
In Hyperthermia Increased ERK and WNT Signaling Suppress Colorectal Cancer Cell Growth


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
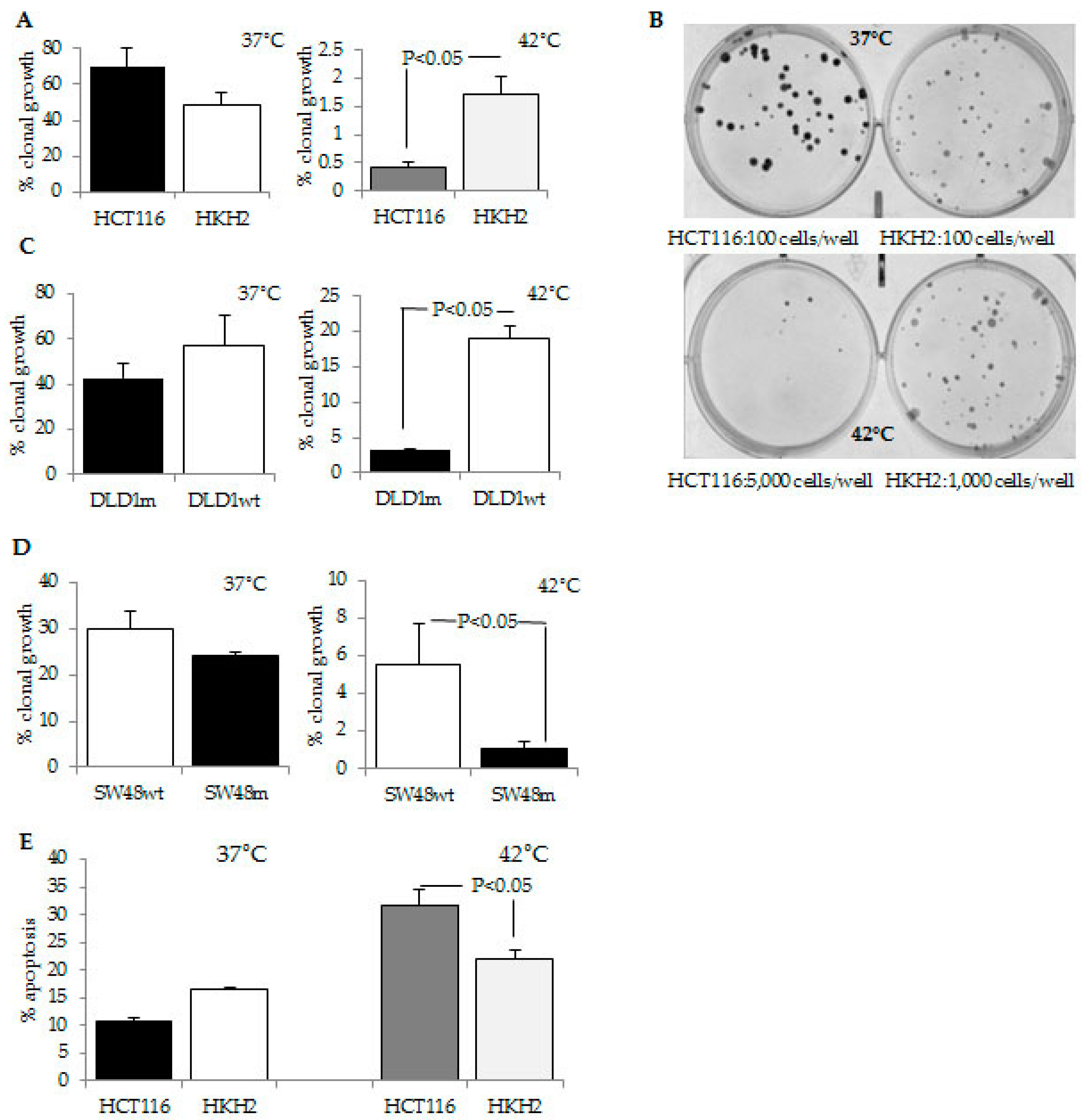
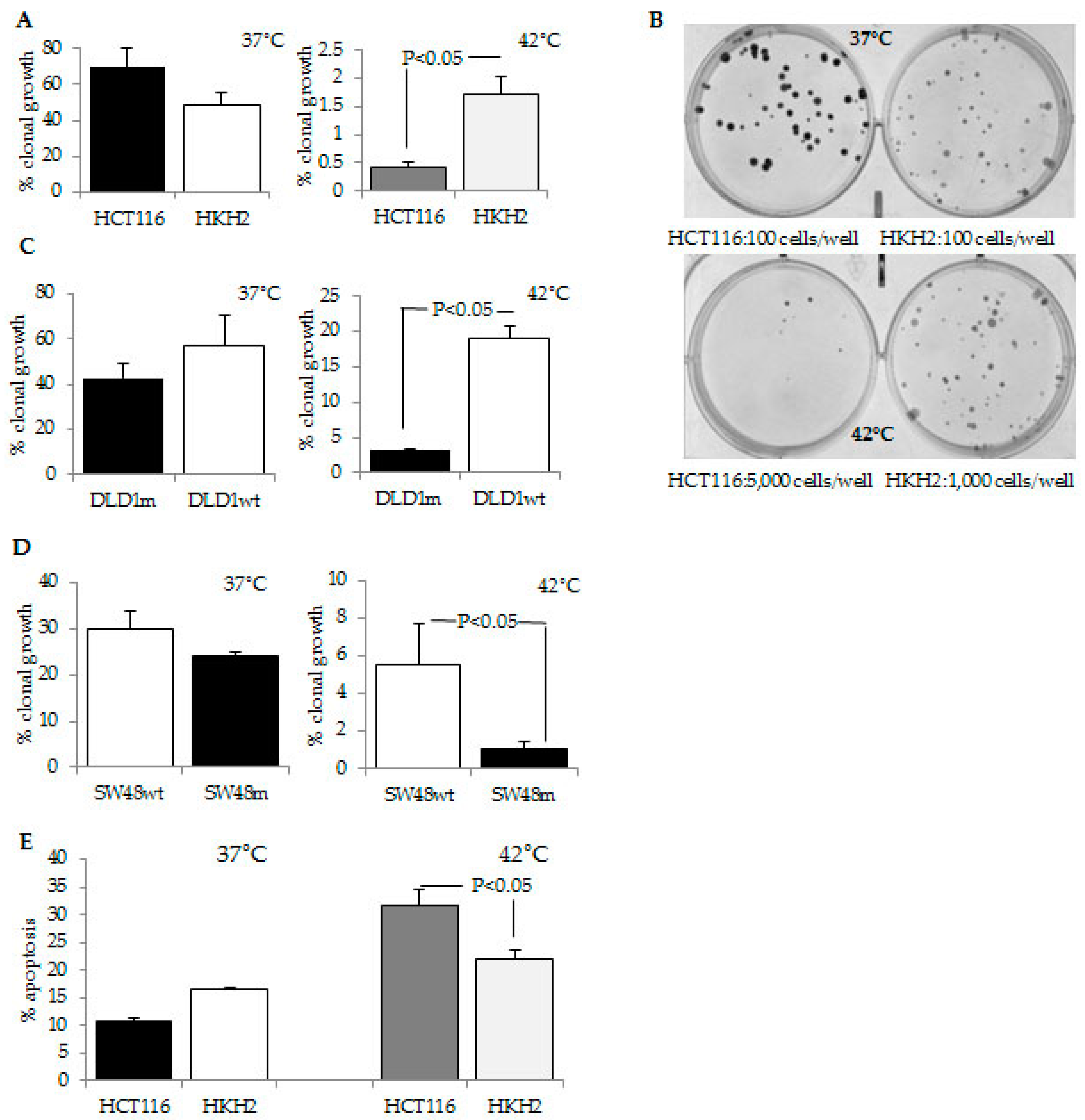
2.1. CRC Cells with a Mutant KRAS Are More Sensitive to Hyperthermia Than CRC Cells with a Wild Type KRAS
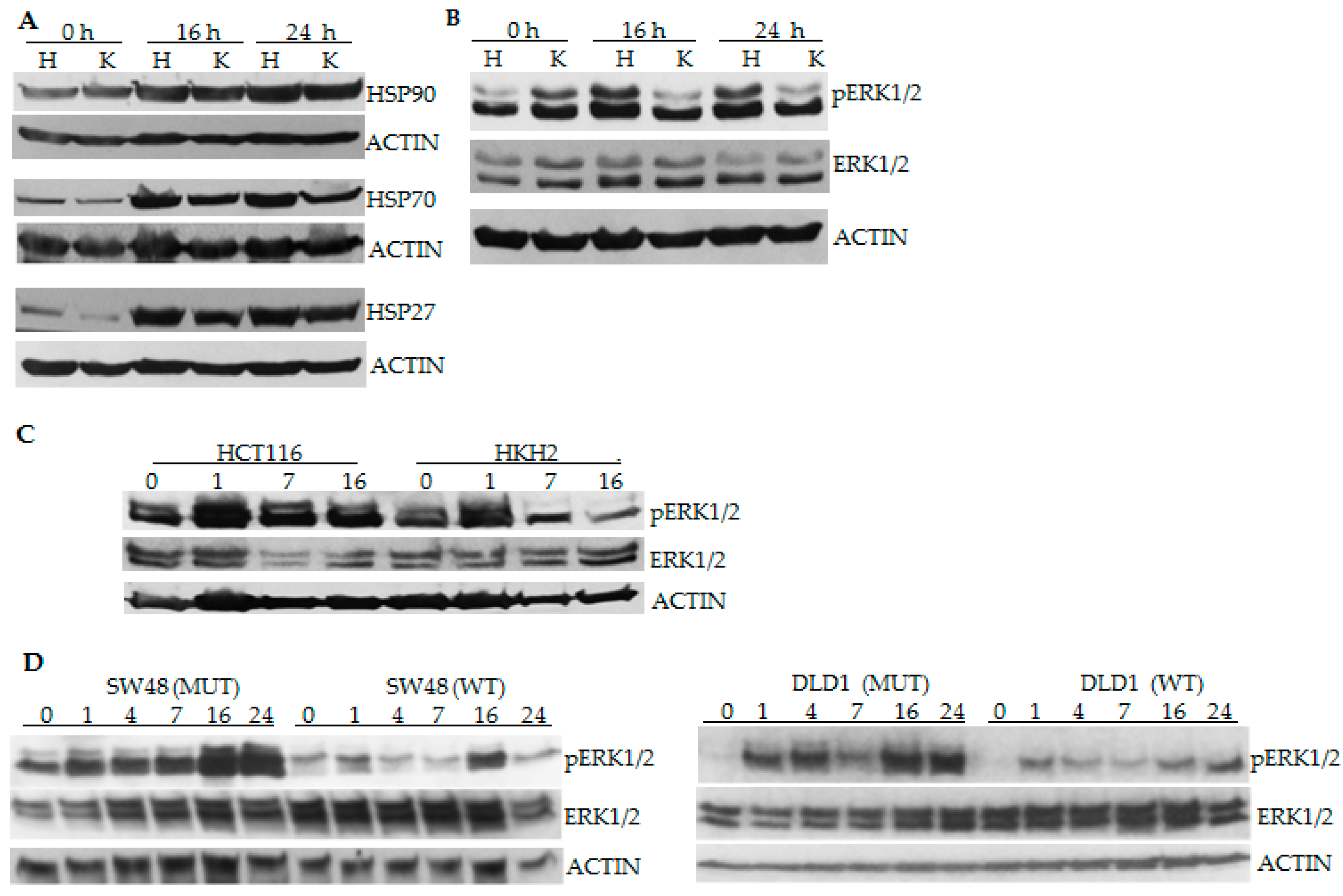
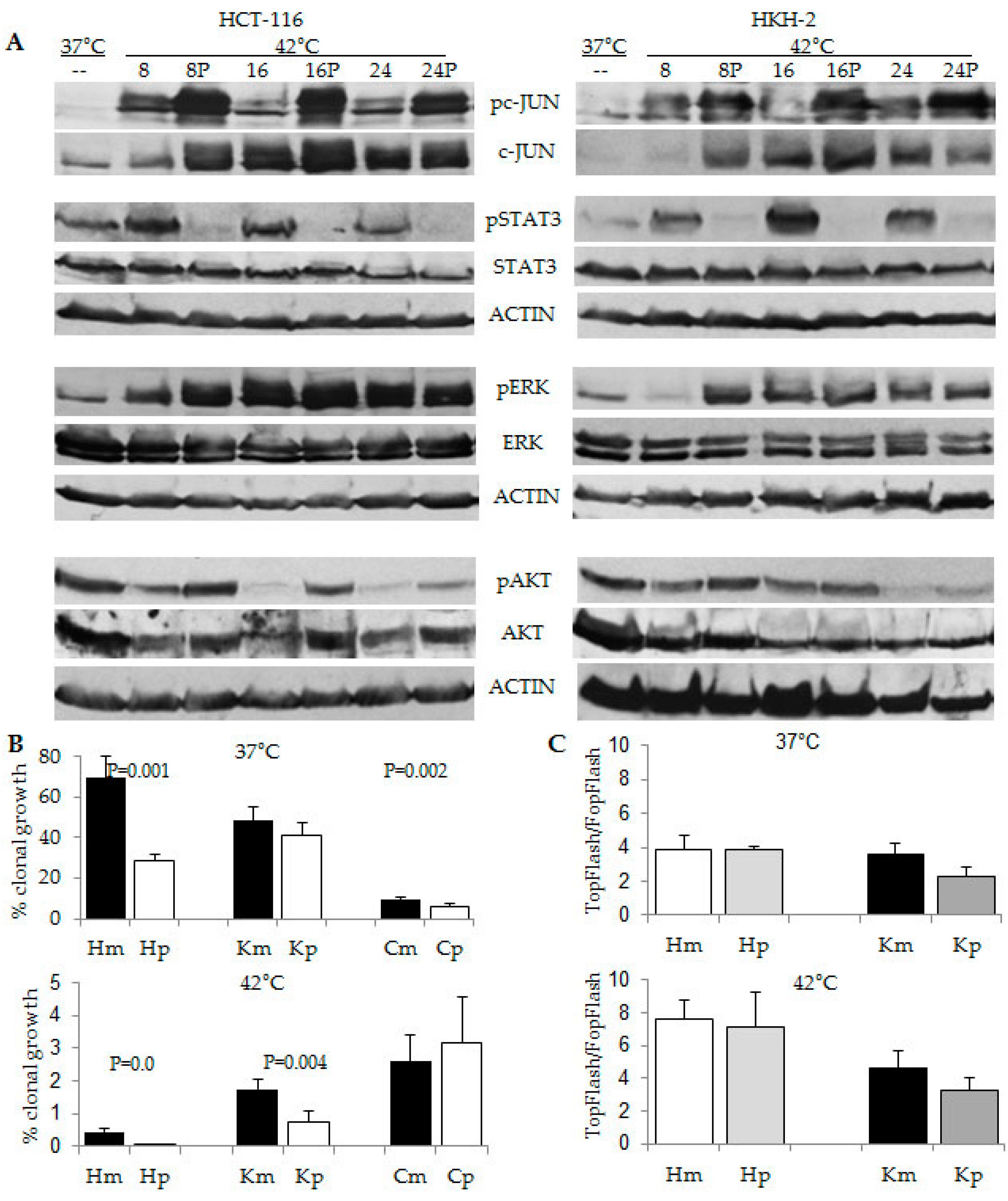
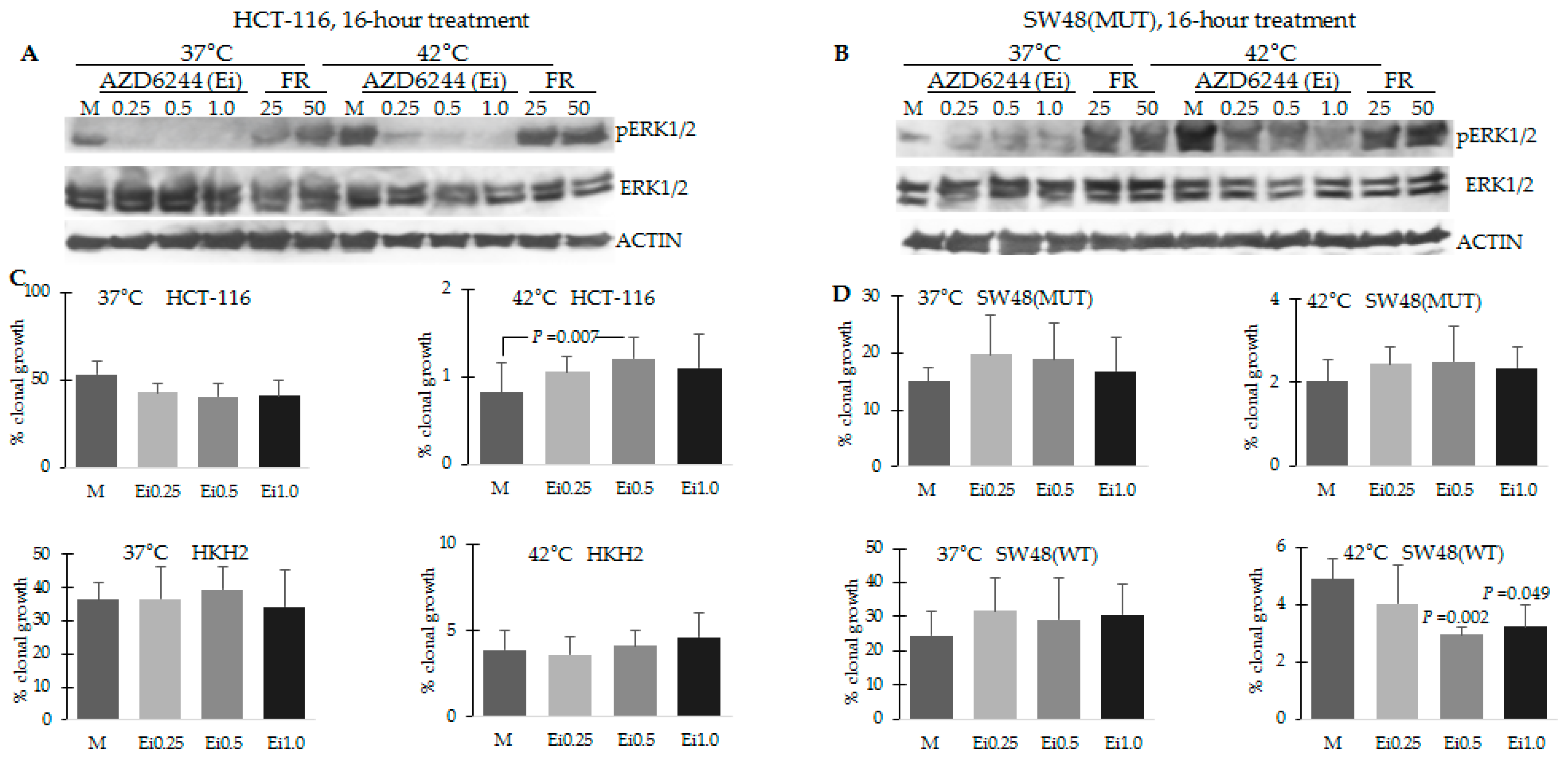
2.2. CRC Cells with Differential Hyperthermia Sensitivity Exhibit Differential Temporal Activation of ERK Signaling
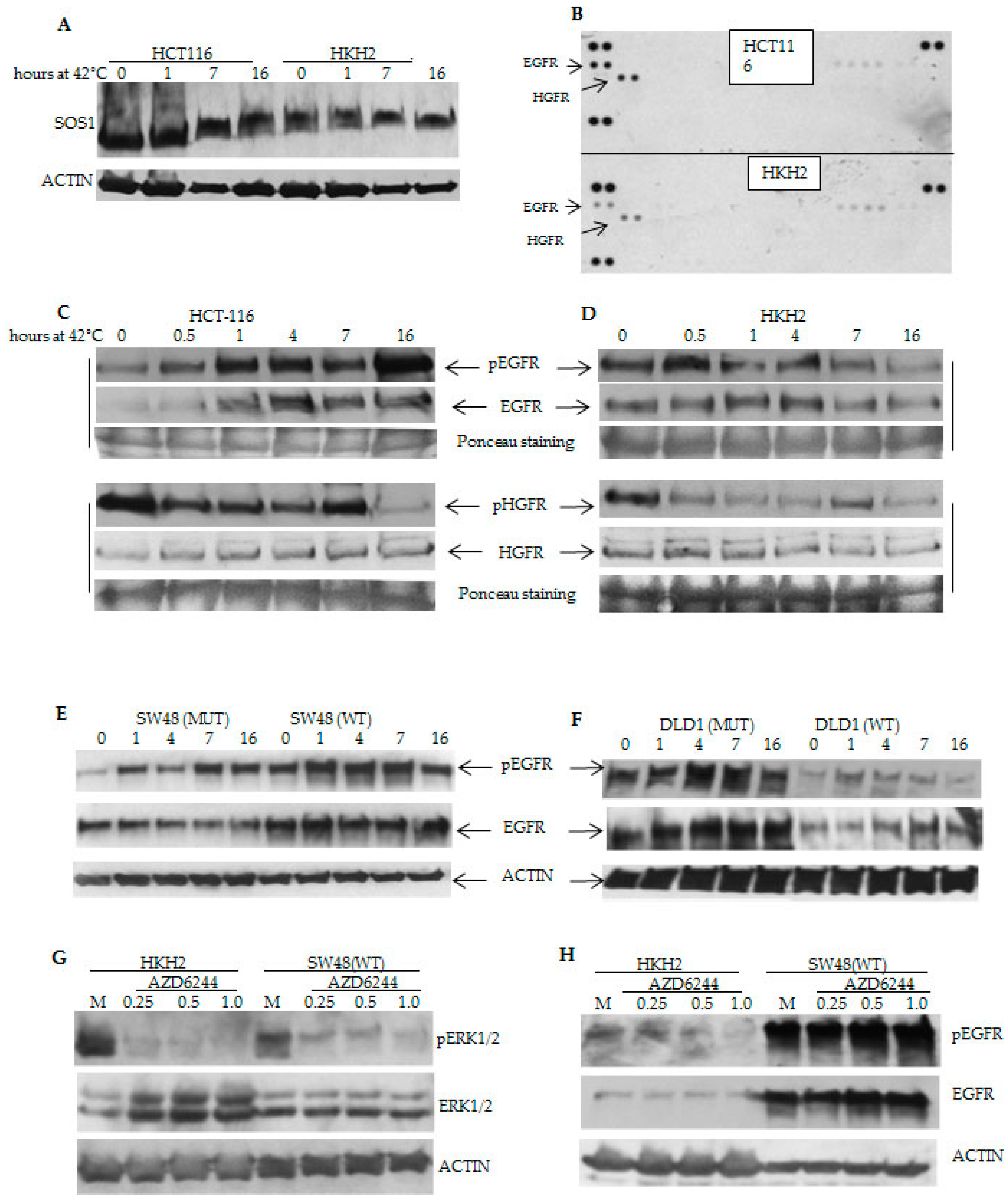
2.3. Differential Temporal Activation of ERK Signaling Correlates with Differential Activation of Epidermal Growth Factor Receptor (EGFR) in CRC Cells with Wild Type and Mutant KRAS
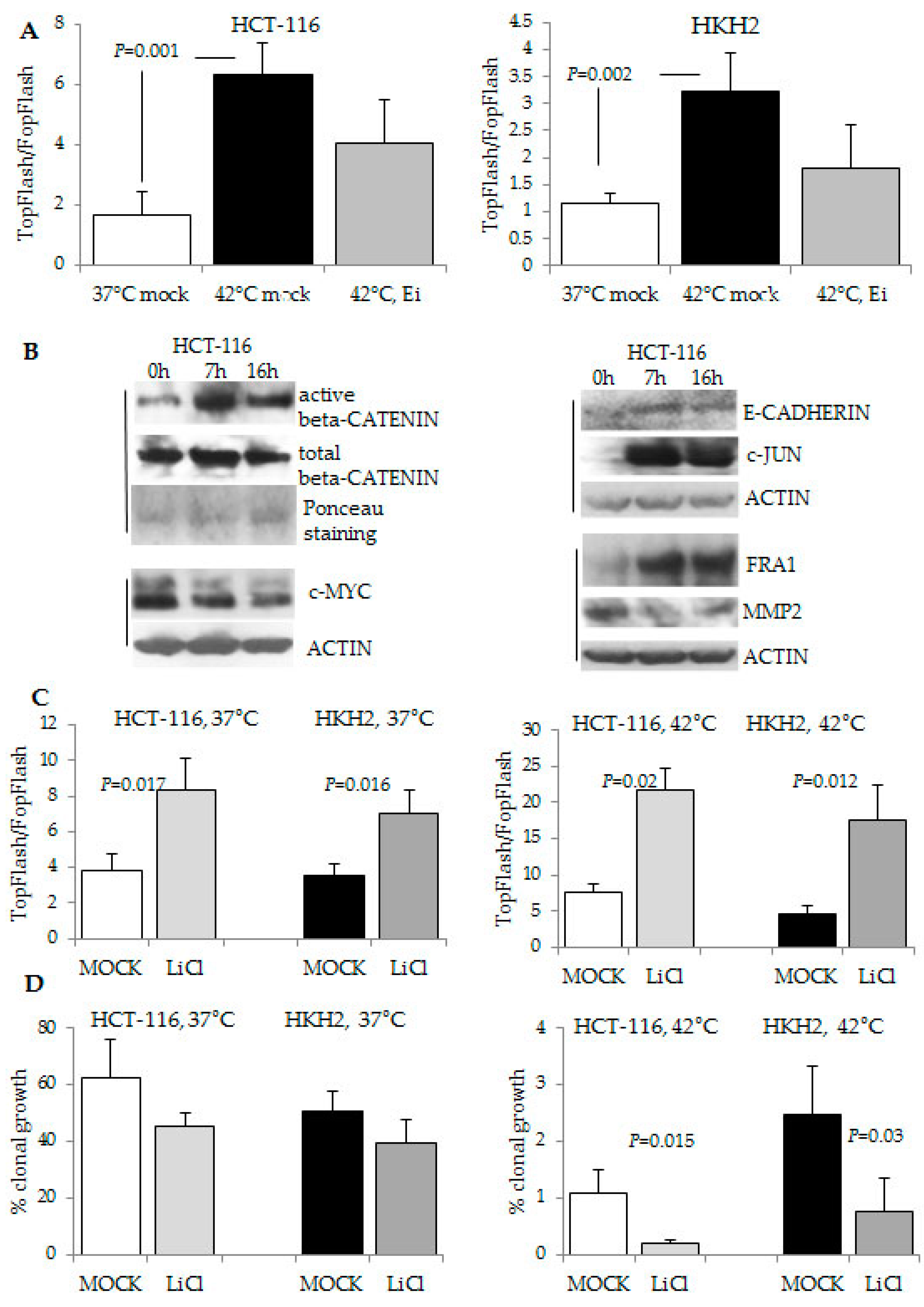
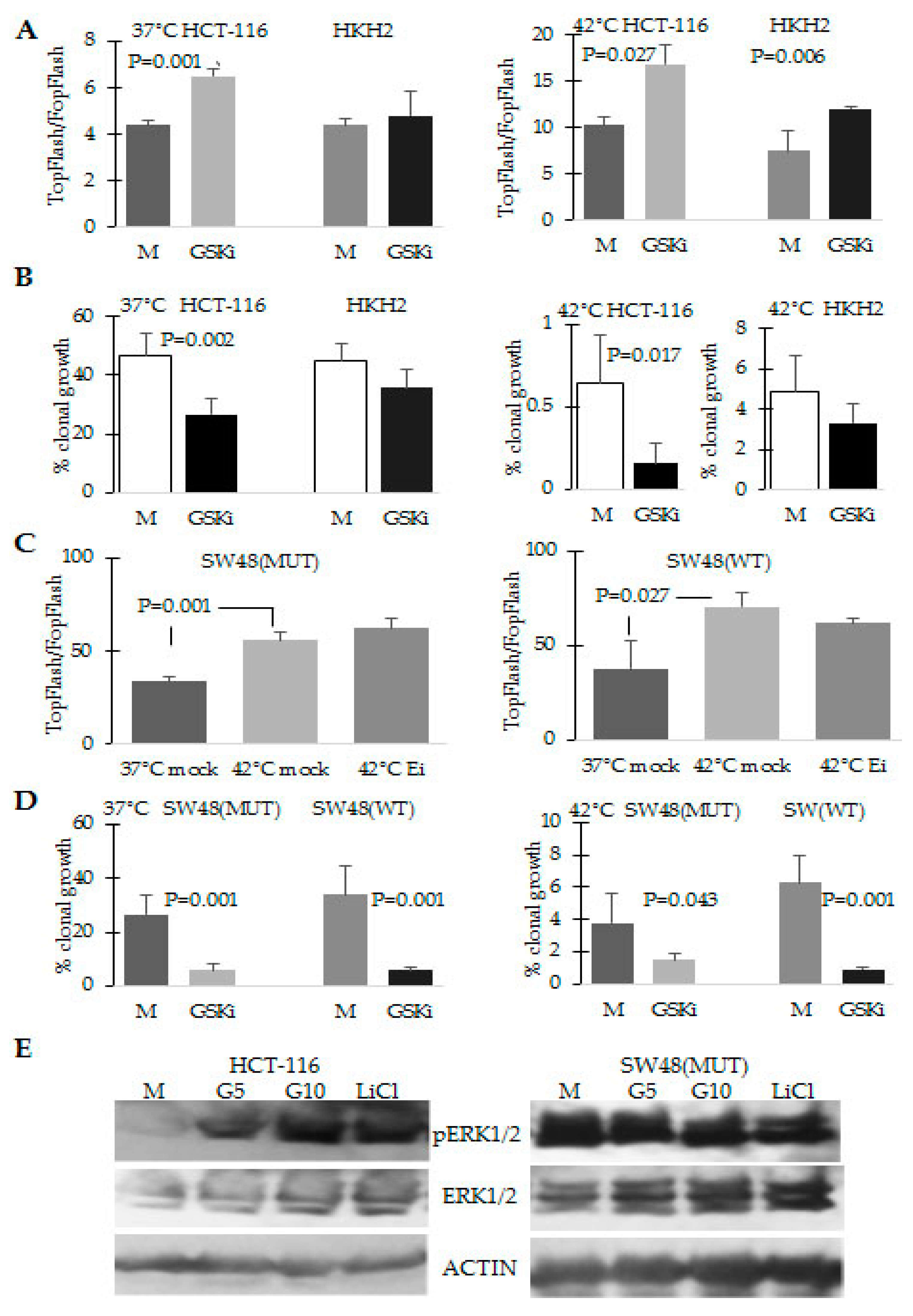
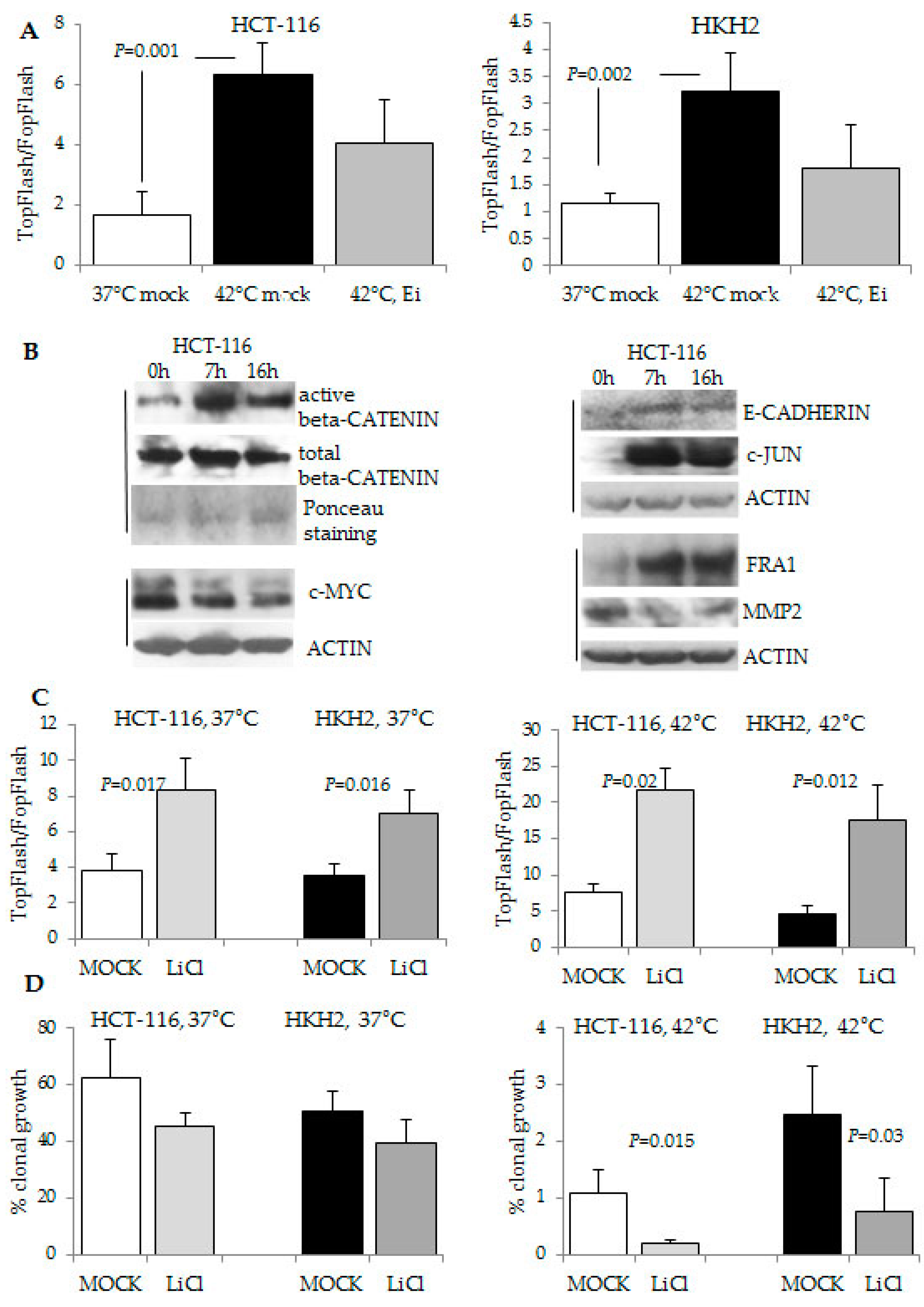
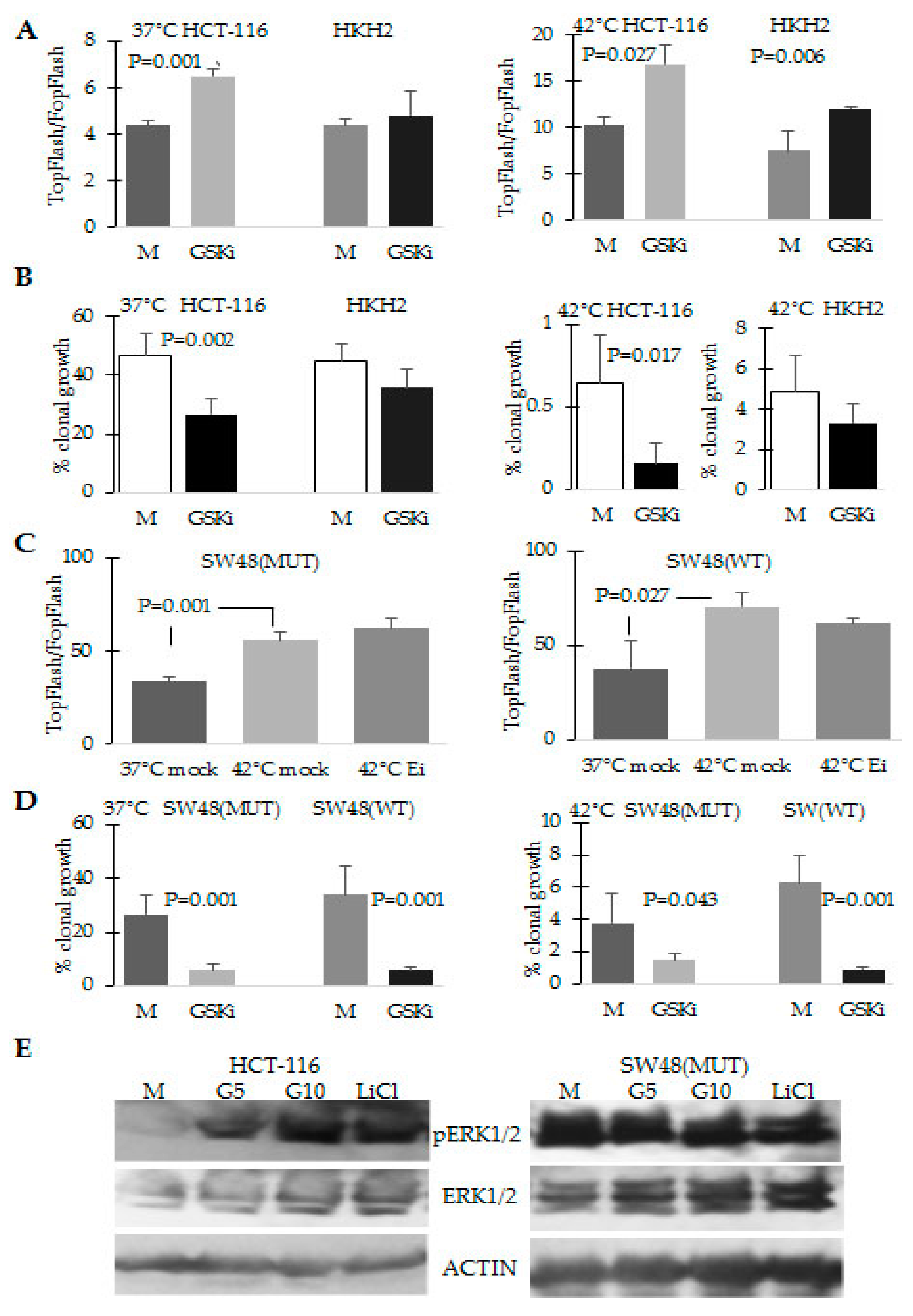
2.4. The Response to Hyperthermia Results in Increased WNT/beta-Catenin Activity
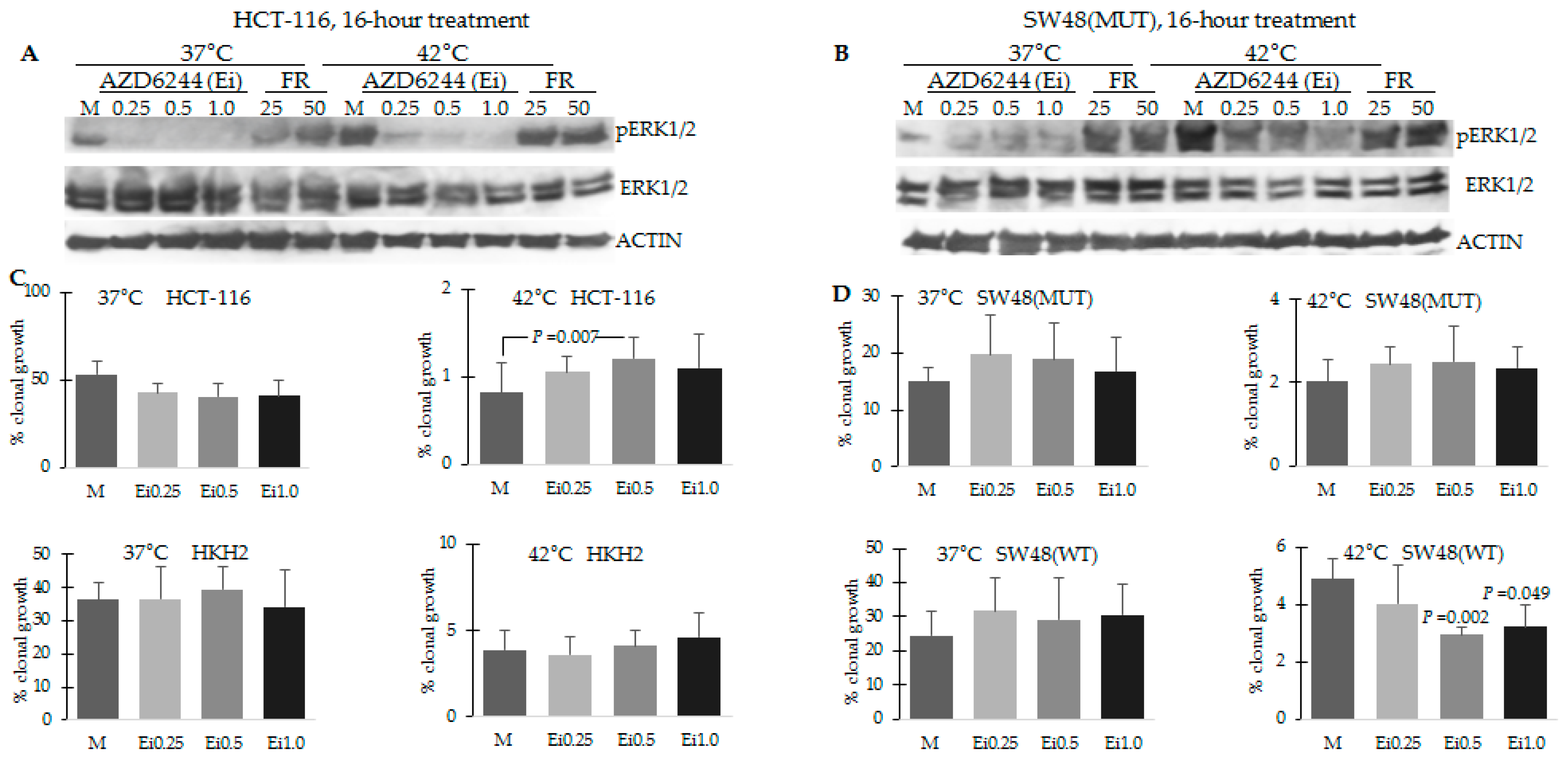
2.5. Combination Treatment of Hyperthermia and Propolis Is Effective in Suppressing CRC Cell Growth
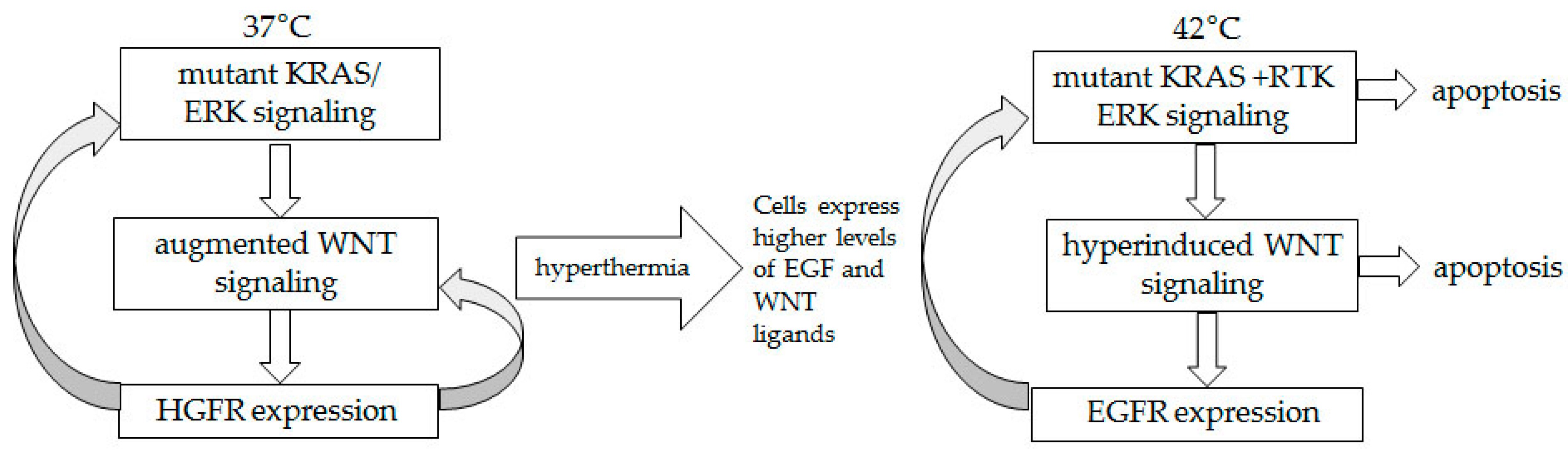
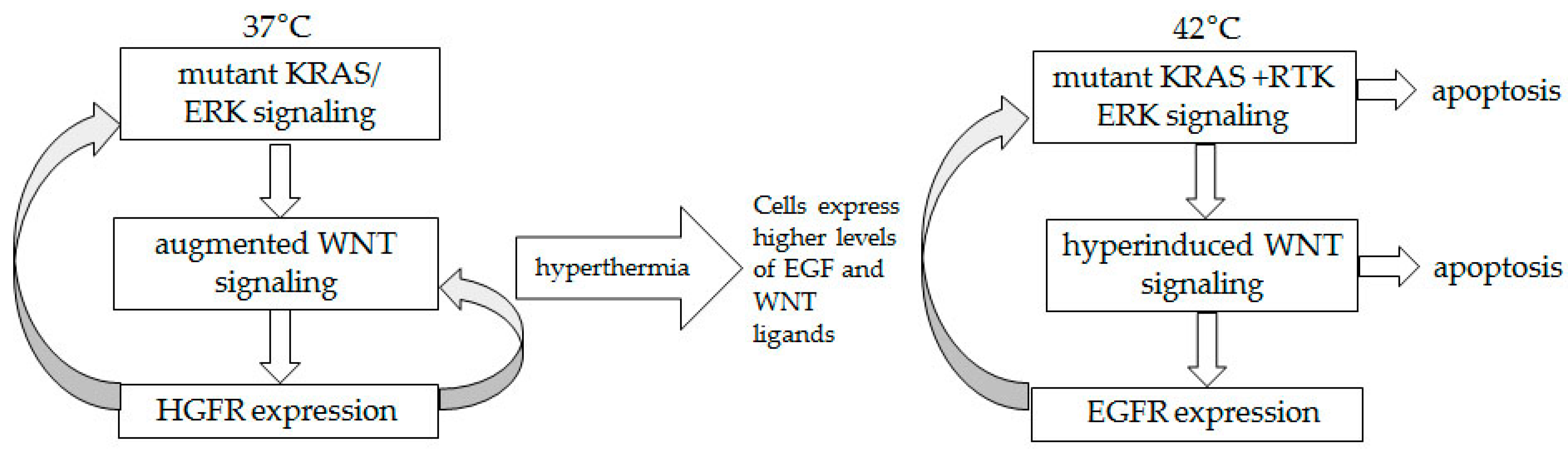
3. Discussion
4. Experimental Section
4.1. Cell Culture and Chemicals
4.2. Western Blot Analyses, Antibodies, RTK Activation Analysis
