
A Second WNT for Old Drugs: Drug Repositioning against WNT-Dependent Cancers


Abstract
:1. Introduction
2. Nonsteroidal Anti-Inflammatory Drugs (NSAIDs)
2.1. Sulindac
2.2. Aspirin
2.3. Indomethacin
2.4. Celecoxib
3. Antiparasitics
3.1. Niclosamide
3.2. Suramin
3.3. Pyrvinium Pamoate
3.4. Ivermectin
4. Antimicrobials
4.1. Salinomycin
4.2. Clofazimine
4.3. Other Antimicrobials
5. Additional Selected Compounds
5.1. Metformin
5.2. Imatinib
5.3. Ethacrynic Acid
5.4. Riluzole
6. Challenges and Future Directions for Repositioning WNT Inhibitors in Cancers
- Frequently, for a novel application the drug is required at a higher dose, for an extended treatment period or with a different formulation as compared to the conventional indication in order to demonstrate a significant effect. This may result in unexpected side effects, jeopardizing the idea of the “fast tracking” of the compound due to necessity of a full-scale preclinical and clinical investigation.
- Intellectual property difficulties due to multitude of patents.
- Drug-drug incompatibility: acceptable levels of adverse effects for one application might make the compound useless or uncompetitive for another purpose, as well as incompatible with other treatments for the purpose.
- Different legal statuses of the drug in various countries, e.g., dependence on the region where the disease is widespread or on the socioeconomical status of the population.
- Multiple and controversial mechanisms of novel action, resulting from superposition of the original drug mechanism with the novel one(s).
- The WNT pathway is complex. Many components of the signaling are shared with other pathways, generating cross-talks of varying intensities. Therefore, it is sometimes difficult to clearly distinguish direct influence of the drug on the WNT pathway from its effects on the intersecting pathways.
- Identification of the molecular target is a complicated process, and it is frequently omitted by researchers. Out of the 16 drugs we reviewed here, only EA, suramin, sulindac, pyrvinium pamoate and indomethacin were shown to directly affect identified components of the WNT pathway. Additionally, metformin is known to affect WNT signaling via a cross-talk from its original target AMPK. Delaying the unequivocal identification of the novel molecular target makes it problematic to optimize the drug and evaluate of the scope of its anticancer applications.
7. Concluding Remarks
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nusse, R. Wnt signaling in disease and in development. Cell Res. 2005, 15, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Krausova, M.; Korinek, V. Wnt signaling in adult intestinal stem cells and cancer. Cell. Signal. 2014, 26, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, S.; Kincade, P.W. Wnt-related molecules and signaling pathway equilibrium in hematopoiesis. Cell Stem Cell 2009, 4, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Whyte, J.L.; Smith, A.A.; Helms, J.A. Wnt signaling and injury repair. Cold Spring Harb. Perspect. Biol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. Drugging Wnt signaling in cancer. EMBO J. 2012, 31, 2737–2746. [Google Scholar] [CrossRef] [PubMed]
- Solis, G.P.; Lüchtenborg, A.M.; Katanaev, V.L. Wnt secretion and gradient formation. Int. J. Mol. Sci. 2013, 14, 5130–5145. [Google Scholar] [CrossRef] [PubMed]
- Koval, A.; Katanaev, V.L. Wnt3a stimulation elicits G-protein-coupled receptor properties of mammalian Frizzled proteins. Biochem. J. 2011, 433, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Schulte, G. Frizzleds and Wnt/β-catenin signaling—The black box of ligand-receptor selectivity, complex stoichiometry and activation kinetics. Eur. J. Pharmacol. 2015, 763, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.H.; van Es, J.H.; Clevers, H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim. Biophys. Acta Rev. Cancer 2003, 1653, 1–24. [Google Scholar] [CrossRef]
- Egger-Adam, D. Trimeric G protein-dependent signaling by Frizzled receptors in animal development. Front. Biosci. 2008, 13, 4740–4755. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Chen, Y.G. Dishevelled: The hub of Wnt signaling. Cell. Signal. 2010, 22, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.; Hamada, F.; Bienz, M. A role of Dishevelled in relocating Axin to the plasma membrane during wingless signaling. Curr. Biol. 2003, 13, 960–966. [Google Scholar] [CrossRef]
- Egger-Adam, D.; Katanaev, V.L. The trimeric G protein Go inflicts a double impact on Axin in the Wnt/Frizzled signaling pathway. Dev. Dyn. 2010, 239, 168–183. [Google Scholar] [CrossRef] [PubMed]
- Kimelman, D.; Xu, W. β-catenin destruction complex: Insights and questions from a structural perspective. Oncogene 2006, 25, 7482–7491. [Google Scholar] [CrossRef] [PubMed]
- Willert, K.; Jones, K.A. Wnt signaling: Is the party in the nucleus? Genes Dev. 2006, 20, 1394–1404. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.J.; Thakur, A.; Wu, J.; Biliran, H.; Sarkar, F.H. Perspectives on c-Myc, Cyclin D1, and their interaction in cancer formation, progression, and response to chemotherapy. Crit. Rev. Oncog. 2007, 13, 93–158. [Google Scholar] [CrossRef] [PubMed]
- Prosperi, J.R.; Luu, H.H.; Goss, K.H. Dysregulation of the Wnt pathway in solid tumors. In Targeting the Wnt Pathway in Cancer; Goss, H.K., Kahn, M., Eds.; Springer: New York, NY, USA, 2011; pp. 81–128. [Google Scholar]
- Fearnhead, N.S.; Britton, M.P.; Bodmer, W.F. The ABC of APC. Hum. Mol. Genet. 2001, 10, 721–733. [Google Scholar] [CrossRef] [PubMed]
- Merle, P.; de la Monte, S.; Kim, M.; Herrmann, M.; Tanaka, S.; von dem Bussche, A.; Kew, M.C.; Trepo, C.; Wands, J.R. Functional consequences of Frizzled-7 receptor overexpression in human hepatocellular carcinoma. Gastroenterology 2004, 127, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wu, X.; Wang, Y.; Zhang, K.; Wu, J.; Yuan, Y.C.; Deng, X.; Chen, L.; Kim, C.C.; Lau, S.; et al. FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene 2011, 30, 4437–4446. [Google Scholar] [CrossRef] [PubMed]
- Wissmann, C.; Wild, P.J.; Kaiser, S.; Roepcke, S.; Stoehr, R.; Woenckhaus, M.; Kristiansen, G.; Hsieh, J.C.; Hofstaedter, F.; Hartmann, A.; et al. WIF1, a component of the Wnt pathway, is down-regulated in prostate, breast, lung, and bladder cancer. J. Pathol. 2003, 201, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Yanaga, F.; Kahn, M. Targeting Wnt signaling: Can we safely eradicate cancer stem cells? Clin. Cancer Res. 2010, 16, 3153–3162. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.C.; Lorenzi, M.V. Drug discovery approaches to target Wnt signaling in cancer stem cells. Oncotarget 2010, 1, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Guzman, M.L.; Noble, M. Cancer stem cells. N. Engl. J. Med. 2006, 355, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Blagodatski, A.; Poteryaev, D.; Katanaev, V.L. Targeting the Wnt pathways for therapies. Mol. Cell. Ther. 2014. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry’s grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [PubMed]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Liu, J.O. Recent advances in drug repositioning for the discovery of new anticancer drugs. Int. J. Biol. Sci. 2014, 10, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Guidance for Industry Expedited Programs for Serious Conditions—Drugs and Biologics. Available online: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM358301.pdf (accessed on 26 June 2016).
