
Stromal Androgen Receptor in Prostate Cancer Development and Progression

Abstract
:1. Introduction
2. Stromal AR in Prostate Cancer Outcome
3. Androgen Signalling
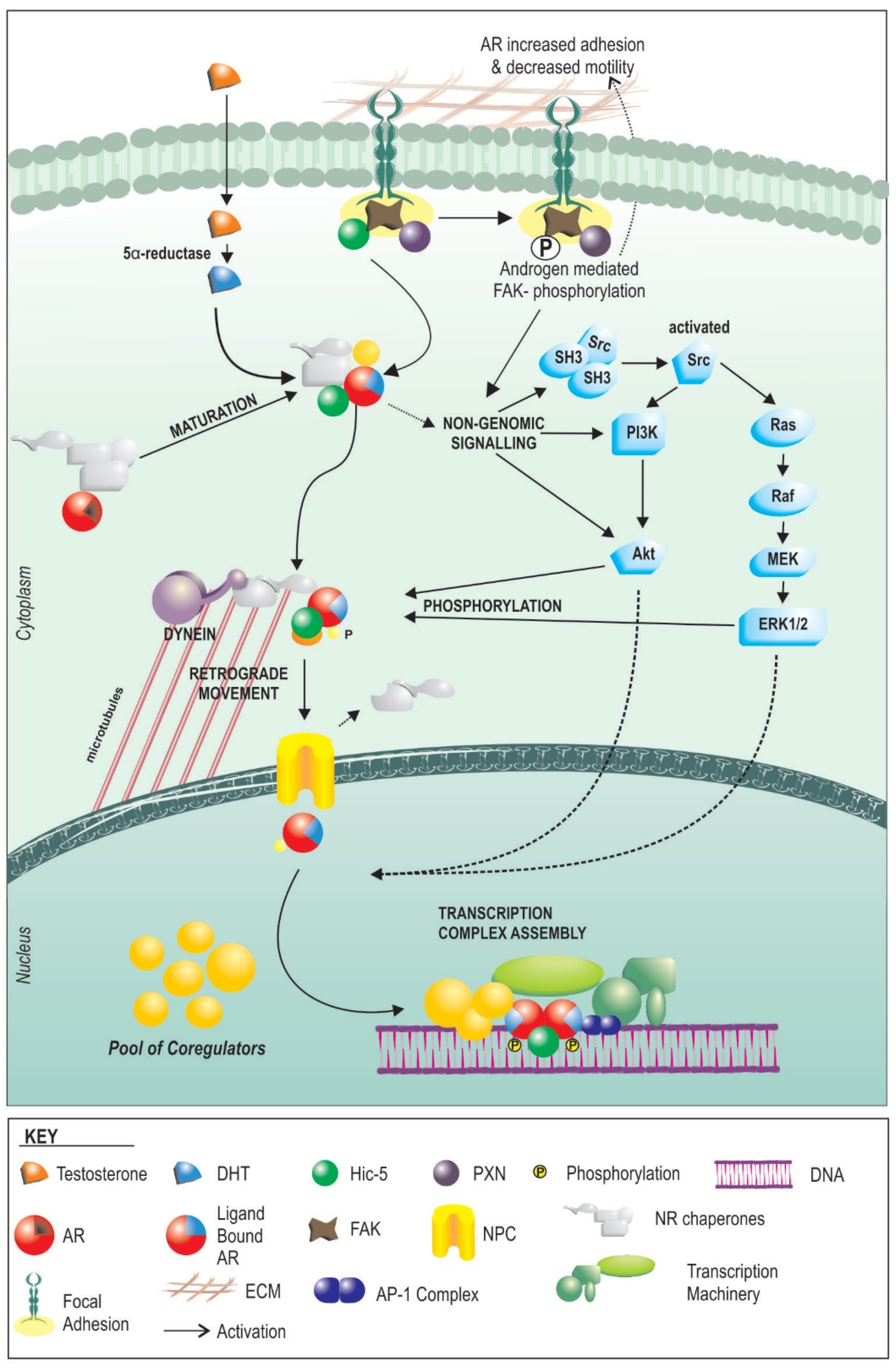
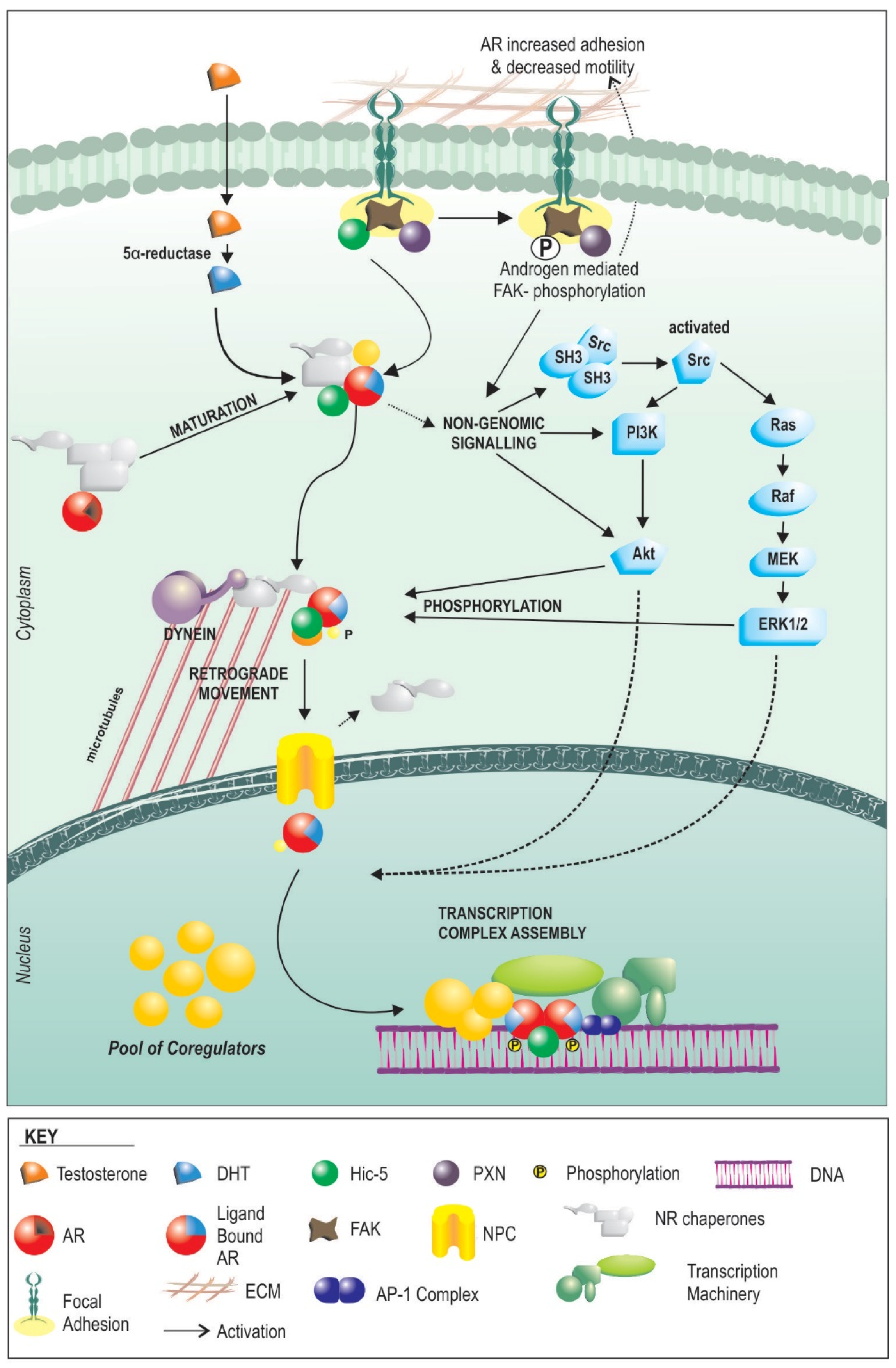
4. How AR Signaling in the Stroma Works
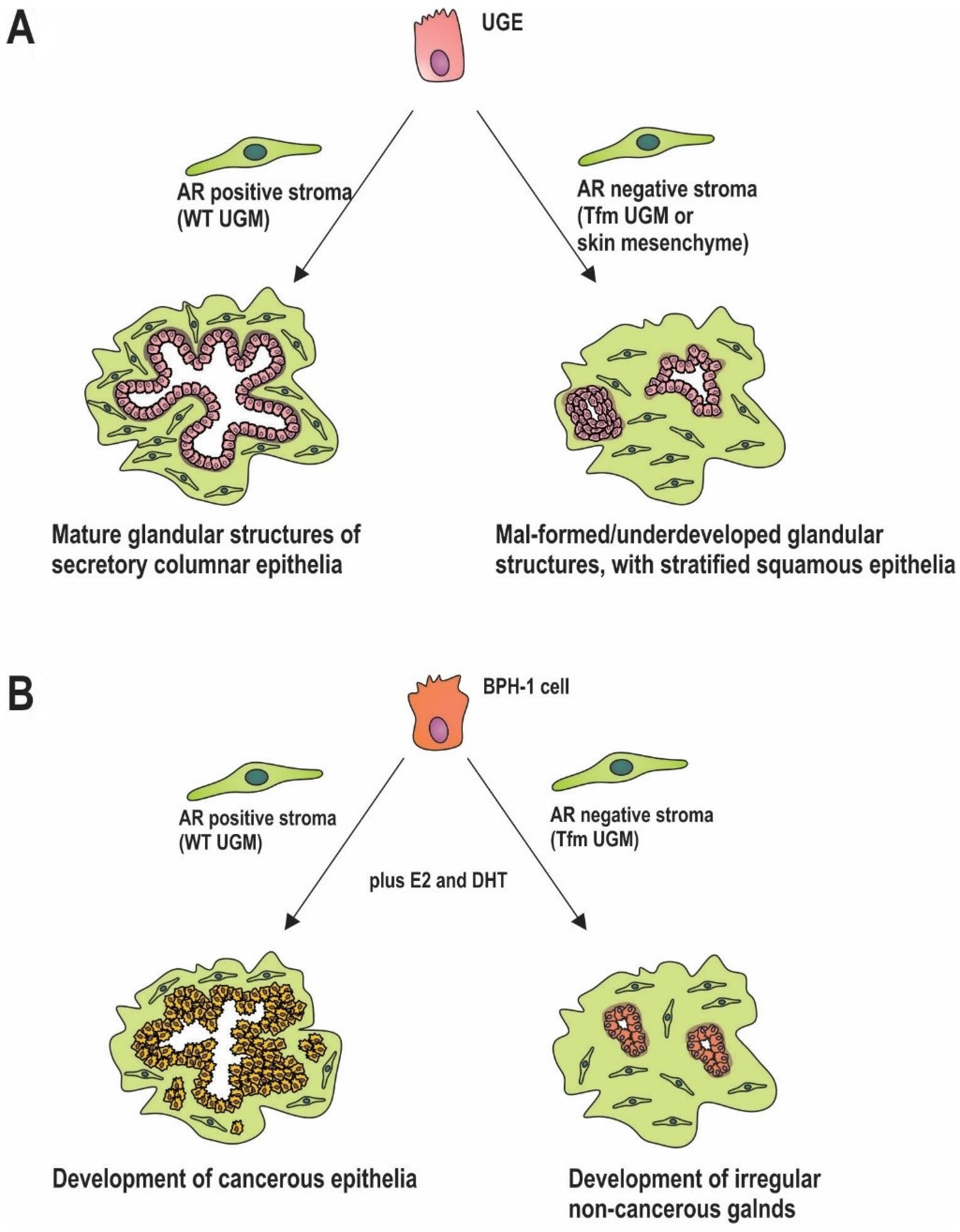
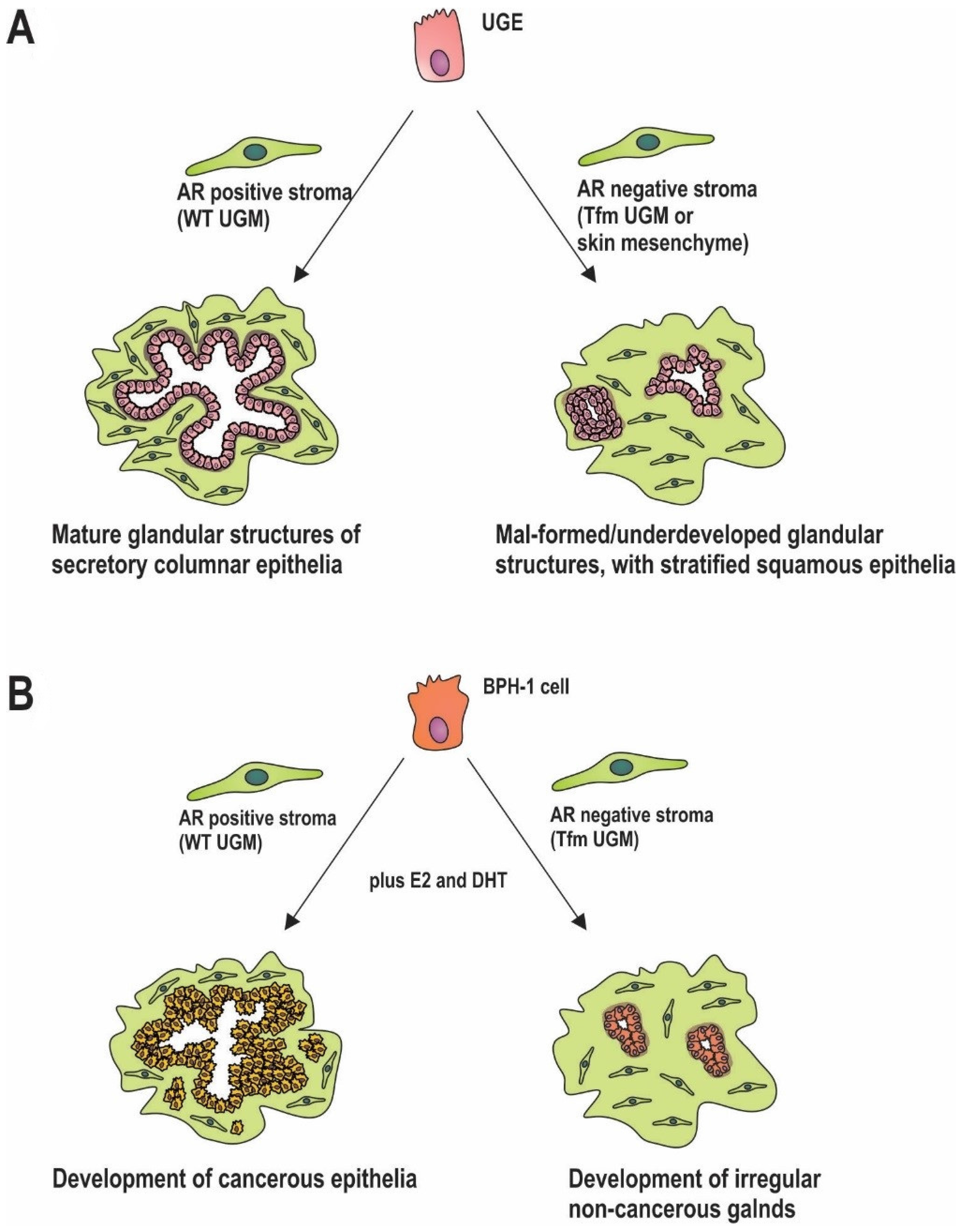
5. Stromal AR in Prostate Development
6. Stromal AR in Carcinogenesis
7. Why Is Stromal AR Lost?
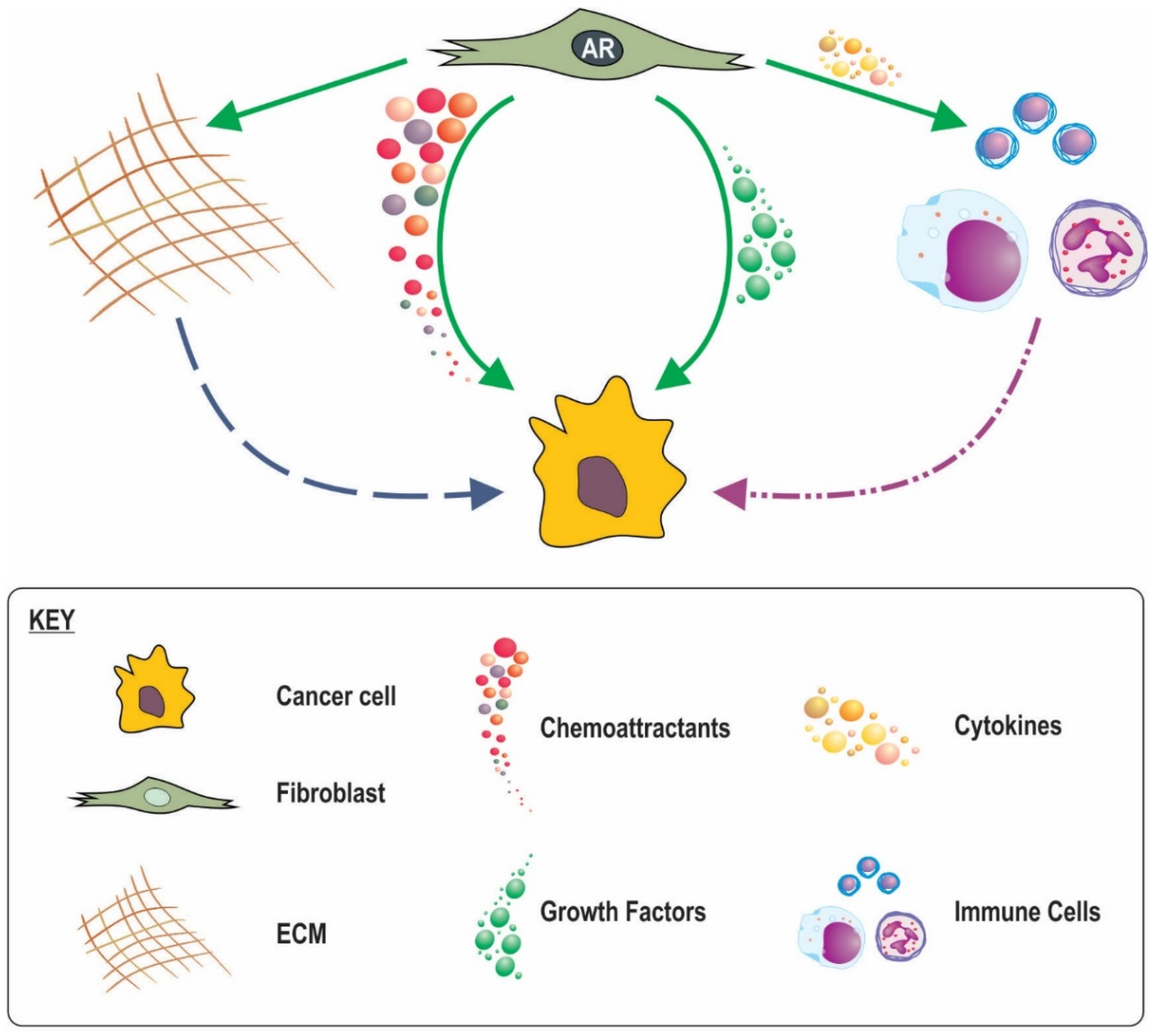
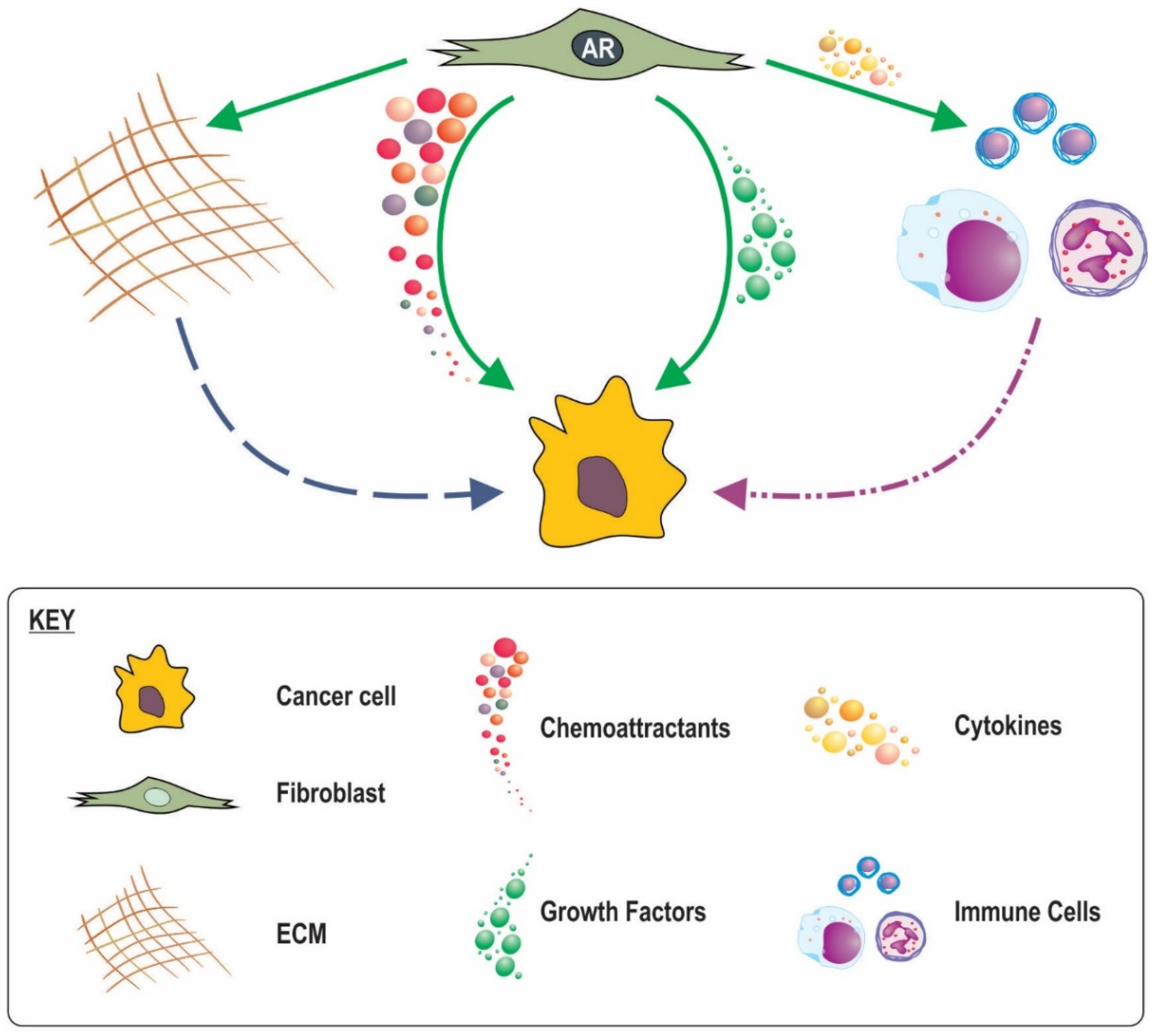
8. Possible Mechanisms for the Involvement of Stromal AR Signalling in Cancer Progression and Outcome
8.1. Loss of Stromal AR Creating Less Favourable Conditions
8.2. A Role for Stromal in AR Inflammatory Processes
8.3. AR in CAF Movement and a Subsequent Role in Cancer Invasion
8.4. Stromal AR Regulation of ECM
9. Potential Importance of Stromal AR in Neoadujant Hormone Therapy
10. Future of Stromal AR
10.1. Prognostic Tool
10.2. Therapeutic Targets
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tuxhorn, J.A.; Ayala, G.E.; Smith, M.J.; Smith, V.C.; Dang, T.D.; Rowley, D.R. Reactive stroma in human prostate cancer: Induction of myofibroblast phenotype and extracellular matrix remodeling. Clin. Cancer Res. 2002, 8, 2912–2923. [Google Scholar] [PubMed]
- Rowley, D.R. What might a stromal response mean to prostate cancer progression? Cancer Metastasis Rev. 1998, 17, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Tuxhorn, J.A.; Ayala, G.E.; Rowley, D.R. Reactive stroma in prostate cancer progression. J. Urol. 2001, 166, 2472–2483. [Google Scholar] [CrossRef]
- Cunha, G.R.; Chung, L.W. Stromal-epithelial interactions—I. Induction of prostatic phenotype in urothelium of testicular feminized (Tfm/y) mice. J. Steroid Biochem. 1981, 14, 1317–1324. [Google Scholar] [CrossRef]
- Donjacour, A.A.; Cunha, G.R. The effect of androgen deprivation on branching morphogenesis in the mouse prostate. Dev. Biol. 1988, 128, 1–14. [Google Scholar] [CrossRef]
- Tamburrino, L.; Salvianti, F.; Marchiani, S.; Pinzani, P.; Nesi, G.; Serni, S.; Forti, G.; Baldi, E. Androgen receptor (AR) expression in prostate cancer and progression of the tumor: Lessons from cell lines, animal models and human specimens. Steroids 2012, 77, 996–1001. [Google Scholar] [CrossRef] [PubMed]
- Mohler, J.L.; Chen, Y.; Hamil, K.; Hall, S.H.; Cidlowski, J.A.; Wilson, E.M.; French, F.S.; Sar, M. Androgen and glucocorticoid receptors in the stroma and epithelium of prostatic hyperplasia and carcinoma. Clin. Cancer Res. 1996, 2, 889–895. [Google Scholar] [PubMed]
- Ricciardelli, C.; Choong, C.S.; Buchanan, G.; Vivekanandan, S.; Neufing, P.; Stahl, J.; Marshall, V.R.; Horsfall, D.J.; Tilley, W.D. Androgen receptor levels in prostate cancer epithelial and peritumoral stromal cells identify non-organ confined disease. Prostate 2005, 63, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Henshall, S.M.; Quinn, D.I.; Lee, C.S.; Head, D.R.; Golovsky, D.; Brenner, P.C.; Delprado, W.; Stricker, P.D.; Grygiel, J.J.; Sutherland, R.L. Altered expression of androgen receptor in the malignant epithelium and adjacent stroma is associated with early relapse in prostate cancer. Cancer Res. 2001, 61, 423–427. [Google Scholar] [PubMed]
- Wikstrom, P.; Marusic, J.; Stattin, P.; Bergh, A. Low stroma androgen receptor level in normal and tumor prostate tissue is related to poor outcome in prostate cancer patients. Prostate 2009, 69, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Olapade-Olaopa, E.O.; MacKay, E.H.; Taub, N.A.; Sandhu, D.P.; Terry, T.R.; Habib, F.K. Malignant transformation of human prostatic epithelium is associated with the loss of androgen receptor immunoreactivity in the surrounding stroma. Clin. Cancer Res. 1999, 5, 569–576. [Google Scholar] [PubMed]
- Leach, D.A.; Need, E.F.; Toivanen, R.; Trotta, A.P.; Palenthorpe, H.M.; Tamblyn, D.J.; Kopsaftis, T.; England, G.M.; Smith, E.; Drew, P.A.; et al. Stromal androgen receptor regulates the composition of the microenvironment to influence prostate cancer outcome. Oncotarget 2015, 6, 16135–16150. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.A.; Trotta, A.P.; Need, E.F.; Risbridger, G.P.; Taylor, R.A.; Buchanan, G. The prognostic value of stromal FK506-binding protein 1 and androgen receptor in prostate cancer outcome. Prostate 2017, 77, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Takeda, H.; Akakura, K.; Masai, M.; Akimoto, S.; Yatani, R.; Shimazaki, J. Androgen receptor content of prostate carcinoma cells estimated by immunohistochemistry is related to prognosis of patients with stage D2 prostate carcinoma. Cancer 1996, 77, 934–940. [Google Scholar] [CrossRef]
- Segawa, N.; Mori, I.; Utsunomiya, H.; Nakamura, M.; Nakamura, Y.; Shan, L.; Kakudo, K.; Katsuoka, Y. Prognostic significance of neuroendocrine differentiation, proliferation activity and androgen receptor expression in prostate cancer. Pathol. Int. 2001, 51, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Pertschuk, L.P.; Schaeffer, H.; Feldman, J.G.; Macchia, R.J.; Kim, Y.D.; Eisenberg, K.; Braithwaite, L.V.; Axiotis, C.A.; Prins, G.; Green, G.L. Immunostaining for prostate cancer androgen receptor in paraffin identifies a subset of men with a poor prognosis. Lab. Investig. 1995, 73, 302–305. [Google Scholar] [PubMed]
- Sweat, S.D.; Pacelli, A.; Bergstralh, E.J.; Slezak, J.M.; Cheng, L.; Bostwick, D.G. Androgen receptor expression in prostate cancer lymph node metastases is predictive of outcome after surgery. J. Urol. 1999, 161, 1233–1237. [Google Scholar] [CrossRef]
- Barboro, P.; Salvi, S.; Rubagotti, A.; Boccardo, S.; Spina, B.; Truini, M.; Carmignani, G.; Introini, C.; Ferrari, N.; Boccardo, F.; et al. Prostate cancer: Prognostic significance of the association of heterogeneous nuclear ribonucleoprotein K and androgen receptor expression. Int. J. Oncol. 2014, 44, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Segawa, N.; Nakamura, M.; Shan, L.; Utsunomiya, H.; Nakamura, Y.; Mori, I.; Katsuoka, Y.; Kakudo, K. Expression and somatic mutation on androgen receptor gene in prostate cancer. Int. J. Urol. 2002, 9, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Rosner, I.L.; Ravindranath, L.; Furusato, B.; Chen, Y.; Gao, C.; Cullen, J.; Sesterhenn, I.A.; McLeod, D.G.; Srivastava, S.; Petrovics, G. Higher tumor to benign ratio of the androgen receptor mRNA expression associates with prostate cancer progression after radical prostatectomy. Urology 2007, 70, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Cordon-Cardo, C.; Kotsianti, A.; Verbel, D.A.; Teverovskiy, M.; Capodieci, P.; Hamann, S.; Jeffers, Y.; Clayton, M.; Elkhettabi, F.; Khan, F.M.; et al. Improved prediction of prostate cancer recurrence through systems pathology. J. Clin. Investig. 2007, 117, 1876–1883. