Liver Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
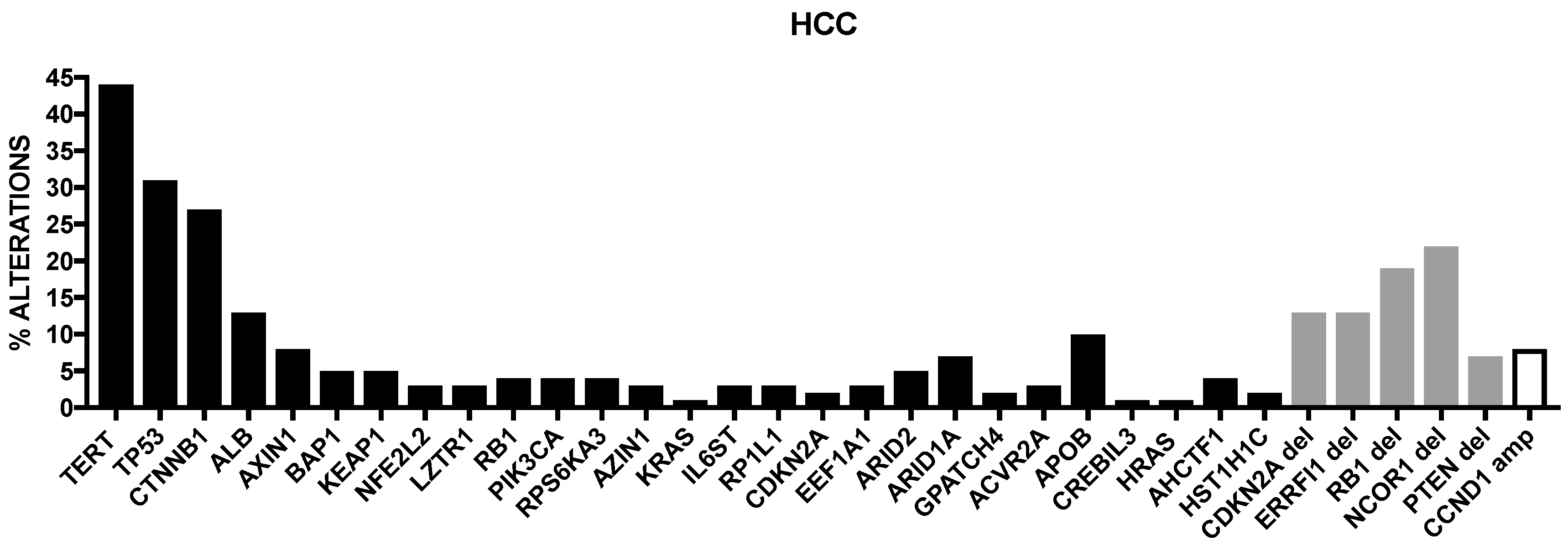
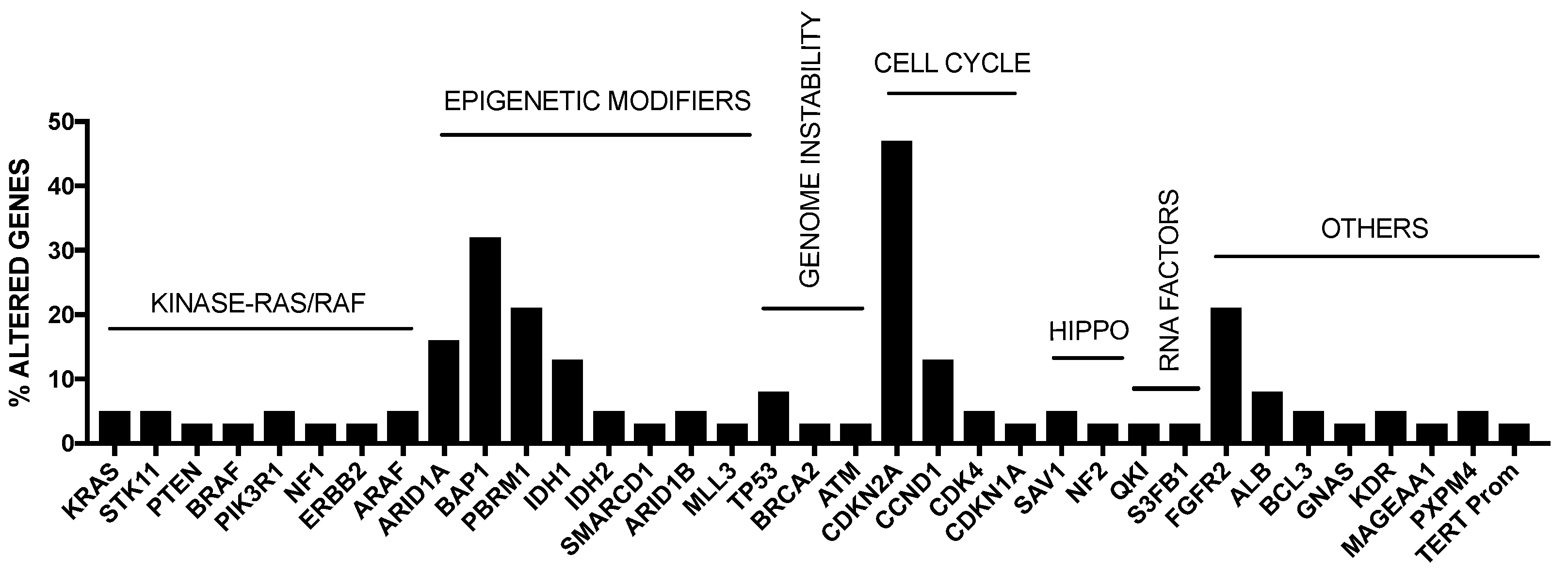
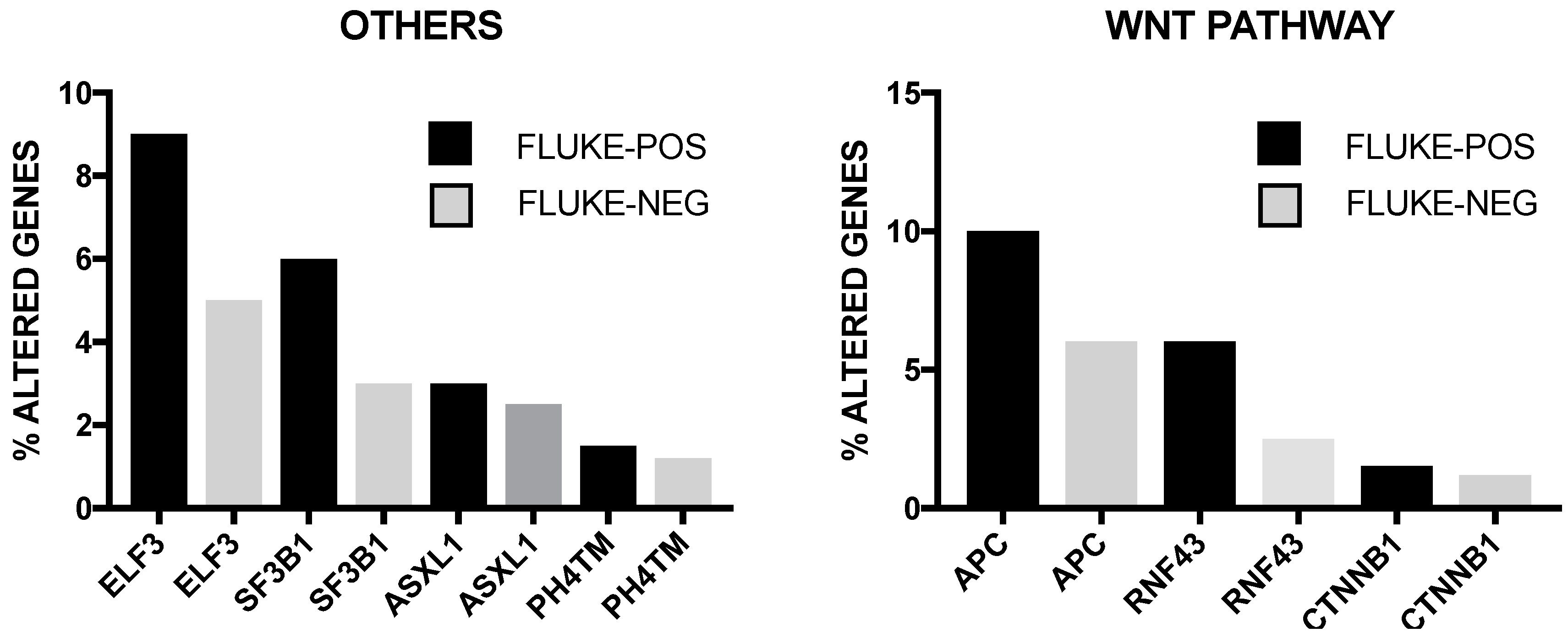
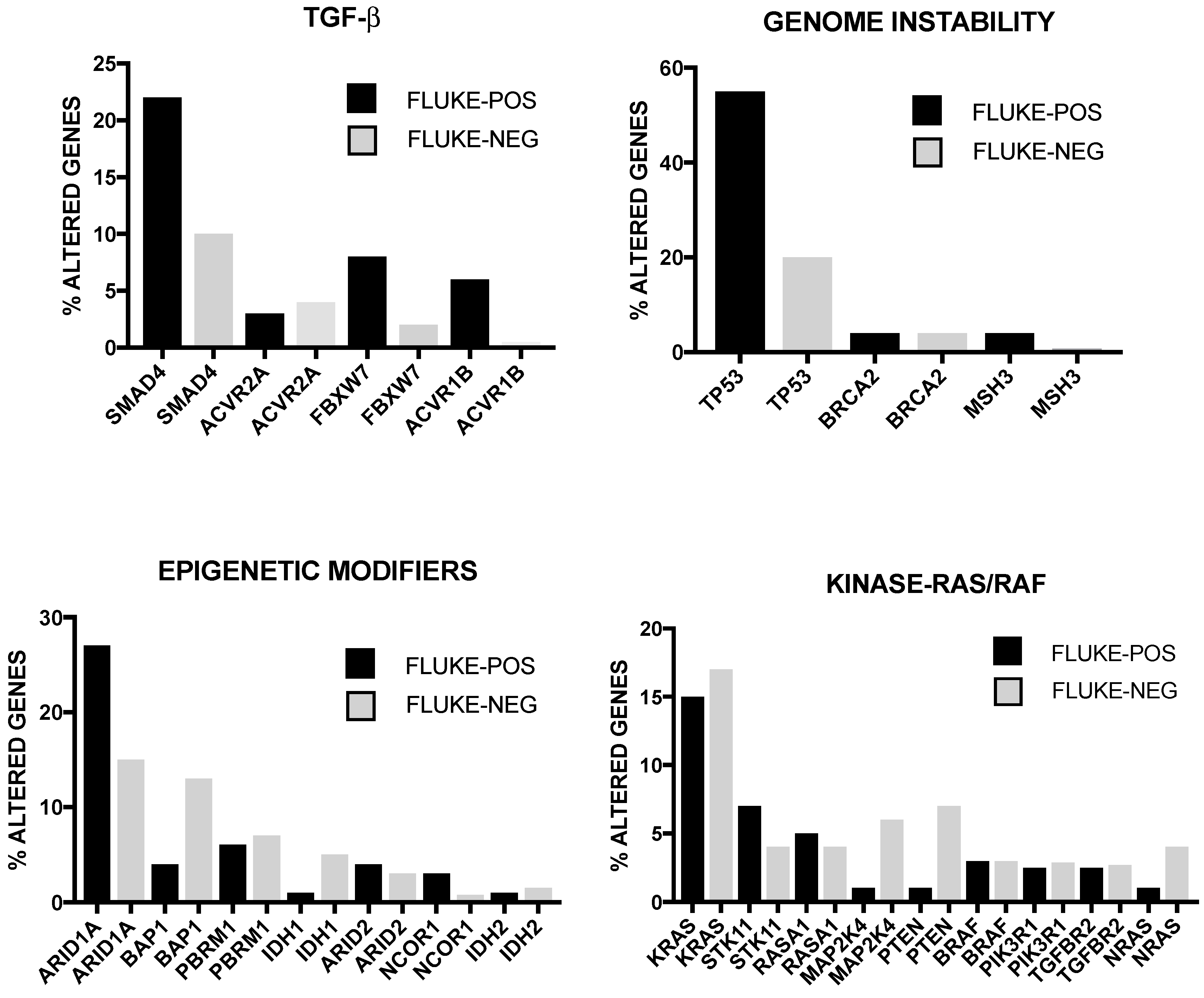
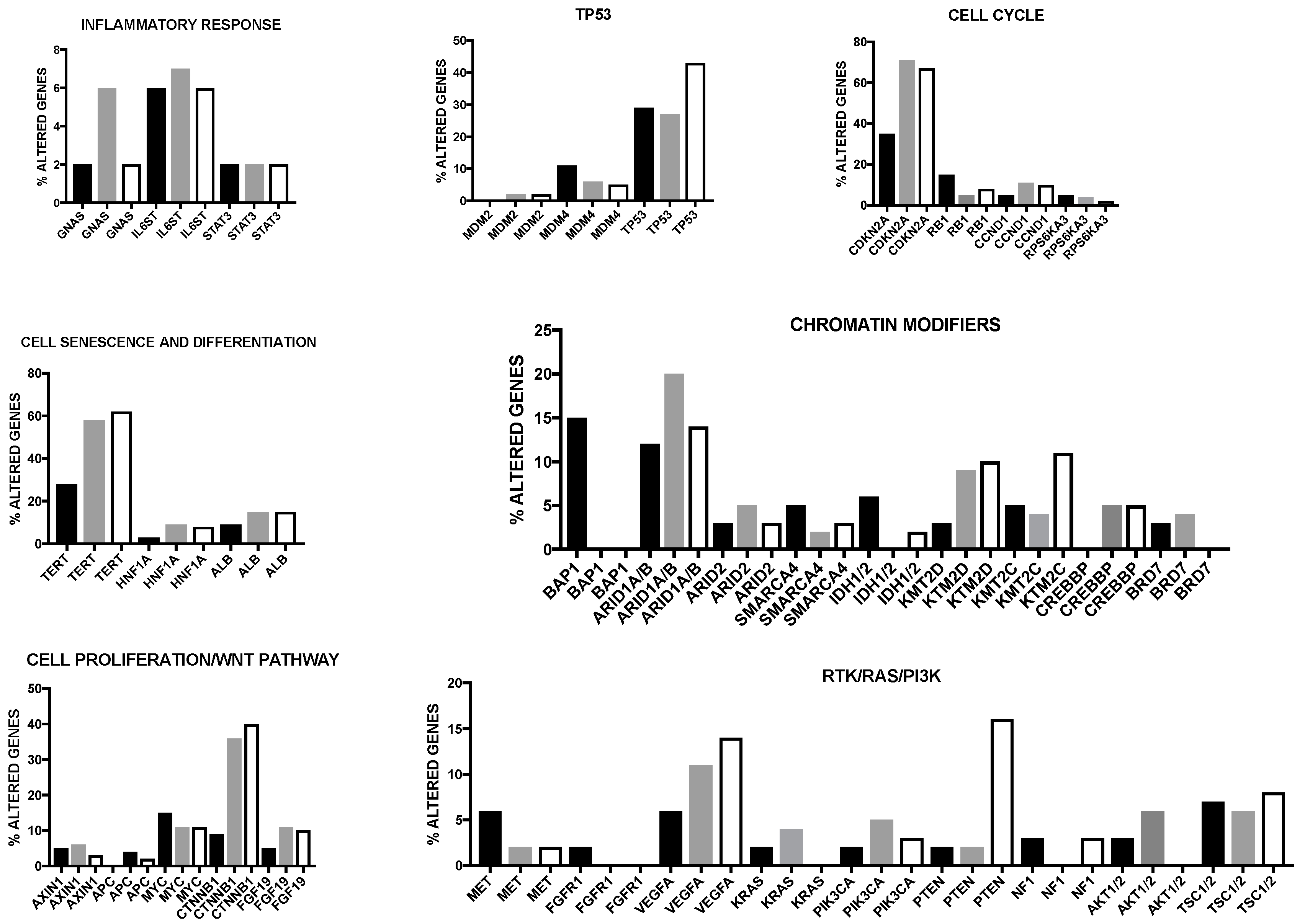
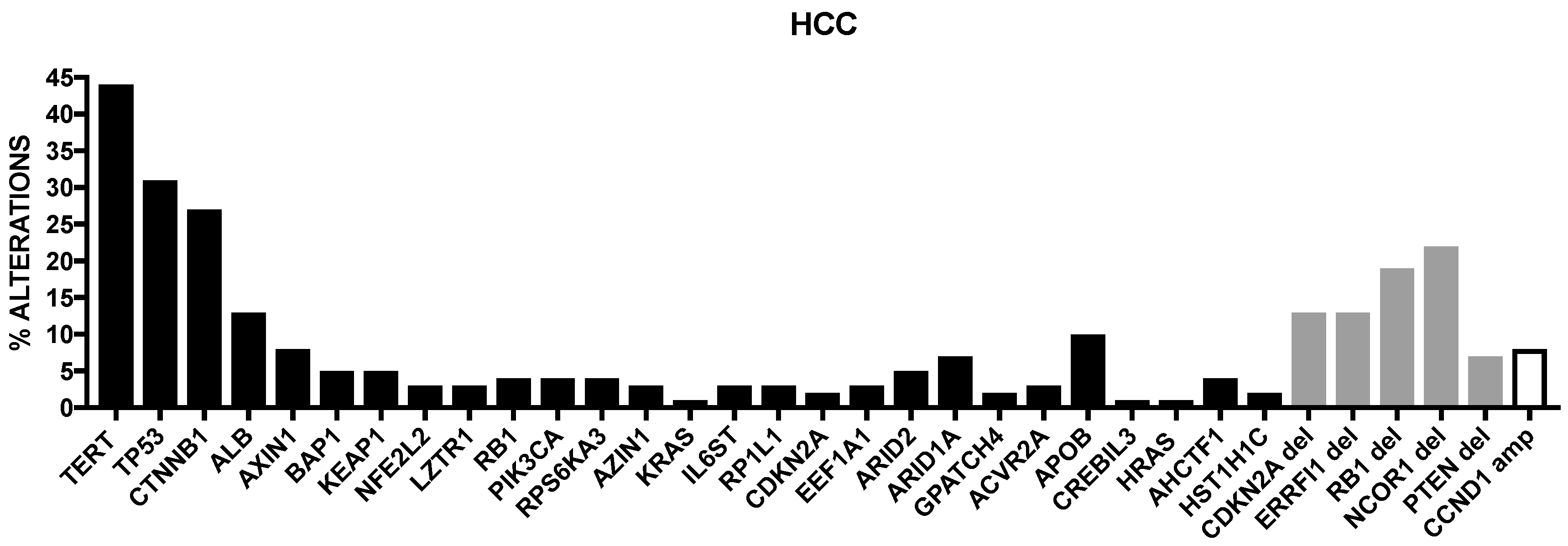
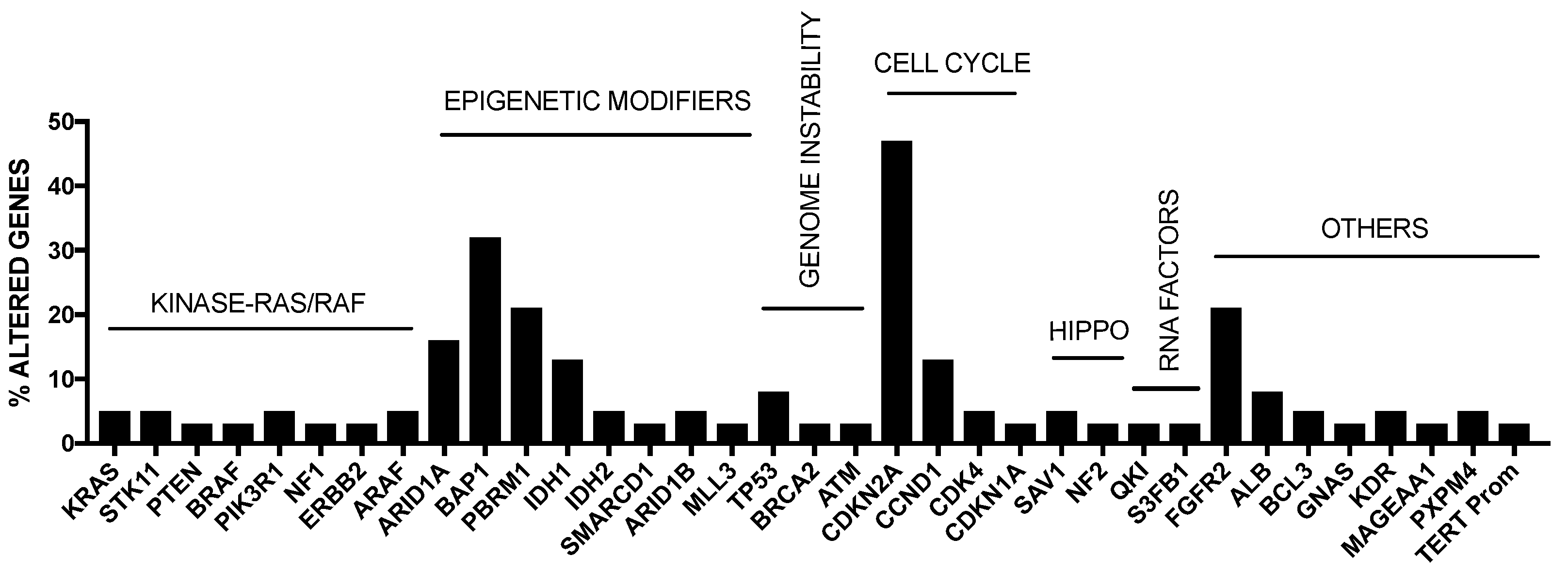
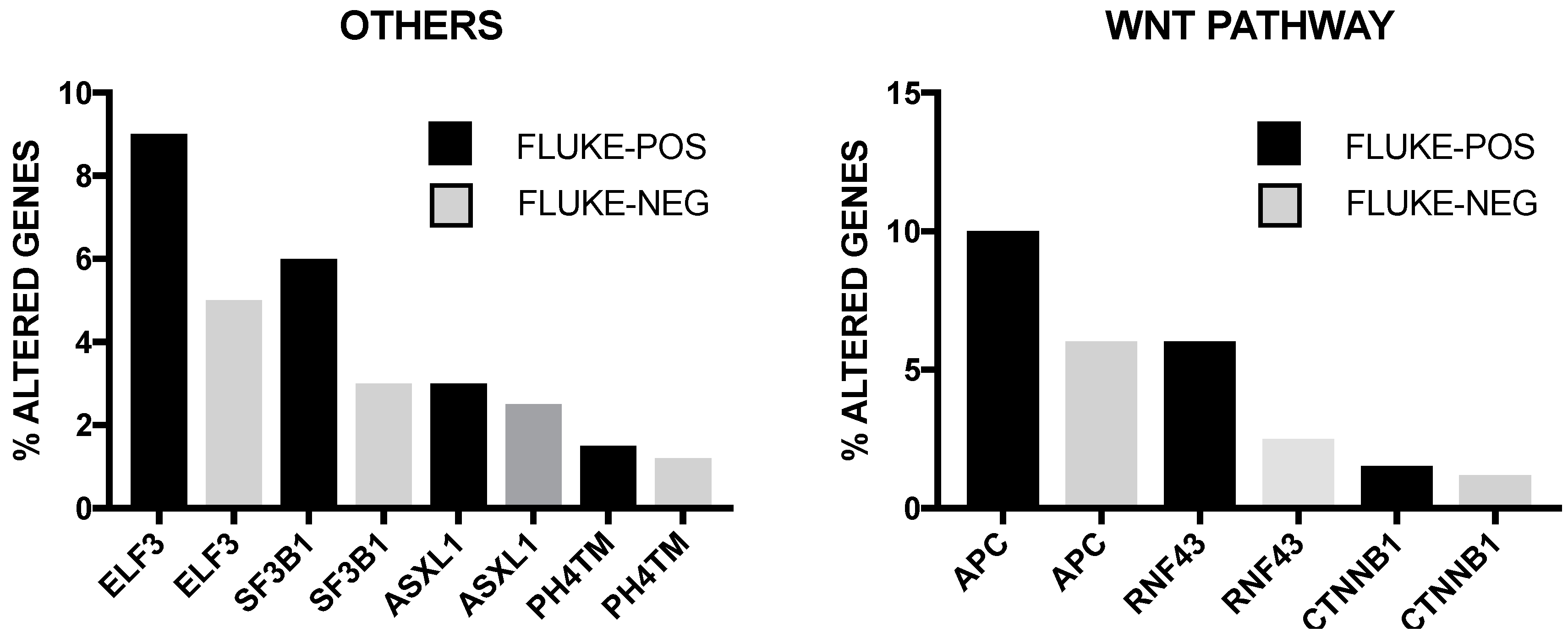
2. Genetic Abnormalities in Liver Cancer
3. Molecular Classification of HCCs
4. Cell Origin of Liver Cancer
5. Liver Cancer Stem Cells
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Ghouri, Y.A.; Misn, I.; Rowe, J.H. Review of hepatocellular carcinoma: Epidemiology, etiology, and carcinogenesis. J. Carcinog. 2017, 16, 1. [Google Scholar]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Villanueva, A.; Friedman, S.L.; Llovet, J.M. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology 2017, 152, 745–761. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Aburatani, H. Exploration of liver cancer genomes. Nat. Rev Gastroenterol. Pathol. 2014, 11, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Andersen, J.B.; Torgeirsson, S.S. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat. Rev. Cancer 2015, 15, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Totoki, Y.; Tatsumo, K.; Covington, K.; Ueda, H.; Creighton, C.J.; Kato, M.; Tsuji, S.; Donehower, L.A.; Slagle, B.L.; Nakamura, H.; et al. Trans-ancestry mutational landscape of hepatocellular carcinoma genomes. Nat. Genet. 2014, 46, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 2017, 169, 1327–1341. [Google Scholar]
- Fujimoto, A.; Furuta, M.; Totaki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakani, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2017, 45, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Dauch, D.; Rudalska, R.; Cossa, G.; Nault, J.C.; Kang, T.W.; Wuestefeld, T.; Hohmeyer, A.; Imbeaud, S.; Yevsa, T.; Hoenicke, L.; et al. A MYC-Aurora kinase a protein complex represents an actionable drug target in p53-altered liver cancer. Nat. Med. 2016, 22, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Niu, Z.S.; Niu, X.J.; Wang, W.H. Genetic alterations in hepatocellular carcinoma: An update. World J. Gastroenterol. 2016, 7, 9069–9095. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, J.U.; Seo, D.; Anderssen, J.B.; Gillen, M.C.; Kim, M.S.; Conner, E.A.; Galle, P.R.; Factor, V.M.; Park, Y.N.; Thorgeirsson, S.S. Sequential transcriptome analysis of human liver cancer indicates late stages acquisition of malignant traits. J. Hepatol. 2014, 60, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Calderaro, G.; Di Tommaso, L.; Balabaud, C.; Zafrani, E.S.; Bioulac-Sage, P.; Roncalli, M.; Zucman-Rossi, J. Telomerase reverse transcriptase promoter mutation is an early somatic genetic alteration in the transformation of premalignant nodules in hepatocellular carcinoma on cirrhosis. Hepatology 2014, 60, 1983–1992. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Mallet, M.; Pilati, C.; Calderaro, J.; Bioulac-Sage, P.; Laurent, C.; Laurent, A.; Cherqui, D.; Balabaud, C.; Zucman-Rossi, J. High frequency of telomerase reverse-transcriptase promoter somatic mutations in hepatocellular carcinoma and preneoplastic lesions. Nat. Commun. 2013, 4, 2218. [Google Scholar] [CrossRef] [PubMed]
- Pilati, C.; Letouzè, E.; Nault, J.C.; Imbeaud, S.; Boulai, A.; Calderaro, J.; Poussin, K.; Franconi, A.; Couchy, G.; Morcrette, G.; et al. Genomic profiling of hepatocellular adenomas reveals recurrent FRK-activating mutations and the mechanism of malignant transformation. Cancer Cell 2014, 25, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Zucman-Rossi, J. TERT promoter mutations in primary liver tumors. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Datta, S.; Imbeaud, S.; Franconi, A.; Mallet, M.; Couchy, G.; Letouzé, E.; Pilati, C.; Verret, B.; Blanc, J.F.; et al. Recurrent AAV2-related insertional mutagenesis in human hepatocellular carcinomas. Nat. Genet. 2015, 47, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; Couchy, G.; Balabaud, C.; Marcrette, G.; Caruso, S.; Blanc, J.F.; Bacq, Y.; Calderaro, J.; Paradis, V.; Ramos, J.; et al. Molecular classification of hepatocellular adenoma associated with risk factors, bleeding, and malignant transformation. Gastroenterology 2017, 152, 880–894. [Google Scholar] [CrossRef] [PubMed]
- Kenmochi, K.; Sugihara, S.; Kojiro, M. Relationship of histologic grade of hepatocellular carcinoma (HCC) to tumor size, and demonstration of tumor cells of multiple different grades in single small HCC. Liver 1987, 7, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Friemel, J.; Rechsteiner, M.; Friek, L.; Böhm, F.; Struckmann, K.; Egger, M.; Moch, H.; Heikenwalder, M.; Weber, A. Intratumor heterogeneity in hepatocellular carcinoma. Clin. Cancer Res. 2014, 21, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Xue, R.; Li, R.; Guo, H.; Guo, L.; Su, Z.; Ni, X.; Qi, L.; Zhang, T.; Li, Q.; Zhang, Z.; et al. Variable intra-tumor genomic heterogeneity of multiple lesions in patients with hepatocellular carcinoma. Gatsroenterology 2016, 150, 998–1008. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.Y.; Xing, Q.; Duang, M.; Wang, Z.C.; Yang, L.X.; Zhao, Y.J.; Wang, X.Y.; Liu, Y.; Deng, M.; Ding, Z.B.; et al. Inferring the progression of multifocal liver cancer from spatial and temporal genomic heterogeneity. Oncotarget 2016, 7, 2867–2877. [Google Scholar] [CrossRef] [PubMed]
- Torrecilla, S.; Sia, D.; Harrington, A.N.; Zhang, Z.; Cabellos, L.; Cornella, H.; Moeini, A.; Camprecios, G.; Leow, W.Q.; Fiel, M.I.; et al. Trunk events present minimal intra- and inter-tumoral heterogeity in hepatocellular carcinoma. J. Hepatol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Han, K.H.; Gores, G.; Llovet, J.M.; Mazzaferro, V. Liver cancer: Approaching a personalized care. J. Hepatol. 2015, 62, 5144–5156. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Hoshida, Y.; Battiston, C.; Tovar, V.; Sia, D.; Alsinet, C.; Cornella, H.; Liberzon, A.; Kobayashi, M.; Kumada, H.; et al. Combining clinical, pathology, and gene expression data to predict recurrence of hepatocellular carcinoma. Gastroenterology 2011, 140, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Yasuchika, K.; Ishii, T.; Katayama, H.; Yoshitoshi, E.Y.; Ogiso, S.; Kita, S.; Yasuda, K.; Fukumitsu, K.; Mizumoto, M.; et al. Keratin 19, a cancer stem cell marker in human hepatocellular carcinoma. Clin. Cancer Res. 2015, 21, 3081–3091. [Google Scholar] [CrossRef] [PubMed]
