
The Impact of Hedgehog Signaling Pathway on DNA Repair Mechanisms in Human Cancer

{kind=link}
{kind=link}
Abstract
:1. Introduction
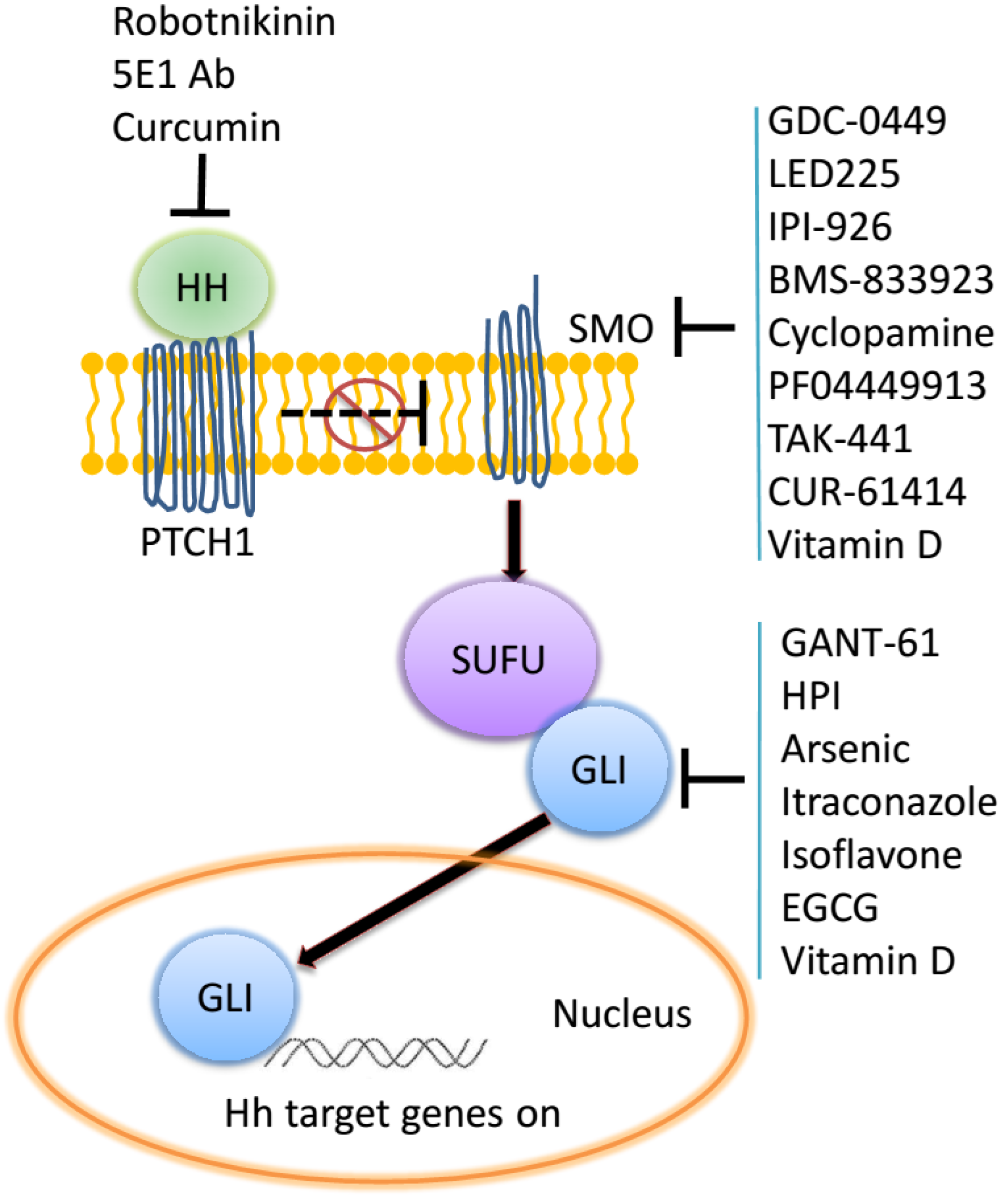
2. The Hedgehog Signaling Pathway
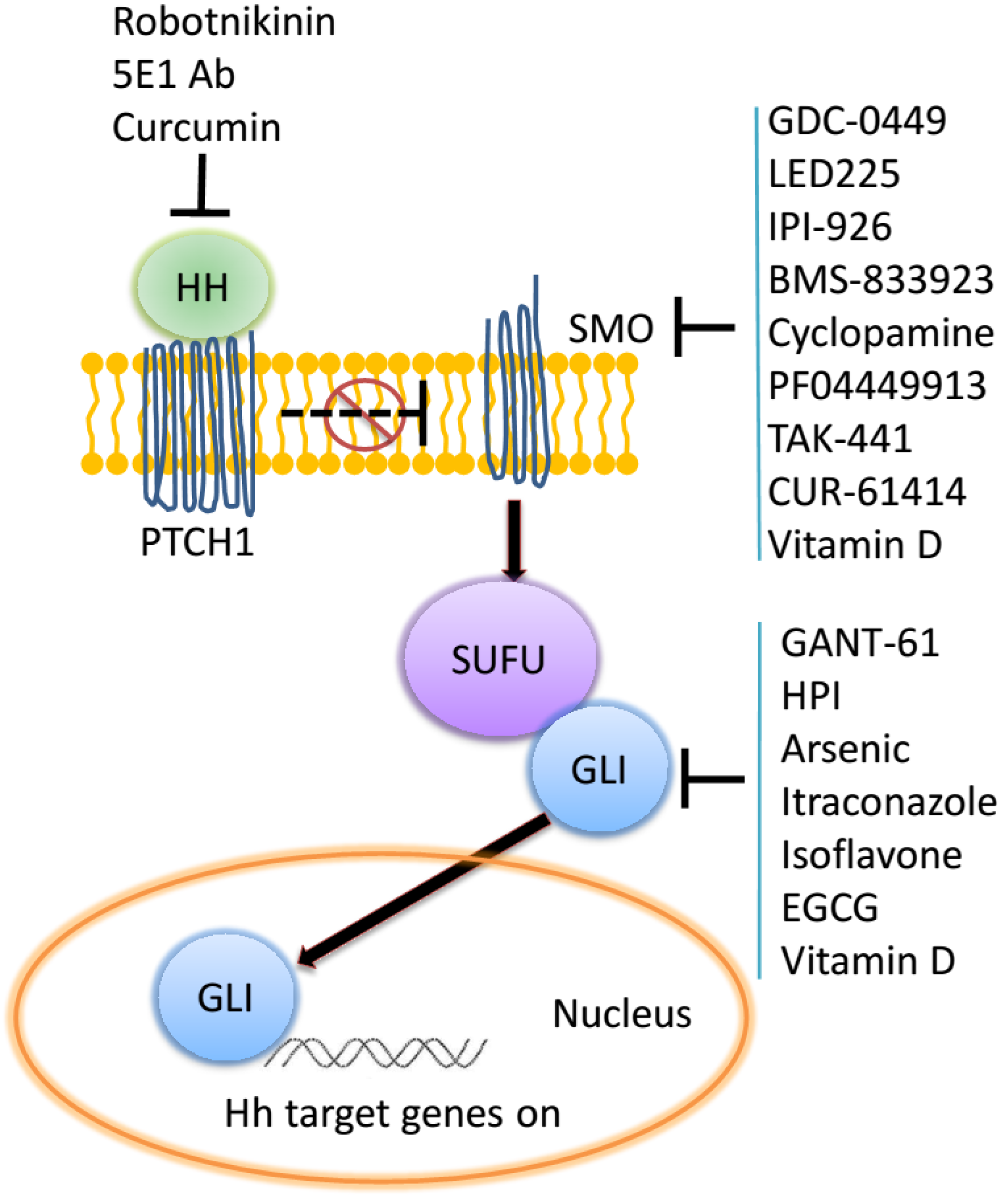
2.1. Mismatch Repair (MMR)
2.2. Nucleotide Excision Repair (NER)
2.3. Direct Repair
2.4. Base Excision Repair (BER)
2.5. DNA Double Strand Break Repair (DSB)
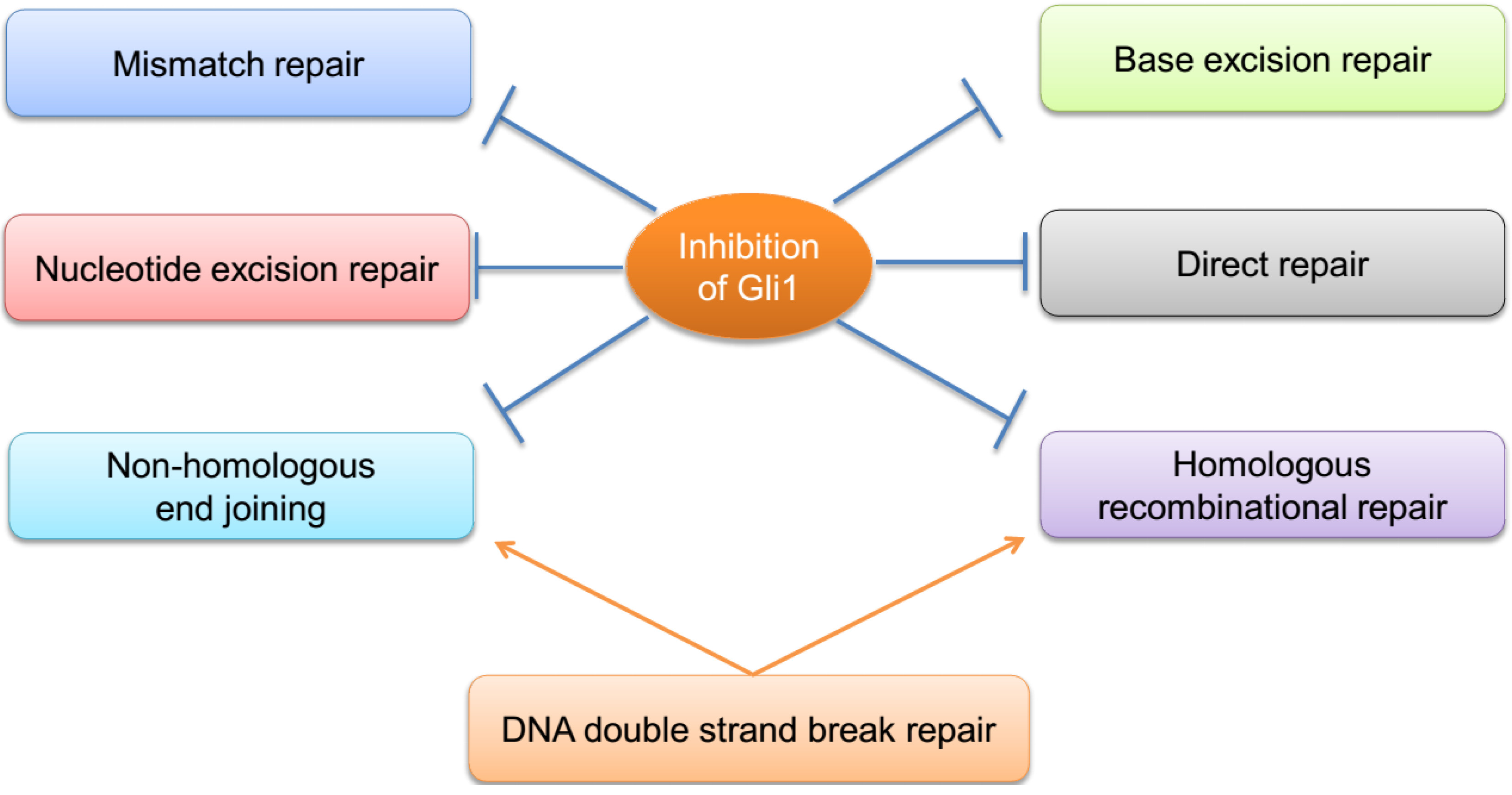
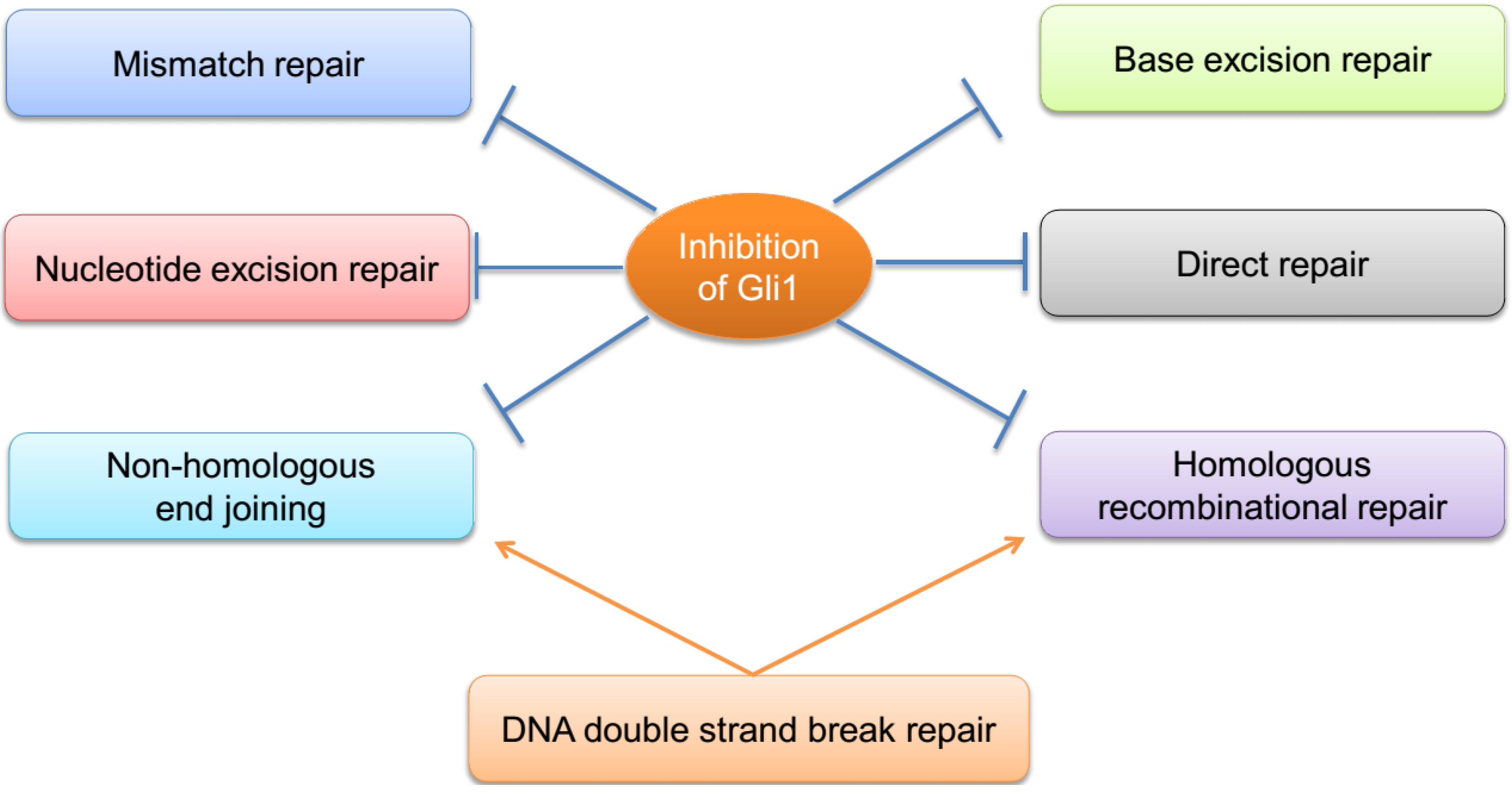
3. Crosstalk among the DNA Repair Pathways
4. Hedgehog Signaling and Escaping Apoptosis—A Fallback Mechanism
5. Conclusion and Perspectives
Acknowledgments
References
- Hansen, W.K.; Kelley, M.R. Review of mammalian DNA repair and translational implications. J. Pharmacol. Exp. 2000, 295, 1–9. [Google Scholar]
- Khanna, A. DNA Damage in Cancer Therapeutics: A Boon or a Curse? Cancer Res. 2015, 75, 2133–2138. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Gao, Z.; Li, H.; Zhang, B.; Wang, G.; Zhang, Q.; Pei, D.; Zheng, J. DNA damage response--a double-edged sword in cancer prevention and cancer therapy. Cancer Lett. 2015, 358, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Lahtz, C.; Pfeifer, G.P. Epigenetic changes of DNA repair genes in cancer. Journal of molecular cell. Biology 2011, 3, 51–58. [Google Scholar]
- Woll, P.S.; Morris, J.K.; Painschab, M.S.; Marcus, R.K.; Kohn, A.D.; Biechele, T.L.; Moon, R.T.; Kaufman, D.S. Wnt signaling promotes hematoendothelial cell development from human embryonic stem cells. Blood 2008, 111, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Takahashi-Yanaga, F.; Kahn, M. Targeting Wnt signaling: Can we safely eradicate cancer stem cells? Clin. Cancer Res. 2010, 16, 3153–3162. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.; Joseph, G.; Wang, Q.; Brennan, S.; Matsui, W. Gli1 regulates the proliferation and differentiation of HSCs and myeloid progenitors. Blood 2010, 115, 2391–2396. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.A.; Matsui, W. Targeting Hedgehog—A cancer stem cell pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz i Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Syn, W.K.; Jung, Y.; Omenetti, A.; Abdelmalek, M.; Guy, C.D.; Yang, L.; Wang, J.; Witek, R.P.; Fearing, C.M.; Pereira, T.A.; et al. Hedgehog-mediated epithelial-to-mesenchymal transition and fibrogenic repair in nonalcoholic fatty liver disease. Gastroenterology 2009, 137, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Malizia, A.P.; Lacey, N.; Walls, D.; Egan, J.J.; Doran, P.P. CUX1/Wnt signaling regulates epithelial mesenchymal transition in EBV infected epithelial cells. Exp. Cell Res. 2009, 315, 1819–1831. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes. Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.C.; Angers, S. Gli proteins in development and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Nishizaki, Y.; Hui, C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar] [PubMed]
- Hassounah, N.B.; Bunch, T.A.; McDermott, K.M. Molecular pathways: The role of primary cilia in cancer progression and therapeutics with a focus on Hedgehog signaling. Clin. Cancer Res. 2012, 18, 2429–2435. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Corcoran, R.B.; Scott, M.P. Hedgehog signal transduction by Smoothened: Pharmacologic evidence for a 2-step activation process. Proc. Natl. Acad. Sci. USA 2009, 106, 3196–3201. [Google Scholar] [CrossRef] [PubMed]
- McMillan, R.; Matsui, W. Molecular pathways: The hedgehog signaling pathway in cancer. Clin. Cancer Res. 2012, 18, 4883–4888. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.L.; Lo, H.W. Hedgehog pathway and GLI1 isoforms in human cancer. Discovery Med. 2012, 13, 105–113. [Google Scholar]
- Shevde, L.A.; Samant, R.S. Non-classical hedgehog-GLI signaling and its clinical implications. Int. J. Cancer 2014, 135, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Samant, R.S.; Shevde, L.A. Nonclassical activation of hedgehog signaling enhances multidrug resistance and makes cancer cells refractory to Smoothened-targeting hedgehog inhibition. J. Biol. Chem. 2013, 288, 11824–11833. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.G.; Samant, R.S.; Shevde, L.A. Hedgehog signaling: Networking to nurture a promalignant tumor microenvironment. Mol. Cancer Res. 2011, 9, 1165–1174. [Google Scholar] [CrossRef] [PubMed]
- Petrova, E.; Rios-Esteves, J.; Ouerfelli, O.; Glickman, J.F.; Resh, M.D. Inhibitors of Hedgehog acyltransferase block Sonic Hedgehog signaling. Nat. Chem. Biol. 2013, 9, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Mas, C.; Ruiz, I.; Altaba, A. Small molecule modulation of HH-GLI signaling: Current leads, trials and tribulations. Biochem. Pharmacol. 2010, 80, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; Lord, C.J.; Ashworth, A. Therapeutic targeting of the DNA mismatch repair pathway. Clin. Cancer Res. 2010, 16, 5107–5113. [Google Scholar] [CrossRef] [PubMed]
- Kinsella, T.J. Coordination of DNA mismatch repair and base excision repair processing of chemotherapy and radiation damage for targeting resistant cancers. Clin. Cancer Res. 2009, 15, 1853–1859. [Google Scholar] [CrossRef] [PubMed]