4.3. Apoptotic and Clonal Growth Assays
4.4. Transcriptional Assays
4.5. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interests
References
- Hobohm, U. Toward general prophylactic cancer vaccination. Bioessays 2009, 31, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Hobohm, U.; Stanford, J.L.; Grange, J.M. Pathogen-associated molecular pattern in cancer immunotherapy. Crit. Rev. Immunol. 2008, 28, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Grange, J.M.; Stanford, J.L.; Stanford, C.A. Campbell De Morgan’s “Observations on cancer”, and their relevance today. J. R. Soc. Med. 2002, 95, 296–299. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, E.F. The toxins of William B. Coley and the treatment of bone and soft-tissue sarcomas. Iowa Orthop. J. 2006, 26, 154–158. [Google Scholar] [PubMed]
- Coley, W.B. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin. Orthop. Relat. Res. 1991, 262, 3–11. [Google Scholar] [PubMed]
- Nauts, H.C.; McLaren, J.R. Coley toxins––The first century. Adv. Exp. Med. Biol. 1990, 267, 483–500. [Google Scholar] [PubMed]
- Wiemann, B.; Starnes, C.O. Coley’s toxins, tumor necrosis factor and cancer research: A historical perspective. Pharmacol. Ther. 1994, 64, 529–564. [Google Scholar] [CrossRef]