- Miltiadous, O.; Sia, D.; Hoshida, Y.; Fiel, M.I.; Harrington, A.N.; Thung, S.N.; Tan, P.S.; Dong, H.; Revill, K.; Chang, C.Y.; et al. Progenitor cell markers predict outcome of patients with hepatocellular carcinoma beyond Milan criteria undergoing liver transplantation. J. Hepatol. 2015, 6, 1368–1377. [Google Scholar] [CrossRef] [PubMed]
- Nault, J.C.; De Reynies, A.; Villanueva, A.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Decaens, T.; Franco, D.; Imbeaud, S.; Rousseau, F.; et al. A hepatocellular carcinoma 5-gene score associated with survival of patients after liver resection. Gastroenterology 2013, 145, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Villanueva, A.; Kobayashi, M.; Peix, J.; Chiang, D.Y.; Camargo, A.; Gupta, S.; Moore, J.; Wrobel, M.J.; Lerner, J.; et al. Gene expression in fixed tissues and outcome in hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Calderaro, J.; Couchy, G.; Imbeaud, S.; Amaddeo, G.; Letouzé, E.; Blanc, J.F.; Laurent, C.; Hajji, Y.; Azoulay, D.; Bioulac-Sage, P.; et al. Histological subtypes of hepatocellular carcinoma are related to gene mutations and molecular tumor classification. J. Hepatol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Moeini, A.; Sia, D.; Bardseesy, N.; Mazzaferro, V.; Liovet, J.M. Molecular pathogenesis and targeted therapies of intrahepatic cholangiocarcinoma. Clin. Cancer Res. 2015, 22, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Chan-on, W.; Nairismagi, M.L.; Ong, C.K.; Lim, W.K.; Dima, S.; Pairojkul, C.; Lim, K.H.; McPherson, J.R.; Cutcutache, I.; Heng, H.L.; et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat. Genet. 2013, 45, 1474–1478. [Google Scholar] [CrossRef] [PubMed]
- Jao, Y.; Pawlick, T.M.; Anders, R.A.; Selaru, F.M.; Streppel, M.M.; Lucas, D.J.; Niknafs, N.; Guthrie, V.B.; Maitra, A.; Argani, P.; et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013, 45, 1470–1473. [Google Scholar] [CrossRef] [PubMed]
- Farshidfar, F.; Zheng, S.; Gingras, M.C.; Newton, Y.; Shih, J.; Robertson, A.G.; Hinoue, T.; Huadley, K.; Gibb, E.; Roszik, J.; et al. Integrative genomic analysis of cholangiocarcinoma identifies distinct IDH-mutant molecular profiles. Cancer Cell 2017, 18, 2780–2794. [Google Scholar]
- Fujimoto, A.; Foruta, M.; Shiraishi, Y.; Gotoh, K.; Kawakami, Y.; Arihiro, K.; Nakamura, T.; Ueno, M.; Ariizumi, S.; Nguyen, H.H.; et al. Whole-genome mutational landscape of liver cancers displaying biliary phenotype reveals hepatitis and molecular diversity. Nat. Commun. 2015, 6, 6120. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Losic, B.; Mocini, A.; Cabellos, L.; Hao, K.; Revill, K.; Bonal, D.; Miltiadous, O.; Zhang, Z.; Hoshida, Y.; et al. Massive parallel sequencing uncovers actionable FGFR2-PPHLN1 fusion and ARAF mutations in intrahepatic cholangiocarcinoma. Nat. Commun. 2015, 6, 6087. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Totoki, Y.; Hosada, F.; Shirota, T.; Hama, N.; Nakamura, H.; Ojima, H.; Furuta, K.; Shimada, K.; Okusaka, T.; et al. Fibroblast growth factor receptor 2 tyrosine kinase fusions define a unique molecular subtype of cholangiocarcinoma. Hepatology 2014, 59, 1427–1434. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Hoshida, Y.; Villanueva, A.; Roayaie, S.; Ferrer, J.; Tabak, B.; Peix, J.; Sole, M.; Tovar, V.; Alsinet, C.; et al. Integrative molecular analysis of intrahepatic cholangiocarcinoma reveals 2 clones that have different outcomes. Gastroenterology 2013, 144, 829–840. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Cutcutache, I.; Yong, C.H.; Lim, J.Q.; Huang, M.N.; Padmannabhan, N.; Nellore, V.; Kongpetch, S.; Ng, A.; Ng, L.M.; et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 2017. [Google Scholar] [CrossRef] [PubMed]
- Chaissaingmongkol, J.; Budhu, A.; Dany, H.; Rabibhadana, S.; Pupacdi, B.; Kwon, S.M.; Forgues, M.; Pomyen, Y.; Bhudisawasdi, V.; Lertprasertsuke, N.; et al. Common molecular subtypes among Asian hepatocellular carcinoma and cholangiocarcinoma. Cancer Cell 2017, 32, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Simbolo, M.; Fassan, M.; Ruzzanente, A.; Mafficini, A.; Wood, L.D.; Corbo, V.; Melisi, D.; Malleo, G.; Vicentini, C.; Malpeli, G.; et al. Multigene mutational profiling of cholangiocarcinomas identifies actionable molecular subgroups. Oncotarget 2014, 5, 2839–2852. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urishidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Gingras, M.C.; Covington, K.R.; Chang, D.K.; Donehower, L.A.; Gill, A.J.; Ittmann, M.M.; Creighton, C.J.; Johns, A.L.; Shinbrot, E.; Dewal, N.; et al. Ampullary cancers harbor ELF3 tumor suppressor gene mutations and exhibit frequent WNT dysregulation. Cell Rep. 2016, 14, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Yachida, S.; Wood, L.D.; Suzuki, M.; Takai, E.; Totoki, Y.; Kato, M.; Luchini, C.; Arai, Y.; Nakamura, H.; Hama, N.; et al. Genomic sequencing identifies ELF3 as a driver of ampullary carcinoma. Cancer Cell 2016, 29, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Malouf, G.G.; Job, S.; Paradis, V.; Fabre, M.; Brugières, L.; Saintigny, P.; Vescovo, L.; Belghiti, J.; Branchereau, S.; Faivre, S.; et al. Transcriptional profiling of pure fibrolamellar hepatocellular carcinoma reveals an endocrine signature. Hepatology 2014, 59, 2228–2237. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Hazard, F.K.; Zmoos, A.F.; Jahchan, N.; Chaib, H.; Garfin, P.M.; Rangaswami, A.; Snyder, M.P.; Sage, J. Genomic analysis of fibrolamellar hepatocellular carcinoma. Hum. Mol. Genet. 2015, 24, 50–63. [Google Scholar] [CrossRef] [PubMed]