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Segawa, T.; Shiraishi, T.; Yoshida, T.; Toda, Y.; Yamada, T.; Kinukawa, N.; Kinoshita, H.; Kamoto, T.; Ogawa, O. Androgen receptor, Ki67, and p53 expression in radical prostatectomy specimens predict treatment failure in Japanese population. Urology 2005, 66, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wheeler, T.; Dai, H.; Frolov, A.; Thompson, T.; Ayala, G. High level of androgen receptor is associated with aggressive clinicopathologic features and decreased biochemical recurrence-free survival in prostate: Cancer patients treated with radical prostatectomy. Am. J. Surg. Pathol. 2004, 28, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Donovan, M.J.; Osman, I.; Khan, F.M.; Vengrenyuk, Y.; Capodieci, P.; Koscuiszka, M.; Anand, A.; Cordon-Cardo, C.; Costa, J.; Scher, H.I. Androgen receptor expression is associated with prostate cancer-specific survival in castrate patients with metastatic disease. BJU Int. 2010, 105, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.Q.; Leuschner, I.; Braun, P.M. Androgen receptor expression in clinically localized prostate cancer: Immunohistochemistry study and literature review. Asian J. Androl. 2008, 10, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Noordzij, M.A.; Bogdanowicz, J.F.; van Krimpen, C.; van der Kwast, T.H.; van Steenbrugge, G.J. The prognostic value of pretreatment expression of androgen receptor and bcl-2 in hormonally treated prostate cancer patients. J. Urol. 1997, 158, 1880–1884. [Google Scholar] [CrossRef]
- Rades, D.; Setter, C.; Dahl, O.; Schild, S.E.; Noack, F. The prognostic impact of tumor cell expression of estrogen receptor-alpha, progesterone receptor, and androgen receptor in patients irradiated for nonsmall cell lung cancer. Cancer 2012, 118, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Sadi, M.V.; Barrack, E.R. Image analysis of androgen receptor immunostaining in metastatic prostate cancer. Heterogeneity as a predictor of response to hormonal therapy. Cancer 1993, 71, 2574–2580. [Google Scholar] [CrossRef]
- Sterbis, J.R.; Gao, C.; Furusato, B.; Chen, Y.; Shaheduzzaman, S.; Ravindranath, L.; Osborn, D.J.; Rosner, I.L.; Dobi, A.; McLeod, D.G.; et al. Higher expression of the androgen-regulated gene PSA/HK3 mRNA in prostate cancer tissues predicts biochemical recurrence-free survival. Clin. Cancer Res. 2008, 14, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Theodoropoulos, V.E.; Tsigka, A.; Mihalopoulou, A.; Tsoukala, V.; Lazaris, A.C.; Patsouris, E.; Ghikonti, I. Evaluation of neuroendocrine staining and androgen receptor expression in incidental prostatic adenocarcinoma: Prognostic implications. Urology 2005, 66, 897–902. [Google Scholar] [CrossRef] [PubMed]
- Fleischmann, A.; Rocha, C.; Schobinger, S.; Seiler, R.; Wiese, B.; Thalmann, G.N. Androgen receptors are differentially expressed in Gleason patterns of prostate cancer and down-regulated in matched lymph node metastases. Prostate 2011, 71, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Minner, S.; Enodien, M.; Sirma, H.; Luebke, A.M.; Krohn, A.; Mayer, P.S.; Simon, R.; Tennstedt, P.; Muller, J.; Scholz, L.; et al. ERG status is unrelated to PSA recurrence in radically operated prostate cancer in the absence of antihormonal therapy. Clin. Cancer Res. 2011, 17, 5878–5888. [Google Scholar] [CrossRef] [PubMed]
- Sweat, S.D.; Pacelli, A.; Bergstralh, E.J.; Slezak, J.M.; Bostwick, D.G. Androgen receptor expression in prostatic intraepithelial neoplasia and cancer. J. Urol. 1999, 161, 1229–1232. [Google Scholar] [CrossRef]
- Ford, O.H., 3rd; Gregory, C.W.; Kim, D.; Smitherman, A.B.; Mohler, J.L. Androgen receptor gene amplification and protein expression in recurrent prostate cancer. J. Urol. 2003, 170, 1817–1821. [Google Scholar] [CrossRef] [PubMed]
- Choucair, K.; Ejdelman, J.; Brimo, F.; Aprikian, A.; Chevalier, S.; Lapointe, J. PTEN genomic deletion predicts prostate cancer recurrence and is associated with low AR expression and transcriptional activity. BMC Cancer 2012. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Coram, M.A.; Nolley, R.; Reese, S.W.; Young, S.R.; Peehl, D.M. Transcript levels of androgen receptor variant AR-V1 or AR-V7 do not predict recurrence in patients with prostate cancer at indeterminate risk for progression. J. Urol. 2012, 188, 2158–2164. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.K.; McSherry, S.A.; Dent, G.A.; Sar, M.; Wilson, E.M.; French, F.S.; Sharief, Y.; Mohler, J.L. Immunohistochemistry of the androgen receptor in human benign and malignant prostate tissue. J. Urol. 1993, 149, 1015–1019. [Google Scholar] [PubMed]
- Schatzl, G.; Madersbacher, S.; Haitel, A.; Gsur, A.; Preyer, M.; Haidinger, G.; Gassner, C.; Ochsner, M.; Marberger, M. Associations of serum testosterone with microvessel density, androgen receptor density and androgen receptor gene polymorphism in prostate cancer. J. Urol. 2003, 169, 1312–1315. [Google Scholar] [CrossRef] [PubMed]
- De Winter, J.A.; Trapman, J.; Brinkmann, A.O.; Boersma, W.J.; Mulder, E.; Schroeder, F.H.; Claassen, E.; van der Kwast, T.H. Androgen receptor heterogeneity in human prostatic carcinomas visualized by immunohistochemistry. J. Pathol. 1990, 160, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Gaston, K.E.; Kim, D.; Singh, S.; Ford, O.H., 3rd; Mohler, J.L. Racial differences in androgen receptor protein expression in men with clinically localized prostate cancer. J. Urol. 2003, 170, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, C.X.; Ye, H.; Chen, F.; Melamed, J.; Peng, Y.; Liu, J.; Wang, Z.; Tsou, H.C.; Wei, J.; et al. Decrease in stromal androgen receptor associates with androgen-independent disease and promotes prostate cancer cell proliferation and invasion. J. Cell. Mol. Med. 2008, 12, 2790–2798. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.M.; Trakul, N.; Thraves, P.S.; Bello-DeOcampo, D.; Chu, W.W.; Storto, P.D.; Huard, T.K.; Rhim, J.S.; Williams, D.E. A human prostatic stromal myofibroblast cell line WPMY-1: A model for stromal-epithelial interactions in prostatic neoplasia. Carcinogenesis 1999, 20, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.A.; Panagopoulos, V.; Nash, C.; Bevan, C.; Thomson, A.A.; Selth, L.A.; Buchanan, G. Cell-lineage specificity and role of AP-1 in the prostate fibroblast androgen receptor cistrome. Mol. Cell. Endocrinol. 2017, 439, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Henke, A.; Franco, O.E.; Stewart, G.D.; Riddick, A.C.; Katz, E.; Hayward, S.W.; Thomson, A.A. Reduced contractility and motility of prostatic cancer-associated fibroblasts after inhibition of heat shock protein 90. Cancers 2016, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.A.; Need, E.F.; Trotta, A.P.; Grubisha, M.J.; DeFranco, D.B.; Buchanan, G. Hic-5 influences genomic and non-genomic actions of the androgen receptor in prostate myofibroblasts. Mol. Cell. Endocrinol. 