- Trieb, K.; Sztankay, A.; Amberger, A.; Lechner, H.; Grubeck-Loebenstein, B. Hyperthermia inhibits proliferation and stimulates the expression of differentiation markers in cultured thyroid carcinoma cells. Cancer Lett. 1994, 87, 65–71. [Google Scholar] [CrossRef]
- Karbach, J.; Neumann, A.; Brand, K.; Wahle, C.; Siegel, E.; Maeurer, M.; Ritter, E.; Tsuji, T.; Gnjatic, S.; Old, L.J.; et al. Phase I clinical trial of mixed bacterial vaccine (Coley’s toxins) in patients with NY-ESO-1 expressing cancers: Immunological effects and clinical activity. Clin. Cancer Res. 2012, 18, 5449–5459. [Google Scholar] [CrossRef] [PubMed]
- Kölmel, K.F.; Pfahlberg, A.; Mastrangelo, G.; Niin, M.; Botev, I.N.; Seebacher, C.; Schneider, D.; Lambert, D.; Shafir, R.; Kokoschka, E.M.; et al. Infections and melanoma risk: Results of a multicentre EORTC case-control study. European Organization for Research and Treatment of Cancer. Melanoma Res. 1999, 9, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Kleef, R.; Hager, E.D. Fever, pyrogens and cancer. In Hyperthermia in Cancer Treatment: A Primer; Baronzio, G.F., Hager, E.D., Eds.; Springer Science+Business Media: Berlin, Germany, 2006. [Google Scholar]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic alterations in colorectal cancer. Gastrointest Cancer Res. 2012, 5, 19–27. [Google Scholar] [PubMed]