- Prolla, T.A.; Baker, S.M.; Harris, A.C.; Tsao, J.L.; Yao, X.; Bronner, C.E.; Zheng, B.; Gordon, M.; Reneker, J.; Arnheim, N.; et al. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat. Genet. 1998, 18, 276–279. [Google Scholar] [CrossRef] [PubMed]
- Li, G.M.; Modrich, P. Restoration of mismatch repair to nuclear extracts of H6 colorectal tumor cells by a heterodimer of human MutL homologs. Proc. Natl. Acad. Sci. USA 1995, 92, 1950–1954. [Google Scholar] [CrossRef] [PubMed]
- Plotz, G.; Raedle, J.; Brieger, A.; Trojan, J.; Zeuzem, S. N-terminus of hMLH1 confers interaction of hMutLalpha and hMutLbeta with hMutSalpha. Nucleic Acids Res. 2003, 31, 3217–3226. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.N.; Hile, S.E.; Eckert, K.A. Defective mismatch repair, microsatellite mutation bias, and variability in clinical cancer phenotypes. Cancer Res. 2010, 70, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; DeVecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. The GLI genes as the molecular switch in disrupting Hedgehog signaling in colon cancer. Oncotarget 2011, 2, 638–645. [Google Scholar] [PubMed]
- Shao, H.; Baitinger, C.; Soderblom, E.J.; Burdett, V.; Modrich, P. Hydrolytic function of Exo1 in mammalian mismatch repair. Nucleic Acids Res. 2014, 42, 7104–7112. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Riku, M.; Hashimoto, M.; Murakami, H.; Saga, S.; Ikeda, H.; Kasai, K. GLI1 interferes with the DNA mismatch repair system in pancreatic cancer through BHLHE41-mediated suppression of MLH1. Cancer Res. 2013, 73, 7313–7323. [Google Scholar] [CrossRef] [PubMed]
- Wogan, G.N.; Hecht, S.S.; Felton, J.S.; Conney, A.H.; Loeb, L.A. Environmental and chemical carcinogenesis. Semin. Cancer Biol. 2004, 14, 473–486. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J. The case for 8,5'-cyclopurine-2'-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience 2007, 145, 1407–1417. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Fink, S.P.; Marnett, L.J. Repair of propanodeoxyguanosine by nucleotide excision repair in vivo and in vitro. J. Biol. Chem. 1997, 272, 11434–11438. [Google Scholar] [PubMed]
- Ariza, R.R.; Keyse, S.M.; Moggs, J.G.; Wood, R.D. Reversible protein phosphorylation modulates nucleotide excision repair of damaged DNA by human cell extracts. Nucleic Acids Res. 1996, 24, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Moggs, J.G.; Yarema, K.J.; Essigmann, J.M.; Wood, R.D. Analysis of incision sites produced by human cell extracts and purified proteins during nucleotide excision repair of a 1,3-intrastrand d(GpTpG)-cisplatin adduct. J. Biol. Chem. 1996, 271, 7177–7186. [Google Scholar] [PubMed]