- Day, R.O.; Graham, G.G. Non-steroidal anti-inflammatory drugs (NSAIDs). BMJ 2013. [Google Scholar] [CrossRef]
- Gurpinar, E.; Grizzle, W.E.; Piazza, G.A. NSAIDs inhibit tumorigenesis, but how? Clin.Cancer Res. 2014, 20, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.J.; Henley, S.J.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst. 2002, 94, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.T.; Ogino, S.; Fuchs, C.S. Aspirin use and survival after diagnosis of colorectal cancer. JAMA 2009, 302, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Dube, C.; Rostom, A.; Lewin, G.; Tsertsvadze, A.; Barrowman, N.; Code, C.; Sampson, M.; Moher, D. The use of aspirin for primary prevention of colorectal cancer: A systematic review prepared for the U.S. Preventive services task force. Ann. Int. Med. 2007, 146, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.M.; Fowkes, F.G.R.; Belch, J.F.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of daily aspirin on long-term risk of death due to cancer: Analysis of individual patient data from randomised trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Jacobs, E.J.; Thun, M.J.; Bain, E.B.; Rodriguez, C.; Henley, S.J.; Calle, E.E. A large cohort study of long-term daily use of adult-strength aspirin and cancer incidence. J. Natl. Cancer Inst. 2007, 99, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Khuder, S.A.; Mutgi, A.B. Breast cancer and NSAIDs use: A meta-analysis. Br. J. Cancer 2001, 84, 1188–1192. [Google Scholar] [CrossRef] [PubMed]
- Yiannakopoulou, E. Aspirin and NSAIDs for breast cancer chemoprevention. Eur. J. Cancer Prev. 2015, 24, 416–421. [Google Scholar] [CrossRef] [PubMed]
- McCormack, V.A.; Hung, R.J.; Brenner, D.R.; Bickeboller, H.; Rosenberger, A.; Muscat, J.E.; Lazarus, P.; Tjonneland, A.; Friis, S.; Christiani, D.C.; et al. Aspirin and NSAID use and lung cancer risk: A pooled analysis in the international lung cancer consortium (ilcco). Cancer Causes Control 2011, 22, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Corley, D.A.; Kerlikowske, K.; Verma, R.; Buffler, P. Protective association of aspirin/ NSAIDs and esophageal cancer: A systematic review and meta-analysis. Gastroenterology 2003, 124, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.C.; Howard, L.E.; Moreira, D.M.; Castro-Santamaria, R.; Andriole, G.L.; Freedland, S.J. Aspirin, nsaids, and risk of prostate cancer: Results from the reduce study. Clin. Cancer Res. 2015, 21, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, S.E.; Pfeiffer, R.M.; Sigurdson, A.J.; Hayes, R.B.; Leitzmann, M.; Schatzkin, A.; Hollenbeck, A.R.; Silverman, D.T. Nonsteroidal antiinflammatory drugs and bladder cancer: A pooled analysis. Am. J. Epidemiol. 2011, 173, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Wan, Y.D.; Sun, Y.L.; Li, J.; Zhu, R.T. Aspirin might reduce the incidence of pancreatic cancer: A meta-analysis of observational studies. Sci. Rep. 2015. [Google Scholar] [CrossRef] [PubMed]
- Castellone, M.D.; Teramoto, H.; Williams, B.O.; Druey, K.M.; Gutkind, J.S. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science 2005, 310, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Jung, C.; Liu, C.; Sheng, H. Prostaglandin E2 stimulates the beta-catenin/T cell factor-dependent transcription in colon cancer. J. Biol. Chem. 2005, 280, 26565–26572. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Dinchuk, J.E.; Kargman, S.L.; Oshima, H.; Hancock, B.; Kwong, E.; Trzaskos, J.M.; Evans, J.F.; Taketo, M.M. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell 1996, 87, 803–809. [Google Scholar] [CrossRef]
- Sheng, H.; Shao, J.; Kirkland, S.C.; Isakson, P.; Coffey, R.J.; Morrow, J.; Beauchamp, R.D.; DuBois, R.N. Inhibition of human colon cancer cell growth by selective inhibition of cyclooxygenase-2. J. Clin. Invest. 1997, 99, 2254–2259. [Google Scholar] [CrossRef] [PubMed]
- Boon, E.M.J.; Keller, J.J.; Wormhoudt, T.A.M.; Giardiello, F.M.; Offerhaus, G.J.A.; van der Neut, R.; Pals, S.T. Sulindac targets nuclear β-catenin accumulation and Wnt signaling in adenomas of patients with familial adenomatous polyposis and in human colorectal cancer cell lines. Br. J. Cancer 2004, 90, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Han, A.; Song, Z.; Tong, C.; Hu, D.; Bi, X.; Augenlicht, L.H.; Yang, W. Sulindac suppresses β-catenin expression in human cancer cells. Eur. J. Pharmacol. 2008, 583, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, H.N.; Gary, B.D.; Keeton, A.B.; Lu, W.; Li, Y.; Piazza, G.A. Inhibition of PDE5 by sulindac sulfide selectively induces apoptosis and attenuates oncogenic Wnt/β-catenin-mediated transcription in human breast tumor cells. Cancer Prev. Res. 2011, 4, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Steinert, G.; Oancea, C.; Roos, J.; Hagemeyer, H.; Maier, T.; Ruthardt, M.; Puccetti, E. Sulindac sulfide reverses aberrant self-renewal of progenitor cells induced by the AML-associated fusion proteins PML/RAR and PLZF/RARalpha. PLoS ONE 2011, 6, e22540. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Tinsley, H.N.; Keeton, A.; Qu, Z.; Piazza, G.A.; Li, Y. Suppression of Wnt/β-catenin signaling inhibits prostate cancer cell proliferation. Eur. J. Pharmacol. 2009, 602, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Wang, N.X.; Shi, D.L.; Zheng, J.J. Sulindac inhibits canonical Wnt signaling by blocking the PDZ domain of the protein Dishevelled. Angew. Chem. 2009, 48, 6448–6452. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Xi, Y.; Tinsley, H.N.; Gurpinar, E.; Gary, B.D.; Zhu, B.; Li, Y.; Chen, X.; Keeton, A.B.; Abadi, A.H.; et al. Sulindac selectively inhibits colon tumor cell growth by activating the cGMP/PKG pathway to suppress Wnt/β-catenin signaling. Mol. Cancer Ther. 2013, 12, 1848–1859. [Google Scholar] [CrossRef] [PubMed]
- Piazza, G.A.; Alberts, D.S.; Hixson, L.J.; Paranka, N.S.; Li, H.; Finn, T.; Bogert, C.; Guillen, J.M.; Brendel, K.; Gross, P.H.; et al. Sulindac sulfone inhibits azoxymethane-induced colon carcinogenesis in rats without reducing prostaglandin levels. Cancer Res. 1997, 57, 2909–2915. [Google Scholar] [PubMed]
- Stein, U.; Arlt, F.; Smith, J.; Sack, U.; Herrmann, P.; Walther, W.; Lemm, M.; Fichtner, I.; Shoemaker, R.H.; Schlag, P.M. Intervening in β-catenin signaling by sulindac inhibits s100a4-dependent colon cancer metastasis. Neoplasia 2011, 13, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Nie, T.; Mackenzie, G.G.; Sun, Y.; Huang, L.; Ouyang, N.; Alston, N.; Zhu, C.; Murray, O.T.; Constantinides, P.P.; et al. The metabolism and pharmacokinetics of phospho-sulindac (OXT-328) and the effect of difluoromethylornithine. Br. J. Pharmacol. 2012, 165, 2152–2166. [Google Scholar] [CrossRef] [PubMed]
- McEntee, M.F.; Chiu, C.H.; Whelan, J. Relationship of beta-catenin and Bcl-2 expression to sulindac-induced regression of intestinal tumors in min mice. Carcinogenesis 1999, 20, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.A.; Skinner, S.A.; Vogiagis, D.; O’Brien, P.E. Inhibition of β-catenin translocation in rodent colorectal tumors: A novel explanation for the protective effect of nonsteroidal antiinflammatory drugs in colorectal cancer. Dig. Dis. Sci. 2001, 46, 2314–2321. [Google Scholar] [CrossRef] [PubMed]
- Kune, G.A.; Kune, S.; Watson, L.F. Colorectal cancer risk, chronic illnesses, operations, and medications: Case control results from the melbourne colorectal cancer study. Cancer Res. 1988, 48, 4399–4404. [Google Scholar] [CrossRef] [PubMed]