- Ward, S.C.; Huang, J.; Tickoo, S.K.; Thung, S.N.; Ladanyi, M.; Klimstra, D.S. Fibrolamellar carcinoma of the liver exhibits immunohistochemical evidence of both hepatocyte and bile duct differentiation. Mod. Pathol. 2010, 23, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Honeyman, J.N.; Simon, E.P.; Robine, N.; Chiaroni-Clarke, R.; Darcy, D.G.; Lim, I.I.; Gleason, C.E.; Murphy, J.M.; Rosenberg, B.R.; Teegan, L.; et al. Detection of recurrent DNAJB1-PKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science 2014, 28, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.P.; Jin, L.; Knutson, D.L.; Kloft-Nelson, S.M.; Greipp, P.T.; Waldburger, N.; Roessler, S.; Longerich, T.; Roberts, L.R.; Oliveira, A.M.; et al. DNAJB1-PRKACA is specific for fibrolamellar carcinoma. Mod. Pathol. 2015, 28, 822–829. [Google Scholar] [CrossRef] [PubMed]
- Darcy, D.; Chiaroni-Clarke, R.; Murphy, J.; Honeyman, J.N.; Bhanot, U.; LaQuaglia, M.; Simon, S.M. The genomic landscape of fibrolamellar hepatocellular carcinoma: Whole genome sequencing of ten patients. Oncotarget 2015, 6, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Riggle, K.M.; Riehle, K.J.; Kenerson, H.L.; Turnham, R.; Homma, M.K.; Kazami, M.; Samelson, B.; Bauer, R.; McKnight, G.S.; Scott, J.D.; et al. Enhanced cAMP-stimulated protein kinase A activity in human fibrolamellar hepatocellular carcinoma. Pediatr. Res. 2016, 80, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Piggle, K.M.; Turnham, R.; Scott, J.D.; Yeung, R.S.; Riehle, K. Fibrolamellar hepatocellular carcinoma: Mechanistic distinction from adult hepatocellular carcinoma. Pediatr. Blood Cancer 2016, 63, 1163–1167. [Google Scholar]
- Griffith, O.L.; Griffith, M.; Krysiak, K.; Magrini, V.; Ramu, A.; Skidmore, Z.L.; Kunisaki, J.; Austin, R.; McGrath, S.; Zhang, J.; et al. A genomic case study of mixed finrolamellar hepatocellular carcinoma. Ann. Oncol. 2016, 27, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.A.; Vitucci, E.C.; Wuathier, E.; Graham, R.P.; Pitman, W.A.; Oikawa, T.; Chen, M.; Silva, G.O.; Greene, K.G.; Torbenson, M.S.; et al. Comprehensive analysis of The Cancer Genome Atlas reveals a unique gene and non-coding RNA signature of fibrolamellar carcinoma. Sci. Rep. 2017, 7, 44653. [Google Scholar] [CrossRef] [PubMed]
- Simon, E.P.; Freije, C.A.; Farber, B.A.; Lalazar, G.; Darcy, D.G.; Honeyman, J.N.; Chiaroni-Clarke, R.; Dill, B.D.; Molina, H.; Bhanot, U.K.; et al. Transcriptomic characterization of fibrolamellar hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, E5916–E5925. [Google Scholar] [CrossRef] [PubMed]
- Cornella, H.; Alsinet, C.; Sayols, S.; Zhang, Z.; Hao, K.; Cabellos, L.; Hoshida, Y.; Villanueva, A.; Thung, S.; Ward, S.C.; et al. Unique genomic profile of fibrolamellar hepatocellular carcinoma. Gastroenterology 2015, 148, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, E.; Kharin, R.; Bamboat, Z.M.; Cavnar, M.J.; Kim, T.S.; Sadot, E.; Zeng, S.; Greer, J.; Seifert, A.M.; Cohen, N.A.; et al. Genome and transcriptome profiling of fibrolamellar hepatocellular carcinoma demonstrates p53 and IGFBP1 dysregulation. PLoS ONE 2017. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Weber, N.; Waha, A.; Hartmann, W.; Denkhaus, D.; Behrens, J.; Birchmeier, W.; von Schweinitz, D.; Pietsch, T. Mutations and elevated transcriptional activity of conductin (AXIN2) in hepatoblastomas. J. Pathol. 2004, 204, 546–554. [Google Scholar] [CrossRef] [PubMed]
- Aretz, S.; Koch, A.; Uhlhaas, S.; Friedl, W.; Propping, P.; von Schweinitz, D.; Pietsch, T. Should children at risk for familial adenomatous polyposis be screened for hepatoblastoma and children with apparently sporadic hepatoblastoma be screened for APC germline mutations? Pediatr. Blood Cancer 2006, 47, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Koch, A.; Waha, A.; Hartmann, W.; Hrychyk, A.; Schuller, U.; Waha, A.; Wharton, K.A.; Fuchs, S.Y.; von Schweinitz, D.; Pietsch, T. Elevated expression of Wnt antagonists is a common event in hepatoblastomas. Clin. Cancer Res. 2005, 11, 4295–4304. [Google Scholar] [CrossRef] [PubMed]
- Eichenmuller, M.; Trippel, F.; Kreuder, M.; Beck, A.; Schwarzmayr, T.; Haberle, B.; Cairo, S.; Leuschner, I.; von Schweinitz, D.; Strom, T.M.; et al. The genomic landscape of hepatoblastoma and their progenies with HCC-like features. J. Hepatol. 2014, 61, 1312–1320. [Google Scholar] [CrossRef] [PubMed]
- Jia, D.; Dong, R.; Jing, Y.; Xu, D.; Wang, Q.; Chen, L.; Li, Q.; Huang, Y.; Zhang, Y.; Zhang, Z.; et al. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology 2014, 60, 1686–1696. [Google Scholar] [CrossRef] [PubMed]
- Cadoret, A.; Ovejero, C.; Souadi-Kheddouci, S.; Souil, E.; Fabre, M.; Romagnolo, B.; Kahn, A.; Perret, C. Hepatomegaly in transgenic mice expressing an oncogenic form of beta-catenin. Cancer Res. 2001, 61, 3245–3249. [Google Scholar] [PubMed]
- Harada, N.; Miyoshi, H.; Murai, N.; Oshima, H.; Tamai, Y.; Oshima, M.; Jaketo, M.M. Lack of tumorigenesis in the mouse liver after adenovirus-mediated expression of a dominant stable mutant of beta-catenin. Cancer Res. 2002, 62, 1971–1977. [Google Scholar] [PubMed]
- Cairo, S.; Armengol, C.; de Reyniès, A.; Wei, Y.; Thomas, E.; Renard, C.A.; Goya, A.; Balekrishnan, A.; Semeraro, M.; Gresh, L.; et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 2008, 14, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Comerford, S.A.; Hinnant, E.; Chen, Y.; Bausal, H.; Klapproth, S.; Rakheja, D.; Finegold, M.; Lopez-Terrada, D.; O’Donnel, K.; Tomlinson, G.; et al. Hepatoblastoma modeling in mice places Nrf2 within a cancer field established by mutant β-catenin. JCI Insight 2016, 1, e88549. [Google Scholar] [CrossRef] [PubMed]
- Sumazin, P.; Chen, Y.; Trevino, L.R.; Sarabia, S.F.; Hampton, O.A.; Parel, K.; Mistretta, T.A.; Zorman, B.; Thompson, P.; Heczey, A.; et al. Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology 2017, 65, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Theise, N.D.; Nakashima, O.; Park, Y.N. Combined hepatocellular-cholangiocarcinoma. In WHO Classification of Tumours of the Digestive System, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2010; pp. 225–227. [Google Scholar]