2014, 384, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Bebermeier, J.H.; Brooks, J.D.; DePrimo, S.E.; Werner, R.; Deppe, U.; Demeter, J.; Hiort, O.; Holterhus, P.M. Cell-line and tissue-specific signatures of androgen receptor-coregulator transcription. J. Mol. Med. (Berl.) 2006, 84, 919–931. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, J.; Montani, M.; Wild, P.; Beer, M.; Huber, F.; Hermanns, T.; Muntener, M.; Kristiansen, G. FOXA1 promotes tumor progression in prostate cancer and represents a novel hallmark of castration-resistant prostate cancer. Am. J. Pathol. 2012, 180, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.J.; Zhao, J.C.; Wu, L.; Kim, J.; Yu, J. Cooperativity and equilibrium with FOXA1 define the androgen receptor transcriptional program. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.L.; Carroll, J.S. FoxA1 is a key mediator of hormonal response in breast and prostate cancer. Front. Endocrinol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Sahu, B.; Laakso, M.; Pihlajamaa, P.; Ovaska, K.; Sinielnikov, I.; Hautaniemi, S.; Janne, O.A. FoxA1 specifies unique androgen and glucocorticoid receptor binding events in prostate cancer cells. Cancer Res. 2013, 73, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.W.; Baskin, L.S.; Haughney, P.C.; Cunha, A.R.; Foster, B.A.; Dahiya, R.; Prins, G.S.; Cunha, G.R. Epithelial development in the rat ventral prostate, anterior prostate and seminal vesicle. Acta Anat. (Basel) 1996, 155, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Hayward, S.W.; Cunha, G.R.; Dahiya, R. Normal development and carcinogenesis of the prostate. A unifying hypothesis. Ann. N. Y. Acad. Sci. 1996, 784, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Bierhoff, E.; Walljasper, U.; Hofmann, D.; Vogel, J.; Wernert, N.; Pfeifer, U. Morphological analogies of fetal prostate stroma and stromal nodules in BPH. Prostate 1997, 31, 234–240. [Google Scholar] [CrossRef]
- Cunha, G.R. Tissue interactions between epithelium and mesenchyme of urogenital and integumental origin. Anat. Rec. 1972, 172, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Chung, L.W.; Cunha, G.R. Stromal-epithelial interactions: II. Regulation of prostatic growth by embryonic urogenital sinus mesenchyme. Prostate 1983, 4, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Zhang, C.; Lin, C.C.; Niu, Y.; Lai, K.P.; Chang, H.C.; Yeh, S. Altered prostate epithelial development and IGF-1 signal in mice lacking the androgen receptor in stromal smooth muscle cells. Prostate 2011, 71, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Donjacour, A.A.; Cunha, G.R. Assessment of prostatic protein secretion in tissue recombinants made of urogenital sinus mesenchyme and urothelium from normal or androgen-insensitive mice. Endocrinology 1993, 132, 2342–2350. [Google Scholar] [PubMed]
- Cunha, G.R. Role of mesenchymal-epithelial interactions in normal and abnormal development of the mammary gland and prostate. Cancer 1994, 74, 1030–1044. [Google Scholar] [CrossRef]
- Hayward, S.W.; Rosen, M.A.; Cunha, G.R. Stromal-epithelial interactions in the normal and neoplastic prostate. Br. J. Urol. 1997, 79, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Cano, P.; Godoy, A.; Escamilla, R.; Dhir, R.; Onate, S.A. Stromal-epithelial cell interactions and androgen receptor-coregulator recruitment is altered in the tissue microenvironment of prostate cancer. Cancer Res. 2007, 67, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.T.; Gray, N.E.; Jacobowitz, K.; Viswanathan, L.; Cheung, P.W.; McFann, K.K.; Le, H.; Blackman, M.R. Human prostate stromal cells stimulate increased PSA production in DHEA-treated prostate cancer epithelial cells. J. Steroid Biochem. Mol. Biol. 2008, 111, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Cooke, P.S.; Young, P.; Hess, R.A.; Cunha, G.R. Estrogen receptor expression in developing epididymis, efferent ductules, and other male reproductive organs. Endocrinology 1991, 128, 2874–2879. [Google Scholar] [CrossRef] [PubMed]
- Cooke, P.S.; Young, P.; Cunha, G.R. Androgen receptor expression in developing male reproductive organs. Endocrinology 1991, 128, 2867–2873. [Google Scholar] [CrossRef] [PubMed]
- Kurita, T.; Young, P.; Brody, J.R.; Lydon, J.P.; O’Malley, B.W.; Cunha, G.R. Stromal progesterone receptors mediate the inhibitory effects of progesterone on estrogen-induced uterine epithelial cell deoxyribonucleic acid synthesis. Endocrinology 1998, 139, 4708–4713. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, Y.; Cunha, G.R.; Bigsby, R.M. Androgenic induction of DNA synthesis in prostatic glands induced in the urothelium of testicular feminized (Tfm/Y) mice. Prostate 1986, 9, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Nieto, C.M.; Rider, L.C.; Cramer, S.D. Influence of stromal-epithelial interactions on androgen action. Endocr. Relat. Cancer 2014, 21, T147–T160. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Chang, H.C.; Tian, J.; Shang, Z.; Niu, Y.; Chang, C. Stromal androgen receptor roles in the development of normal prostate, benign prostate hyperplasia, and prostate cancer. Am. J. Pathol. 2015, 185, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Jha, R.; Melamed, J.; Shapiro, E.; Hayward, S.W.; Lee, P. Stromal androgen receptor in prostate development and cancer. Am. J. Pathol. 2014, 184, 2598–2607. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sudilovsky, D.; Zhang, B.; Haughney, P.C.; Rosen, M.A.; Wu, D.S.; Cunha, T.J.; Dahiya, R.; Cunha, G.R.; Hayward, S.W. A human prostatic epithelial model of hormonal carcinogenesis. Cancer Res. 2001, 61, 6064–6072. [Google Scholar] [PubMed]
- Ricke, E.A.; Williams, K.; Lee, Y.F.; Couto, S.; Wang, Y.; Hayward, S.W.; Cunha, G.R.; Ricke, W.A. Androgen hormone action in prostatic carcinogenesis: Stromal androgen receptors mediate prostate cancer progression, malignant transformation and metastasis. Carcinogenesis 2012, 33, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.P.; Yamashita, S.; Huang, C.K.; Yeh, S.; Chang, C. Loss of stromal androgen receptor leads to suppressed prostate tumourigenesis via modulation of pro-inflammatory cytokines/chemokines. EMBO Mol. Med. 2012, 4, 791–807. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Altuwaijri, S.; Yeh, S.; Lai, K.P.; Yu, S.; Chuang, K.H.; Huang, S.P.; Lardy, H.; Chang, C. Targeting the stromal androgen receptor in primary prostate tumors at earlier stages. Proc. Natl. Acad. Sci. USA 2008, 105, 12188–12193. [Google Scholar] [CrossRef] [PubMed]