- Guan, J.; Stavridi, E.; Leeper, D.B.; Iliakis, G. Effects of hyperthermia on p53 protein expression and activity. J. Cell Physiol. 2002, 190, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Shinoura, N.; Egawa, N.; Asai, A.; Kirino, T.; Shibasaki, F.; Shitara, N. Adenovirus-mediated transfer of p53 augments hyperthermia-induced apoptosis in U251 glioma cells. Int. J. Radiat. Oncol. Biol. Phys. 2001, 50, 525–531. [Google Scholar] [CrossRef]
- Bordonaro, M.; Lazarova, D. Hypothesis: Obesity is associated with a lower mutation threshold in colon cancer. J. Cancer 2015, 6, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Marchionni, L.; Nowak, M.A.; Parmigiani, G.; Vogelstein, B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.N.; Goel, A.; Blum, H.E.; Boland, C.R. Molecular pathogenesis of colorectal cancer, implications for molecular diagnosis. Cancer 2005, 104, 2035–2047. [Google Scholar] [CrossRef] [PubMed]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. British J. Cancer 2004, 91, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Forrester, K.; Almoguera, C.; Han, K.; Grizzle, W.E.; Perucho, M. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature 1987, 327, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Matos, P.; Oliveira, C.; Velho, S.; Goncalves, V.; da Costa, L.T.; Moyer, M.P.; Seruca, R.; Jordan, P. B-Raf (V600E) cooperates with alternative spliced Rac1b to sustain colorectal cancer cell survival. Gastroenterology 2008, 135, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Sugio, K.; Sasazuki, T. K-ras activation in colorectal tumors from patients with familial polyposis coli. Cancer 1990, 65, 2576–2579. [Google Scholar] [CrossRef]
