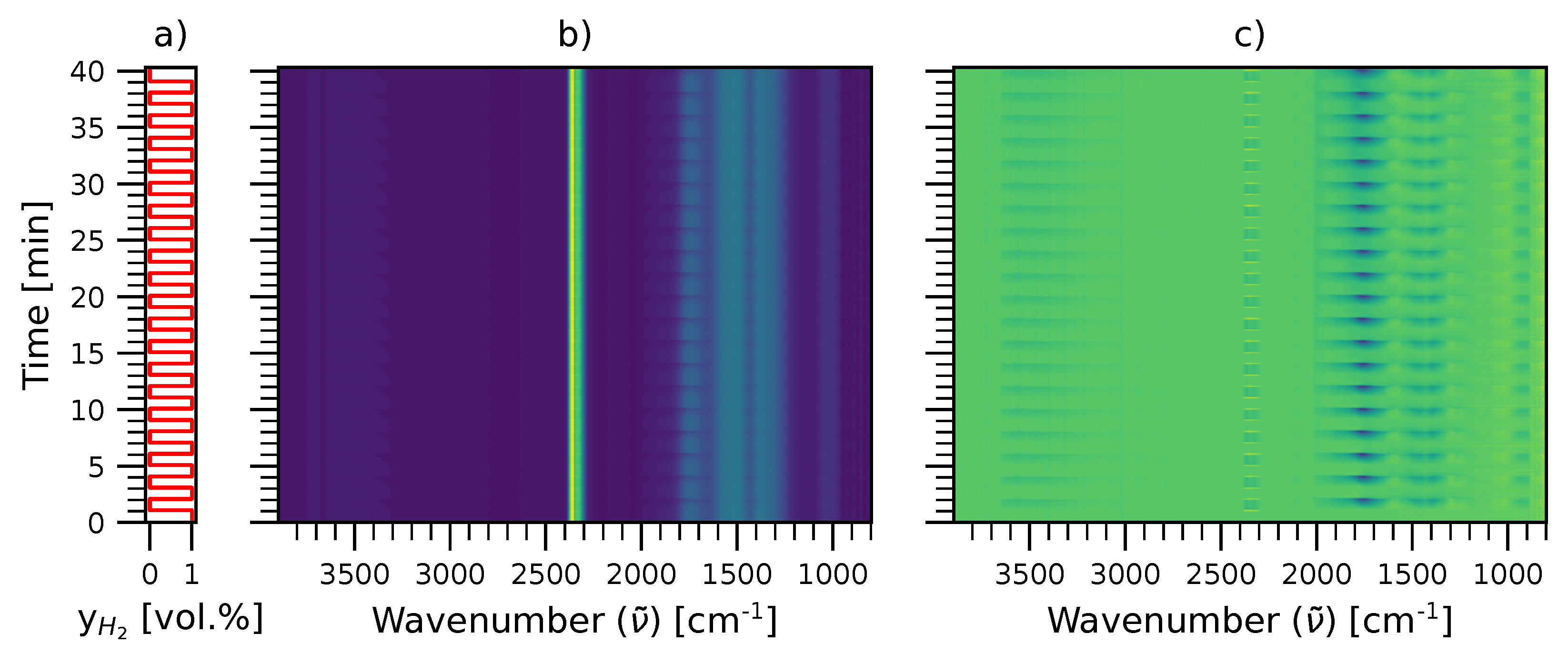
2.2. Time-Resolved DRIFTS and MES-PSD
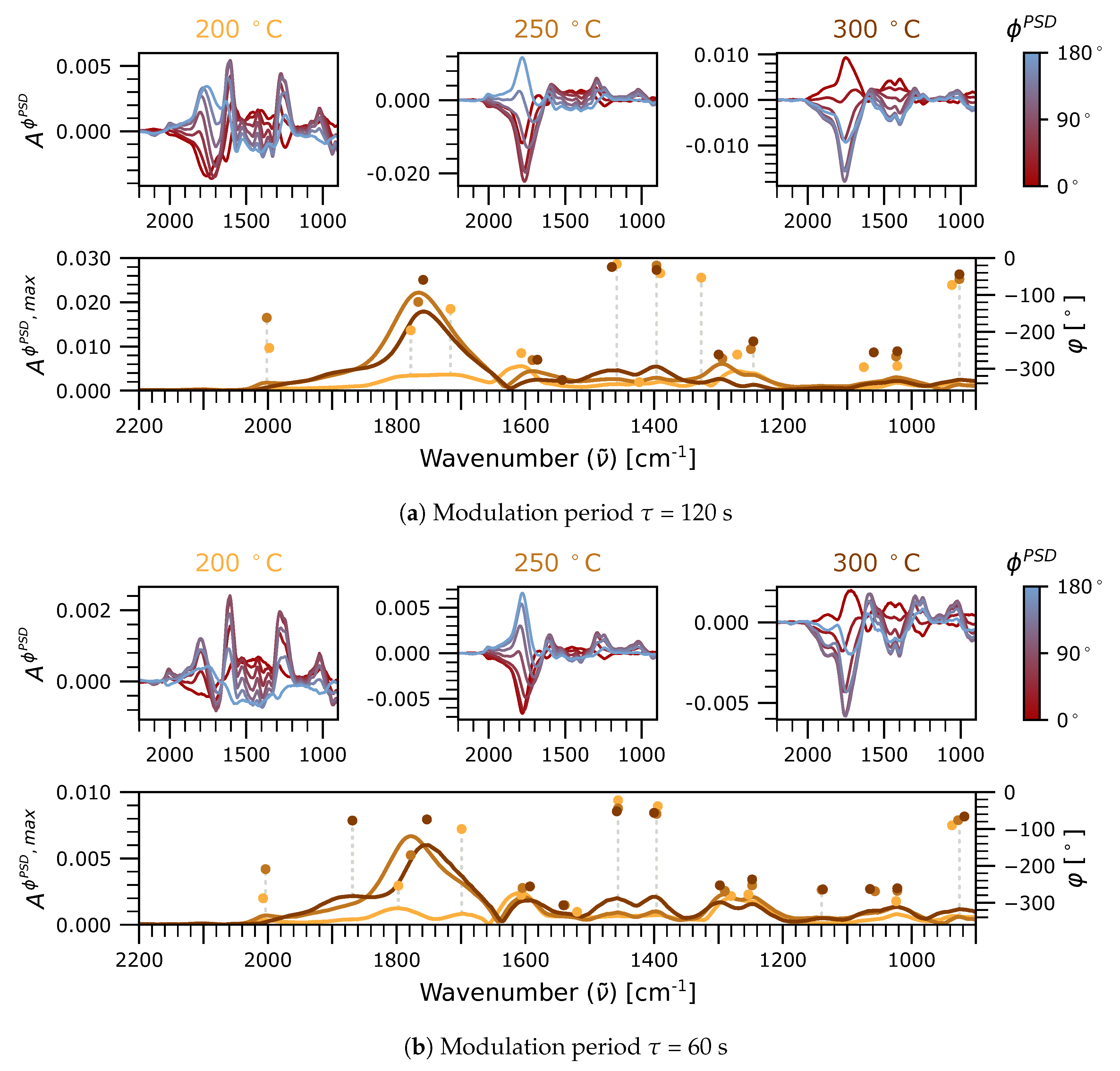
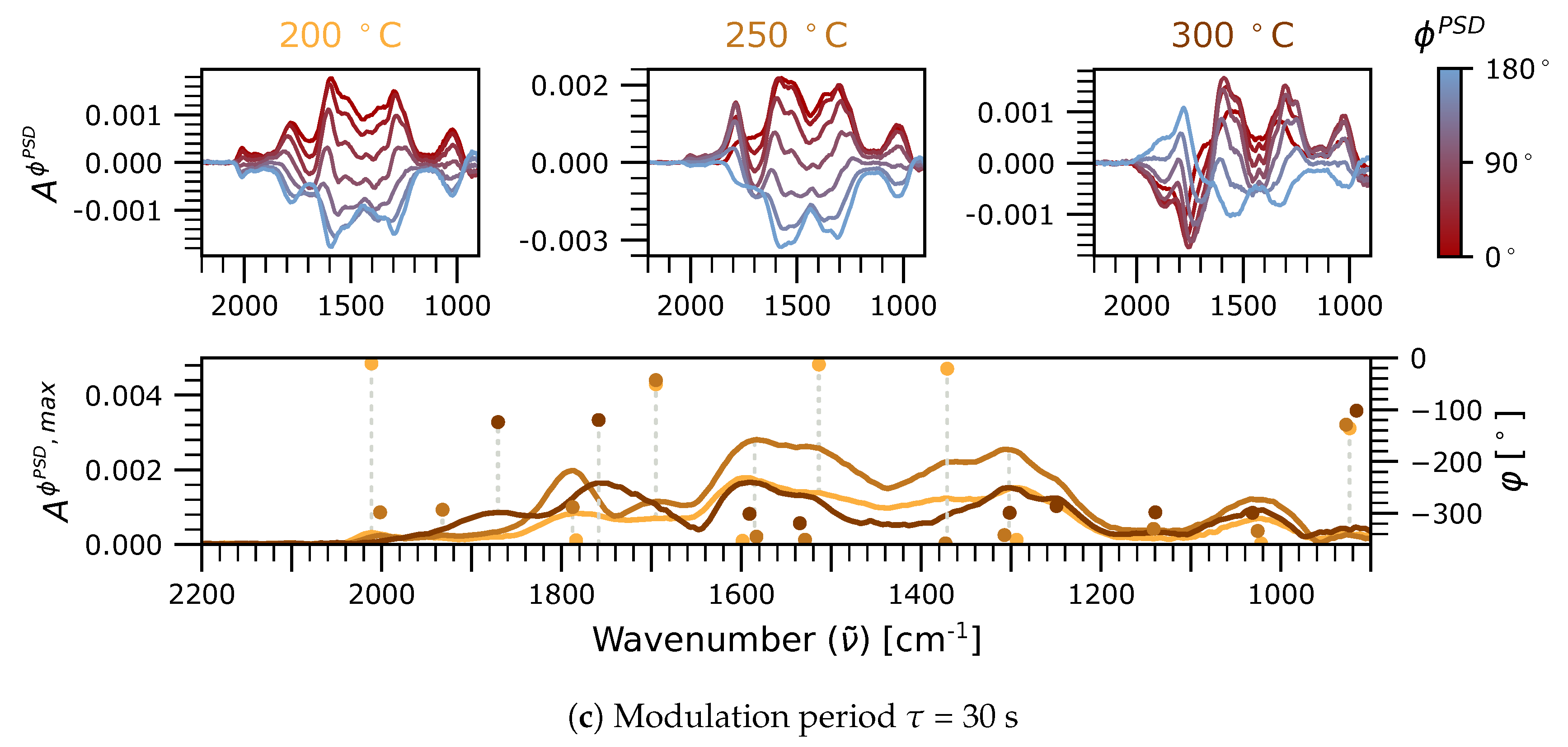
2.2.1. Phase-Resolved Spectra in the 2100–1700 cm Region: Carbonyls
2.2.2. Phase-Resolved Spectra in the 1700–800 and 3700–2700 cm Regions: Formates, Hydroxyls, and Carbonates
2.3. Significance of Adsorbed HCOO, CO, and CO as Reaction Intermediates
3. Materials and Methods
3.1. Catalyst Preparation and Characterization
3.2. Modulation Excitation Spectroscopy (MES) Measurements
3.3. Data Analysis by Phase Sensitive Detection (PSD)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Orecchini, F. The era of energy vectors. Int. J. Hydrog. Energy 2006, 31, 1951–1954. [Google Scholar] [CrossRef]
- Utgikar, V.P.; Thiesen, T. Safety of compressed hydrogen fuel tanks: Leakage from stationary vehicles. Technol. Soc. 2005, 27, 315–320. [Google Scholar] [CrossRef]
- Pasman, H.J.; Rogers, W.J. Safety challenges in view of the upcoming hydrogen economy: An overview. J. Loss Prev. Process Ind. 2010, 23, 697–704. [Google Scholar] [CrossRef]
- San Marchi, C.; Hecht, E.; Ekoto, I.; Groth, K.; LaFleur, C.; Somerday, B.; Mukundan, R.; Rockward, T.; Keller, J.; James, C. Overview of the DOE hydrogen safety, codes and standards program, part 3: Advances in research and development to enhance the scientific basis for hydrogen regulations, codes and standards. Int. J. Hydrog. Energy 2017, 42, 7263–7274. [Google Scholar]
- Vogt, C.; Monai, M.; Kramer, G.J.; Weckhuysen, B.M. The renaissance of the Sabatier reaction and its applications on Earth and in space. Nat. Catal. 2019, 2, 188–197. [Google Scholar]