- Olapade-Olaopa, E.O.; Muronda, C.A.; MacKay, E.H.; Danso, A.P.; Sandhu, D.P.; Terry, T.R.; Habib, F.K. Androgen receptor protein expression in prostatic tissues in Black and Caucasian men. Prostate 2004, 59, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Hay, C.W.; McEwan, I.J. The impact of point mutations in the human androgen receptor: Classification of mutations on the basis of transcriptional activity. PLoS ONE 2012, 7, e32514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keil, K.P.; Abler, L.L.; Laporta, J.; Altmann, H.M.; Yang, B.; Jarrard, D.F.; Hernandez, L.L.; Vezina, C.M. Androgen receptor DNA methylation regulates the timing and androgen sensitivity of mouse prostate ductal development. Dev. Biol. 2014, 396, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Shenk, J.L.; Fisher, C.J.; Chen, S.Y.; Zhou, X.F.; Tillman, K.; Shemshedini, L. p53 represses androgen-induced transactivation of prostate-specific antigen by disrupting hAR amino- to carboxyl-terminal interaction. J. Biol. Chem. 2001, 276, 38472–38479. [Google Scholar] [CrossRef] [PubMed]
- Jia, Z.; Rahmatpanah, F.B.; Chen, X.; Lernhardt, W.; Wang, Y.; Xia, X.Q.; Sawyers, A.; Sutton, M.; McClelland, M.; Mercola, D. Expression changes in the stroma of prostate cancer predict subsequent relapse. PLoS ONE 2012, 7, e41371. [Google Scholar] [CrossRef]
- Lee, S.O.; Chun, J.Y.; Nadiminty, N.; Lou, W.; Gao, A.C. Interleukin-6 undergoes transition from growth inhibitor associated with neuroendocrine differentiation to stimulator accompanied by androgen receptor activation during LNCaP prostate cancer cell progression. Prostate 2007, 67, 764–773. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Choong, C.S.; Ricciardelli, C.; Kim, J.; Tilley, W.D.; Coetzee, G.A. Androgen receptor signaling: Mechanism of interleukin-6 inhibition. Cancer Res. 2004, 64, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Khaleghzadegan, S.; Mears, B.; Hatano, K.; Kudrolli, T.A.; Chowdhury, W.H.; Yeater, D.B.; Ewing, C.M.; Luo, J.; Isaacs, W.B.; et al. Identification of miR-30b-3p and miR-30d-5p as direct regulators of Androgen Receptor Signaling in Prostate Cancer by complementary functional microRNA library screening. Oncotarget 2016, 7, 72593–72607. [Google Scholar] [CrossRef] [PubMed]
- Ostling, P.; Leivonen, S.K.; Aakula, A.; Kohonen, P.; Makela, R.; Hagman, Z.; Edsjo, A.; Kangaspeska, S.; Edgren, H.; Nicorici, D.; et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011, 71, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.E.; Dart, D.A.; Bevan, C.L. Interplay between steroid signalling and microRNAs: Implications for hormone-dependent cancers. Endocr. Relat. Cancer 2014, 21, R409–R429. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, R.; Girotti, A.W. Pro-survival and pro-growth effects of stress-induced nitric oxide in a prostate cancer photodynamic therapy model. Cancer Lett. 2014, 343, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Fahey, J.M.; Girotti, A.W. Accelerated migration and invasion of prostate cancer cells after a photodynamic therapy-like challenge: Role of nitric oxide. Nitric Oxide 2015, 49, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Cronauer, M.V.; Ince, Y.; Engers, R.; Rinnab, L.; Weidemann, W.; Suschek, C.V.; Burchardt, M.; Kleinert, H.; Wiedenmann, J.; Sies, H.; et al. Nitric oxide-mediated inhibition of androgen receptor activity: Possible implications for prostate cancer progression. Oncogene 2007, 26, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Shigemura, K.; Isotani, S.; Wang, R.; Fujisawa, M.; Gotoh, A.; Marshall, F.F.; Zhau, H.E.; Chung, L.W. Soluble factors derived from stroma activated androgen receptor phosphorylation in human prostate LNCaP cells: Roles of ERK/MAP kinase. Prostate 2009, 69, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Tanner, M.J.; Welliver, R.C., Jr.; Chen, M.; Shtutman, M.; Godoy, A.; Smith, G.; Mian, B.M.; Buttyan, R. Effects of androgen receptor and androgen on gene expression in prostate stromal fibroblasts and paracrine signaling to prostate cancer cells. PLoS ONE 2011, 6, e16027. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Xia, S.; Yang, D.; Wang, K.; Yeh, S.; Gao, Z.; Chang, C. Androgen receptor in human prostate cancer-associated fibroblasts promotes prostate cancer epithelial cell growth and invasion. Med. Oncol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Altuwaijri, S.; Lai, K.P.; Wu, C.T.; Ricke, W.A.; Messing, E.M.; Yao, J.; Yeh, S.; Chang, C. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc. Natl. Acad. Sci. USA 2008, 105, 12182–12187. [Google Scholar] [CrossRef] [PubMed]
- Aprelikova, O.; Palla, J.; Hibler, B.; Yu, X.; Greer, Y.E.; Yi, M.; Stephens, R.; Maxwell, G.L.; Jazaeri, A.; Risinger, J.I.; et al. Silencing of miR-148a in cancer-associated fibroblasts results in WNT10B-mediated stimulation of tumor cell motility. Oncogene 2013, 32, 3246–3253. [Google Scholar] [CrossRef] [PubMed]
- Placencio, V.R.; Sharif-Afshar, A.R.; Li, X.; Huang, H.; Uwamariya, C.; Neilson, E.G.; Shen, M.M.; Matusik, R.J.; Hayward, S.W.; Bhowmick, N.A. Stromal transforming growth factor-beta signaling mediates prostatic response to androgen ablation by paracrine Wnt activity. Cancer Res. 2008, 68, 4709–4718. [Google Scholar] [CrossRef] [PubMed]
- Murashima, A.; Kishigami, S.; Thomson, A.; Yamada, G. Androgens and mammalian male reproductive tract development. Biochim. Biophys. Acta 2015, 1849, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yeh, C.R.; Niu, Y.; Chang, H.C.; Tsai, Y.C.; Moses, H.L.; Shyr, C.R.; Chang, C.; Yeh, S. Altered prostate epithelial development in mice lacking the androgen receptor in stromal fibroblasts. Prostate 2012, 72, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Saylor, P.J.; Kozak, K.R.; Smith, M.R.; Ancukiewicz, M.A.; Efstathiou, J.A.; Zietman, A.L.; Jain, R.K.; Duda, D.G. Changes in biomarkers of inflammation and angiogenesis during androgen deprivation therapy for prostate cancer. Oncologist 2012, 17, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Saylor, P.J.; Karoly, E.D.; Smith, M.R. Prospective study of changes in the metabolomic profiles of men during their first three months of androgen deprivation therapy for prostate cancer. Clin. Cancer Res. 2012, 18, 3677–3685. [Google Scholar] [CrossRef] [PubMed]
- Ohlson, N.; Bergh, A.; Stattin, P.; Wikstrom, P. Castration-induced epithelial cell death in human prostate tissue is related to locally reduced IGF-1 levels. Prostate 2007, 67, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Neilson, E.G.; Moses, H.L. Stromal fibroblasts in cancer initiation and progression. Nature 2004, 432, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Diener, K.R.; Need, E.F.; Buchanan, G.; Hayball, J.D. TGF-beta signalling and immunity in prostate tumourigenesis. Expert Opin. Ther. Targets 2010, 14, 179–192. [Google Scholar] [CrossRef] [PubMed]
- Berry, P.A.; Maitland, N.J.; Collins, A.T. Androgen receptor signalling in prostate: Effects of stromal factors on normal and cancer stem cells. Mol. Cell. Endocrinol. 2008, 288, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Kwabi-Addo, B.; Ozen, M.; Ittmann, M. The role of fibroblast growth factors and their receptors in prostate cancer. Endocr. Relat. Cancer 2004, 11, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Tidehag, V.; Hammarsten, P.; Egevad, L.; Granfors, T.; Stattin, P.; Leanderson, T.; Wikstrom, P.; Josefsson, A.; Hagglof, C.; Bergh, A. High density of S100A9 positive inflammatory cells in prostate cancer stroma is associated with poor outcome. Eur. J. Cancer 2014, 50, 1829–1835. [Google Scholar] [CrossRef] [PubMed]
