
The Role of Hedgehog Signaling in Tumor Induced Bone Disease

1
Department of Veterans Affairs, Tennessee Valley Healthcare System, Nashville, TN 37235, USA
2
Vanderbilt Center for Bone Biology, Department of Medicine, Division of Clinical Pharmacology Vanderbilt University, Nashville, TN 372335, USA
3
Department of Cancer Biology, Vanderbilt University, Nashville, TN 37235, USA
*
Author to whom correspondence should be addressed.
Cancers 2015, 7(3), 1658-1683; https://doi.org/10.3390/cancers7030856
Submission received: 9 July 2015
/
Revised: 13 August 2015
/
Accepted: 18 August 2015
/
Published: 26 August 2015
(This article belongs to the Special Issue Hedgehog Signaling Pathway in Cancer: Smoothened and GLI Take Center Stage)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Despite significant progress in cancer treatments, tumor induced bone disease continues to cause significant morbidities. While tumors show distinct mutations and clinical characteristics, they behave similarly once they establish in bone. Tumors can metastasize to bone from distant sites (breast, prostate, lung), directly invade into bone (head and neck) or originate from the bone (melanoma, chondrosarcoma) where they cause pain, fractures, hypercalcemia, and ultimately, poor prognoses and outcomes. Tumors in bone secrete factors (interleukins and parathyroid hormone-related protein) that induce RANKL expression from osteoblasts, causing an increase in osteoclast mediated bone resorption. While the mechanisms involved varies slightly between tumor types, many tumors display an increase in Hedgehog signaling components that lead to increased tumor growth, therapy failure, and metastasis. The work of multiple laboratories has detailed Hh signaling in several tumor types and revealed that tumor establishment in bone can be controlled by both canonical and non-canonical Hh signaling in a cell type specific manner. This review will explore the role of Hh signaling in the modulation of tumor induced bone disease, and will shed insight into possible therapeutic interventions for blocking Hh signaling in these tumors.
1. Introduction
Hedgehog (Hh) signaling is a complex signaling pathway that was initially identified as a developmental signaling pathway. Since its original discovery in 1980, its role in disease has been better defined. Aberrant Hh signaling has been demonstrated by numerous groups to contribute to multiple cancer types both through contributing to changes in tumor growth directly or altering the tumor microenvironment. Because of its role in multiple tumor types, Hh signaling is an attractive pathway for both basic research and drug development to identify new targets and new therapeutic approaches for inhibiting tumor growth and metastasis.
2. Hedgehog Signaling in Skeletal Development and Bone Homeostasis
2.1. Hedgehog Signal Transduction
Hh signaling, first identified by its developmental role in Drosophila melanogaster, is an evolutionary conserved signaling pathway that is now known to have additional important roles in tissue homeostasis as well as tumorigenesis [1,2,3,4]. In the late 1960s, the field of embryonic development had no dogma to explain how the relatively simple body plan of drosophila embryos gave rise to the complex body structure of adult fruit flies. In the 1970s, Christiane Nüsslein-Volhard and Eric Wieschaus made ground breaking progress in understanding drosophila tissue patterning and polarity, which they were later awarded for with the Nobel Bell Prize in physiology or medicine. Utilizing embryonic lethal screening techniques, the pair introduced random mutations into the genome of fruit flies