- Elwood, P.C.; Gallagher, A.M.; Duthie, G.G.; Mur, L.A.J.; Morgan, G. Aspirin, salicylates, and cancer. Lancet 2009, 373, 1301–1309. [Google Scholar] [CrossRef]
- Dihlmann, S.; Siermann, A.; von Knebel Doeberitz, M. The nonsteroidal anti-inflammatory drugs aspirin and indomethacin attenuate β-catenin/TCF-4 signaling. Oncogene 2001, 20, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Dihlmann, S.; Klein, S.; Doeberitz, M.V. Reduction of beta-catenin/T-cell transcription factor signaling by aspirin and indomethacin is caused by an increased stabilization of phosphorylated β-catenin. Mol. Cancer Ther. 2003, 2, 509–516. [Google Scholar] [PubMed]
- Bos, C.L.; Kodach, L.L.; van den Brink, G.R.; Diks, S.H.; van Santen, M.M.; Richel, D.J.; Peppelenbosch, M.P.; Hardwick, J.C. Effect of aspirin on the Wnt/β-catenin pathway is mediated via protein phosphatase 2a. Oncogene 2006, 25, 6447–6456. [Google Scholar] [CrossRef] [PubMed]
- Ratcliffe, M.J.; Itoh, K.; Sokol, S.Y. A positive role for the PP2A catalytic subunit in Wnt signal transduction. J. Biol. Chem. 2000, 275, 35680–35683. [Google Scholar] [CrossRef] [PubMed]
- Gala, M.K.; Chan, A.T. Molecular pathways: Aspirin and Wnt signaling-a molecularly targeted approach to cancer prevention and treatment. Clin.Cancer Res. 2015, 21, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, N.N.; Dannenberg, A.J.; Mestre, J.; Bilinski, R.T.; Churchill, M.R.; Martucci, C.; Newmark, H.; Bertagnolli, M.M. Aspirin prevents tumors in a murine model of familial adenomatous polyposis. Surgery 1998, 124, 225–231. [Google Scholar] [CrossRef]
- Alfonso, L.; Ai, G.; Spitale, R.C.; Bhat, G.J. Molecular targets of aspirin and cancer prevention. Br. J. Cancer 2014, 111, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.E. Clinical evidence for the use of aspirin in the treatment of cancer. Ecancermedicalscience 2013. [Google Scholar] [CrossRef]
- Stark, L.A.; Reid, K.; Sansom, O.J.; Din, F.V.; Guichard, S.; Mayer, I.; Jodrell, D.I.; Clarke, A.R.; Dunlop, M.G. Aspirin activates the NF-kappaB signaling pathway and induces apoptosis in intestinal neoplasia in twoin vivo models of human colorectal cancer. Carcinogenesis 2007, 28, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Din, F.V.; Theodoratou, E.; Farrington, S.M.; Tenesa, A.; Barnetson, R.A.; Cetnarskyj, R.; Stark, L.; Porteous, M.E.; Campbell, H.; Dunlop, M.G. Effect of aspirin and NSAIDs on risk and survival from colorectal cancer. Gut 2010, 59, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Cerletti, C.; Bonati, M.; del Maschio, A.; Galletti, F.; Dejana, E.; Tognoni, G.; de Gaetano, G. Plasma levels of salicylate and aspirin in healthy volunteers: Relevance to drug interaction on platelet function. J. Lab. Clin. Med. 1984, 103, 869–877. [Google Scholar] [PubMed]
- Grosser, T.; Smyth, E.; FitzGerald, G.A. Chapter 34. Anti-inflammatory, antipyretic, and analgesic agents; pharmacotherapy of gout. In Goodman & Amp; Gilman’s the Pharmacological Basis of Therapeutics, 12e; Brunton, L.L., Chabner, B.A., Knollmann, B.C., Eds.; The McGraw-Hill Companies: New York, NY, USA, 2011. [Google Scholar]
- Smith, M.L.; Hawcroft, G.; Hull, M.A. The effect of non-steroidal anti-inflammatory drugs on human colorectal cancer cells: Evidence of different mechanisms of action. Eur. J. Cancer 2000, 36, 664–674. [Google Scholar] [CrossRef]
- Kapitanovic, S.; Cacev, T.; Antica, M.; Kralj, M.; Cavric, G.; Pavelic, K.; Spaventi, R. Effect of indomethacin on E-cadherin and beta-catenin expression in HT-29 colon cancer cells. Exp. Mol. Pathol. 2006, 80, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Hawcroft, G.; D’Amico, M.; Albanese, C.; Markham, A.F.; Pestell, R.G.; Hull, M.A. Indomethacin induces differential expression of beta-catenin, gamma-catenin and T-cell factor target genes in human colorectal cancer cells. Carcinogenesis 2002, 23, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.A.; Skinner, S.A.; Malcontenti-Wilson, C.; Vogiagis, D.; O’Brien, P.E. Non-steroidal anti-inflammatory drugs with activity against either cyclooxygenase 1 or cyclooxygenase 2 inhibit colorectal cancer in a dmh rodent model by inducing apoptosis and inhibiting cell proliferation. Gut 2001, 48, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Guissou, P.; Cuisinaud, G.; Legheand, J.; Sassard, J. Chronopharmacokinetics of indomethacin in rats. Arzneim. Forsch. 1987, 37, 1034–1037. [Google Scholar]
- Sakoguchi-Okada, N.; Takahashi-Yanaga, F.; Fukada, K.; Shiraishi, F.; Taba, Y.; Miwa, Y.; Morimoto, S.; Iida, M.; Sasaguri, T. Celecoxib inhibits the expression of survivin via the suppression of promoter activity in human colon cancer cells. Biochem. Pharmacol. 2007, 73, 1318–1329. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Yanaga, F.; Yoshihara, T.; Jingushi, K.; Miwa, Y.; Morimoto, S.; Hirata, M.; Sasaguri, T. Celecoxib-induced degradation of T-cell factors-1 and -4 in human colon cancer cells. Biochem. Biophys. Res. Commun. 2008, 377, 1185–1190. [Google Scholar] [CrossRef] [PubMed]
- Tuynman, J.B.; Vermeulen, L.; Boon, E.M.; Kemper, K.; Zwinderman, A.H.; Peppelenbosch, M.P.; Richel, D.J. Cyclooxygenase-2 inhibition inhibits c-met kinase activity and Wnt activity in colon cancer. Cancer Res. 2008, 68, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Sareddy, G.R.; Kesanakurti, D.; Kirti, P.B.; Babu, P.P. Nonsteroidal anti-inflammatory drugs diclofenac and celecoxib attenuates Wnt/β-catenin/TCF signaling pathway in human glioblastoma cells. Neurochem. Res. 2013, 38, 2313–2322. [Google Scholar] [CrossRef] [PubMed]
- Behari, J.; Zeng, G.; Otruba, W.; Thompson, M.D.; Muller, P.; Micsenyi, A.; Sekhon, S.S.; Leoni, L.; Monga, S.P. R-etodolac decreases β-catenin levels along with survival and proliferation of hepatoma cells. J. Hepatol. 2007, 46, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.J.; Pei, L.B.; Zhuang, J.P.; Ji, Y.; Xu, G.P.; Zhang, Z.P.; Li, N.; Yan, J.L. Celecoxib inhibits β-catenin-dependent survival of the human osteosarcoma MG-63 cell line. J. Int. Med. Res. 2010, 38, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Su, Q.; Mo, J.; Fu, X.; Zhang, Y.; Lin, E.H. Celecoxib downregulates CD133 expression through inhibition of the Wnt signaling pathway in colon cancer cells. Cancer Invest. 2013, 31, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Dharmapuri, G.; Doneti, R.; Philip, G.H.; Kalle, A.M. Celecoxib sensitizes imatinib-resistant K562 cells to imatinib by inhibiting MRP1-5, ABCA2 and ABCG2 transporters via Wnt and Ras signaling pathways. Leuk. Res. 2015, 39, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Yoshimi, N.; Hirose, Y.; Hara, A.; Shimizu, M.; Kuno, T.; Katayama, M.; Qiao, Z.; Mori, H. Suppression of occurrence and advancement of β-catenin-accumulated crypts, possible premalignant lesions of colon cancer, by selective cyclooxygenase-2 inhibitor, celecoxib. Jpn. J. Cancer Res. Gann 2001, 92, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Da, L.; Yang, X.; Feng, D.; Yin, R.; Li, M.; Zhang, Z.; Jiang, F.; Xu, L. Celecoxib potentially inhibits metastasis of lung cancer promoted by surgery in mice, via suppression of the PGE2-modulated beta-catenin pathway. Toxicol. Lett. 2014, 225, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Oh, Y.T.; Han, J.H.; Pyo, H. Antitumor enhancement of celecoxib, a selective cyclooxygenase-2 inhibitor, in a lewis lung carcinoma expressing cyclooxygenase-2. J. Exp. Clin. Cancer Res. 2008. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaka, R.; Shibata, M.A.; Morimoto, J.; Tanigawa, N.; Otsuki, Y. Cox-2 inhibitor celecoxib suppresses tumor growth and lung metastasis of a murine mammary cancer. Anticancer Res. 2006, 26, 4245–4254. [Google Scholar] [PubMed]