- Bernoulli, J.; Yatkin, E.; Konkol, Y.; Talvitie, E.M.; Santti, R.; Streng, T. Prostatic inflammation and obstructive voiding in the adult Noble rat: Impact of the testosterone to estradiol ratio in serum. Prostate 2008, 68, 1296–1306. [Google Scholar] [CrossRef] [PubMed]
- Bernoulli, J.; Yatkin, E.; Laakso, A.; Anttinen, M.; Bosland, M.; Vega, K.; Kallajoki, M.; Santti, R.; Pylkkanen, L. Histopathological evidence for an association of inflammation with ductal pin-like lesions but not with ductal adenocarcinoma in the prostate of the noble rat. Prostate 2008, 68, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.M.; Vermeulen, A. The decline of androgen levels in elderly men and its clinical and therapeutic implications. Endocr. Rev. 2005, 26, 833–876. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.L.; Liu, X.; Yan, J.Y.; Chong, L.M.; Li, L.; Ma, A.C.; Zhou, L.; Sun, Z.Y. The alteration of inflammatory markers and apoptosis on chronic prostatitis induced by estrogen and androgen. Int. Urol. Nephrol. 2015, 47, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Comito, G.; Giannoni, E.; Segura, C.P.; Barcellos-de-Souza, P.; Raspollini, M.R.; Baroni, G.; Lanciotti, M.; Serni, S.; Chiarugi, P. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene 2014, 33, 2423–2431. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, K.; Balachandran, C.; Manohar, B.M.; Puvanakrishnan, R. Effects of testosterone, estrogen and progesterone on TNF-alpha mediated cellular damage in rat arthritic synovial fibroblasts. Rheumatol. Int. 2012, 32, 3181–3188. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Itoh, Y.; Hayashi, H.; Takii, T.; Miyazawa, K.; Onozaki, K. Dihydrotestosterone inhibits interleukin-1alpha or tumor necrosis factor alpha-induced proinflammatory cytokine production via androgen receptor-dependent inhibition of nuclear factor-kappaB activation in rheumatoid fibroblast-like synovial cell line. Biol. Pharm. Bull. 2011, 34, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- De Wever, O.; Nguyen, Q.D.; van Hoorde, L.; Bracke, M.; Bruyneel, E.; Gespach, C.; Mareel, M. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004, 18, 1016–1018. [Google Scholar] [PubMed]
- Denys, H.; Derycke, L.; Hendrix, A.; Westbroek, W.; Gheldof, A.; Narine, K.; Pauwels, P.; Gespach, C.; Bracke, M.; De Wever, O. Differential impact of TGF-beta and EGF on fibroblast differentiation and invasion reciprocally promotes colon cancer cell invasion. Cancer Lett. 2008, 266, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xie, R.L.; Croce, C.M.; Stein, J.L.; Lian, J.B.; van Wijnen, A.J.; Stein, G.S. A program of microRNAs controls osteogenic lineage progression by targeting transcription factor Runx2. Proc. Natl. Acad. Sci. USA 2011, 108, 9863–9868. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Tang, H.; Xu, L.; Wang, X.; Yang, C.; Ruan, S.; Guo, J.; Hu, S.; Wang, Z. Fibroblasts in omentum activated by tumor cells promote ovarian cancer growth, adhesion and invasiveness. Carcinogenesis 2012, 33, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Fuyuhiro, Y.; Yashiro, M.; Noda, S.; Matsuoka, J.; Hasegawa, T.; Kato, Y.; Sawada, T.; Hirakawa, K. Cancer-associated orthotopic myofibroblasts stimulates the motility of gastric carcinoma cells. Cancer Sci. 2012, 103, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Alcoser, T.A.; Bordeleau, F.; Carey, S.P.; Lampi, M.C.; Kowal, D.R.; Somasegar, S.; Varma, S.; Shin, S.J.; Reinhart-King, C.A. Probing the biophysical properties of primary breast tumor-derived fibroblasts. Cell. Mol. Bioeng. 2015, 8, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar] [CrossRef] [PubMed]
- Coulson-Thomas, V.J.; Gesteira, T.F.; Coulson-Thomas, Y.M.; Vicente, C.M.; Tersariol, I.L.; Nader, H.B.; Toma, L. Fibroblast and prostate tumor cell cross-talk: Fibroblast differentiation, TGF-beta, and extracellular matrix down-regulation. Exp. Cell Res. 2010, 316, 3207–3226. [Google Scholar] [CrossRef] [PubMed]
- Dang, T.T.; Prechtl, A.M.; Pearson, G.W. Breast cancer subtype-specific interactions with the microenvironment dictate mechanisms of invasion. Cancer Res. 2011, 71, 6857–6866. [Google Scholar] [CrossRef] [PubMed]
- Shieh, A.C.; Rozansky, H.A.; Hinz, B.; Swartz, M.A. Tumor cell invasion is promoted by interstitial flow-induced matrix priming by stromal fibroblasts. Cancer Res. 2011, 71, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Yin, T.; Wu, Y.I.; Inoue, T.; Levchenko, A. Interplay between chemotaxis and contact inhibition of locomotion determines exploratory cell migration. Nat. Commun. 2015. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cheng, G.; Hao, M.; Zheng, J.; Zhou, X.; Zhang, J.; Taichman, R.S.; Pienta, K.J.; Wang, J. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010, 29, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.; Jennek, S.; Singh, R.; Enkelmann, A.; Junker, K.; Rippaus, N.; Berndt, A.; Friedrich, K. Malignancy of bladder cancer cells is enhanced by tumor-associated fibroblasts through a multifaceted cytokine-chemokine loop. Exp. Cell Res. 2015, 335, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sterling, J.A.; Fan, K.H.; Vessella, R.L.; Shyr, Y.; Hayward, S.W.; Matrisian, L.M.; Bhowmick, N.A. Loss of TGF-beta responsiveness in prostate stromal cells alters chemokine levels and facilitates the development of mixed osteoblastic/osteolytic bone lesions. Mol. Cancer Res. 2012, 10, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.H.; Tsuang, Y.H.; Sun, J.S.; Sun, M.G.; Chen, M.H. Centrifugal force induces human ligamentum flavum fibroblasts inflammation through activation of JNK and p38 pathways. Connect. Tissue Res. 2012, 53, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.H.; Yang, H.S.; Sun, M.G.; Sun, J.S.; Chen, M.H. Elastin-derived peptides induce inflammatory responses through the activation of NF-kappaB in human ligamentum flavum cells. Connect. Tissue Res. 2012, 53, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Maller, O.; DuFort, C.C.; Weaver, V.M. YAP forces fibroblasts to feel the tension. Nat. Cell Biol. 2013, 15, 570–572. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, P.P.; Inman, D.R.; Eliceiri, K.W.; Knittel, J.G.; Yan, L.; Rueden, C.T.; White, J.G.; Keely, P.J. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Ngoc, K.V.; Cheung, K.J.; Brenot, A.; Shamir, E.R.; Gray, R.S.; Hines, W.C.; Yaswen, P.; Werb, Z.; Ewald, A.J. ECM microenvironment regulates collective migration and local dissemination in normal and malignant mammary epithelium. Proc. Natl. Acad. Sci. USA 2012, 109, E2595–E2604. [Google Scholar] [CrossRef] [PubMed]