using ethyl methanosulfonate (EMS) and identified 15 loci important for segment number and polarity in drosophila larvae [5]. The resulting developmental defects were mainly named after the phenotypic observations of the larvae. In the case of Hh, the phenotype affected the denticles, which are bristle hairs used for gripping surfaces and locomotion. While normal embryos displayed discrete bands of denticles, Hh mutants showed an unorganized “denticle lawn” reminiscent of the spines of the hedgehog, hence the name Hh. Further research in the field identified Hh signaling as an important morphogen, where levels of secreted ligand creates a short span gradient that directly controls genes important for cell proliferation, polarity, and differentiation [6,7,8,9,10].
Additionally, in context specific settings, Hh signaling has been found to function in tandem with other important developmental signaling pathways such as Wnt, BMP and TGFβ signaling [10,11,12]. Crosstalk between these pathways facilitates multiple levels of transcriptional control over target genes and as such, Hh signaling can both positively and negatively regulate other signaling pathways. Hh signaling has an active and inactive state, which is controlled by the presence of Hh ligand. In insects, when there is no Hh ligand bound to the receptor Patched (Ptch), the membrane protein Smoothened (Smo) is unable to accumulate and its effector molecule, the zinc finger protein, cubitus interruptus is proteasomally cleaved into a transcriptional repressor (CiR). In contrast, Hh ligand bound to Ptch leads to Smo accumulation which prevents Ci cleavage, allowing the full length protein to function as a transcriptional activator [13].
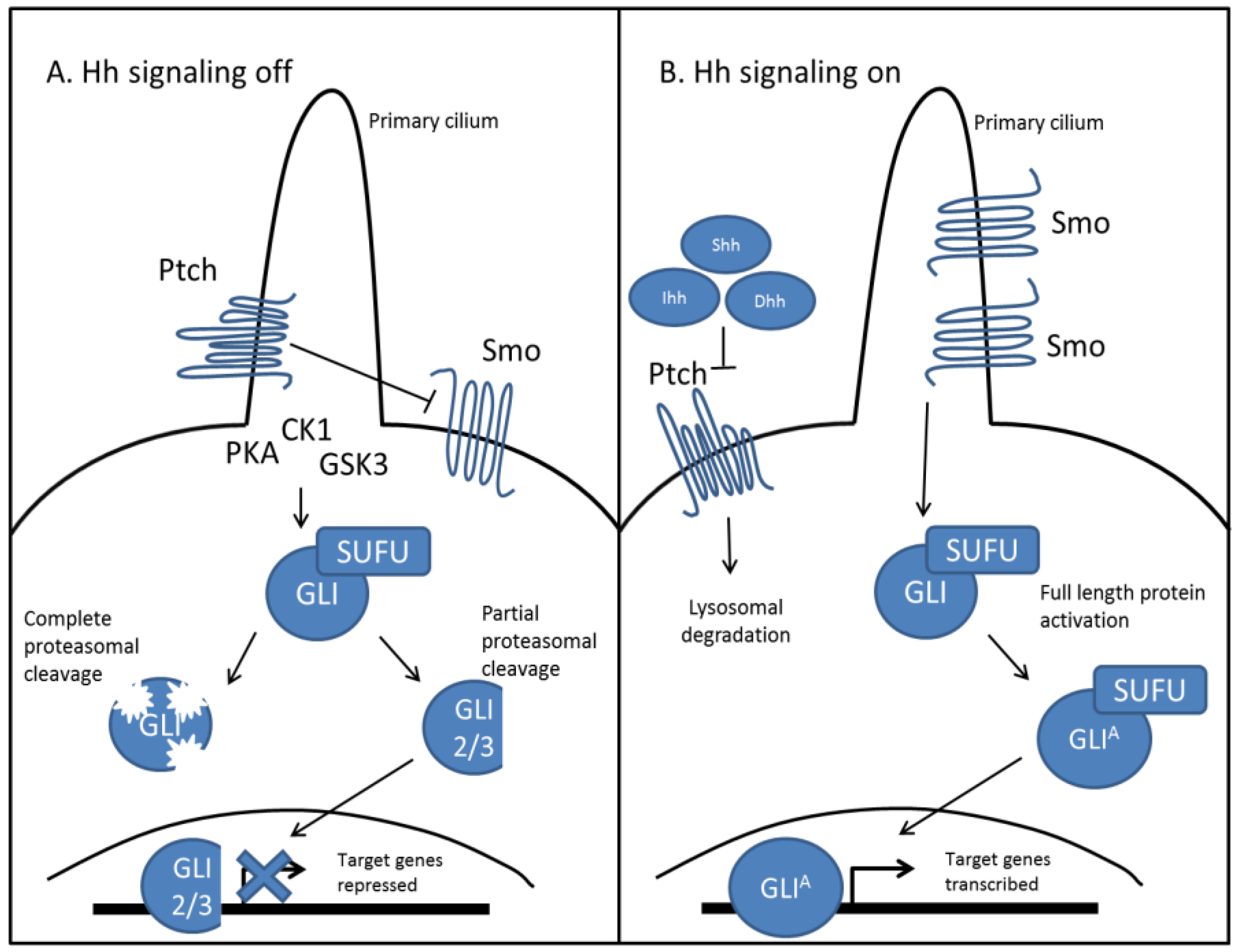
As an evolutionarily conserved signaling pathway, Hh signaling works similarly in vertebrates, as outlined in Figure 1 [14]. Of the three mammalian Hh ligands, Sonic Hh (Shh), Indian Hh (Ihh), and Desert Hh (Dhh), the most studied and well understood ligand remains Shh. Binding of the Hh ligand to Ptch relieves its inhibitory effect on Smo, which then accumulates in the primary cilium and through a complex signaling cascade, facilitates the recruitment and activation of Gli proteins, which are the mammalian homologues of Ci [15]. Once active, these proteins are translocated to the nucleus, where they function as transcription factors and upregulate their target genes. Importantly, activation of Hh signaling leads to expression of both Ptch and Gli1, where Ptch expression creates a negative feedback loop to regulate both levels and duration of Hh signaling [16]. Gli1 expression is used to amplify target genes, as it functions exclusively as a transcriptional activator [17]. Conversely, in the absence of Hh ligand, Gli proteins are complexed with several binding proteins in the cytosol. Ptch mediated activation of several kinases (CK1, PKA, GSK3) lead to Gli phosphorylation, which in the case of Gli1, targets it for complete proteasomal degradation. Gli3 and to some extent, Gli2 are processed into repressors via proteasome-mediated carboxyl cleavage [18,19]. These repressors are translocated to the nucleus where they downregulate Hh target genes, which include genes important for proliferation (Cyclin D1/D20), cell survival (BCL2), and epithelial to mesenchymal transition (EMT) (Snail1) [20,21]. Hh target genes include a myriad of regulators which include both oncogenes and tumor suppressors, giving rise to a diverse and interconnected signaling system, that when dysregulated can lead to a host of diseases including cancer [22,23,24,25].
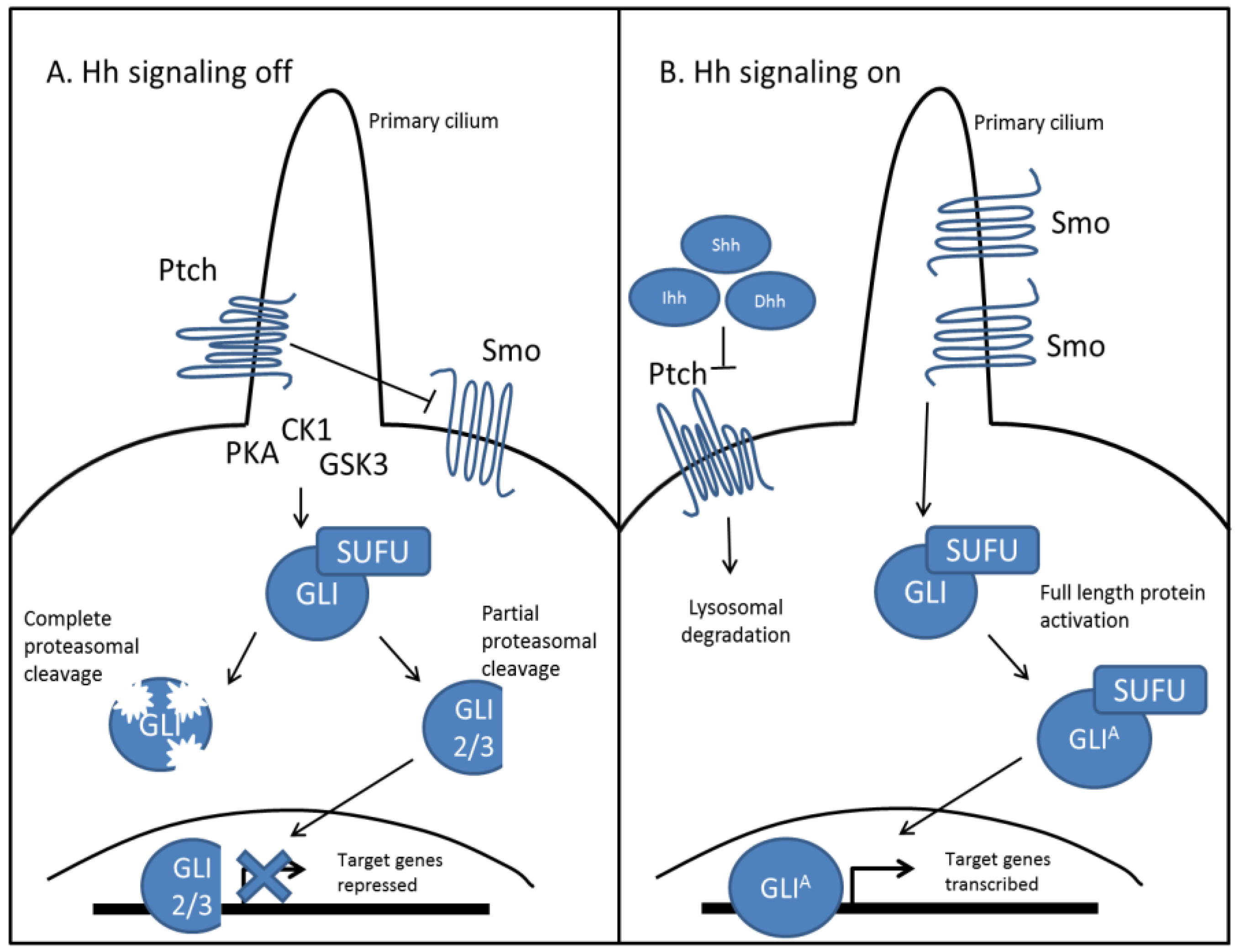
Figure 1.