- Lindahl, T.; Barnes, D.E.; Klungland, A.; Mackenney, V.J.; Schar, P. Repair and processing events at DNA ends. Ciba Found. Symp. 1997, 211, 198–205. [Google Scholar] [PubMed]
- Lindahl, T.; Karran, P.; Wood, R.D. DNA excision repair pathways. Curr. Opin. Genet. Dev. 1997, 7, 158–169. [Google Scholar] [CrossRef]
- He, Z.; Henricksen, L.A.; Wold, M.S.; Ingles, C.J. RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature 1995, 374, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, M.; Takehara, A.; Matsuda, K.; Eguchi, H.; Ohigashi, H.; Ishikawa, O.; Shinomura, Y.; Imai, K.; Nakamura, Y.; Nakagawa, H. Oncogenic role of KIAA0101 interacting with proliferating cell nuclear antigen in pancreatic cancer. Cancer Res. 2007, 67, 2568–2576. [Google Scholar] [CrossRef] [PubMed]
- Kudo, K.; Gavin, E.; Das, S.; Amable, L.; Shevde, L.A.; Reed, E. Inhibition of Gli1 results in altered c-Jun activation, inhibition of cisplatin-induced upregulation of ERCC1, XPD and XRCC1, and inhibition of platinum-DNA adduct repair. Oncogene 2012, 31, 4718–4724. [Google Scholar] [CrossRef] [PubMed]
- Kreklau, E.L.; Limp-Foster, M.; Liu, N.; Xu, Y.; Kelley, M.R.; Erickson, L.C. A novel fluorometric oligonucleotide assay to measure O( 6)-methylguanine DNA methyltransferase, methylpurine DNA glycosylase, 8-oxoguanine DNA glycosylase and abasic endonuclease activities: DNA repair status in human breast carcinoma cells overexpressing methylpurine DNA glycosylase. Nucleic Acids Res. 2001, 29, 2558–2566. [Google Scholar] [PubMed]
- Marathi, U.K.; Dolan, M.E.; Erickson, L.C. Extended depletion of O6-methylguanine-DNA methyltransferase activity following O6-benzyl-2'-deoxyguanosine or O6-benzylguanine combined with streptozotocin treatment enhances 1,3-bis(2-chloroethyl)-1-nitrosourea cytotoxicity. Cancer Res. 1994, 54, 4371–4375. [Google Scholar] [PubMed]
- Erickson, L.C. The role of O-6 methylguanine DNA methyltransferase (MGMT) in drug resistance and strategies for its inhibition. Semin. Cancer Biol. 1991, 2, 257–265. [Google Scholar] [PubMed]
- Pieper, R.O.; Futscher, B.W.; Dong, Q.; Ellis, T.M.; Erickson, L.C. Comparison of O-6-methylguanine DNA methyltransferase (MGMT) mRNA levels in Mer+ and Mer-human tumor cell lines containing the MGMT gene by the polymerase chain reaction technique. Cancer Commun. 1990, 2, 13–20. [Google Scholar] [PubMed]