- Kakkad, S.M.; Solaiyappan, M.; O’Rourke, B.; Stasinopoulos, I.; Ackerstaff, E.; Raman, V.; Bhujwalla, Z.M.; Glunde, K. Hypoxic tumor microenvironments reduce collagen I fiber density. Neoplasia 2010, 12, 608–617. [Google Scholar] [CrossRef] [PubMed]
- Carey, S.P.; Kraning-Rush, C.M.; Williams, R.M.; Reinhart-King, C.A. Biophysical control of invasive tumor cell behavior by extracellular matrix microarchitecture. Biomaterials 2012, 33, 4157–4165. [Google Scholar] [CrossRef] [PubMed]
- Carey, S.P.; D'Alfonso, T.M.; Shin, S.J.; Reinhart-King, C.A. Mechanobiology of tumor invasion: Engineering meets oncology. Crit. Rev. Oncol. Hematol. 2012, 83, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Wolf, K.; Te Lindert, M.; Krause, M.; Alexander, S.; Te Riet, J.; Willis, A.L.; Hoffman, R.M.; Figdor, C.G.; Weiss, S.J.; Friedl, P. Physical limits of cell migration: Control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J. Cell Biol. 2013, 201, 1069–1084. [Google Scholar] [CrossRef] [PubMed]
- Bruni-Cardoso, A.; Augusto, T.M.; Pravatta, H.; Damas-Souza, D.M.; Carvalho, H.F. Stromal remodelling is required for progressive involution of the rat ventral prostate after castration: Identification of a matrix metalloproteinase-dependent apoptotic wave. Int. J. Androl. 2010, 33, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Justulin, L.A., Jr.; Delella, F.K.; Felisbino, S.L. Doxazosin reduces cell proliferation and increases collagen fibers in rat prostatic lobes. Cell Tissue Res. 2008, 332, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Justulin, L.A., Jr.; Acquaro, C.; Carvalho, R.F.; Silva, M.D.; Felisbino, S.L. Combined effect of the finasteride and doxazosin on rat ventral prostate morphology and physiology. Int. J. Androl. 2010, 33, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Delella, F.K.; Justulin, L.A., Jr.; Felisbino, S.L. Finasteride treatment alters MMP-2 and -9 gene expression and activity in the rat ventral prostate. Int. J. Androl. 2010, 33, e114–e122. [Google Scholar] [CrossRef] [PubMed]
- Li, S.C.; Chen, G.F.; Chan, P.S.; Choi, H.L.; Ho, S.M.; Chan, F.L. Altered expression of extracellular matrix and proteinases in Noble rat prostate gland after long-term treatment with sex steroids. Prostate 2001, 49, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Charras, G.; Sahai, E. Physical influences of the extracellular environment on cell migration. Nat. Rev. Mol. Cell Biol. 2014, 15, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Sieh, S.; Taubenberger, A.V.; Rizzi, S.C.; Sadowski, M.; Lehman, M.L.; Rockstroh, A.; An, J.; Clements, J.A.; Nelson, C.C.; Hutmacher, D.W. Phenotypic characterization of prostate cancer LNCaP cells cultured within a bioengineered microenvironment. PLoS ONE 2012, 7, e40217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaman, M.H.; Trapani, L.M.; Sieminski, A.L.; Mackellar, D.; Gong, H.; Kamm, R.D.; Wells, A.; Lauffenburger, D.A.; Matsudaira, P. Migration of tumor cells in 3D matrices is governed by matrix stiffness along with cell-matrix adhesion and proteolysis. Proc. Natl. Acad. Sci. USA 2006, 103, 10889–10894. [Google Scholar] [CrossRef] [PubMed]
- Sabeh, F.; Shimizu-Hirota, R.; Weiss, S.J. Protease-dependent versus -independent cancer cell invasion programs: Three-dimensional amoeboid movement revisited. J. Cell Biol. 2009, 185, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Tozluoglu, M.; Tournier, A.L.; Jenkins, R.P.; Hooper, S.; Bates, P.A.; Sahai, E. Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat. Cell Biol. 2013, 15, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E. Mechanisms of cancer cell invasion. Curr. Opin. Genet. Dev. 2005, 15, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Cooperberg, M.R.; Broering, J.M.; Kantoff, P.W.; Carroll, P.R. Contemporary trends in low risk prostate cancer: Risk assessment and treatment. J. Urol. 2007, 178, S14–S19. [Google Scholar] [CrossRef] [PubMed]
- Zelefsky, M.J.; Leibel, S.A.; Burman, C.M.; Kutcher, G.J.; Harrison, A.; Happersett, L.; Fuks, Z. Neoadjuvant hormonal therapy improves the therapeutic ratio in patients with bulky prostatic cancer treated with three-dimensional conformal radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 755–761. [Google Scholar] [CrossRef]
- Henderson, A.; Laing, R.W.; Langley, S.E. Identification of pubic arch interference in prostate brachytherapy: Simplifying the transrectal ultrasound technique. Brachytherapy 2003, 2, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Cooperberg, M.R.; Vickers, A.J.; Broering, J.M.; Carroll, P.R. Comparative risk-adjusted mortality outcomes after primary surgery, radiotherapy, or androgen-deprivation therapy for localized prostate cancer. Cancer 2010, 116, 5226–5234. [Google Scholar] [CrossRef] [PubMed]
- Lu-Yao, G.L.; Albertsen, P.C.; Moore, D.F.; Shih, W.; Lin, Y.; DiPaola, R.S.; Yao, S.L. Survival following primary androgen deprivation therapy among men with localized prostate cancer. JAMA 2008, 300, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.N.; Freedland, S.J.; Egleston, B.; Vapiwala, N.; Uzzo, R.; Armstrong, K. The role of primary androgen deprivation therapy in localized prostate cancer. Eur. Urol. 2009, 56, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Studer, U.E.; Whelan, P.; Albrecht, W.; Casselman, J.; de Reijke, T.; Hauri, D.; Loidl, W.; Isorna, S.; Sundaram, S.K.; Debois, M.; et al. Immediate or deferred androgen deprivation for patients with prostate cancer not suitable for local treatment with curative intent: European Organisation for Research and Treatment of Cancer (EORTC) Trial 30891. J. Clin. Oncol. 2006, 24, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- Lu-Yao, G.L.; Albertsen, P.C.; Li, H.; Moore, D.F.; Shih, W.; Lin, Y.; Dipaola, R.S.; Yao, S.L. Does primary androgen-deprivation therapy delay the receipt of secondary cancer therapy for localized prostate cancer? Eur. Urol. 2012, 62, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Labrie, F.; Candas, B.; Gomez, J.L.; Cusan, L. Can combined androgen blockade provide long-term control or possible cure of localized prostate cancer? Urology 2002, 60, 115–119. [Google Scholar] [CrossRef]
- Akaza, H.; Homma, Y.; Usami, M.; Hirao, Y.; Tsushima, T.; Okada, K.; Yokoyama, M.; Ohashi, Y.; Aso, Y. Efficacy of primary hormone therapy for localized or locally advanced prostate cancer: Results of a 10-year follow-up. BJU Int. 2006, 98, 573–579. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.J.; Eisenberger, M.A.; Pili, R.; Denmeade, S.R.; Rathkopf, D.; Slovin, S.F.; Farrelly, J.; Chudow, J.J.; Vincent, M.; Scher, H.I.; et al. A phase I/IIA study of AGS-PSCA for castration-resistant prostate cancer. Ann. Oncol. 2012, 23, 2714–2719. [Google Scholar] [CrossRef] [PubMed]
- Byar, D.P.; Corle, D.K. Hormone therapy for prostate cancer: Results of the Veterans Administration Cooperative Urological Research Group studies. NCI Monogr. 1988, 7, 165–170. [Google Scholar]