Canonical Hedgehog signaling in mammals is ligand dependent. (A) In the absence of Hh ligands, Ptch accumulates in the primary cilium and inhibits the function of Smo. Ptch facilitates the activation of several kinases (CK1, PKA, GSK3), at the base of the primary cilium, which differentially phosphorylate Gli protein. This can lead to complete degradation of Gli protein by the proteasome, as well as partial cleavage of Gli2 and Gli3. Partially cleaved Gli is translocated to the nucleus and functions as a transcriptional repressor for Hh target genes; (B) In the presence of Hh ligands, Shh, Ihh, or Dhh binds to Ptch, which induces its lysosomal degradation. This relieves its inhibitory effect on Smo, which accumulates in the primary cilium and prevents degradation of Gli proteins. Activated Gli protein is translocated to the nucleus and functions as a transcriptional activator for Hh target genes.
Figure 1.
Canonical Hedgehog signaling in mammals is ligand dependent. (A) In the absence of Hh ligands, Ptch accumulates in the primary cilium and inhibits the function of Smo. Ptch facilitates the activation of several kinases (CK1, PKA, GSK3), at the base of the primary cilium, which differentially phosphorylate Gli protein. This can lead to complete degradation of Gli protein by the proteasome, as well as partial cleavage of Gli2 and Gli3. Partially cleaved Gli is translocated to the nucleus and functions as a transcriptional repressor for Hh target genes; (B) In the presence of Hh ligands, Shh, Ihh, or Dhh binds to Ptch, which induces its lysosomal degradation. This relieves its inhibitory effect on Smo, which accumulates in the primary cilium and prevents degradation of Gli proteins. Activated Gli protein is translocated to the nucleus and functions as a transcriptional activator for Hh target genes.
2.2. Hedgehog Signaling in Skeletal Development
In vertebrates, both growth and ossification of the skeletal system is achieved via Hh signaling. Early in development, cellular patterning of the limb bud is controlled predominantly by Shh. Acting as a classical morphogen; secretion of Shh forms a spatial and temporal gradient which controls cellular differentiation, polarization, and proliferation [26]. In contrast, Ihh is the predominant ligand governing Hh signaling for its role in bone formation. Proper endochondral ossification is essential for long bone formation. Ihh controls endochondral ossification through a dynamic feedback loop that also controls growth plate development [27]. In this process, Ihh secretion from prehypertrophic chondrocytes at the ends of the bones induces expression of parathyroid hormone-related protein (PTHrP) in periarticular chondrocytes [28,29,30]. PTHrP is secreted and diffuses along the growth plate, which increases chondrocyte proliferation in the growth plate region, and induces them to deposits large amounts of collagenous extra-cellular matrix (ECM), extending the length of the developing bone. Beyond this region, levels of PTHrP drop and chondrocytes no longer proliferate but instead undergo hypertrophy and then apoptosis [31]. Following, the resulting matrix and empty space is invaded by vasculature which is followed by osteoblast mediated mineralization, completing ossification.
Loss of Ihh during bone development has been shown to have detrimental effects on bone elongation and ossification [27]. One group demonstrated that loss of Ihh leads to premature chondrocyte hypertrophy, resulting in significantly shortened limbs lacking ossification. Additionally, levels of osteoblasts were also significantly reduced [32,33]. Several groups have demonstrated that Hh signaling is critical for osteoblast differentiation, which is important for chondrogenesis and cartilage vascularization and thus endochondral ossification [34,35,36]. Intramembranous ossification also requires Hh signaling. Ihh null mice present with underdeveloped calvaria that show reduced ossification [27,37]. Work done to understand this phenotype has demonstrated that Ihh regulates intramembranous ossification via osteogenic differentiation, and to some extent, proliferation [38]. Additionally, in these models, Ihh levels regulate BMP2/4 expression, suggesting that these pathways cooperate to control intramembranous ossification [38]. These findings appear to be central to Hh signaling, as partial loss of Gli3, which mainly functions as a repressor for Hh signaling, leads to increased ossification of calvarial bone. In these mouse models it was observed that loss of one Gli3 allele led to craniosynostosis of the lambdoid sutures in the skull [39,40]. Collectively, this information supports the essential role of Hh signaling in bone development.
2.3. Hedgehog Signaling in Bone Homeostasis
Post-pubescent skeletons undergo constant, albeit low levels of remodeling. The two main cell types that maintain bone homeostasis are osteoblasts and osteoclasts. Osteoblasts are differentiated bone forming cells derived from mesenchymal stem cells, while osteoclasts are differentiated bone resorbing cells derived from monocytes. These cell populations have directly opposing functions that regulate one another in a tightly controlled feedback loop [41]. Osteoblasts express receptor activator of NFkB ligand (RANKL) on their cell surface which binds to RANK on pre-osteoclasts and facilitates their differentiation. As these cells differentiate, they fuse into large multinucleated cells. Differentiated osteoclasts secrete metalloprotein enzymes that are tartate resistant, and are known as tartate resistant acid phosphatase (TRAP) positive cells [42]. Other hallmarks of a functional osteoclast include a “zone of attachment” or sealing zone, where the plasma membrane of the cell adheres to the bone surface through use of integrin mediated podosomes and a resorption pit, the space directly under the osteoclast. The ruffled border, which increases the surface area of the cell that can contact the bone, as well as Cathepsin K secretion are also required for resorption. Cathepsin K is a protease known to catabolize collagen as well as elastin. Combined with the acidic gradient produced in the resorption pit, TRAP, Cathepsin K, and other secreted enzymes, such as matrix metalloproteinases (MMPs) degrade the bone ECM, allowing dissolution of the inorganic component of bone, hydroxyapatite [43]. Importantly, bone resorption releases and activates large amounts of growth factors and cytokines, which are normally sequestered in the ECM. These released signaling molecules have several effects, including contributing to osteoblastogenesis and inducing osteoprotegerin (OPG) expression. OPG is a soluble receptor for RANKL, known as a decoy receptor because it binds free RANKL and therefore blocks RANK/RANKL binding, thus preventing further osteoclastogenesis. This coupling of osteoblast activity to osteoclast activity allows for balanced levels of bone remodeling, where bone is resorbed at about the same rate that it is made [44].