- Paulson, S.K.; Zhang, J.Y.; Breau, A.P.; Hribar, J.D.; Liu, N.W.; Jessen, S.M.; Lawal, Y.M.; Cogburn, J.N.; Gresk, C.J.; Markos, C.S.; et al. Pharmacokinetics, tissue distribution, metabolism, and excretion of celecoxib in rats. Drug Metab. Dispos. Biol. Fate Chem. 2000, 28, 514–521. [Google Scholar] [PubMed]
- Steinbach, G.; Lynch, P.M.; Phillips, R.K.; Wallace, M.H.; Hawk, E.; Gordon, G.B.; Wakabayashi, N.; Saunders, B.; Shen, Y.; Fujimura, T.; et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N. Engl. J. Med. 2000, 342, 1946–1952. [Google Scholar] [CrossRef] [PubMed]
- Katz, M. Anthelmintics. Drugs 1977, 13, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Hu, Z.; Sun, S.-Y.; Chen, Z.G.; Owonikoko, T.K.; Sica, G.L.; Ramalingam, S.S.; Curran, W.J.; Khuri, F.R.; Deng, X. Niclosamide overcomes acquired resistance to erlotinib through suppression of STAT3 in non-small cell lung cancer. Mol. Cancer Ther. 2013, 12, 2200–2212. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Lu, Z.; Ding, K.; Li, J.; Du, X.; Chen, C.; Sun, X.; Wu, Y.; Zhou, J.; Pan, J. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: Inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer Res. 2010, 70, 2516–2527. [Google Scholar] [CrossRef] [PubMed]
- Tomizawa, M.; Shinozaki, F.; Motoyoshi, Y.; Sugiyama, T.; Yamamoto, S.; Sueishi, M.; Yoshida, T. Niclosamide suppresses hepatoma cell proliferation via the Wnt pathway. Onco Targets Ther. 2013, 6, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Zhang, L.; Zhang, Y.; Chelluri, R.; Boufraqech, M.; Nilubol, N.; Patel, D.; Shen, M.; Kebebew, E. Identification of niclosamide as a novel anticancer agent for adrenocortical carcinoma. Clin. Cancer Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Arend, R.C.; Londoño-Joshi, A.I.; Samant, R.S.; Li, Y.; Conner, M.; Hidalgo, B.; Alvarez, R.D.; Landen, C.N.; Straughn, J.M.; Buchsbaum, D.J. Inhibition of Wnt/β-catenin pathway by niclosamide: A therapeutic target for ovarian cancer. Gynecol. Oncol. 2014, 134, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Wieland, A.; Trageser, D.; Gogolok, S.; Reinartz, R.; Höfer, H.; Keller, M.; Leinhaas, A.; Schelle, R.; Normann, S.; Klaas, L. Anticancer effects of niclosamide in human glioblastoma. Clin. Cancer Res. 2013, 19, 4124–4136. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.-X.; Ding, K.; Wang, C.-Y. Niclosamide, an old antihelminthic agent, demonstrates antitumor activity by blocking multiple signaling pathways of cancer stem cells. Chin. J. Cancer 2012, 31, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Londoño-Joshi, A.I.; Arend, R.C.; Aristizabal, L.; Lu, W.; Samant, R.S.; Metge, B.J.; Hidalgo, B.; Grizzle, W.E.; Conner, M.; Forero-Torres, A. Effect of niclosamide on basal-like breast cancers. Mol. Cancer Ther. 2014, 13, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Lin, C.; Roberts, M.J.; Waud, W.R.; Piazza, G.A.; Li, Y. Niclosamide suppresses cancer cell growth by inducing Wnt co-receptor LRP6 degradation and inhibiting the Wnt/β-catenin pathway. PLoS ONE 2011, 6, e29290. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Wang, J.; Lu, J.; Bond, M.C.; Ren, X.-R.; Lyerly, H.K.; Barak, L.S.; Chen, W. The anti-helminthic niclosamide inhibits Wnt/Frizzled1 signaling. Biochemistry 2009, 48, 10267–10274. [Google Scholar] [CrossRef] [PubMed]
- Osada, T.; Chen, M.; Yang, X.Y.; Spasojevic, I.; Vandeusen, J.B.; Hsu, D.; Clary, B.M.; Clay, T.M.; Chen, W.; Morse, M.A.; et al. Anti-helminth compound niclosamide downregulates Wnt signaling and elicits antitumor responses in tumors with activating APC mutations. Cancer Res. 2011, 71, 4172–4182. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.M.; Ku, H.H.; Liang, Y.C.; Chen, Y.C.; Hwu, Y.M.; Yeh, T.S. The autonomous notch signal pathway is activated by baicalin and baicalein but is suppressed by niclosamide in K562 cells. J. Cell. Biochem. 2009, 106, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.D.; Diering, G.H.; Bidinosti, M.A.; Dalal, K.; Alain, T.; Balgi, A.D.; Forestieri, R.; Nodwell, M.; Rajadurai, C.V.; Gunaratnam, C. Structure-activity analysis of niclosamide reveals potential role for cytoplasmic pH in control of mammalian target of rapamycin complex 1 (mTORC1) signaling. J. Biol. Chem. 2012, 287, 17530–17545. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Xiong, Y.; Yan, Y.; Xia, Y.; Song, X.; Liu, L.; Li, D.; Wang, N.; Zhang, L.; Zhu, Y.; et al. The anthelmintic drug niclosamide induces apoptosis, impairs metastasis and reduces immunosuppressive cells in breast cancer model. PLoS ONE 2014, 9, e85887. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.; Thyssen, J.; Lorke, D. The biology and toxicology of molluscicides, bayluscide. Pharmacol. Ther. 1982, 19, 245–295. [Google Scholar] [CrossRef]
- Voogd, T.E.; Vansterkenburg, E.L.; Wilting, J.; Janssen, L.H. Recent research on the biological activity of suramin. Pharmacol. Rev. 1993, 45, 177–203. [Google Scholar] [PubMed]
- Babokhov, P.; Sanyaolu, A.O.; Oyibo, W.A.; Fagbenro-Beyioku, A.F.; Iriemenam, N.C. A current analysis of chemotherapy strategies for the treatment of human african trypanosomiasis. Pathog. Glob. Health 2013, 107, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.W.; Lui, R.; Fanta, P.; Salmon, S.E. Effects of suramin on in vitro growth of fresh human tumors. J. Natl. Cancer Inst. 1992, 84, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.K. Suramin: An anticancer drug with unique biological effects. Cancer Chemother. Pharmacol. 1993, 32, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, H.; Qu, H.; Zhao, M.; Yuan, B.; Cao, M.; Cui, J. Suramin inhibits cell proliferation in ovarian and cervical cancer by downregulating heparanase expression. Cancer Cell Int. 2015, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Coffey, R.J., Jr.; Leof, E.B.; Shipley, G.D.; Moses, H.L. Suramin inhibition of growth factor receptor binding and mitogenicity in akr-2b cells. J. Cell. Physiol. 1987, 132, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Pesenti, E.; Sola, F.; Mongelli, N.; Grandi, M.; Spreafico, F. Suramin prevents neovascularisation and tumour growth through blocking of basic fibroblast growth factor activity. Br. J. Cancer 1992, 66, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J.; Mayr, U.; Frank, H.; Hombach, V. Suramin is a potent inhibitor of vascular endothelial growth factor. A contribution to the molecular basis of its antiangiogenic action. J. Mol. Cell. Cardiol. 1996, 28, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Rideout, D.C.; Bustamante, A.; Patel, R.; Henderson, G.B. Suramin sodium: Pronounced effects on methotrexate transport and anti-folate activity in cultured tumor cells. Int. J. Cancer 1995, 61, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Koval, A.; Ahmed, K.; Katanaev, V.L. Inhibition of Wnt signaling and breast tumour growth by the multi-purpose drug suramin through suppression of heterotrimeric G proteins and Wnt endocytosis. Biochem. J. 2016, 473, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Koval, A.; Katanaev, V.L. Platforms for high-throughput screening of Wnt/Frizzled antagonists. Drug Discov. Today 2012, 17, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Katanaev, V.L. Prospects of targeting Wnt signaling in cancer. J. Pharmacol. Toxicol. Res. 2014, 1, 1–3. [Google Scholar]