- Frontera, P.; Macario, A.; Ferraro, M.; Antonucci, P. Supported catalysts for CO2 methanation: A review. Catalysts 2017, 7, 59. [Google Scholar] [CrossRef]
- Barrientos, J.; Lualdi, M.; Suárez París, R.; Montes, V.; Boutonnet, M.; Järås, S. CO methanation over TiO2-supported nickel catalysts: A carbon formation study. Appl. Catal. A Gen. 2015, 502, 276–286. [Google Scholar]
- Karelovic, A.; Ruiz, P. CO2 hydrogenation at low temperature over Rh/γ-Al2O3 catalysts: Effect of the metal particle size on catalytic performances and reaction mechanism. Appl. Catal. B Environ. 2012, 113–114, 237–249. [Google Scholar] [CrossRef]
- Martin, N.M.; Velin, P.; Skoglundh, M.; Bauer, M.; Carlsson, P.A. Catalytic hydrogenation of CO2 to methane over supported Pd, Rh and Ni catalysts. Catal. Sci. Technol. 2017, 7, 1086–1094. [Google Scholar] [CrossRef] [Green Version]
- Jacquemin, M.; Beuls, A.; Ruiz, P. Catalytic production of methane from CO2 and H2 at low temperature: Insight on the reaction mechanism. Catal. Today 2010, 157, 462–466. [Google Scholar]
- Luo, L.; Li, H.; Peng, Y.; Feng, C.; Zeng, J. Rh-Based Nanocatalysts for Heterogeneous Reactions. ChemNanoMat 2018, 4, 451–466. [Google Scholar] [CrossRef]
- Martin, N.M.; Hemmingsson, F.; Wang, X.; Merte, L.R.; Hejral, U.; Gustafson, J.; Skoglundh, M.; Meira, D.M.; Dippel, A.C.; Gutowski, O.; et al. Structure–function relationship during CO2 methanation over Rh/Al2O3 and Rh/SiO2 catalysts under atmospheric pressure conditions. Catal. Sci. Technol. 2018, 8, 2686–2696. [Google Scholar] [CrossRef] [Green Version]