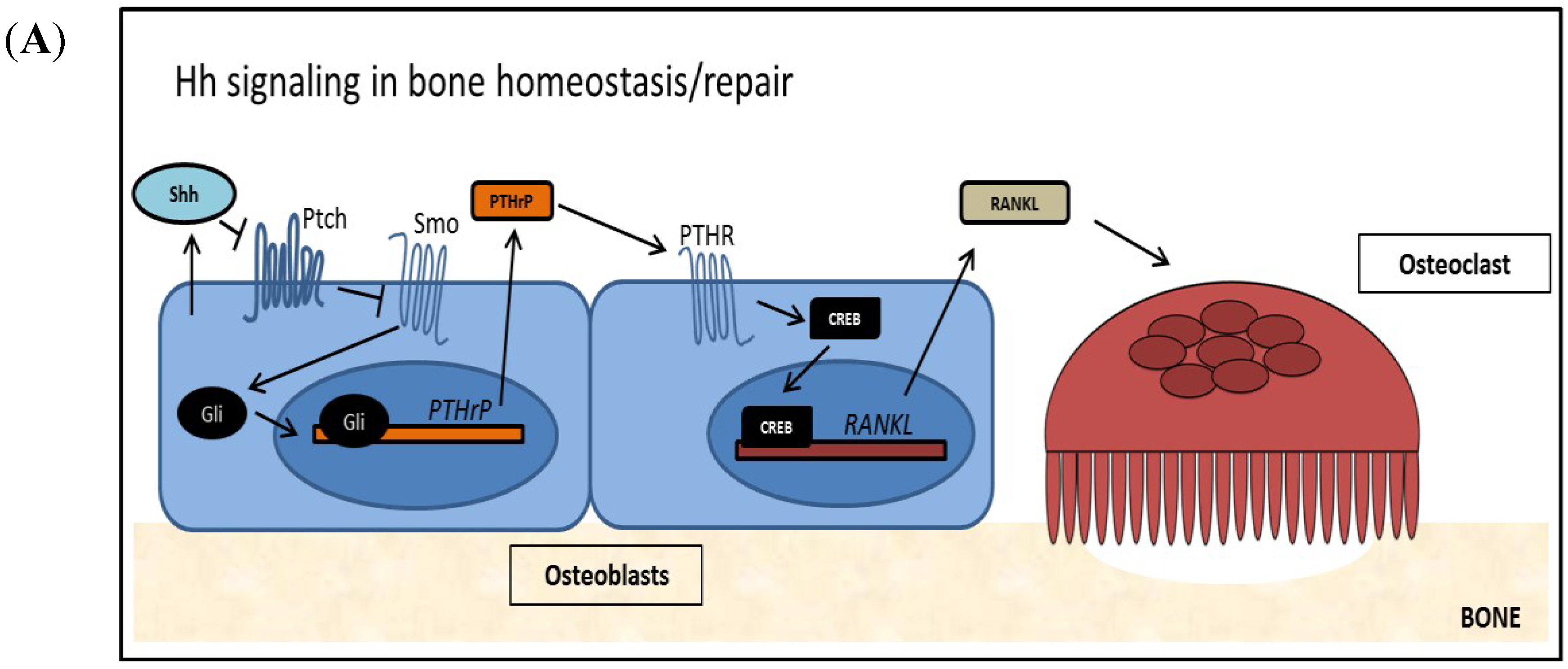
Defects in Hh signaling have been shown to play an important role in bone remodeling. Evidence for this includes the finding that Ptch1 deficient patients show increased bone mass due to increased osteoblast differentiation [45]. Conversely, in a mouse model utilizing activated Hh signaling in mature osteoblasts, bone density and quality were shown to be significantly reduced. This was found to be caused indirectly; in mature osteoblasts Hh signaling induces PTHrP expression which upregulates RANKL, increasing osteoclastogenesis [46]. This finding is physiologically relevant, as Shh is found to be upregulated in mature osteoblasts at fracture sites. Hh signaling in mature osteoblasts has been shown to be key for several processes, such as osteoblast proliferation and differentiation, as well as healing by mediating vascularization [47,48,49]. As outlined in Figure 2A, Hh signaling in mature osteoblasts leads to expression and secretion of PTHrP, which induces expression and secretion of RANKL. RANKL is required for osteoclastogenesis. Mature osteoclasts cause an increase in osteoclast mediated resorption of old and/or damaged bone, which is required to facilitate healing and normal bone turnover.
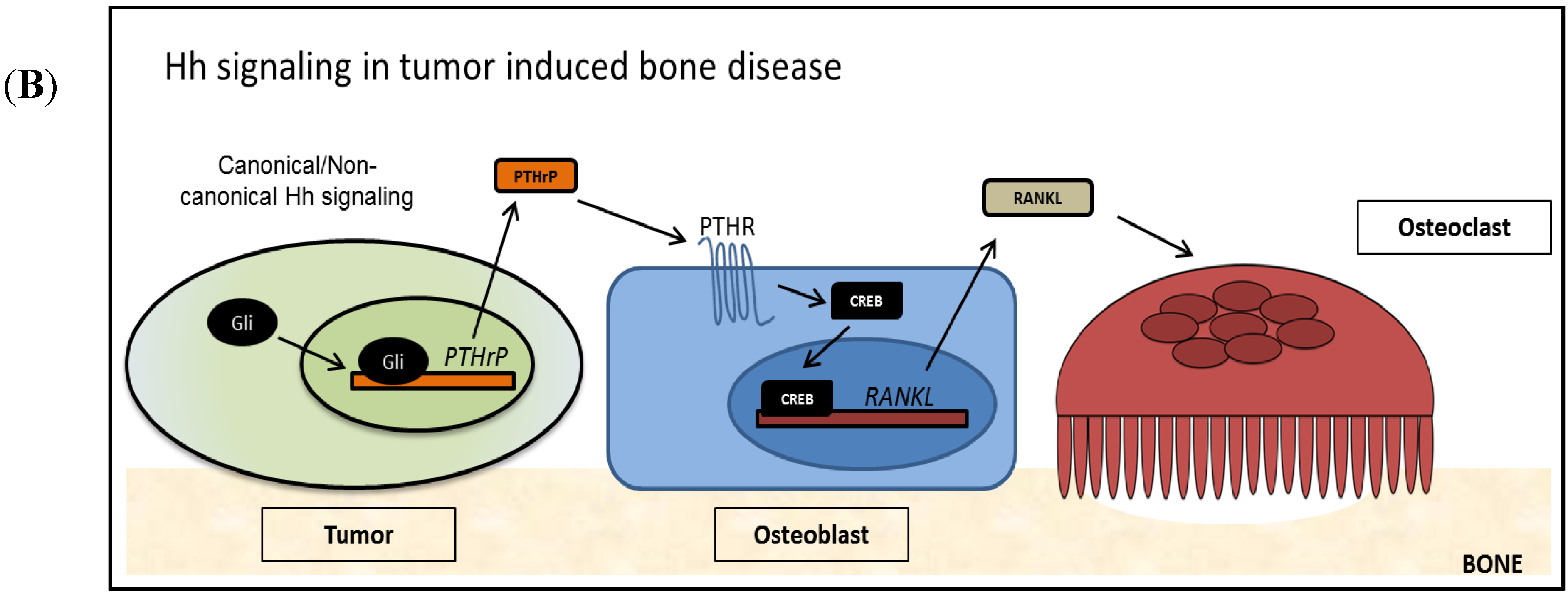
Figure 2.