- Shirasawa, S.; Furuse, M.; Yokoyama, N.; Sasazuki, T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science 1993, 260, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Kraybill, W.G.; Olenki, T.; Evans, S.S.; Ostberg, J.R.; O’Leary, K.A.; Gibbs, J.F.; Repasky, E.A. A phase I study of fever-range whole body hyperthermia (FR-WBH) in patients with advanced solid tumours: Correlation with mouse models. Int. J. Hyperthermia 2002, 18, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Waters, S.B.; Holt, K.H.; Pessin, J.E. SOS phosphorylation and disassociation of the Grb2-SOS complex by the ERK and JNK signaling pathways. J. Biol. Chem. 1996, 271, 6328–6332. [Google Scholar]
- Kamioka, Y.; Yasuda, S.; Fujita, Y.; Aoki, K.; Matsuda, M. Multiple decisive phosphorylation sites for the negative feedback regulation of SOS1 via ERK. J. Biol. Chem. 2010, 285, 33540–33548. [Google Scholar] [CrossRef] [PubMed]
- Sparks, A.B.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998, 58, 1130–1134. [Google Scholar] [PubMed]
- Janssen, K.P.; Alberici, P.; Fsihi, H.; Gaspar, C.; Breukel, C.; Franken, P.; Rosty, C.; Abal, M.; El Marjou, F.; Smits, R.; et al. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology 2006, 131, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Drago, E.; Bordonaro, M.; Lee, S.; Atamna, W.; Lazarova, D.L. Propolis augments apoptosis induced by butyrate via targeting cell survival pathways. PLoS ONE 2013, 8, e73151. [Google Scholar] [CrossRef] [PubMed]
- Bordonaro, M.; Drago, E.; Atamna, W.; Lazarova, D.L. Comprehensive suppression of all apoptosis-induced proliferation pathways as a proposed approach to colorectal cancer prevention and therapy. PLoS ONE 2014, 9, e115068. [Google Scholar] [CrossRef] [PubMed]
- Marshall, C.J. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef]
- Albeck, J.G.; Mills, G.B.; Brugge, J.S. Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol. Cell. 2013, 49, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death––Apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.J.; Routh, E.D.; Rubinas, T.; Peacock, J.; Martin, T.D.; Shen, X.J.; Sandler, R.S.; Kim, H.J.; Keku, T.O.; Der, C.J. KRAS/BRAF mutation status and ERK1/2 activation as biomarkers for MEK1/2 inhibitor therapy in colorectal cancer. Mol. Cancer Ther. 2009, 8, 834–843. [Google Scholar] [CrossRef] [PubMed]
- Zavadil, J.; Böttinger, E.P. TGF-β and epithelial-to-mesenchymal transitions. Oncogene 2005, 24, 5764–5774. [Google Scholar] [CrossRef] [PubMed]