- Koval, A.; Purvanov, V.; Egger-Adam, D.; Katanaev, V.L. Yellow submarine of the Wnt/Frizzled signaling: Submerging from the G protein harbor to the targets. Biochem. Pharmacol. 2011, 82, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Purvanov, V.; Koval, A.; Katanaev, V.L. A direct and functional interaction between Go and Rab5 during G protein-coupled receptor signaling. Sci. Signal. 2010. [Google Scholar] [CrossRef] [PubMed]
- Lustberg, M.B.; Pant, S.; Ruppert, A.S.; Shen, T.; Wei, Y.; Chen, L.; Brenner, L.; Shiels, D.; Jensen, R.R.; Berger, M.; et al. Phase I/II trial of non-cytotoxic suramin in combination with weekly paclitaxel in metastatic breast cancer treated with prior taxanes. Cancer Chemother. Pharmacol. 2012, 70, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Gradishar, W.J.; Soff, G.; Liu, J.; Cisneros, A.; French, S.; Rademaker, A.; Benson Iii, A.B.; Bouck, N. A pilot trial of suramin in metastatic breast cancer to assess antiangiogenic activity in individual patients. Oncology 2000, 58, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Falcone, A.; Pfanner, E.; Cianci, C.; Danesi, R.; Brunetti, I.; Del Tacca, M.; Conte, P.F. Suramin in patients with metastatic colorectal cancer pretreated with fluoropyrimidine-based chemotherapy. A phase II study. Cancer 1995, 75, 440–443. [Google Scholar] [CrossRef]
- Falcone, A.; Pfanner, E.; Brunetti, I.; Allegrini, G.; Lencioni, M.; Galli, C.; Masi, G.; Danesi, R.; Antonuzzo, A.; Del Tacca, M.; et al. Suramin in combination with 5-fluorouracil (5-FU) and leucovorin (LV) in metastatic colorectal cancer patients resistant to 5-FU+LV-based chemotherapy. Tumori 1998, 84, 666–668. [Google Scholar] [PubMed]
- Lam, E.T.; Au, J.L.; Otterson, G.A.; Guillaume Wientjes, M.; Chen, L.; Shen, T.; Wei, Y.; Li, X.; Bekaii-Saab, T.; Murgo, A.J.; et al. Phase I trial of non-cytotoxic suramin as a modulator of docetaxel and gemcitabine therapy in previously treated patients with non-small cell lung cancer. Cancer Chemother. Pharmacol. 2010, 66, 1019–1029. [Google Scholar] [CrossRef] [PubMed]
- Hutson, P.R.; Tutsch, K.D.; Rago, R.; Arzoomanian, R.; Alberti, D.; Pomplun, M.; Church, D.; Marnocha, R.; Cheng, A.L.; Kehrli, N.; et al. Renal clearance, tissue distribution, and CA-125 responses in a phase I trial of suramin. Clin. Cancer Res. 1998, 4, 1429–1436. [Google Scholar] [PubMed]
- Bowden, C.J.; Figg, W.D.; Dawson, N.A.; Sartor, O.; Bitton, R.J.; Weinberger, M.S.; Headlee, D.; Reed, E.; Myers, C.E.; Cooper, M.R. A phase I/II study of continuous infusion suramin in patients with hormone-refractory prostate cancer: Toxicity and response. Cancer Chemother. Pharmacol. 1996, 39, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Figg, W.D.; Cooper, M.R.; Thibault, A.; Headlee, D.; Humphrey, J.; Bergan, R.C.; Reed, E.; Sartor, O. Acute renal toxicity associated with suramin in the treatment of prostate cancer. Cancer 1994, 74, 1612–1614. [Google Scholar] [CrossRef]
- Sridhara, R.; Eisenberger, M.A.; Sinibaldi, V.J.; Reyno, L.M.; Egorin, M.J. Evaluation of prostate-specific antigen as a surrogate marker for response of hormone-refractory prostate cancer to suramin therapy. J. Clin. Oncol. 1995, 13, 2944–2953. [Google Scholar] [PubMed]
- Kaur, M.; Reed, E.; Sartor, O.; Dahut, W.; Figg, W.D. Suramin’s development: What did we learn? Invest. New Drugs 2002, 20, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Ord, J.J.; Streeter, E.; Jones, A.; Le Monnier, K.; Cranston, D.; Crew, J.; Joel, S.P.; Rogers, M.A.; Banks, R.E.; Roberts, I.S.; et al. Phase I trial of intravesical suramin in recurrent superficial transitional cell bladder carcinoma. Br. J. Cancer 2005, 92, 2140–2147. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, E.; Mazzitelli, S.; Fabbri, J.; Rohayem, J.; Ruokolainen, J.; Nykanen, A.; Milani, M.; Pezzullo, M.; Nastruzzi, C.; Bolognesi, M. Delivery of suramin as an antiviral agent through liposomal systems. Chemmedchem 2014, 9, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Sadashiva, M.P.; Basappa, S.; Nanjundaswamy, S.; Li, F.; Manu, K.A.; Sengottuvelan, M.; Prasanna, D.S.; Anilkumar, N.C.; Sethi, G.; Sugahara, K.; et al. Anti-cancer activity of novel dibenzo[b,f]azepine tethered isoxazoline derivatives. BMC Chem. Biol. 2012. [Google Scholar] [CrossRef]
- Baghdiguian, S.; Nickel, P.; Fantini, J. Double screening of suramin derivatives on human colon cancer cells and on neural cells provides new therapeutic agents with reduced toxicity. Cancer Lett. 1991, 60, 213–219. [Google Scholar] [CrossRef]
- McCain, D.F.; Wu, L.; Nickel, P.; Kassack, M.U.; Kreimeyer, A.; Gagliardi, A.; Collins, D.C.; Zhang, Z.Y. Suramin derivatives as inhibitors and activators of protein-tyrosine phosphatases. J. Biol. Chem. 2004, 279, 14713–14725. [Google Scholar] [CrossRef] [PubMed]
- Beck, J.W.; Saavedra, D.; Antell, G.J.; Tejeiro, B. The treatment of pinworm infections in humans (enterobiasis) with pyrvinium chloride and pyrvinium pamoate. Am. J. Trop. Med. Hyg. 1959, 8, 349–352. [Google Scholar] [PubMed]
- Wiegering, A.; Uthe, F.W.; Huttenrauch, M.; Muhling, B.; Linnebacher, M.; Krummenast, F.; Germer, C.T.; Thalheimer, A.; Otto, C. The impact of pyrvinium pamoate on colon cancer cell viability. Int. J. Colorectal. Dis. 2014, 29, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Venerando, A.; Girardi, C.; Ruzzene, M.; Pinna, L.A. Pyrvinium pamoate does not activate protein kinase CK1, but promotes Akt/PKB down-regulation and GSK3 activation. Biochem. J. 2013, 452, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Thorne, C.A.; Hanson, A.J.; Schneider, J.; Tahinci, E.; Orton, D.; Cselenyi, C.S.; Jernigan, K.K.; Meyers, K.C.; Hang, B.I.; Waterson, A.G.; et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol. 2010, 6, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Tomitsuka, E.; Kita, K.; Esumi, H. An anticancer agent, pyrvinium pamoate inhibits the NADH-fumarate reductase system—A unique mitochondrial energy metabolism in tumour microenvironments. J. Biochem. 2012, 152, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Ishii, I.; Hatake, K.; Kasahara, T. Pyrvinium pamoate inhibits proliferation of myeloma/erythroleukemia cells by suppressing mitochondrial respiratory complex I and STAT3. Cancer Lett. 2012, 319, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.H.; Macdonald, J.; Liu, G.; Lee, A.S.; Ly, M.; Davis, T.; Ke, N.; Zhou, D.; Wong-Staal, F.; Li, Q.X. Pyrvinium targets the unfolded protein response to hypoglycemia and its anti-tumor activity is enhanced by combination therapy. PLoS ONE 2008, 3, e3951. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Lei, Y.; Liu, R.; Li, J.; Yuan, K.; Li, Y.; Chen, Y.; Liu, Y.; Lu, Y.; Edwards, C.K., 3rd; et al. Pyrvinium targets autophagy addiction to promote cancer cell death. Cell Death Dis. 2013. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.C.; Kinkel, A.W.; Gryczko, C.M.; Goulet, J.R. Absorption of pyrvinium pamoate. Clin. Pharmacol. Ther. 1976, 19, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.O.; Bolton, E.C.; Huang, Y.; Feau, C.; Guy, R.K.; Yamamoto, K.R.; Hann, B.; Diamond, M.I. Non-competitive androgen receptor inhibition in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 7233–7238. [Google Scholar] [CrossRef] [PubMed]