The Role of Hh Signaling in Bone (A) Hh signaling is active in normal bone remodeling and repair. Osteoblast derived Hh ligand activates Hh signaling cell autonomously as well as non-cell autonomously. PTHrP, a target of Hh signaling, is transcribed and PTHrP is secreted from the cell. Secreted PTHrP binds to its receptor PTHR on the surface of osteoblasts and activates the transcription factor CREB. RANKL, a target of PTHrP/PTHR signaling is expressed and RANKL is presented at the cell surface of osteoblasts, where it induces osteoclastogenesis and supports bone resorption; (B) Tumor-derived Hh signaling disrupts normal bone remodeling and induces TIBD. Tumor cells re-activate Hh signaling by canonical and/or non-canonical mechanisms, increasing Gli activity and Hh target genes. PTHrP is over expressed in these cells, and a large amount of PTHrP is secreted, which activates the PTHR receptor on osteoblasts. Following, large amounts of RANKL are produced by the osteoblasts, which lead to increased osteoclastogenesis and excessive bone resorption.
Figure 2.
The Role of Hh Signaling in Bone (A) Hh signaling is active in normal bone remodeling and repair. Osteoblast derived Hh ligand activates Hh signaling cell autonomously as well as non-cell autonomously. PTHrP, a target of Hh signaling, is transcribed and PTHrP is secreted from the cell. Secreted PTHrP binds to its receptor PTHR on the surface of osteoblasts and activates the transcription factor CREB. RANKL, a target of PTHrP/PTHR signaling is expressed and RANKL is presented at the cell surface of osteoblasts, where it induces osteoclastogenesis and supports bone resorption; (B) Tumor-derived Hh signaling disrupts normal bone remodeling and induces TIBD. Tumor cells re-activate Hh signaling by canonical and/or non-canonical mechanisms, increasing Gli activity and Hh target genes. PTHrP is over expressed in these cells, and a large amount of PTHrP is secreted, which activates the PTHR receptor on osteoblasts. Following, large amounts of RANKL are produced by the osteoblasts, which lead to increased osteoclastogenesis and excessive bone resorption.


3. Hedgehog Signaling in Tumorigenesis and Tumor Induced Bone Disease (TIBD)
3.1. Hedgehog Signaling in Tumorigenesis
Hedgehog signaling has been implicated in several stages of tumorigenesis [50]. The first major finding supporting this came from studying Gorlin syndrome [51,52,53,54]. Patients with Gorlin syndrome present with several skeletal abnormalities, including intracranial calcification, abnormal rib and spine curvature, and cranio-facial defects [55]. This condition is also known as nevoid basal cell carcinoma syndrome as patients often develop basal cell carcinoma (BCC). Research in genetics has identified the cause of this syndrome. A microdeletion of chromosome 9q causes a loss-of-function mutation in the PTCH1 gene [56,57]. This leads to inadequate levels and/or a mutated non-functional version of Ptch, both of which causes haploinsufficiency. In mouse models heterozygous for PTCH1, the murine skeletal abnormalities mirror those of humans [58]. In addition, UV damage is sufficient to induce BCC in mice heterozygous for PTCH1 [59]. It is thought that the ultra violet radiation causes mutations that render the remaining PTCH1 allele non-functional. This supports the finding that sequenced BCC tumors often show PTCH1 deletions, as well as the clinical observations of BCC being most commonly diagnosed on the face, arms, back and chest; areas commonly exposed to sunlight [60,61]. Patients heterozygous for PTCH1 have an increased risk for other tumors as well [62]. Keratocystic odontogenic tumors of the jaw are most common but are benign and are rarely life-threatening. Other tumors, including rhabdomyosarcoma and fibromas can occur. While Gorlin syndrome is an inherited autosomal dominant disorder that causes loss of the tumor suppressor gene PTCH1, it only facilitates tumor development if the function of the other allele is disrupted. However, sporadic BCC and medulloblastoma (MB) frequently occur in patients with both PTCH1 alleles because discrete cellular alterations cause Hh signaling to become constitutively active. This often occurs genetically through mutations that directly inactivate Ptch or activate Smo, but can also occur indirectly if mutations affecting downstream regulatory proteins cause an upregulation of Hh target genes [53,63,64,65,66]. Several studies in MB have supported this finding by identifying mutations in SUFU, GLI1, and GLI2 in tumors, but not in normal surrounding stroma [67,68].
There are several other tumors types that show dysregulation in Hh signaling. For example, GLI1 is named glioma-associated oncogene homolog1 for its association with glioblastoma multiforme (GBM) [69]. Although aberrant Hh signaling has not been found to be a driver mutation in forming these tumors, there remains a role for Hh signaling in other stages of GBM tumorigenesis [70,71,72]. Several groups have shown activated Hh signaling in gliomas, which sustains survival and stemness of glioma cells [73,74,75]. Additionally, Hh inhibition using cyclopamine has been shown to reduce glioma tumorigenicity [73,75,76]. Together these data support the role of Hh signaling in maintaining the tumorigenic potential of glioblastoma.
In breast and prostate cancer, activated Hh signaling is correlated with recurrence, metastasis, and ultimately lower overall survival [77,78,79,80,81,82]. In breast cancer, Hh signaling components play several roles. Over-expression of Hh ligands is associated with a basal-like phenotype that leads to poor prognosis in patients [77]. Similarly, activated Gli1 protein correlates with higher risk of recurrence after surgery [77,78,79]. Importantly, Hh signaling can be activated as a result of signaling cooperativity with a number of other signaling pathways [83,84,85,86,87,88,89,90,91]. For example, in breast cancer cells responsive to estrogen (ER+), estrogen stimulation induces GLI1 expression, which is important for invasion and cell renewal [92]. In breast cancer cells that acquire resistance to anti-estrogen therapies, Hh signaling remains activated downstream of the receptor via the PI3K/AKT pathway [93]. Additionally, Hh ligand secreted from the tumors can stimulate Hh signaling in the tumor cells, but the microenvironment also plays an important part in responding to secreted Hh ligand [94]. The stimulated stromal cells in the microenvironment cause dynamic changes that can facilitate tumor progression by inducing vascularization and migration associated genes [95]. Thus in breast cancer, activated Hh signaling can support tumorigenesis by facilitating tumor progression as well as modulating the microenvironment to a pro-tumorigenic state.