- Coulouarn, C.; Cavard, C.; Rubbia-Brandt, L. Combined hepatocellular-cholangiocarcinomas exhibit progenitor features and activation of Wnt and TGFbeta signaling pathways. Carcinogenesis 2012, 33, 1791–1796. [Google Scholar] [CrossRef] [PubMed]
- Moeini, A.; Sia, D.; Zhang, Z.; Compacios, G.; Stueck, A.; Dong, H.; Montal, R.; Torrens, L.; Martinez-Quetglas, I.; Fiel, M.I.; et al. Mixed hepatocellular-cholangiocarcinoma tumors: Cholangiocellular carcinoma is distinct molecular entity. J. Hepatol. 2017, 66, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, M.; Batur, T.; Ekin, U.; Erdogan, A.; Iscan, E.; Keles, U.; Oz, O.; Ozen, C. Molecular pathogenesis of liver cancer. J Gastrointest. Cancer 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Villanueva, A.; Lachenmayer, A.; Finn, R.S. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol. 2015, 12, 408–424. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Llovet, J.M. Translating “-omics” results into precision medicine for hepatocellular carcinoma. Nat. Rev. Gastroenterol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Nijman, S.M.B.; Kobayashi, M.; Chan, J.A.; Brunet, J.P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative transcriptomic analysis reveals common molecular subclasses of human hepatocellular carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef] [PubMed]
- Goossesns, N.; Sun, X.; Hoshida, Y. Molecular classification of hepatocellular carcinoma: Potential therapeutic implications. Hepatol. Oncol. 2015, 2, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.X.; Zhu, Q.G.; Zhang, S.M.; Guan, L.; Li, T.; Zhang, L.; Wang, S.Y.; Ren, W.L.; Chen, X.M.; Zhao, J.; et al. Precision medicine for hepatocellular carcinoma: driver mutations and targeted therapy. Oncotarget 2017, 8, 55715–55730. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Xu, S.H.; Wang, H.Q.; Cai, Y.J.; Ying, L.; Song, M.; Wang, Y.Q.; Du, S.J.; Shi, K.Q.; Zhou, M.T. Prognostic value of DNA repair based stratification of hepatocellular carcinoma. Sci. Rep. 2016, 6, 25999. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Heo, J.; Lebbrecht, L.; Chu, I.S.; Kaposi-Novak, P.; Calvisi, D.F.; Mikaelyan, A.; Roberts, L.R.; Demetris, A.J.; Sun, Z.; et al. A novel prognostic subtype of human hepatocellular carcinoma derived from he3patic progenitor cells. Nat. Med. 2006, 12, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; Castro de Moura, M.; Putra, J.; Camprecios, J.; Bassaganyas, L.; Akers, N.; et al. Identification of an immune-specific class of hepatocellular carcinoma, based on molecular features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef] [PubMed]
- El-Khoueiry, A.; Sangro, B.; Yau, T.; Crocenzi, T.; Kuda, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H., 3rd; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): An open-label, non-comparative, phase ½ dose escalation and expansion trial. Lancet 2017, 389, 2492–2502. [Google Scholar] [CrossRef]
- Tarlow, B.D.; Finegold, M.J.; Grompe, M. Clonal tracing of Sox9+ liver progenitors in mouse oval cell injury. Hepatology 2014, 60, 278–289. [Google Scholar] [CrossRef] [PubMed]
- Schaub, J.R.; Malato, Y.; Gormond, C.; Willenbring, H. Evidence against a stem cell origin of new hepatocytes in a common mouse model of chronic liver injury. Cell Rep. 2014, 8, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Yanger, K.; Knigin, D.; Zong, Y.; Maggs, L.; Gu, G.; Akiyama, H.; Pikarsky, E.; Stanger, B.Z. Adult hepatocytes are generated by self-duplication rather than stem cell differentiation. Cell Stem Cell 2014, 15, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Tanimizu, N.; Nishikawa, Y.; Ichinohe, N.; Akiyama, H.; Mitaka, T. Sry HMG box protein p-positive (Sox9+) epithelial cell adhesion molecule-negative (EpCAM)-biphenotypic cells derived from hepatocytes are involved in mouse liver regeneration. J. Biol. Chem. 2014, 289, 7589–7598. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, S.; Suzuki, A. Hepatocytes, rather than cholangiocytes, can be the target source of primitive ductules in the chronically injured mouse liver. Am. J. Pathol. 2014, 184, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Yanger, K.; Zong, Y.; Maggs, L.R.; Shapira, S.N.; Maddipati, R.; Aiello, N.M.; Thung, S.N.; Wells, R.G.; Greenbaum, L.E.; Stanger, B.Z. Robust cellular reprogramming occurs spontaneously during liver regeneration. Genes. Dev. 2013, 27, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Yimlamai, D.; Christodoulou, C.; Galli, G.G.; Yanger, K.; Pepe-Mooney, B.; Gurung, B.; Shrestha, K.; Cahan, P.; Stanger, B.Z.; Camargo, F.D. Hippo pathway activity influences liver cell fate. Cell 2014, 157, 1324–1338. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Chen, H.L.; Chien, C.S.; Wu, S.H.; Ho, Y.T.; Yu, C.H.; Chang, M.H. Contribution of mature hepatocytes to biliary regeneration in rats with acute and chronic biliary injury. PLoS ONE 2015, 10, e013427. [Google Scholar] [CrossRef] [PubMed]