- Martin, N.M.; Hemmingsson, F.; Schaefer, A.; Ek, M.; Merte, L.R.; Hejral, U.; Gustafson, J.; Skoglundh, M.; Dippel, A.C.; Gutowski, O.; et al. Structure–function relationship for CO2 methanation over ceria supported Rh and Ni catalysts under atmospheric pressure conditions. Catal. Sci. Technol. 2019, 9, 1644–1653. [Google Scholar] [CrossRef] [Green Version]
- Müller, P.; Hermans, I. Applications of Modulation Excitation Spectroscopy in Heterogeneous Catalysis. Ind. Eng. Chem. Res. 2017, 56, 1123–1136. [Google Scholar] [CrossRef]
- Li, C.; Sakata, Y.; Arai, T.; Domen, K.; Maruya, K.i.; Onishi, T. Carbon monoxide and carbon dioxide adsorption on cerium oxide studied by Fourier-transform infrared spectroscopy. Part 1.—Formation of carbonate species on dehydroxylated CeO2, at room temperature. J. Chem. Soc. Faraday Trans. 1 1989, 85, 929–943. [Google Scholar] [CrossRef]
- Kiennemann, A.; Breault, R.; Hindermann, J.P.; Laurin, M. Ethanol promotion by the addition of cerium to rhodium–silica catalysts. J. Chem. Soc. Faraday Trans. 1 1987, 83, 2119–2128. [Google Scholar]
- Trautmann, S.; Baerns, M. Infrared Spectroscopic Studies of CO Adsorption on Rhodium Supported by SiO2, Al2O3, and TiO2. J. Catal. 1994, 150, 335–344. [Google Scholar] [CrossRef]
- Araiza, D.G.; Gómez-Cortés, A.; Díaz, G. Reactivity of methanol over copper supported on well-shaped CeO2: A TPD-DRIFTS study. Catal. Sci. Technol. 2017, 7, 5224–5235. [Google Scholar] [CrossRef]
- Beutler, A.; Lundgren, E.; Nyholm, R.; Andersen, J.; Setlik, B.; Heskett, D. Coverage- and temperature-dependent site occupancy of carbon monoxide on Rh(111) studied by high-resolution core-level photoemission. Surf. Sci. 1998, 396, 117–136. [Google Scholar] [CrossRef]