Hedgehog pathway activation is also observed in prostate cancer, where activation can be detected in both the tumor and the stroma [96,97,98]. Hh signaling has been found to become upregulated in prostate cancer cells when they are deprived of androgen for long periods of time [99]. Additionally, Hh signaling remains activated in androgen-independent cells [100,101]. Research done by Chen and colleagues demonstrate that upregulated Hh signaling supports androgen signaling by enhancing androgen specific gene expression [101,102]. Other tumor types that use cooperative signaling of Hh signaling and other pathways to promote tumorigenesis include pancreatic cancer, malignant melanoma, gastric/colon cancer, lung and ovarian cancer, which are well outlined in this recent review [103].
3.2. Hedgehog Signaling in Tumor Induced Bone Disease
Advanced stage tumors, as well as aggressive tumors have an increased propensity to metastasize to distant sites of the body. Along with the lung and the liver, the bone is a common site of metastasis, where tumors in the bone disrupt normal bone remodeling and cause TIBD [104]. In patients, TIBD causes bone pain, hypercalcemia, and an increased risk of fracture, along with reduced skeletal mass and bone lesions [105]. Many tumor types, including breast, prostate, and lung, metastasize to bone via the circulatory system, while other tumors such as head and neck cancer invade directly into the facial bones from surrounding tissues. Primary bone tumors, such as osteosarcoma and multiple myeloma, also cause bone disease. While the molecular mechanisms controlling bone disease varies by tumor type, all of the above mentioned tumors dysregulate normal bone remodeling and induce excessive bone resorption in a process coined “The Vicious Cycle” [106]. In the vicious cycle, tumor cells that arrive at the bone microenvironment respond to chemical and physical cues by secreting factors that stimulate bone resorption. Cytokines such TGFβ and BMP present due to normal bone remodeling can stimulate tumor cells [107]. Additionally, our group has shown that the rigidity of the bone matrix can stimulate gene expression via mechanotransduction signaling [108]. As outlined in Figure 2B, stimulated tumor cells, in a cell type specific manner, re-activate Hh signaling, which can be achieved by activating mutations in canonical Hh signaling receptor proteins Smo and Ptch, or non-canonically through other signaling pathways that lead to Gli activation such as TGFβ, PI3K/AKT, ERK and others. Increased Gli levels lead to expression and secretion of parathyroid hormone-related protein (PTHrP), which increases RANKL production in osteoblasts, stimulating osteoclastogenesis. Additional signaling factors such as interleukins 6/8/11, and TNF-α, also increase osteoclastogenesis in a similar manner, but have not been shown to be directly dependent on Hh signaling [109,110,111,112]. Ultimately, as an increased number of osteoclasts mature and resorb bone, the released growth factors from the ECM further stimulate tumor cells, which causes a positive feedback loop. Tumor cells in the bone microenvironment uncouple osteoblast/osteoclast signaling, and lead to increased bone resorption as well as impair bone quality.
In breast cancer metastases to bone, this process is mediated by Hh signaling. Using bone metastatic MDA-MB-231 breast cancer cells, we have shown that TGFβ stimulation increases expression of Gli2 and PTHrP, which controls bone destruction [112]. In an experimental model of metastasis, tumor cells injected into the left cardiac ventricle home to the skeleton where they proliferate and induce bone destruction. We show that when Gli2 expression is repressed using a plasmid over-expressing Gli2 repressor, both PTHrP expression and bone destruction are significantly reduced [113]. In this system, Gli activity does not seem to be regulated canonically, as bone tropic MDA-MB-231 cells do not express the Smo receptor nor do they respond to cyclopamine treatment [112]. A similar observation can be seen in malignant melanoma. Interestingly, while the melanoma cell lines used showed Smo and Ptch gene expression, Gli2 was found to be a transcriptional target of TGFβ signaling. Using 1205Lu melanoma cells that express high levels of Gli2, the authors showed that loss of Gli2 using shRNA led to a decreased incidence of bone metastases as well as smaller lesions [114].
Not surprisingly, primary bone tumors such as chondrosarcoma and osteosarcoma also dysregulate bone remodeling. Chondrosarcoma tumors originate from cartilage and are thought to arise due to activating mutations in Hh signaling [115]. Studies have shown that many primary chondrosarcomas express all canonical Hh signaling proteins, as well as PTHrP [115,116,117,118,119]. Using these tissues in primary organ cultures, one group showed that Hh signaling was constitutively active due to loss of PTCH1/2, activation of Smo or a combination of both. Additionally, using the Hh inhibitor, triparanol, in a xenograph mouse model, the authors observed decreased proliferation, cellularity, and tumor size [115].
Aberrant Hh signaling inducing TIBD is also observed in head and neck tumors. Oral squamous cell carcinoma (OSCC), which accounts for almost 90% of all head and neck tumors show activated Hh signaling as measured by immunohistochemical staining of Hh proteins in patient biopsies [120]. Levels of Hh proteins also correlate with poor prognosis [121]. A subset of OSCC patients are affected by mandibular invasion, where the tumor directly invades the mandible through underlying tissue and begins to proliferate in the bone, stimulating bone destruction [122,123]. One group in Japan demonstrated that loss of Shh in OSCC cells decreased tumor volume as well as bone destruction in an in vivo model of bone destruction [124]. Here tumor cells stably expressing shRNA against Shh were inoculated into the tibia of immunocompromised mice. Our group has also observed OSCC dependency on Hh signaling. We utilized an orthotopic model of mandibular invasion and bone destruction, where tumor cells are injected into the masseter muscle and invade directly through underlying tissues into the mandible. Using this model, we injected human OSCC cells stably expressing shRNA against Gli2 and observed that loss of Gli2 decreased both bony invasion and bone destruction in male athymic mice. These studies highlight the importance of Hh signaling on TIBD and suggest that Hh signaling components are viable targets to prevent TIBD.