- Font-Burgada, J.; Shalapour, S.; Ramaswamy, S.; Hsueh, B.; Rossell, D.; Umemura, A.; Taniguchi, K.; Nakagawa, H.; Valasek, M.A.; Ye, L.; et al. Hybri periportal hepatocytes regenerate the injured liver without giving rise to cancer. Cell 2015, 162, 766–779. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, A.; Tanaka, M.; Itoh, T. Stem/progenitor cells in liver development, homeostasis, regeneration and reprogramming. Cell Stem Cell 2014, 14, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhao, L.; Fish, M.; Logan, C.Y.; Nusse, R. Self-renewing diploid Axin2+ cells fuel homeostatic renewal of the liver. Nature 2015, 254, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Tarlow, B.D.; Pelz, C.; Naugler, W.E.; Wakefield, L.; Wilson, E.M.; Fineglod, M.J.; Grompe, M. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell 2014, 15, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Boulter, L.; Govaere, O.; Bird, T.G.; Radulescu, S.; Ramachandran, P.; Pellicoro, A.; Ridgway, R.A.; Seo, S.S.; Spee, B.; Van Rooijen, N.; et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat. Med. 2012, 18, 572–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulter, L.; Lu, W.Y.; Farbes, S.J. Differentiation of progenitors in the liver: A matter of local choice. J. Clin. Investig. 2013, 123, 1867–1873. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Dorrell, C.; Boj, S.F.; van Es, J.H.; Li, V.S.; van de Wetering, M.; Sato, T.; Hamer, K.; Sasaki, N.; Finegold, M.J.; et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013, 494, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Gehart, H.; Bourtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Raven, A.; Lu, W.Y.; Man, T.Y.; Ferreira-Gonzalez, S.; O’Duibhir, E.; Dwer, B.J.; Thomson, J.P.; Meehan, R.; Bogorad, R.; Kotelinasky, Y.; et al. Cholangiocytes acr as facultative liver stem cells during impaired hepatocyte regeneration. Nature 2017, 547, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Kordes, C.; Sawitza, I.; Gotze, S.; Herebian, D.; Haussinger, D. Hepatic stellate cells contribute to progenitor cells and liver regeneration. J. Clin. Investig. 2014, 124, 5503–5515. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Sawitza, I.; Kordes, C.; Gotze, S.; Herbian, D.; Haussinger, D. Bile acids induce hepatic differentiation of mesenchymal stem cells. Sci. Rep. 2015, 5, 13320. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Calvisi, D.F. Hydrodynamic transfection for generation of models for liver cancer research. Am. J. Pathol. 2014, 184, 912–923. [Google Scholar] [CrossRef] [PubMed]
- Tschaharganeh, D.F.; Xue, W.; Calvisi, D.F.; Evert, M.; Michurina, T.V.; Dow, L.E.; Banito, A.; Katz, S.F.; Kastenhuber, E.R.; Weissmueller, S.; et al. p53-dependent nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 2014, 158, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Fitamant, J.; Kottakis, F.; Benhamouche, S.; Tian, H.S.; Chuvin, N.; Parachoniak, C.A.; Nagle, J.M.; Perera, R.M.; Lapouge, M.; Deshpande, V.; et al. YAP inhibition restores hepatocyte differentiation in advanced HCC, leading to tumor regression. Cell Rep. 2015, 10, 1692–1707. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Espanol-Suner, R.; Mederacke, I.; Affò, S.; Manco, R.; Sempoux, C.; Lemaigre, F.P.; Adili, A.; Yuan, D.; Weber, A.; et al. Hepatocellular carcinoma originates from hepatocytes and not from the progenitor/biliary compartment. J. Clin. Investig. 2015, 125, 3891–3902. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A. Evidence of cell fate conversion from hepatocytes to cholangiocytes in the injured liver: In vivo genetic lineage-tracing approaches. Curr. Opin. Gastroenterol. 2015, 31, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Zender, S.; Nickeleit, I.; Wuestefeld, T.; Sorensen, I.; Dauch, D.; Bozko, P.; El-Khatib, M.; Geffers, R.; Berktas, H.; Manns, M.P.; et al. A critical role for Notch signaling in the formation of cholangiocellular carcinomas. Cancer Cell 2013, 23, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Guest, R.V.; Boulter, L.; Kendall, T.J.; Minnis-Lyons, S.E.; Walker, R.; Wigmore, S.J.; Sanson, O.J.; Forbes, S.J. Cell lineage tracing reveals a biliary origin of intrahepatic cholangiocarcinoma. Cancer Res. 2014, 74, 1005–1010. [Google Scholar] [CrossRef]
- Lu, W.Y.; Bird, T.; Boulter, L.; Tsuchiya, A.; Cole, A.M.; Hay, T.; Guest, R.V.; Wojtacha, D.; Man, T.Y.; Mackinnon, A.; et al. Hepatic progenitor cells of biliary origin with liver repopulation capacity. Nat. Cell Biol. 2015, 17, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Boulter, L.; Guest, R.V.; Kendall, T.J.; Wilson, D.H.; Wojtacha, D.; Robson, A.J.; Ridgway, R.A.; Samuel, K.; Van Rooijen, N.; Barry, S.T.; et al. WNT signaling drives oligocarcinoma growth and can be pharmacologically inhibited. J. Clin. Investig. 2015, 125, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Zheng, Y.W.; Kita, K.; Yokosuka, O.; Saisho, H.; Onodera, M.; Miyoshi, H.; Nakano, M.; Zen, Y.; Nakanuma, Y.; et al. Enhanced self-renewal capability in hepatic stem/progenitor cells drives cancer initiation. Gatsroenterology 2007, 133, 937–950. [Google Scholar] [CrossRef] [PubMed]
- Mokkapati, S.; Niopek, K.; Huang, L.; Cunniff, K.J.; Ruteshouser, E.C.; deCaestecker, M.; Finegold, M.J.; Huff, V. Beta-catenin activation in a novel liver progenitor cell type is sufficient to cause hepatocellular carcinoma and hepatoblastoma. Cancer Res. 2014, 74, 4515–4525. [Google Scholar] [CrossRef] [PubMed]
- Zender, L.; Spector, M.S.; Xue, W.; Flemming, P.; Cordon-Cardo, C.; Silke, J.; Fan, S.T.; Luk, J.M.; Wigler, M.; Hannon, G.J.; et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006, 125, 1253–1267. [Google Scholar] [CrossRef] [PubMed]
- Saborowski, A.; Saborowski, M.; Davare, M.A.; Druker, B.J.; Klimstra, D.S.; Lowe, S.W. Mouse model of intrahepatic cholangiocarcinoma validates FIG-ROS as a potent fusion oncogene and therapeutic target. Proc. Natl. Acad. Sci. USA 2013, 110, 19513–19518. [Google Scholar] [CrossRef] [PubMed]
- Tummala, K.S.; Brandt, M.; Teijeiro, A.; Grana, O.; Schwabe, R.; Perna, C.; Djouder, N. Hepatocellular carcinomas originate predominantly from hepatocytes and benign lesions from hepatic progenitor cells. Cell Rep. 2017, 19, 584–600. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Parachoniak, C.A.; Ghanta, K.S.; Fitamant, J.; Ross, K.N.; Najem, M.S.; Gurumurthy, S.; Akbay, E.A.; Sia, D.; Cornella, H.; et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014, 513, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Dayton, T.L.; Gocheva, V.; Miller, K.M.; Israelsen, W.J.; Bhutkar, A.; Clish, C.B.; Davidson, S.M.; Luengo, A.; Bronson, R.T.; Jacks, T.; et al. Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma. Genes. Dev. 2016, 30, 1020–1033. [Google Scholar] [CrossRef] [PubMed]