- Geary, T.G. Ivermectin 20 years on: Maturation of a wonder drug. Trends Parasitol. 2005, 21, 530–532. [Google Scholar] [CrossRef] [PubMed]
- Baraka, O.Z.; Mahmoud, B.M.; Marschke, C.K.; Geary, T.G.; Homeida, M.M.; Williams, J.F. Ivermectin distribution in the plasma and tissues of patients infected with onchocerca volvulus. Eur. J. Clin. Pharmacol. 1996, 50, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Guzzo, C.A.; Furtek, C.I.; Porras, A.G.; Chen, C.; Tipping, R.; Clineschmidt, C.M.; Sciberras, D.G.; Hsieh, J.Y.; Lasseter, K.C. Safety, tolerability, and pharmacokinetics of escalating high doses of ivermectin in healthy adult subjects. J. Clin. Pharmacol. 2002, 42, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Lynagh, T.; Lynch, J.W. Molecular mechanisms of cys-loop ion channel receptor modulation by ivermectin. Front. Mol. Neurosci. 2012. [Google Scholar] [CrossRef] [PubMed]
- Melotti, A.; Mas, C.; Kuciak, M.; Lorente-Trigos, A.; Borges, I.; Ruiz i Altaba, A. The river blindness drug ivermectin and related macrocyclic lactones inhibit Wnt-TCF pathway responses in human cancer. EMBO Mol. Med. 2014, 6, 1263–1278. [Google Scholar] [CrossRef] [PubMed]
- Sharmeen, S.; Skrtic, M.; Sukhai, M.A.; Hurren, R.; Gronda, M.; Wang, X.; Fonseca, S.B.; Sun, H.; Wood, T.E.; Ward, R.; et al. The antiparasitic agent ivermectin induces chloride-dependent membrane hyperpolarization and cell death in leukemia cells. Blood 2010, 116, 3593–3603. [Google Scholar] [CrossRef] [PubMed]
- Dadarkar, S.S.; Deore, M.D.; Gatne, M.M. Comparative evaluation of acute toxicity of ivermectin by two methods after single subcutaneous administration in rats. Regul. Toxicol. Pharmacol. 2007, 47, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wang, F.; Dai, W.-Q.; Wu, D.; Lin, C.-L.; Wu, S.-M.; Cheng, P.; Zhang, Y.; Shen, M.; Wang, C.-F.; et al. Mechanism of action of salinomycin on growth and migration in pancreatic cancer cell lines. Pancreatology 2013, 13, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Kusunoki, S.; Kato, K.; Tabu, K.; Inagaki, T.; Okabe, H.; Kaneda, H.; Suga, S.; Terao, Y.; Taga, T.; Takeda, S. The inhibitory effect of salinomycin on the proliferation, migration and invasion of human endometrial cancer stem-like cells. Gynecol. Oncol. 2013, 129, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Choi, M.Y.; Yu, J.; Castro, J.E.; Kipps, T.J.; Carson, D.A. Salinomycin inhibits Wnt signaling and selectively induces apoptosis in chronic lymphocytic leukemia cells. Proc. Natl. Acad. Sci. USA 2011, 108, 13253–13257. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Li, Y. Salinomycin suppresses LRP6 expression and inhibits both Wnt/beta-catenin and mTORC1 signaling in breast and prostate cancer cells. J. Cell. Biochem. 2014, 115, 1799–1807. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.L.; Zhao, Z.Q.; Li, J.C.; Liang, Y.; Yin, J.Q.; Zou, C.Y.; Xie, X.B.; Zeng, Y.X.; Shen, J.N.; Kang, T.; et al. Salinomycin inhibits osteosarcoma by targeting its tumor stem cells. Cancer Lett. 2011, 311, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; He, L.; Dai, W.Q.; Xu, Y.P.; Wu, D.; Lin, C.L.; Wu, S.M.; Cheng, P.; Zhang, Y.; Shen, M.; et al. Salinomycin inhibits proliferation and induces apoptosis of human hepatocellular carcinoma cells in vitro andin vivo. PLoS ONE 2012, 7, e50638. [Google Scholar]
- Wu, D.; Zhang, Y.; Huang, J.; Fan, Z.; Shi, F.; Wang, S. Salinomycin inhibits proliferation and induces apoptosis of human nasopharyngeal carcinoma cell in vitro and suppresses tumor growthin vivo. Biochem. Biophys. Res. Commun. 2014, 443, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Fan, S.; Ma, W.; Fan, P.; Wang, B.; Zhang, J.; Wang, H.; Tang, B.; Zhang, Q.; Yu, X.; et al. Roles of Wnt/β-catenin signaling in the gastric cancer stem cells proliferation and salinomycin treatment. Cell Death Dis. 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, C.; Xue, F.; Chen, W.; Zhi, X.; Feng, X.; Bai, X.; Liang, T. Salinomycin decreases doxorubicin resistance in hepatocellular carcinoma cells by inhibiting the beta-catenin/TCF complex association via FOXO3a activation. Oncotarget 2015, 6, 10350–10365. [Google Scholar] [CrossRef] [PubMed]
- Naujokata, C.; Lauferc, S. Targeting cancer stem cells with defined compounds and drugs. J. Cancer Res. Updates 2013, 2, 36–67. [Google Scholar] [CrossRef]
- Lagas, J.S.; Sparidans, R.W.; van Waterschoot, R.A.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. P-glycoprotein limits oral availability, brain penetration, and toxicity of an anionic drug, the antibiotic salinomycin. Antimicrob. Agents Chemother. 2008, 52, 1034–1039. [Google Scholar] [CrossRef] [PubMed]
- Naujokat, C.; Steinhart, R. Salinomycin as a drug for targeting human cancer stem cells. J. Biomed. Biotechnol. 2012. [Google Scholar] [CrossRef] [PubMed]
- Koval, A.V.; Vlasov, P.; Shichkova, P.; Khunderyakova, S.; Markov, Y.; Panchenko, J.; Volodina, A.; Kondrashov, F.A.; Katanaev, V.L. Anti-leprosy drug clofazimine inhibits growth of triple-negative breast cancer cells via inhibition of canonical Wnt signaling. Biochem. Pharmacol. 2014, 87, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Van Rensburg, C.E.; van Staden, A.M.; Anderson, R. The riminophenazine agents clofazimine and B669 inhibit the proliferation of cancer cell lines in vitro by phospholipase A2-mediated oxidative and nonoxidative mechanisms. Cancer Res. 1993, 53, 318–323. [Google Scholar] [PubMed]
- Sri-Pathmanathan, R.M.; Plumb, J.A.; Fearon, K.C. Clofazimine alters the energy metabolism and inhibits the growth rate of a human lung-cancer cell line in vitro and in vivo. Int. J. Cancer 1994, 56, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Nix, D.E.; Adam, R.D.; Auclair, B.; Krueger, T.S.; Godo, P.G.; Peloquin, C.A. Pharmacokinetics and relative bioavailability of clofazimine in relation to food, orange juice and antacid. Tuberculosis 2004, 84, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Vanrensburg, C.; Durandt, C.; Garlinski, P.; Osullivan, J. Evaluation of the antineoplastic activities of the riminophenazine agents clofazimine and B669 in tumor-bearing rats and mice. Int. J. Oncol. 1993, 3, 1011–1013. [Google Scholar] [CrossRef] [PubMed]
- Ruff, P.; Chasen, M.R.; Long, J.E.; van Rensburg, C.E. A phase II study of oral clofazimine in unresectable and metastatic hepatocellular carcinoma. Ann. Oncol. 1998, 9, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Falkson, C.I.; Falkson, G. A phase II evaluation of clofazimine plus doxorubicin in advanced, unresectable primary hepatocellular carcinoma. Oncology 1999, 57, 232–235. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, R.; O’Sullivan, J.F.; O’Kennedy, R. The pharmacology, metabolism, and chemistry of clofazimine. Drug Metab. Rev. 1995, 27, 591–614. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jiao, S.; Li, X.; Banu, H.; Hamal, S.; Wang, X. Therapeutic effects of antibiotic drug tigecycline against cervical squamous cell carcinoma by inhibiting Wnt/β-catenin signaling. Biochem. Biophys. Res. Commun. 2015, 467, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Bolzan, A.D.; Bianchi, M.S. Genotoxicity of streptonigrin: A review. Mutat. Res. 2001, 488, 25–37. [Google Scholar] [CrossRef]
- Harris, M.N.; Medrek, T.J.; Golomb, F.M.; Gumport, S.L.; Postel, A.H.; Wright, J.C. Chemotherapy with streptonigrin in advanced cancer. Cancer 1965, 18, 49–57. [Google Scholar] [CrossRef]