- Krenn, G.; Bako, I.; Schennach, R. CO adsorption and CO and O coadsorption on Rh(111) studied by reflection absorption infrared spectroscopy and density functional theory. J. Chem. Phys. 2006, 124, 144703. [Google Scholar] [CrossRef]
- Kusama, H.; Bando, K.K.; Okabe, K.; Arakawa, H. Effect of metal loading on CO2 hydrogenation reactivity over Rh/SiO2 catalysts. Appl. Catal. A Gen. 2000, 197, 255–268. [Google Scholar] [CrossRef]
- Fisher, I.A.; Bell, A.T. A Comparative Study of CO and CO2 Hydrogenation over Rh/SiO2. J. Catal. 1996, 162, 54–65. [Google Scholar] [CrossRef]
- Li, C.; Sakata, Y.; Arai, T.; Domen, K.; Maruya, K.i.; Onishi, T. CO disproportionation at mild temperatures over partially reduced cerium oxide. J. Chem. Soc. Chem. Commun. 1991, 410–411. [Google Scholar] [CrossRef]
- Jin, T.; Okuhara, T.; Mains, G.J.; White, J. Temperature-programmed desorption of carbon monoxide and carbon dioxide from platinum/ceria: An important role for lattice oxygen in carbon monoxide oxidation. J. Phys. Chem. 1987, 91, 3310–3315. [Google Scholar] [CrossRef]
- Li, C.; Sakata, Y.; Arai, T.; Domen, K.; Maruya, K.i.; Onishi, T. Adsorption of carbon monoxide and carbon dioxide on cerium oxide studied by Fourier-transform infrared spectroscopy. Part 2.—Formation of formate species on partially reduced CeO2 at room temperature. J. Chem. Soc. Faraday Trans. 1 1989, 85, 1451–1461. [Google Scholar] [CrossRef]