- Orsulic, S.; Huber, O.; Aberle, H.; Arnold, S.; Kemler, R. E-cadherin binding prevents beta-catenin nuclear localization and beta-catenin/LEF-1-mediated transactivation. J. Cell Sci. 1999, 112, 1237–1245. [Google Scholar] [PubMed]
- Kinch, M.S.; Clark, G.J.; Der, C.J.; Burridge, K. Tyrosine phosphorylation regulates the adhesions of ras-transformed breast epithelia. J. Cell Biol. 1995, 130, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N. Functional crosstalk between Wnt signaling and tyrosine kinase signaling in cancer. Semin. Oncol. 2015, 42, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.T.; Holderfield, M.; Galeas, J.; Delrosario, R.; To, M.D.; Balmain, A.; McCormick, F. K-Ras promotes tumorigenicity through suppression of non-canonical Wnt signaling. Cell 2015, 163, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, D.L.; Bordonaro, M.; Carbone, R.; Sartorelli, A.C. Linear relationship between Wnt activity levels and apoptosis in colorectal carcinoma cells exposed to butyrate. Int. J. Cancer 2004, 110, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Bordonaro, M.; Lazarova, D.L.; Sartorelli, A.C. The activation of beta-catenin by Wnt signaling mediates the effects of histone deacetylase inhibitors. Exp. Cell Res. 2007, 313, 1652–1666. [Google Scholar] [CrossRef] [PubMed]