- Park, S.; Chun, S. Streptonigrin inhibits β-catenin/tcf signaling and shows cytotoxicity in beta-catenin-activated cells. Biochim. Biophys. Acta 2011, 1810, 1340–1345. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Gwak, J.; Cho, M.; Song, T.; Won, J.; Kim, D.E.; Shin, J.G.; Oh, S. Hexachlorophene inhibits Wnt/β-catenin pathway by promoting Siah-mediated β-catenin degradation. Mol. Pharmacol. 2006, 70, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Min, H.J.; Cho, I.R.; Srisuttee, R.; Park, E.H.; Cho, D.H.; Ahn, J.H.; Lee, I.S.; Johnston, R.N.; Oh, S.; Chung, Y.H. Hexachlorophene suppresses β-catenin expression by up-regulation of Siah-1 in EBV-infected B lymphoma cells. Cancer Lett. 2009, 276, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Bowker, S.L.; Majumdar, S.R.; Veugelers, P.; Johnson, J.A. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care 2006, 29, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.; Zhang, S.; Zhang, X.; Wang, P.; Hao, X.; Zhang, J. Clinical pathological characteristics and prognostic analysis of 1,013 breast cancer patients with diabetes. Breast Cancer Res. Treat. 2013, 137, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Kwan, H.T.; Chan, D.W.; Cai, P.C.H.; Mak, C.S.L.; Yung, M.M.H.; Leung, T.H.Y.; Wong, O.G.W.; Cheung, A.N.Y.; Ngan, H.Y.S. Ampk activators suppress cervical cancer cell growth through inhibition of Dvl3 mediated Wnt/β-catenin signaling activity. PLoS ONE 2013, 8, e53597. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.C.; Myers, R.; Li, Y.; Chen, Y.L.; Shen, X.L.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Chae, Y.K.; Arya, A.; Malecek, M.K.; Shin, D.S.; Carneiro, B.; Chandra, S.; Kaplan, J.; Kalyan, A.; Altman, J.K.; Platanias, L.; et al. Repurposing metformin for cancer treatment: Current clinical studies. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Eikawa, S.; Nishida, M.; Mizukami, S.; Yamazaki, C.; Nakayama, E.; Udono, H. Immune-mediated antitumor effect by type 2 diabetes drug, metformin. Proc. Natl. Acad. Sci. USA 2015, 112, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Ashinuma, H.; Takiguchi, Y.; Kitazono, S.; Kitazono-Saitoh, M.; Kitamura, A.; Chiba, T.; Tada, Y.; Kurosu, K.; Sakaida, E.; Sekine, I.; et al. Antiproliferative action of metformin in human lung cancer cell lines. Oncol. Rep. 2012, 28, 8–14. [Google Scholar] [PubMed]
- Gong, J.; Robbins, L.A.; Lugea, A.; Waldron, R.T.; Jeon, C.Y.; Pandol, S.J. Diabetes, pancreatic cancer, and metformin therapy. Front. Physiol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Gong, J.; Iwama, H.; Kitanaka, A.; Tani, J.; Miyoshi, H.; Nomura, K.; Mimura, S.; Kobayashi, M.; Aritomo, Y.; et al. The antidiabetic drug metformin inhibits gastric cancer cell proliferation in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Shank, J.J.; Yang, K.; Ghannam, J.; Cabrera, L.; Johnston, C.J.; Reynolds, R.K.; Buckanovich, R.J. Metformin targets ovarian cancer stem cells in vitro and in vivo. Gynecol. Oncol. 2012, 127, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Chen, J.; Yi, G.; Deng, M.; Liu, H.; Liang, M.; Shi, B.; Fu, X.; Chen, Y.; Chen, L.; et al. Metformin suppresses hypoxia-induced stabilization of HIF-1α through reprogramming of oxygen metabolism in hepatocellular carcinoma. Oncotarget 2016, 7, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Pardanani, A.; Tefferi, A. Imatinib targets other than bcr/abl and their clinical relevance in myeloid disorders. Blood 2004, 104, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.S.; Kremenevskaja, N.; von Wasielewski, R.; Jakubcakova, V.; Kant, S.; Resch, J.; Brabant, G. Wnt/β-catenin signaling mediates antineoplastic effects of imatinib mesylate (gleevec) in anaplastic thyroid cancer. J. Clin. Endocrinol. Metab. 2006, 91, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; An, N.; Haydon, R.C.; Zhou, Q.; Cheng, H.; Peng, Y.; Jiang, W.; Luu, H.H.; Vanichakarn, P.; Szatkowski, J.P.; et al. Tyrosine kinase inhibitor STI-571/gleevec down-regulates the β-catenin signaling activity. Cancer Lett. 2003, 193, 161–170. [Google Scholar] [CrossRef]
- Lu, D.; Liu, J.X.; Endo, T.; Zhou, H.; Yao, S.; Willert, K.; Schmidt-Wolf, I.G.; Kipps, T.J.; Carson, D.A. Ethacrynic acid exhibits selective toxicity to chronic lymphocytic leukemia cells by inhibition of the Wnt/β-catenin pathway. PLoS ONE 2009, 4, e8294. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Kim, Y.; Gast, S.M.; Endo, T.; Lu, D.; Carson, D.; Schmidt-Wolf, I.G. Increased in vivo efficacy of lenalidomide and thalidomide by addition of ethacrynic acid. In Vivo 2011, 25, 325–333. [Google Scholar] [PubMed]
- Kim, Y.; Gast, S.M.; Endo, T.; Lu, D.; Carson, D.; Schmidt-Wolf, I.G. In vivo efficacy of the diuretic agent ethacrynic acid against multiple myeloma. Leuk. Res. 2012, 36, 598–600. [Google Scholar] [CrossRef] [PubMed]
- Lacreta, F.P.; Brennan, J.M.; Nash, S.L.; Comis, R.L.; Tew, K.D.; O’Dwyer, P.J. Pharmakokinetics and bioavailability study of ethacrynic acid as a modulator of drug resistance in patients with cancer. J. Pharmacol. Exp. Ther. 1994, 270, 1186–1191. [Google Scholar] [PubMed]
- Webster, M.R.; Weeraratna, A.T. A Wnt-er migration: The confusing role of β-catenin in melanoma metastasis. Sci. Signal. 2013. [Google Scholar] [CrossRef] [PubMed]
- Chien, A.J.; Moore, E.C.; Lonsdorf, A.S.; Kulikauskas, R.M.; Rothberg, B.G.; Berger, A.J.; Major, M.B.; Hwang, S.T.; Rimm, D.L.; Moon, R.T. Activated Wnt/β-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc. Natl. Acad. Sci. USA 2009, 106, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Biechele, T.L.; Camp, N.D.; Fass, D.M.; Kulikauskas, R.M.; Robin, N.C.; White, B.D.; Taraska, C.M.; Moore, E.C.; Muster, J.; Karmacharya, R.; et al. Chemical-genetic screen identifies riluzole as an enhancer of Wnt/β-catenin signaling in melanoma. Chem. Biol. 2010, 17, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Yip, D.; Le, M.N.; Chan, J.L.; Lee, J.H.; Mehnert, J.A.; Yudd, A.; Kempf, J.; Shih, W.J.; Chen, S.; Goydos, J.S. A phase 0 trial of riluzole in patients with resectable stage III and IV melanoma. Clin.Cancer Res. 2009, 15, 3896–3902. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Loh, K.M.; Nusse, R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014. [Google Scholar] [CrossRef] [PubMed]
- Novac, N. Challenges and opportunities of drug repositioning. Trends Pharmacol. Sci. 2013, 34, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Mook, R.A., Jr.; Wang, J.; Ren, X.R.; Chen, M.; Spasojevic, I.; Barak, L.S.; Lyerly, H.K.; Chen, W. Structure-activity studies of Wnt/β-catenin inhibition in the niclosamide chemotype: Identification of derivatives with improved drug exposure. Bioorg. Med. Chem. 2015, 23, 5829–5838. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Lu, D.; Yao, S.; Wu, C.C.; Liu, J.X.; Carson, D.A.; Cottam, H.B. Amide derivatives of ethacrynic acid: Synthesis and evaluation as antagonists of Wnt/β-catenin signaling and cll cell survival. Bioorg. Med. Chem. Lett. 2009, 19, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Huczynski, A.; Janczak, J.; Antoszczak, M.; Wietrzyk, J.; Maj, E.; Brzezinski, B. Antiproliferative activity of salinomycin and its derivatives. Bioorg. Med. Chem. Lett. 2012, 22, 7146–7150. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Borgstrom, B.; Kempengren, S.; Persson, L.; Hegardt, C.; Strand, D.; Oredsson, S. Breast cancer stem cell selectivity of synthetic nanomolar-active salinomycin analogs. BMC Cancer 2016. [Google Scholar] [CrossRef] [PubMed]