- Lin, L.; Yao, S.; Liu, Z.; Zhang, F.; Li, N.; Vovchok, D.; Martínez-Arias, A.; Castañeda, R.; Lin, J.; Senanayake, S.D.; et al. In Situ Characterization of Cu/CeO2 Nanocatalysts for CO2 Hydrogenation: Morphological Effects of Nanostructured Ceria on the Catalytic Activity. J. Phys. Chem. C 2018, 122, 12934–12943. [Google Scholar] [CrossRef]
- Binet, C.; Daturi, M.; Lavalley, J.C. IR study of polycrystalline ceria properties in oxidised and reduced states. Catal. Today 1999, 50, 207–225. [Google Scholar] [CrossRef]
- King, J.; Liu, C.; Chuang, S.S.C. In situ infrared approach to unravel reaction intermediates and pathways on catalyst surfaces. Res. Chem. Intermed. 2019, 45, 5831–5847. [Google Scholar] [CrossRef]
- Li, C.; Domen, K.; ichi Maruya, K.; Onishi, T. Spectroscopic identification of adsorbed species derived from adsorption and decomposition of formic acid, methanol, and formaldehyde on cerium oxide. J. Catal. 1990, 125, 445–455. [Google Scholar] [CrossRef]
- Ye, J.; Liu, C.; Mei, D.; Ge, Q. Active Oxygen Vacancy Site for Methanol Synthesis from CO2 Hydrogenation on In2O3(110): A DFT Study. ACS Catal. 2013, 3, 1296–1306. [Google Scholar] [CrossRef]
- Eilers, P.H.; Boelens, H.F. Baseline correction with asymmetric least squares smoothing. Leiden Univ. Med. Cent. Rep. 2005, 1, 5. [Google Scholar]
- Baurecht, D.; Fringeli, U.P. Quantitative modulated excitation Fourier transform infrared spectroscopy. Rev. Sci. Instrum. 2001, 72, 3782–3792. [Google Scholar]
- Baurecht, D.; Porth, I.; Fringeli, U. A new method of phase sensitive detection in modulation spectroscopy applied to temperature induced folding and unfolding of RNase A. Vib. Spectrosc. 2002, 30, 85–92. [Google Scholar]