- Cui, D.; Xu, Q.; Wang, K.; Che, X. Gli1 is a potential target for alleviating multidrug resistance of gliomas. J. Neurol. Sci. 2010, 288, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Gilbertson, R.; Iannaccone, S.; Iannaccone, P.; Walterhouse, D. Defining a role for Sonic hedgehog pathway activation in desmoplastic medulloblastoma by identifying GLI1 target genes. Int. J. Cancer 2009, 124, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Lo, H.W. The Human Glioma-Associated Oncogene Homolog 1 (GLI1) Family of Transcription Factors in Gene Regulation and Diseases. Curr. Genomics 2010, 11, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Dianov, G.L.; Hubscher, U. Mammalian base excision repair: The forgotten archangel. Nucleic Acids Res. 2013, 41, 3483–3490. [Google Scholar] [CrossRef] [PubMed]
- David, S.S.; O’Shea, V.L.; Kundu, S. Base-excision repair of oxidative DNA damage. Nature 2007, 447, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Taverna, P.; Hwang, H.S.; Schupp, J.E.; Radivoyevitch, T.; Session, N.N.; Reddy, G.; Zarling, D.A.; Kinsella, T.J. Inhibition of base excision repair potentiates iododeoxyuridine-induced cytotoxicity and radiosensitization. Cancer Res. 2003, 63, 838–846. [Google Scholar] [PubMed]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. The mechanism of vertebrate nonhomologous DNA end joining and its role in V(D)J recombination. DNA Repair 2004, 3, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; Devecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. Blocking Hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res. 2011, 71, 5904–5914. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.Y.; Che, J.; Sun, K.K.; Shen, X.J.; Yang, D.; Zhong, N.; Zhao, H. Cyclopamine increases the radiosensitivity of human pancreatic cancer cells by regulating the DNA repair signal pathway through an epidermal growth factor receptordependent pathway. Mol. Med. Rep. 2013, 8, 979–983. [Google Scholar] [PubMed]
- Aparicio, T.; Baer, R.; Gautier, J. DNA double-strand break repair pathway choice and cancer. DNA Repair 2014, 19, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.H.; Schild, D. Recombinational DNA repair and human disease. Mutat. Res. 2002, 509, 49–78. [Google Scholar] [CrossRef]