4. Potential Therapeutics to Target Hh Signaling Components
Due to its role in tumorigenesis, Hh signaling inhibitors are a promising potential therapy to inhibit tumor growth and disease, and, thus, have been tested as anti-tumor agents extensively [125,126,127]. The natural plant alkaloid cyclopamine was the first compound identified as an inhibitor of Hh signaling [128]. In animal models, cyclopamine demonstrated great potential and was found to inhibit tumor growth in several tumor types, including BCC, MB, breast, and pancreatic cancer [81,90,129,130,131,132,133]. Unfortunately, use of cyclopamine in vivo revealed that the drug had low oral bioavailability, a short elimination half-life, and serious adverse effects due to toxicity [134]. However, with the development of synthetic and semi-synthetic compounds, as well as the identification of additional inhibitors using high throughput screening, Smo has been shown to be an excellent druggable target. Vismodegid, TAK-441, IPI-926, Saridegib, Sonidegib/Erismodegib, BMS-833923/XL139, PF-04449913, Taladegib/LY2940680, and CUR61414 represent a newer class of small molecule inhibitors with increased potency for Smo and an improved pharmacokinetic profile [135,136,137,138,139,140,141,142,143,144,145,146,147,148]. Several clinical trials have demonstrated the efficacy of these compounds, especially in tumor types with activating mutations in Hh signaling due to Smo [142,143,144,147,149,150,151,152]. In January 2012, the FDA approved Vismodegib for use in locally advanced and metastatic BCC, after a Phase II clinical trial showed response rates over 30% [149]. Even so, patients on Smo inhibitors relapse due to acquired mutations in Smo or reactivation of Hh signaling downstream through other signaling pathways affecting Gli activity [88,91,141,153,154,155,156,157,158]. Unfortunately, these patients relapse in a matter of months and fail to respond to additional Smo inhibition. While therapies to target drug-resistant tumors are being developed, inhibition of Hh signaling by targeting downstream modulators is actively being investigated [159]. Importantly, tumors that show resistance to Smo inhibitors retain sensitivity to Gli inhibitors and when treated, significant reductions in Hh target genes are observed [160].
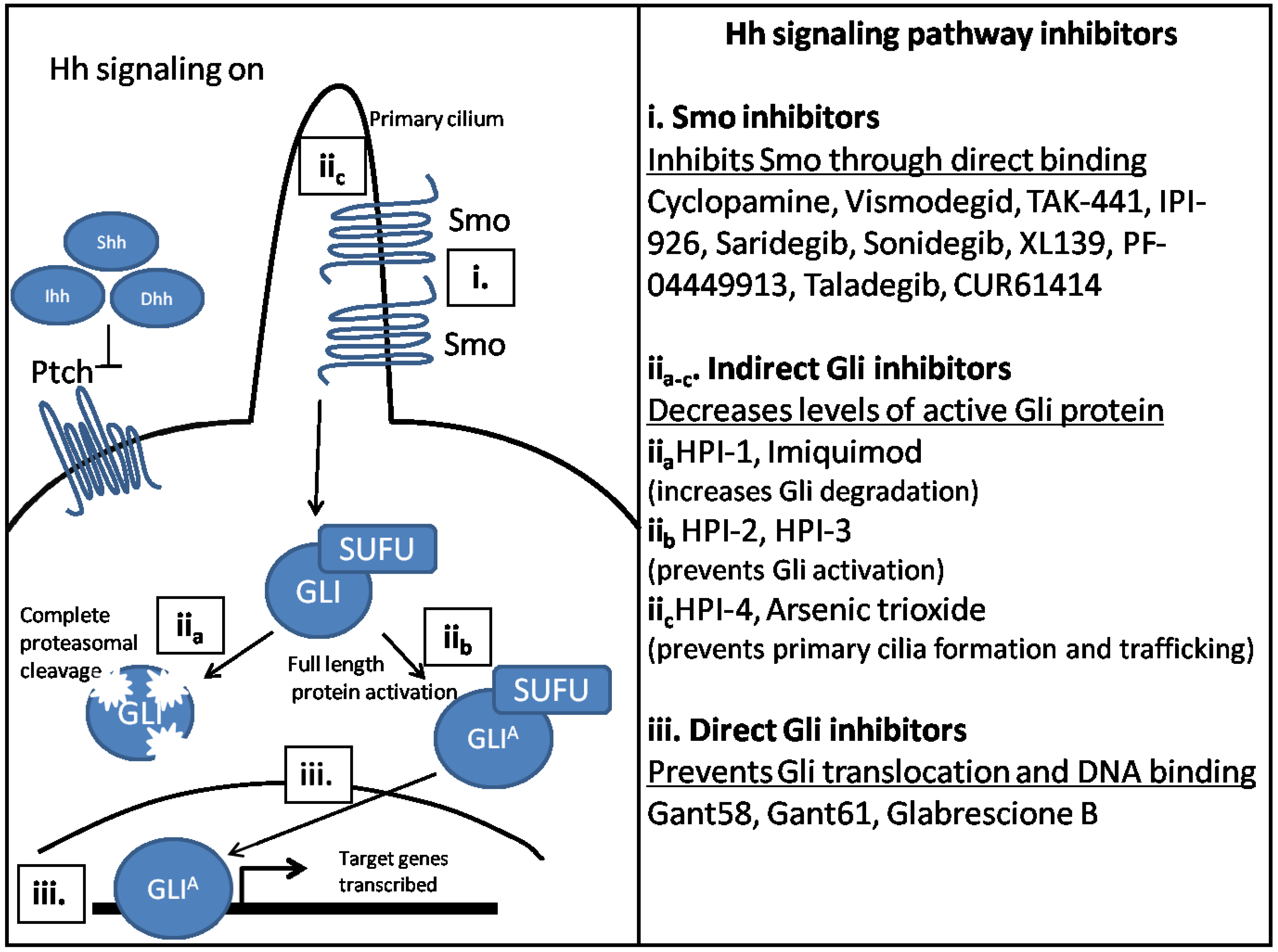
Thus, the field has now transitioned to developing Hh inhibitors that target Hh signaling downstream of Smo. Gli inhibition has been proposed as the ultimate target, as it is the effector molecule for Hh signaling. Inhibitors of Hh signaling that indirectly affect Gli are also being investigated. Gli inhibition would enable downregulation of Hh signaling in non-canonical mechanisms of Hh signaling activation, such as PI3K activation. Inhibitors that directly and indirectly inhibit Gli have been identified, and are described in Figure 3. Gli inhibitors are the most attractive as drug targets because they are most downstream on the Hh signaling pathway, decreasing the likelihood of Hh reactivation.