- Urakawa, A.; Bürgi, T.; Baiker, A. Sensitivity enhancement and dynamic behavior analysis by modulation excitation spectroscopy: Principle and application in heterogeneous catalysis. Chem. Eng. Sci. 2008, 63, 4902–4909. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Site | Vibration | Wavenumber | Reference |
|---|---|---|---|---|
| [cm] | ||||
| RhCe-CO | Rh-CeO | (C=O) | 1700 | [16] |
| h-CO | Rh | (C=O) | 1800 | [20] |
| br-CO | Rh | (C=O) | 1875 | [22] |
| m-CO | Rh | (C=O) | 2010 | [22] |
| Species | Site | Vibration | Wavenumber | Reference |
|---|---|---|---|---|
| [cm] | ||||
| p-CO | CeO | (OCO) | 1460 | [27] |
| (OCO) | 1390 | [27] | ||
| b-CO | CeO | (OCO) | 1590 | [15] |
| (OCO) | 1280 | [15] | ||
| b-HCOO | CeO | (OCO) | 1550 | [18] |
| (OCO) | 1300 | [18] | ||
| (CH) | 2850 | [18] | ||
| (CH)+(OCO) | 2950 | [18,27,29] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hemmingsson, F.; Schaefer, A.; Skoglundh, M.; Carlsson, P.-A. CO2 Methanation over Rh/CeO2 Studied with Infrared Modulation Excitation Spectroscopy and Phase Sensitive Detection. Catalysts 2020, 10, 601. https://doi.org/10.3390/catal10060601
Hemmingsson F, Schaefer A, Skoglundh M, Carlsson P-A. CO2 Methanation over Rh/CeO2 Studied with Infrared Modulation Excitation Spectroscopy and Phase Sensitive Detection. Catalysts. 2020; 10(6):601. https://doi.org/10.3390/catal10060601
Chicago/Turabian StyleHemmingsson, Felix, Andreas Schaefer, Magnus Skoglundh, and Per-Anders Carlsson. 2020. "CO2 Methanation over Rh/CeO2 Studied with Infrared Modulation Excitation Spectroscopy and Phase Sensitive Detection" Catalysts 10, no. 6: 601. https://doi.org/10.3390/catal10060601