- Liu, C.; Srihari, S.; Cao, K.A.; Chenevix-Trench, G.; Simpson, P.T.; Ragan, M.A.; Khanna, K.K. A fine-scale dissection of the DNA double-strand break repair machinery and its implications for breast cancer therapy. Nucleic Acids Res. 2014, 42, 6106–6127. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Chen, D.J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 2006, 5, 1042–1048. [Google Scholar] [CrossRef] [PubMed]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Gautier, J. The Fanconi anemia pathway and ICL repair: Implications for cancer therapy. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 424–439. [Google Scholar] [CrossRef] [PubMed]
- Geiman, T.M.; Durum, S.K.; Muegge, K. Characterization of gene expression, genomic structure, and chromosomal localization of Hells (Lsh). Genomics 1998, 54, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Mazumdar, T.; Devecchio, J.; Duan, Z.H.; Agyeman, A.; Aziz, M.; Khanna, K.K. cDNA microarray gene expression profiling of hedgehog signaling pathway inhibition in human colon cancer cells. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.M.; Ye, H.; Wetmore, C.; Karnitz, L.M. Sonic Hedgehog signaling impairs ionizing radiation-induced checkpoint activation and induces genomic instability. J. Cell Biol. 2008, 183, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.; Nievera, C.J.; Lee, A.Y.; Chen, L.; Wu, X. The Mre11-Rad50-Nbs1 complex acts both upstream and downstream of ataxia telangiectasia mutated and Rad3-related protein (ATR) to regulate the S-phase checkpoint following UV treatment. J. Biol. Chem. 2007, 282, 22939–22952. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Luo, K.; Lou, Z.; Chen, J. MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 2008, 105, 11200–11205. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.J.; Williams, R.S.; Williams, J.S.; Moncalian, G.; Arvai, A.S.; Limbo, O.; Khanna, K.K. ABC ATPase signature helices in Rad50 link nucleotide state to Mre11 interface for DNA repair. Nat. Struct. Mol. Biol. 2011, 18, 423–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agyeman, A.; Mazumdar, T.; Houghton, J.A. Regulation of DNA damage following termination of Hedgehog (HH) survival signaling at the level of the GLI genes in human colon cancer. Oncotarget 2012, 3, 854–868. [Google Scholar] [PubMed]
- Glassner, B.J.; Rasmussen, L.J.; Najarian, M.T.; Posnick, L.M.; Samson, L.D. Generation of a strong mutator phenotype in yeast by imbalanced base excision repair. Proc. Natl. Acad. Sci. USA 1998, 95, 9997–10002. [Google Scholar] [CrossRef] [PubMed]
- Drotschmann, K.; Aronshtam, A.; Fritz, H.J.; Marinus, M.G. The Escherichia coli MutL protein stimulates binding of Vsr and MutS to heteroduplex DNA. Nucleic Acids Res. 1998, 26, 948–953. [Google Scholar] [CrossRef] [PubMed]