- Holczbauer, A.; Factor, V.M.; Andersen, J.B.; Marquardt, J.U.; Kleiner, D.E.; Raggi, C.; Kitade, M.; Seo, D.; Akita, H.; Durkin, M.E.; et al. Modeling pathogenesis of primary liver cancer in lineage-specific mouse cell types. Gastroenterology 2013, 145, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Yang, H.; Learned, M.; Tian, H.; Learned, M.; Tiang, H.; Ling, L. Non-cell-autonomous activation of IL-6/STAT3 activation of IL-6/STAT3 signaling mediates FGF19-driven hepatocarcinogenesis. Nat. Commun. 2017, 8, 15433. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, N.; Utsunomiya, I.; Inoue, H.; Tanaka, F.; Mimori, K.; Barnard, G.F.; Mori, M. Characterization of a side population of cancer cells from human gastrointestinal system. Stem Cells 2006, 24, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Chiba, T.; Kita, K.; Zheng, I.W.; Yokosuka, O.; Saisho, H.; Iwama, A.; Nakauchi, H.; Taniguchi, H. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology 2006, 44, 240–251. [Google Scholar] [CrossRef] [PubMed]
- Suetsugu, A.; Nagaki, M.; Aoki, H.; Motohashi, T.; Kunisada, T.; Moriwaki, H. Characterization of CD133+ hepatocellular carcinoma cells as cancer stem/progenitor cells. Biochem. Biophys. Res. Commun. 2006, 351, 820–824. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Li, J.; Hu, C.; Chen, X.; Yao, M.; Yan, M.; Jiang, G.; Ge, C.; Xie, H.; Wan, D.; et al. CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int. J. Cancer. 2007, 120, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chan, K.W.; Hu, L.; Lee, T.K.; Wo, J.Y.; Ng, I.O.; Zheng, B.J.; Guan, X.Y. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Tang, K.H.; Chan, Y.P.; Lee, T.K.; Kwan, P.S.; Castilho, A.; Ng, I.; Man, K.; Wong, N.; To, K.F.; et al. miR-130b promotes CD133+ liver tumor-initiating cell growth and self-renewal via tumor protein 53-induced nuclear protein 1. Cell Stem Cell 2010, 7, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Ma, S.; Lu, T.; Chan, Y.P.; Kwan, P.S.; Tong, C.M.; Ng, I.O.; Man, K.; To, K.F.; Lai, P.B.; et al. CD133+ liver tumor-initiating cells promote tumor angiogenesis, growth, and self-renewal through neurotensin/interleukin-8/CXCL1 signaling. Hepatology 2012, 55, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Lee, T.K.; Zheng, B.J.; Chan, K.W.; Guan, X.Y. CD133+ HCC cancer stem cells confer chemoresistance by preferential expression of the AKT/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Lau, C.K.; Yang, Z.F.; Ho, D.W.; Ng, M.N.; Yeoh, G.C.; Poon, R.T.; Fan, S.T. An AKT/hypoxia inducible factor-1alpha/platelet-derived growth factor-BB autocrine loop mediates hypoxia-induced chemoresistance in liver cancer cells and tyumorigenic hepatic progenitor cells. Clin. Cancer. Res. 2009, 15, 3462–3471. [Google Scholar] [CrossRef] [PubMed]
- Piao, L.S.; Hur, W.; Kim, T.K.; Hong, S.W.; Kim, S.W.; Choi, J.E.; Sung, P.S.; Song, M.J.; Lee, B.C.; Hwang, D.; et al. CD133+ liver cancer stem cells modulate radioresistance in human hepatocellular carcinoma. Cancer Lett. 2012, 315, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Lau, X.; Wu, Y.Z.; Wang, Y.; Wu, F.R.; Zang, C.B.; Tang, C.; Cao, S.; Li, S.L. CD133 silencing inhibits stemness properties and enhanced chemoradiosensitivity in CD133-positive liver cancer stem cells. Int. J. Mol. Med. 2013, 31, 315–324. [Google Scholar]
- Mima, K.; Okabe, H.; Ishimoto, T.; Hayashi, H.; Nakagawa, H.K.; Watanabe, M.; Beppu, T.; Tamada, M.; Nagano, O.; et al. CD44s regulates the TGF-beta-mediated mesenchymal phenotype and is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res. 2012, 72, 3414–3423. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Hao, X.; Yan, M.; Yao, M.; Ge, C.; Gu, J.; Li, J. Cancer stem/ progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int. J. Cancer 2010, 126, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zou, Q.; Ge, R.; Shen, F.; Wang, Y. The critical role of CD133(+)CD44+/high tumor cells in hematogenous metastasis of liver cancers. Cell Res. 2012, 22, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.W.; Tsuchida, T.; Shimao, T.; Li, B.; Takebe, T.; Zhang, R.R.; Sakurai, Y.; Ueno, Y.; Sekine, K.; Ishibashi, N.; et al. The CD133+CD44+ precancerous subpopulation of oval cells is a therapeutic target for hepatocellular carcinoma. Stem Cell Dev. 2014, 23, 2237–2249. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Zhao, E.; Kryczek, I.; Vatan, L.; Sadovskaya, A.; Ludema, G.; Simeone, D.M.; Zou, W.; Welling, T.H. Tumor-associated macrophages interleukin 6 and signal via STAT3 to promote expansion of human hepatocarcinoma stem cells. Gastroenterology 2014, 147, 1393–1404. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Li, H.; Tao, K.; Li, R.; Song, Z.; Zhao, Q.; Zhang, F.; Dou, K. Expression and clinical significance of the stem cell marker CD133 in hepatocellular carcinoma. Int. J. Clin. Pract. 2008, 62, 1212–1218. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.C.; Yang, J.Y.; Yan, L.N. Relevant markers of cancer stem cells indicate a poor prognosis in hepatocellular carcinoma patients: A meta-analysis. Eur. J. Gastroenterol. Hepatol. 2013, 25, 1007–1016. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.K.; Castilho, A.; Cheung, V.C.; Tang, K.H.; Ma, S.; Ng, I.O. CD24+ liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 2011, 9, 50–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, A.Y.; Cai, Y.; Mao, Y.; Lin, Y.; Zheng, H.; Wu, T.; Huang, Y.; Fang, X.; Lin, S.; Feng, Q.; et al. Twist2 promotes self-renewal of liver cancer stem-like cells by regulating CD24. Carcinogenesis 2014, 35, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Imrich, S.; Hochmeister, M.; Gires, O. EpCAM and ist potential role in tumor-initiating cells. Cell Adhes. Migr. 2012, 6, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Forgues, M.; Wang, W.; Kim, J.W.; Ye, Q.; Jia, H.; Budhu, A.; Zanetti, K.A.; Chen, Y.; Qin, L.X.; et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res. 2008, 68, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Budhu, A.; Forgues, M.; Wang, X.W. Activation of hepatic stem cell marker EpCAM by Wnt-beta-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007, 67, 10831–10839. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Ji, L.; Budhu, A.; Forgues, M.; Yang, W.; Wang, H.Y.; Jia, H.; Ye, Q.; Qin, L.X.; Wauthier, E.; et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 2009, 136, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Tenis, B.; Cavard, C.; Perret, C. EpCAM, a new marker for cancer stem cells in hepatocellular carcinoma. J. Hepatol. 2010, 52, 280–281. [Google Scholar]