- Sheffield, L.G. Epidermal growth factor as an autocrine modulator of stress response in mammary epithelial cells. J. Endocrinol. 1998, 159, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Olkku, A.; Leskinen, J.J.; Lammi, M.J.; Hynynen, K.; Mahonen, A. Ultrasound-induced activation of Wnt signaling in human MG-63 osteoblastic cells. Bone 2010, 47, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, T.; Terracina, K.P.; Raza, A.; Takabe, K. Current treatment options for colon cancer peritoneal carcinomatosis. World J. Gastroenterol. 2014, 20, 12493–12500. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, D.L.; Bordonaro, M.; Sartorelli, A.C. Transcriptional regulation of the vitamin D3 receptor gene by ZEB. Cell Growth Differ. 2001, 12, 319–326. [Google Scholar] [PubMed]



© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bordonaro, M.; Shirasawa, S.; Lazarova, D.L. In Hyperthermia Increased ERK and WNT Signaling Suppress Colorectal Cancer Cell Growth. Cancers 2016, 8, 49. https://doi.org/10.3390/cancers8050049
Bordonaro M, Shirasawa S, Lazarova DL. In Hyperthermia Increased ERK and WNT Signaling Suppress Colorectal Cancer Cell Growth. Cancers. 2016; 8(5):49. https://doi.org/10.3390/cancers8050049
Chicago/Turabian StyleBordonaro, Michael, Senji Shirasawa, and Darina L. Lazarova. 2016. "In Hyperthermia Increased ERK and WNT Signaling Suppress Colorectal Cancer Cell Growth" Cancers 8, no. 5: 49. https://doi.org/10.3390/cancers8050049