- Merglen, A.; Schmidlin, F.; Fioretta, G.; Verkooijen, H.M.; Rapiti, E.; Zanetti, R.; Miralbell, R.; Bouchardy, C. Short- and long-term mortality with localized prostate cancer. Arch. Intern. Med. 2007, 167, 1944–1950. [Google Scholar] [CrossRef] [PubMed]
- Zhou, E.H.; Ellis, R.J.; Cherullo, E.; Colussi, V.; Xu, F.; Chen, W.D.; Gupta, S.; Whalen, C.C.; Bodner, D.; Resnick, M.I.; et al. Radiotherapy and survival in prostate cancer patients: A population-based study. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Schulman, C.C.; Debruyne, F.M.; Forster, G.; Selvaggi, F.P.; Zlotta, A.R.; Witjes, W.P. 4-Year follow-up results of a European prospective randomized study on neoadjuvant hormonal therapy prior to radical prostatectomy in T2-3N0M0 prostate cancer. European Study Group on Neoadjuvant Treatment of Prostate Cancer. Eur. Urol. 2000, 38, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Gleave, M.E.; Goldenberg, S.L.; Chin, J.L.; Warner, J.; Saad, F.; Klotz, L.H.; Jewett, M.; Kassabian, V.; Chetner, M.; Dupont, C.; et al. Randomized comparative study of 3 versus 8-month neoadjuvant hormonal therapy before radical prostatectomy: Biochemical and pathological effects. J. Urol. 2001, 166, 500–506. [Google Scholar] [CrossRef]
- Prezioso, D.; Lotti, T.; Polito, M.; Montironi, R. Neoadjuvant hormone treatment with leuprolide acetate depot 3.75 mg and cyproterone acetate, before radical prostatectomy: A randomized study. Urol. Int. 2004, 72, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Selli, C.; Montironi, R.; Bono, A.; Pagano, F.; Zattoni, F.; Manganelli, A.; Selvaggi, F.P.; Comeri, G.; Fiaccavento, G.; Guazzieri, S.; et al. Effects of complete androgen blockade for 12 and 24 weeks on the pathological stage and resection margin status of prostate cancer. J. Clin. Pathol. 2002, 55, 508–513. [Google Scholar] [CrossRef] [PubMed]
- Gravina, G.L.; Festuccia, C.; Galatioto, G.P.; Muzi, P.; Angelucci, A.; Ronchi, P.; Costa, A.M.; Bologna, M.; Vicentini, C. Surgical and biologic outcomes after neoadjuvant bicalutamide treatment in prostate cancer. Urology 2007, 70, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Soloway, M.S.; Pareek, K.; Sharifi, R.; Wajsman, Z.; McLeod, D.; Wood, D.P., Jr.; Puras-Baez, A. Lupron Depot Neoadjuvant Prostate Cancer Study Group. Neoadjuvant androgen ablation before radical prostatectomy in cT2bNxMo prostate cancer: 5-year results. J. Urol. 2002, 167, 112–116. [Google Scholar] [CrossRef]
- Aus, G.; Abrahamsson, P.A.; Ahlgren, G.; Hugosson, J.; Lundberg, S.; Schain, M.; Schelin, S.; Pedersen, K. Three-month neoadjuvant hormonal therapy before radical prostatectomy: A 7-year follow-up of a randomized controlled trial. BJU Int. 2002, 90, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Yee, D.S.; Lowrance, W.T.; Eastham, J.A.; Maschino, A.C.; Cronin, A.M.; Rabbani, F. Long-term follow-up of 3-month neoadjuvant hormone therapy before radical prostatectomy in a randomized trial. BJU Int. 2010, 105, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Klotz, L.H.; Goldenberg, S.L.; Jewett, M.A.; Fradet, Y.; Nam, R.; Barkin, J.; Chin, J.; Chatterjee, S. Canadian Uro-Oncology Group. Long-term followup of a randomized trial of 0 versus 3 months of neoadjuvant androgen ablation before radical prostatectomy. J. Urol. 2003, 170, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Ayala, G.E.; Muezzinoglu, B.; Hammerich, K.H.; Frolov, A.; Liu, H.; Scardino, P.T.; Li, R.; Sayeeduddin, M.; Ittmann, M.M.; Kadmon, D.; et al. Determining prostate cancer-specific death through quantification of stromogenic carcinoma area in prostatectomy specimens. Am. J. Pathol. 2011, 178, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Tomas, D.; Spajic, B.; Milosevic, M.; Demirovic, A.; Marusic, Z.; Kruslin, B. Intensity of stromal changes predicts biochemical recurrence-free survival in prostatic carcinoma. Scand. J. Urol. Nephrol. 2010, 44, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, N.; Li, R.; Rowley, D.; Liu, H.; Kadmon, D.; Miles, B.J.; Wheeler, T.M.; Ayala, G.E. Stromogenic prostatic carcinoma pattern (carcinomas with reactive stromal grade 3) in needle biopsies predicts biochemical recurrence-free survival in patients after radical prostatectomy. Hum. Pathol. 2007, 38, 1611–1620. [Google Scholar] [CrossRef] [PubMed]
- Kinseth, M.A.; Jia, Z.; Rahmatpanah, F.; Sawyers, A.; Sutton, M.; Wang-Rodriguez, J.; Mercola, D.; McGuire, K.L. Expression differences between African American and Caucasian prostate cancer tissue reveals that stroma is the site of aggressive changes. Int. J. Cancer 2014, 134, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Planche, A.; Bacac, M.; Provero, P.; Fusco, C.; Delorenzi, M.; Stehle, J.C.; Stamenkovic, I. Identification of prognostic molecular features in the reactive stroma of human breast and prostate cancer. PLoS ONE 2011, 6, e18640. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Berriguete, G.; Sanchez-Espiridion, B.; Cansino, J.R.; Olmedilla, G.; Martinez-Onsurbe, P.; Sanchez-Chapado, M.; Paniagua, R.; Fraile, B.; Royuela, M. Clinical significance of both tumor and stromal expression of components of the IL-1 and TNF-alpha signaling pathways in prostate cancer. Cytokine 2013, 64, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wiseman, G.; Welt, S.; Adjei, A.; Lee, F.T.; Hopkins, W.; Divgi, C.R.; Hanson, L.H.; Mitchell, P.; Gansen, D.N.; et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 2003, 9, 1639–1647. [Google Scholar] [PubMed]
- LeBeau, A.M.; Brennen, W.N.; Aggarwal, S.; Denmeade, S.R. Targeting the cancer stroma with a fibroblast activation protein-activated promelittin protoxin. Mol. Cancer Ther. 2009, 8, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Brennen, W.N.; Rosen, D.M.; Wang, H.; Isaacs, J.T.; Denmeade, S.R. Targeting carcinoma-associated fibroblasts within the tumor stroma with a fibroblast activation protein-activated prodrug. J. Natl. Cancer Inst. 2012, 104, 1320–1334. [Google Scholar] [CrossRef] [PubMed]
- Sluka, P.; Davis, I.D. Cell mates: Paracrine and stromal targets for prostate cancer therapy. Nat. Rev. Urol. 2013, 10, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Killian, P.H.; Kronski, E.; Michalik, K.M.; Barbieri, O.; Astigiano, S.; Sommerhoff, C.P.; Pfeffer, U.; Nerlich, A.G.; Bachmeier, B.E. Curcumin inhibits prostate cancer metastasis in vivo by targeting the inflammatory cytokines CXCL1 and -2. Carcinogenesis 2012, 33, 2507–2519. [Google Scholar] [CrossRef] [PubMed]
- Yeung, T.L.; Leung, C.S.; Li, F.; Wong, S.S.; Mok, S.C. Targeting stromal-cancer cell crosstalk networks in ovarian cancer treatment. Biomolecules 2016. [Google Scholar] [CrossRef] [PubMed]
- Aigner, A.; Renneberg, H.; Bojunga, J.; Apel, J.; Nelson, P.S.; Czubayko, F. Ribozyme-targeting of a secreted FGF-binding protein (FGF-BP) inhibits proliferation of prostate cancer cells in vitro and in vivo. Oncogene 2002, 21, 5733–5742. [Google Scholar] [CrossRef] [PubMed]