- Chen, Y.; Yu, D.; Zhang, H.; He, H.; Zhang, C.; Zhao, W.; Shao, R. CD133+EpCAM+ phenotype possesses more characteristics of tumor initiating cells in hepatocellular carcinoma Huh7 cells. Int. J. Biol. Sci. 2012, 8, 992–1004. [Google Scholar] [CrossRef] [PubMed]
- Armeanu-Ebinger, S.; Hoh, A.; Wenz, J.; Fuchs, J. Targeting EpCAM (CD326) for immunotherapy of hepatoblastoma. Oncoimmunology 2013, 2, e22620. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.F.; Ho, D.; Ng, M.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.; Lam, C.T.; Poon, R.T.; Fan, S.T.; et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Jia, Q.; Zhang, X.; Deng, T.; Gao, J. Positive correlation of Oct4 and ABCG2 to chemotherapeutic resistance in CD90+CD133+ cancer stem cells. Cell Reprogram. 2013, 15, 143–150. [Google Scholar]
- Yamashita, T.; Honda, M.; Nakamoto, Y.; Baba, M.; Nio, K.; Hara, Y.; Zeng, S.S.; Hayashi, T.; Kondo, M.; Takatori, H.; et al. Discrete nature of EpCAM+ and CD90+ cancer stem cells in human hepatocellular carcinoma. Hepatology 2013, 57, 1484–1497. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Lo, C.M.; Poon, R.; Cheung, T.T.; Chan, A.; Chen, L.; Yang, S.; Tsao, G.; Wang, X.Q. Smad inhibitor induces CSC differentiation for effective chemosensitization in cyclin D1- and TGF-β/Smad-regulated liver cancer stem cell-like cells. Oncotarget 2017, 8, 38818–38824. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.W.; Yang, Z.F.; Yi, K.; Lam, C.T.; Ng, M.N.; Yu, W.C.; Lau, J.; Wan, T.; Wang, X.; Yan, Z.; et al. Gene expression profiling of liver cancer stem cells by RNA-sequencing. PLoS ONE 2012, 7, e37159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraguchi, N.; Ishii, H.; Mimori, K.; Tanaka, F.; Ohkuma, M.; Kim, H.M.; Akita, H.; Takiuchi, D.; Hatano, H.; Nagano, H.; et al. CD13 is a therapeutic target in human liver cancer stem cells. J. Clin. Investig. 2010, 120, 3326–3339. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Haraguchi, N.; Ishii, H.; Ohkuma, M.; Okano, M.; Mimori, K.; Eguchi, H.; Yamamoto, H.; Nagano, H.; Sekimoto, M.; et al. Increased CD13 expression reduces reactive oxygen species, promoting survival of liver cancer stem cells via an epithelial-mesenchymal transition-like phenomenon. Ann. Surg. Oncol. 2012, 19, SS39–SS48. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Yan, H.X.; Chen, L.; Liu, Q.; He, Y.Q.; Yu, L.X.; Zhang, S.H.; Huang, D.D.; Tang, L.; Kong, X.N.; et al. Wnt/beta-catenin signaling contributes to activation of normal and tumorigenic liver progenitor cells. Cancer Res. 2008, 68, 4287–4295. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Wang, C.; Lin, Y.; Liu, Q.; Yu, L.X.; Tang, L.; Yan, H.X.; Fu, J.; Chen, Y.; Zhang, H.L.; et al. OV6+ tumor-initiating cells contribute to tumor progression and invasion in human hepatocellular carcinoma. J. Hepatol. 2012, 57, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chan, K.W.; Lee, T.; Tang, K.H.; Wo, J.Y.; Zheng, B.J.; Guan, X.Y. Aldehyde dehydrogenase discriminates the CD133 liver cancer stem cell populations. Mol. Cancer. Res. 2008, 6, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.S.; Hu, Z.; Duan, W.; Tian, A.; Wang, X.M.; McLeod, D.; Lam, V.; George, J.; Qiao, L. Efficacy of using cancer stem cell markers in isolating and characterizing liver cancer stem cells. Stem Cells Dev. 2013, 22, 2655–2664. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wang, L.; Han, H.; Jin, K.; Lin, N.; Guo, T.; Chen, Y.; Cheng, H.; Lu, F.; Fang, W.; et al. 1B50-1, a mAb raised against recurrent tumor cells, targets liver tumor-initiating cells by binding to the calcium channel α2δ1 subunit. Cancer Cell 2013, 23, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wang, Y.; He, L.; Huang, G.; Du, Y.; Zhang, G.; Yan, X.; Xia, P.; Ye, B.; Wang, S.; et al. ZIC2-dependent OCT4 activation drives self-renewal of human liver cancer stem cells. J. Clin. Investig. 2015, 125, 3795–3806. [Google Scholar] [CrossRef] [PubMed]
- Yong, K.Y.; Kiao, C.; Lim, J.; Yan, B.; Yang, H.; Dimitrov, T.; Kawasaki, A.; Ong, C.W.; Wong, K.F.; Lee, S.; et al. Oncofetal gene SLL4 in aggressive hepatocellular carcinoma. N. Engl. J. Med. 2013, 368, 2266–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, S.S.; Yamashita, T.; Kondo, M.; Nio, K.; Hayashi, T.; Hara, Y.; Nomura, Y.; Yoshida, M.; Hayashi, T.; Oishi, N.; et al. The transcription factor SALL4 regulates stemness of EpCAM-positive hepatocellular carcinoma. J. Hepatol. 2014, 60, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Lee, H.; Seo, A.V.; Cho, J.Y.; Choi, Y.R.; Yoon, Y.S.; Han, H.S.; Park, Y.N.; Kim, H. SALL4 expression in hepatocellular carcinomas is associated with EpCAM positivity and poor prognosis. J. Pathol. Transl. Med. 2015, 49, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Zhu, L.; Huang, F.; Nie, W.; Huang, W.; Xu, H.; Zheng, S.; Yi, Z.; Wan, T. SALL4 is a novel therapeutic target in intrahepatic cholangiocarcinoma. Oncotarget 2015, 6, 27416–27426. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, T.; Wauthier, E.; Dinh, T.A.; Selitsky, S.R.; Reyna-Neyra, A.; Carpino, G.; Levine, R.; Cardinale, V.; Klimstra, D.; Gaudio, E.; et al. Model of fibrolamellar hepatocellular carcinomas reveals striking enrichment in cancer stem cells. Nat. Commun. 2015, 6, 8070. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wang, Y.; Huang, G.; Ye, B.; Liu, B.; Wu, J.; Du, J.; He, L.; Fan, Z. Lnc-β-Catm elicits EZH2-dependent β-catenin stabilization and sustains liver CSC self-renewal. Nat. Struct. Mol. Biol. 2016, 23, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; He, L.; Du, Y.; Zhu, P.; Huang, G.; Luo, J.; Yan, X.; Ye, B.; Li, C.; Xia, P.; et al. The long noncoding RNA lncTCF7 promotes self-renewal of human liver cancer stem cells through activation of Wnt signaling. Cell Stem Cell 2015, 16, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Liu, C.; Liu, L.; Chen, X.; Shan, J.; Shen, J.; Zhu, W.; Qian, C. MEK1 signaling promotes self-renewal and tumorigenicity of liver cancer stem cells via maintaining SIRT1 protein stabilization. Oncotarget 2016, 7, 20597–20611. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, P.; Wang, R.; Wang, J.; Liu, M.; Xiong, S.; Li, Y.; Cheng, B. The Notch pathway promotes the cancer stem cell characteristics of CD90+ cells in hepatocellular carcinoma. Oncotarget 2016, 7, 9525–9537. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, L.; Chen, X.; Cheng, J.; Zhang, H.; Shen, J.; Xu, Y.; Yang, Z.; Lai, M.; Qian, C. Sox9 regulates self-renewal and tumorigenicity by promoting symmetrical cell division of cancer stem cells in hepatocellular carcinoma. Hepatology 2016, 64, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Wang, Y.; Wu, J.; Huang, G.; Liu, B.; Ye, B.; Du, Y.; Guo, G.; Tian, Y.; Fan, Z. Lnc BRM initiates YAP1 signalling activation to drive self-renewal of liver cancer stem cells. Nat. Commun. 2017, 7, 13608. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 2016, 29, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Inoue, K.I.; Haciya, H.; Shibuya, N.; Aoki, T.; Kubota, K. Accumulation of phosphorylated p62 is associated with NF-E2-related factor 2 activation in hepatocellular carcinoma. J. Hepatobiliary Pancreat. Sci. 2016, 23, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, M.A.; Guzzo, G.; Morandi, A.; Perra, A.; Menegon, S.; Masgras, I.; Trevisan, E.; Angioni, M.M.; Fornari, F.; Quagliata, L.; et al. Metabolic reprogramming identifies the most aggressive lesions at early phases of hepatic carcinogenesis. Oncotarget 2016, 7, 32375–32393. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef] [PubMed]
- Towsend, E.C.; Murakami, M.A.; Christodoulou, A.; Christie, A.L.; Koster, J.; DeSouza, T.; Morgan, E.A.; Kallgren, S.P.; Liu, H.; Whu, S.C.; et al. The public repository of xenografts enalbles discovery and randomized phase II-like trials in mice. Cancer Cell 2016, 29, 574–586. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Cheung, P.; Yip, C.W.; Ng, L.; Lo, K.W.; Chow, C.; Chan, K.F.; Cheung, T.T.; Cheung, S.T. Comprehensive characterization of the patient-derived xenograft and the paralleled primary hepatocellular carcinoma cell line. Cancer Cell Int. 2016, 16, 41. [Google Scholar] [CrossRef] [PubMed]
- Nicolle, D.; Fabre, M.; Simon-Coma, M.; Gorse, A.; Kappler, R.; Nonell, L.; Mallo, M.; Haidar, H.; Deas, O.; Mussini, C.; et al. Patients-derived xenografts from pediatric liver cancer preditc tumor recurrence and advise clinical management. Hepatology 2016, 64, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Bissig-Choisat, B.; Kettlun-Leyton, C.; Legras, X.D.; Zozman, B.; Barzi, M.; Chen, L.L.; Amin, M.D.; Huang, Y.H.; Paufler, R.G.; Hampton, O.A.; et al. Novel patient-derived xenograft and cell line models for therapeutic testing of pediatric liver cancer. J. Hepatol. 2016, 65, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Low, X.C.; Hou, W.; Abdu llah, L.N.; Toh, T.B.; Mohd Abdul Rashid, M.; Chow, E.K. Epirubicin-adsorbed nanodiamonds kill chemoresistant hepatic cancer stem cells. ACS Nano 2014, 8, 12151–12166. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tan, J.; McConville, C.; Kannappan, V.; Tawari, P.E.; Brown, J.; Ding, J.; Armesilla, A.L.; Irache, J.M.; Mei, Q.B.; et al. Poly-lactic co-glycolic acid controlled delivery of disulfiram to target liver cancer stem-like cells. Nanomedicine 2017, 13, 641–657. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Li, K.; Tu, H.; Pan, X.; Jiang, H.; Shi, B.; Kong, J.; Wang, H.; Yang, S.; Gu, J.; Li, Z. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin. Cancer Res. 2014, 20, 6418–6428. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.Z.; Pan, K.; Wang, Q.J.; Weng, D.S.; Zhao, J.J.; Zheng, H.X.; Zhang, X.F.; Jiang, S.S.; Lv, L.; Tang, Y.; et al. Annexin A3 as a potential target for immunotherapy of liver cancer stem-like cells. Stem Cells 2015, 33, 354–366. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, A.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2017, 3879, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomized, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Nakagawa, S.; Wei, L.; Song, W.M.; Higashi, T.; Ghoshal, S.; Kim, R.S.; Bian, C.B.; Yamada, S.; Sun, X.; Venkatesh, A.; et al. Molecular liver cancer prevention in cirrhosis by organ transcriptome analysis and lysophosphatidic acid pathway inhibition. Cancer Cell 2016, 30, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Cheng, A.L.; Meinhard, G.; Nakajima, K.; De Sancris, Y.; Llovet, J. Prognostic factors and predictors of sorafenib benefit in patients with hepatocellular carcinoma: Analysis of two phase 3 studies. J. Hepatol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castelli, G.; Pelosi, E.; Testa, U. Liver Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Cancers 2017, 9, 127. https://doi.org/10.3390/cancers9090127
Castelli G, Pelosi E, Testa U. Liver Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Cancers. 2017; 9(9):127. https://doi.org/10.3390/cancers9090127
Chicago/Turabian StyleCastelli, Germana, Elvira Pelosi, and Ugo Testa. 2017. "Liver Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells" Cancers 9, no. 9: 127. https://doi.org/10.3390/cancers9090127
APA StyleCastelli, G., Pelosi, E., & Testa, U. (2017). Liver Cancer: Molecular Characterization, Clonal Evolution and Cancer Stem Cells. Cancers, 9(9), 127. https://doi.org/10.3390/cancers9090127