- Lieb, M.; Rehmat, S.; Bhagwat, A.S. Interaction of MutS and Vsr: Some dominant-negative mutS mutations that disable methyladenine-directed mismatch repair are active in very-short-patch repair. J. Bacteriol. 2001, 183, 6487–6490. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernandez-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes. Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Fu, J.; Srivastava, R.K.; Shankar, S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: Molecular mechanisms. PLoS ONE 2011, 6, e27306. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Siu, M.K.; Au, C.W.; Wong, E.S.; Chan, H.Y.; Ip, P.P.; Ngan, H.Y.; Cheung, A.N. Aberrant activation of hedgehog signaling pathway in ovarian cancers: Effect on prognosis, cell invasion and differentiation. Carcinogenesis 2009, 30, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Horiuchi, A.; Kikuchi, N.; Osada, R.; Yoshida, J.; Shiozawa, T.; Konishi, I. Hedgehog signal pathway is activated in ovarian carcinomas, correlating with cell proliferation: It’s inhibition leads to growth suppression and apoptosis. Cancer Sci. 2007, 98, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Pan, L.; Che, X.; Cui, D.; Li, C. Gli1 inhibition induces cell-cycle arrest and enhanced apoptosis in brain glioma cell lines. J. Neurooncol. 2010, 98, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Das, T.; Gulati, P.; Biswas, N.K.; Rote, S.; Chatterjee, U. Hedgehog signaling pathway is active in GBM with GLI1 mRNA expression showing a single continuous distribution rather than discrete high/low clusters. PLoS ONE 2015, 10, e0116390. [Google Scholar] [CrossRef] [PubMed]
- Bigelow, R.L.; Chari, N.S.; Unden, A.B.; Spurgers, K.B.; Lee, S.; Roop, D.R.; Toftgard, R.; McDonnell, T.J. Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. J. Biol. Chem. 2004, 279, 1197–1205. [Google Scholar] [CrossRef] [PubMed]
- Sims-Mourtada, J.; Izzo, J.G.; Ajani, J.; Chao, K.S. Sonic Hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene 2007, 26, 5674–5679. [Google Scholar] [CrossRef] [PubMed]
- Amable, L.; Fain, J.; Gavin, E.; Reed, E. Gli1 contributes to cellular resistance to cisplatin through altered cellular accumulation of the drug. Oncol Rep. 2014, 32, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Moukharskaya, J.; Verschraegen, C. Topoisomerase 1 inhibitors and cancer therapy. Hematol./Oncol. Clin. N. Am. 2012, 26, 507–525. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Chen, P.M.; Chen, Y.J. DNA topoisomerase I drugs and radiotherapy for lung cancer. J. Thorac. Dis. 2012, 4, 390–397. [Google Scholar] [PubMed]