- Herbert, C.; Schieborr, U.; Saxena, K.; Juraszek, J.; De Smet, F.; Alcouffe, C.; Bianciotto, M.; Saladino, G.; Sibrac, D.; Kudlinzki, D.; et al. Molecular mechanism of SSR128129E, an extracellularly acting, small-molecule, allosteric inhibitor of fgf receptor signaling. Cancer Cell. 2016, 30, 176–178. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, F.; Bottaro, D.P. Novel antagonists of heparin binding growth factors. Oncotarget 2012, 3, 911–912. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, F.X.; Luo, H.L.; Liu, J.J.; Luo, T.; Bai, T.; Li, L.Q.; Fan, X.H. Berberine upregulates miR-22-3p to suppress hepatocellular carcinoma cell proliferation by targeting Sp1. Am. J. Transl. Res. 2016, 8, 4932–4941. [Google Scholar] [PubMed]
- Foley, C.; Mitsiades, N. Moving beyond the androgen receptor (AR): Targeting AR-interacting proteins to treat prostate cancer. Horm. Cancer 2016, 7, 84–103. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Authors | Specimens | Cohort Size | Methods | Effect on Prostate Cancer Outcome |
|---|---|---|---|---|
| [14] | Biopsies | 62 | IHC | Higher AR, better prognosis |
| [15] | Biopsy, RP and TURP | 42 | IHC | Higher AR, better prognosis |
| [16] | Biopsies | 90 | IHC | Higher AR, better prognosis |
| [17] | RP | 197 | IHC | Higher AR, better prognosis |
| [18] | RP | 105 | IHC | Higher AR, better prognosis |
| [19] | mixed RP, TURP, Biopsy | 42 | IHC | Higher AR, better prognosis |
| [9] | RP | 96 | IHC | Higher AR, biochemical relapse |
| [20] | RP | 115 | RT-PCR | Higher AR, biochemical relapse |
| [21] | RP | 340 | IHC | Higher AR, biochemical relapse |
| [22] | RP | 52 | IHC | Higher AR, biochemical relapse |
| [8] | RP | 53 | IHC | Higher AR, biochemical relapse |
| [22] | RP | 52 | IHC | Higher AR, worse prognosis |
| [23] | RP | 640 | IHC | Higher AR, worse prognosis |
| [24] | mixed RP/biopsy | 66 | IF | Higher AR, worse prognosis |
| [11] | RP | 56 | IHC | Not prognostic |
| [25] | RP | 232 | IHC | Not prognostic |
| [26] | TURP | 68 | IHC | Not prognostic |
| [27] | RP | 64 | IHC | Not prognostic |
| [28] | Biopsies | 17 | IHC | Not prognostic |
| [29] | RP | 121 | RT-PCR | Not prognostic |
| [30] | TURP and RP | 81 | IHC | Not prognostic |
| [31] | RP and metastases | 119 | IHC | Not prognostic |
| [32] | RP | 2805 | IHC and RT-PCR | Not prognostic |
| [33] | RP | 172 | IHC | Not prognostic |
| [34] | TURP | 24 | IHC | Not prognostic |
| [10] | TURP + biopsy | 154 | IHC | Not prognostic |
| [35] | RP | 43 | IHC | Not prognostic |
| [7] | RP | 20 | IHC | Not prognostic |
| [12] | TURP | 64 | IHC | Not prognostic |
| [36] | RP | 53 | branched chain DNA | Not prognostic |
| [37] | RP | 10 | IHC | Unavailable |
| [38] | Biopsies | 39 | IHC | Unavailable |
| [39] | RP | 26 | IHC | Unavailable |
| [40] | RP | 50 | IHC | Unavailable |
| Authors | Specimens | Cohort Size | Methods | Effect on Prostate Cancer Outcome |
|---|---|---|---|---|
| [41] | RP | 44 | IHC | Low AR, biochemical relapse |
| [8] | RP | 53 | IHC | Low AR, biochemical relapse |
| [9] | RP | 96 | IHC | Low AR, biochemical relapse |
| [12] | TURP | 64 | IHC | Low AR, PCSM |
| [10] | TURP + biopsy | 152 | IHC | Low AR, worse prognosis |
| [11] | RP | 56 | IHC | Low AR, worse prognosis |
| [7] | RP | 20 | IHC | (low AR, no association with Gleason) |
| Paracrine Factor | Effect | Androgen Regulation |
|---|---|---|
| CTGF | P | Y |
| FGF (2, 5, 7, 8, 9, 10) | P, D | Y (2, 5, 7), N (8), N/A (9, 10) |
| HGF | P, D | Y |
| IGF (1, 2) | P, D | Y (1, 2) |
| IL-6 | P | Y |
| PDGF | P, D | Y |
| TGFb (1, 2, 3) | P, D | Y (1, 2, 3) |
| VEGF (A, B, C) | P | Y (A,C), N (B) |
| WNT | P | Y |
| CXCL12 | P | N |
| EGF | P, D | N/A |
| TGFa | P, D | N/A |
| References | N | Pca Staging | ADT Use | Comparison | Outcome |
|---|---|---|---|---|---|
| [149] | 7538 | T1–T3 | Neo | ADT vs. surgery or radiation | ADT increases hazard ratio |
| [150] | 19,271 | T1–T2 | Neo (<180 days) | ADT vs. conservative management | Decreased PCSS |
| [151] | 16,000 | T1–T2 | Neo (<first 6 months) | ADT in first 6months vs. no ADT in first 6 months | Increased PCSM |
| [153] | 29,775 | Localized | Neo | ADT vs. noADT | ADT increases need for subsequent treatments |
| [158] | 844 | Neo (<first 6 months) | Neo compared to WW, RP, radiotherapy | Neo had worse 10 year PCSS of all treatments | |
| [159] | 10,179 | Localised | Neo | Neo compared to no treatment, RP, BT, ERBT | ADT worse PCSS |
| [160] | 402 | Localised | Neo (<first 3 months) | Neo compared to RP alone | Neo = pathological downstaging and lowers % of patients with positive margins |
| [161] | 547 | Localised | Neo | 3-month vs. 8-month neo | Positive margin rates were significantly lower in the 8 than 3-month group |
| [162] | 167 | T1a–T2b | Neo (<first 3 months) | 3-month neo vs. RP alone | Neo had less lymph node involvement, less positive margins |
| [163] | 393 | T2–T3 | Neo (3–6 months) | Neo vs. RP alone | Neo had better positive margin rates |
| [164] | 119 | T2–T3a | Neo (<first 4 months) | 4-month neo vs. RP alone | Neo had better positive margin rates |
| [154] | 176 | B2/T2–T3 | Neo | 1-year ADT vs. long term ADT | No measurable significant benefit |
| [155] | 57 | Neo | No benefit | ||
| [156] | 1006 | Low-intermediate | Neo ADT + LDB | ADT prior to or after LDB | No effect of PCSS |
| [165] | 282 | T2b | Neo (<first 3 months) | 3-month neo vs. RP alone | No difference in 5 year BCR |
| [166] | 126 | T1b–T3aNXM0 | Neo (<first 3 months) | 3-month neo vs. RP alone | No difference in PSA progression-free survival (7 year follow up) |
| [167] | 148 | T1b–T3 | Neo (<first 3 months) | 3-month neo vs. RP alone | No significant difference in BCR-free (8 year followup) |
| [152] | 985 | Localized | ADT Immediately or upon symptoms of progression | Immediate ADT vs. delayed ADT | Delayed ADT increased risk of mortality |
| [157] | 1903 | T1–T2 | Neo (diethylstilbesterol) | ADT in T1 vs. ADT in T2 | benefit T2, deletrious in T1 |
| [168] | 213 | T1b/c–T2c | Neo | Neo prior to surgery vs. surgery alone | Neo = less organ confinement, lower 7-year survival |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leach, D.A.; Buchanan, G. Stromal Androgen Receptor in Prostate Cancer Development and Progression. Cancers 2017, 9, 10. https://doi.org/10.3390/cancers9010010
Leach DA, Buchanan G. Stromal Androgen Receptor in Prostate Cancer Development and Progression. Cancers. 2017; 9(1):10. https://doi.org/10.3390/cancers9010010
Chicago/Turabian StyleLeach, Damien A., and Grant Buchanan. 2017. "Stromal Androgen Receptor in Prostate Cancer Development and Progression" Cancers 9, no. 1: 10. https://doi.org/10.3390/cancers9010010
APA StyleLeach, D. A., & Buchanan, G. (2017). Stromal Androgen Receptor in Prostate Cancer Development and Progression. Cancers, 9(1), 10. https://doi.org/10.3390/cancers9010010