- Goluboff, E.T.; Shabsigh, A.; Saidi, J.A.; Weinstein, I.B.; Mitra, N.; Heitjan, D.; Piazza, G.A.; Pamukcu, R.; Buttyan, R.; Olsson, C.A. Exisulind (sulindac sulfone) suppresses growth of human prostate cancer in a nude mouse xenograft model by increasing apoptosis. Urology 1999, 53, 440–445. [Google Scholar] [CrossRef]
- Piazza, G.A.; Keeton, A.B.; Tinsley, H.N.; Gary, B.D.; Whitt, J.D.; Mathew, B.; Thaiparambil, J.; Coward, L.; Gorman, G.; Li, Y.; et al. A novel sulindac derivative that does not inhibit cyclooxygenases but potently inhibits colon tumor cell growth and induces apoptosis with antitumor activity. Cancer Prev. Res. 2009, 2, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Rigas, B. Novel agents for cancer prevention based on nitric oxide. Biochem. Soc. Trans. 2007, 35, 1364–1368. [Google Scholar] [CrossRef] [PubMed]
- Rigas, B.; Tsioulias, G.J. The evolving role of nonsteroidal anti-inflammatory drugs in colon cancer prevention: A cause for optimism. J. Pharmacol. Exp. Ther. 2015, 353, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Gerhart, J. 1998 warkany lecture: Signaling pathways in development. Teratology 1999, 60, 226–239. [Google Scholar] [CrossRef]
- Sekulic, A.; von Hoff, D. Hedgehog pathway inhibition. Cell 2016. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Clevers, H. Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 997–1014. [Google Scholar] [CrossRef] [PubMed]
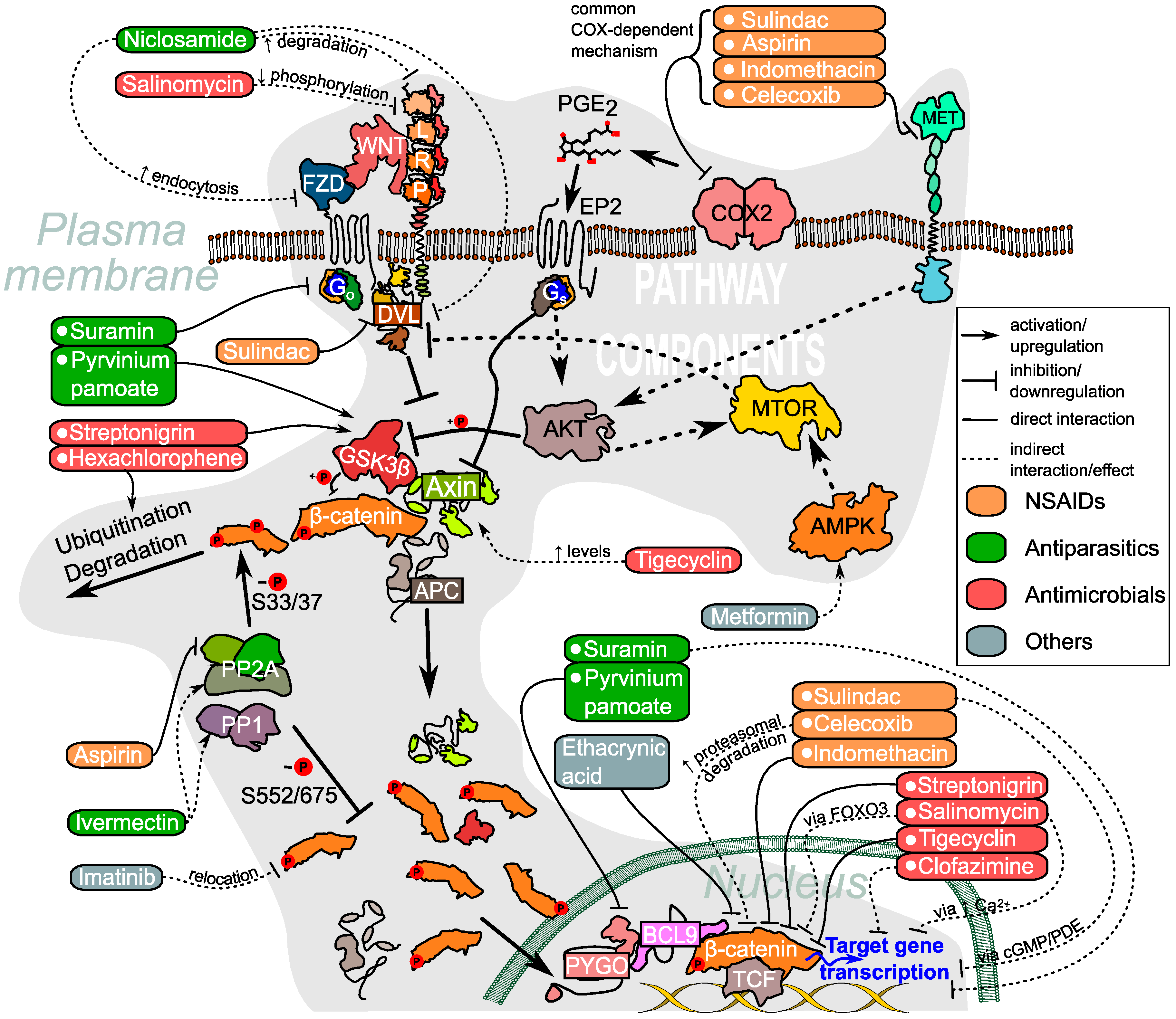

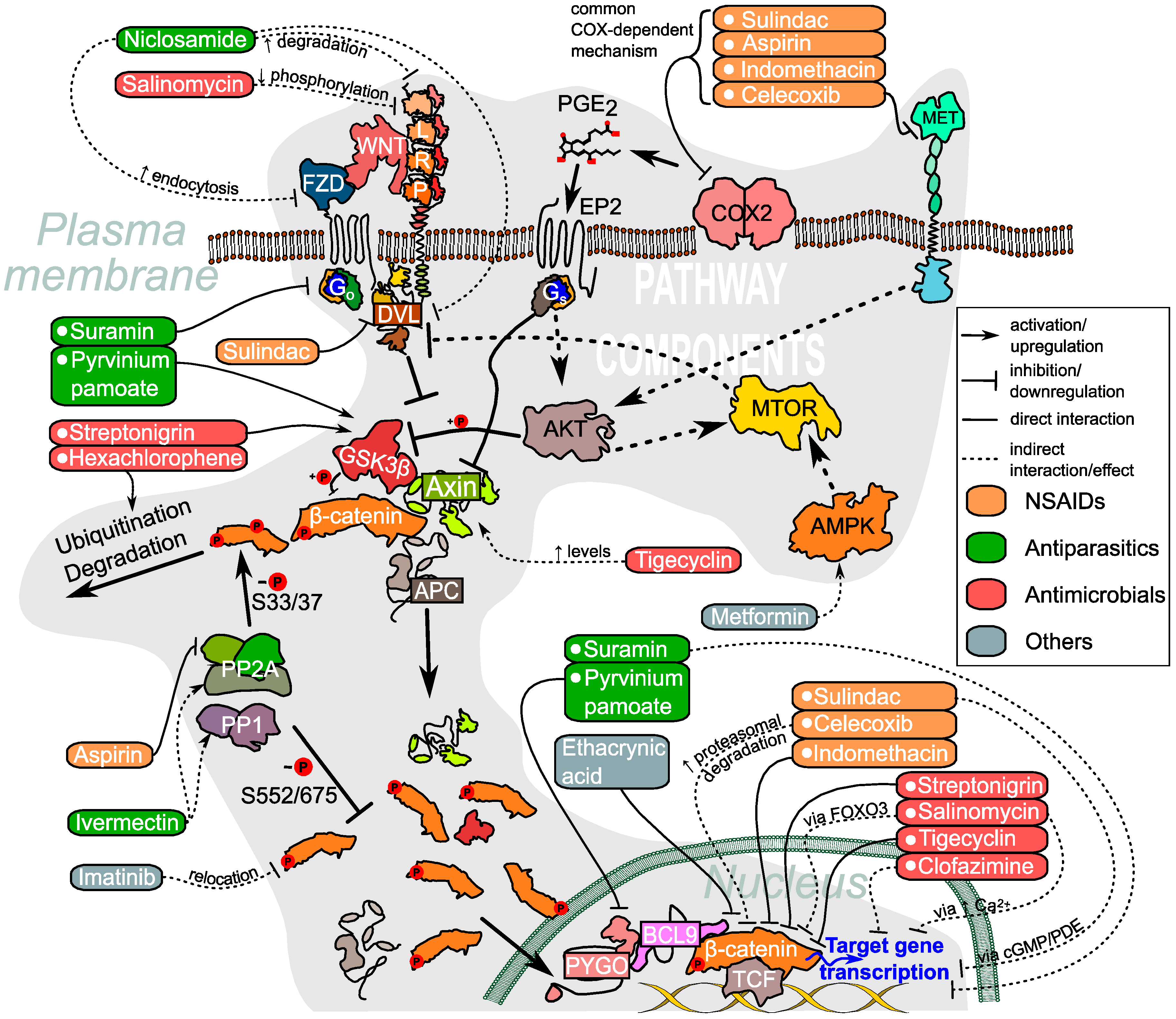{kind=link}
| Drug Category | Drug Name | Mode(-s) of Action of WNT Inhibition | Outcome in Vitro | Outcome in Animal Models | Clinical Results |
|---|---|---|---|---|---|
| NSAID | Sulindac |
| Inhibition of proliferation in breast, lung and colon cancer cell lines | Reduction in tumor growth and metastasis in colon (xenograft and chemically-induced and intestine) while decreasing β-catenin levels | Reduction in β-catenin nuclear staining of adenomas in familial adenomatous polyposis (FAP) patients treated for 6 months |
| |||||
| |||||
| Aspirin |
| Proliferation inhibition in virtually any WNT-dependent cancer | Decreased tumor formation in FAP murine model with concomitant decrease in tumor β-catenin levels | Retrospective studies, especially for colon cancer prevention | |
| Multiple trials for combination therapy and chemoprevention | ||||
| Recommended for CRC prevention in people between 50–69 years old | ||||
| Indomethacin |
| Inhibition of growth in colorectal cancer cell lines | Reduced tumor burden in chemically induced colon cancer; reduced β-catenin nuclear staining | No data available yet | |
| |||||
| |||||
| Celecoxib |
| Impaired proliferation in colorectal cancer, hepatoma, osteosarcoma, glioblastoma and prostate cells lines; Reduction of CD133+ colon cancer stem cells; sensitization of imatinib-resistant leukaemia cells | Inhibition of β-catenin-positive premalignant lesions in the mice colon and in rat colon cancer model | Reduction of polyps in FAP patients after 6 months of treatment | |
| Prevention of lung cancer metastasis in mice | FDA approval for the prevention of cancer in FAP patients retracted due to lacking proof of clinical benefit | |||
| Suppression of mammary carcinoma and Lewis lung tumor | ||||
| Antiparasitics | Niclosamide |
| WNT pathway inhibition is associated with reduction of cell numbers in osteosarcoma, colorectal, breast and lung cancer cell lines; also effective against hepatoma, glioblastoma, andrenocortical and ovarian cancers | Lowers β-catenin levels in mice models of colorectal and basal-like breast cancers | No data available yet |
| |||||
| |||||
| Suramin |
| Tested and found effective against virtually all WNT-dependent in vitro cancer models | Extensive record of in vivo studies involving WNT-dependent cancers | Enrolled in multiple trials; mildly effective or ineffective in a combination therapy against breast cancer; reported multiple toxicities when used in doses comparable to WNT-inhibitory ones | |
| |||||
| Pyrvinium pamoate |
| Efficient against colon cancer | Inhibits tumor growth in colon cancer model | No data available yet | |
| |||||
| |||||
| Ivermectin |
| Anti-proliferative for colon (including stem cells) and lung cancers | Reduction of tumor growth in the xenograft models of the colon cancer with reduced WNT markers levels in the tumors | No data available yet | |
| Antimicrobials | Salinomycin |
| Reduction of cancer stem cells in osteosarcoma and breast and endometrial cancers. Anti-proliferative for many WNT-dependent cancer cell lines, e.g., hepatocellular carcinoma, CLL, pancreatic, nasopharyngeal, breast and prostate cancers. | Inhibition of growth of gastric tumors, osteosarcoma, hepatocellular carcinoma and nasopharyngeal carcinoma with signatures of WNT signaling deficiency (reduction of LRP-6 and β-catenin; decreased GSK3β phosphorylation) | Clinical uncontrolled pilot study on several cases with metastatic cancers with positive dynamics such as metastasis regression observed. Minor acute toxicity reported (tachycardia and mild tremors) |
| |||||
| |||||
| Clofazimine |
| Growth inhibition of squamous hepatocellular carcinoma and lung cancer | Growth inhibition of lung and mammary cancer growth | Several combination and monotherapy studies on hepatocellular carcinoma with mild positive results. | |
| Tigecyclin |
| Cervical cancer cell growth inhibition | Cervical cancer xenografts growth inhibition | No data available yet | |
| |||||
| Streptonigrin |
| Growth inhibition of β-catenin-dependent colorectal and gastric cancer cell lines | No data available yet | No data available yet | |
| |||||
| Hexachlorophene |
| Inhibition of colon cancer and B lymphoma cells growth | No data available yet | No data available yet | |
| Others | Metformin |
| Anti-proliferative in lung, pancreatic, gastric cancer, hepatoma and ovarian cancers | Inhibit tumor growth in hepatocellular carcinoma and ovarian xenografts | Retrospective study of more than 5000 breast cancer patients showing clear survival benefits |
| 55 trial launched, no conclusive data yet | |||||
| Imatinib |
| Anti-proliferative in thyroid carcinoma cells and colon cancer | No in vivo WNT effects were reported yet | Approved for use for multiple cancers | |
| |||||
| Ethacrynic acid |
| Anti-proliferative in CLL and myeloma cells | Reduced tumor growth for myeloma in mice | No data available yet | |
| |||||
| Riluzole |
| Induces melanoma cells differentiation and reduces proliferation | Inhibits metastases | A pilot study assessed safety and efficacy of the compound through biomarkers (pERK and pAKT). |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmed, K.; Shaw, H.V.; Koval, A.; Katanaev, V.L. A Second WNT for Old Drugs: Drug Repositioning against WNT-Dependent Cancers. Cancers 2016, 8, 66. https://doi.org/10.3390/cancers8070066
Ahmed K, Shaw HV, Koval A, Katanaev VL. A Second WNT for Old Drugs: Drug Repositioning against WNT-Dependent Cancers. Cancers. 2016; 8(7):66. https://doi.org/10.3390/cancers8070066
Chicago/Turabian StyleAhmed, Kamal, Holly V. Shaw, Alexey Koval, and Vladimir L. Katanaev. 2016. "A Second WNT for Old Drugs: Drug Repositioning against WNT-Dependent Cancers" Cancers 8, no. 7: 66. https://doi.org/10.3390/cancers8070066
APA StyleAhmed, K., Shaw, H. V., Koval, A., & Katanaev, V. L. (2016). A Second WNT for Old Drugs: Drug Repositioning against WNT-Dependent Cancers. Cancers, 8(7), 66. https://doi.org/10.3390/cancers8070066