- Pommier, Y. DNA topoisomerase I inhibitors: Chemistry, biology, and interfacial inhibition. Chem. Rev. 2009, 109, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, K.; Mani, C.; Barnett, R.; Nalluri, S.; Bachaboina, L.; Rocconi, R.P.; Athar, M.; Owen, L.B.; Palle, K. Gli1 protein regulates the S-phase checkpoint in tumor cells via Bid protein, and its inhibition sensitizes to DNA topoisomerase 1 inhibitors. J. Biol. Chem. 2014, 289, 31513–31525. [Google Scholar] [CrossRef] [PubMed]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. Gli1/DNA interaction is a druggable target for Hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernandez-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes de Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Maitra, A. The hedgehog pathway and pancreatic cancer. N. Engl. J. Med. 2009, 361, 2094–2096. [Google Scholar] [CrossRef] [PubMed]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Lin, T.; Zhu, J.; Tong, W.; Huo, Y.; Jia, H.; Zhang, Y.; Liu, X.S.; Cengel, K.; Xia, B.; et al. PTH1–34 blocks radiation-induced osteoblast apoptosis by enhancing DNA repair through canonical Wnt pathway. J. Biol. Chem. 2015, 290, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.R.; Lee, J.; An, Y.S.; Jin, Y.B.; Park, I.C.; Chung, E.; Shin, I.; Barcellos-Hoff, M.H.; Yi, J.Y. TGFbeta1 protects cells from gamma-IR by enhancing the activity of the NHEJ repair pathway. Mol. Cancer Res. 2015, 13, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wang, Y.; Ren, X.; Tsuyada, A.; Li, A.; Liu, L.J.; Wang, S.E. Context-dependent bidirectional regulation of the MutS homolog 2 by transforming growth factor beta contributes to chemoresistance in breast cancer cells. Mol. Cancer Res. 2010, 8, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, E.; Hanna, A.; Samant, R.S.; Shevde, L.A. The Impact of Hedgehog Signaling Pathway on DNA Repair Mechanisms in Human Cancer. Cancers 2015, 7, 1333-1348. https://doi.org/10.3390/cancers7030839
Meng E, Hanna A, Samant RS, Shevde LA. The Impact of Hedgehog Signaling Pathway on DNA Repair Mechanisms in Human Cancer. Cancers. 2015; 7(3):1333-1348. https://doi.org/10.3390/cancers7030839
Chicago/Turabian StyleMeng, Erhong, Ann Hanna, Rajeev S. Samant, and Lalita A. Shevde. 2015. "The Impact of Hedgehog Signaling Pathway on DNA Repair Mechanisms in Human Cancer" Cancers 7, no. 3: 1333-1348. https://doi.org/10.3390/cancers7030839