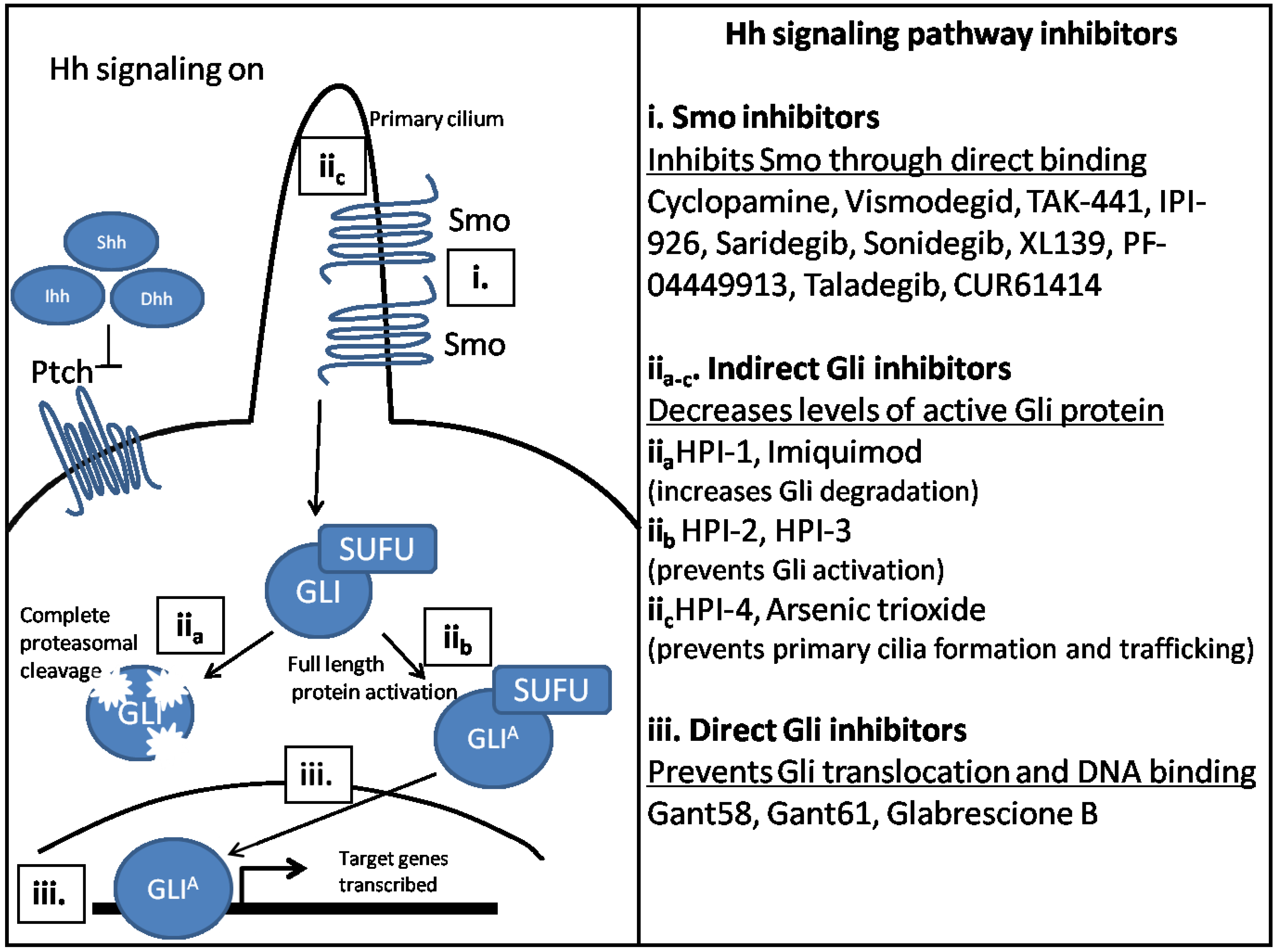
Figure 3.
Inhibitors of the Hh signaling pathway target different Hh signaling components. (i) Smo is the first identified target for Hh inhibition and remains the best studied. Smo inhibitors function by binding directly to Smo and preventing downstream activation; (ii) Indirect Gli inhibitors inhibit Hh signaling downstream of Smo, but have differing mechanisms. HPI-1 and Imiquimod increase Gli degradation (iia), while HPI-2 and HPI-3 prevent Gli protein activation (iib). HPI-4 inhibits ciliogenesis, while arsenic trioxide prevents Gli trafficking to the primary cilium (iic); (iii) Direct Gli inhibitors inhibit Hh signaling by binding directly to Gli protein and inhibiting its transcriptional activity. GANT58 prevents Gli translocation to the nucleus. GANT61 and Glabrescione B inhibit transcription of Hh target genes, by interfering with the DNA binding pocket of Gli protein.
Figure 3.
Inhibitors of the Hh signaling pathway target different Hh signaling components. (i) Smo is the first identified target for Hh inhibition and remains the best studied. Smo inhibitors function by binding directly to Smo and preventing downstream activation; (ii) Indirect Gli inhibitors inhibit Hh signaling downstream of Smo, but have differing mechanisms. HPI-1 and Imiquimod increase Gli degradation (iia), while HPI-2 and HPI-3 prevent Gli protein activation (iib). HPI-4 inhibits ciliogenesis, while arsenic trioxide prevents Gli trafficking to the primary cilium (iic); (iii) Direct Gli inhibitors inhibit Hh signaling by binding directly to Gli protein and inhibiting its transcriptional activity. GANT58 prevents Gli translocation to the nucleus. GANT61 and Glabrescione B inhibit transcription of Hh target genes, by interfering with the DNA binding pocket of Gli protein.
The Gli antagonists, GANT58 and GANT61, were the first small molecule inhibitors determined to inhibit Hh signaling at the level of Gli protein [161]. Recently, another Gli inhibitor, Glabrescione B has been identified [162]. Glabrescione B binds directly to the DNA binding site of Gli1 while GANT61 binds indirectly, but still prevents Gli mediated transcription [162,163]. GANT58 in contrast, prevents Gli translocation to the nucleus [161]. GANT61 has been shown to inhibit tumor growth in several mouse models of tumor burden [163,164,165]. Additionally, Glabrescione B has been shown to be efficacious in decreasing tumor burden in tumors over-expressing Hh signaling components [162]. While these drugs show great promise both in vitro and in pre-clinical studies, it remains unclear if any are potential candidates for clinical trials.
Four Hh pathway inhibitors (HPI-1-4) have been identified by Hyman and colleagues [166]. These compounds inhibit processing and trafficking of Gli protein and each inhibitor has been found to have a distinct mechanism of action. HPI-1 causes an increase in repressor forms of Gli, while HPI-2 and HPI-3 both prevent Gli protein activation [166]. HPI-4 inhibits ciliogenesis, preventing both Gli accumulation and processing in the primary cilium [166]. Imiquimod, which is used to treat BCC, also inhibits Hh signaling downstream of Smo [4,167]. Known as an agonist of Toll-like receptor 7/8, imiquimod activates PKA, which induces increased degradation of Gli proteins. Arsenic trioxide is another inhibitor of Hh signaling, which is used for treatment of acute promyelocytic leukemia [168]. This drug prevents Gli trafficking to the primary cilium, thus preventing its activation. Newly emerging studies, investigating the use of epigenetic silencing and indirect inhibitors to target Hh signaling have been conducted [169,170,171,172,173,174,175].
Currently, there have been several murine studies exploring the efficacy of a variety of Gli inhibitors in different tumor models [176,177,178,179]. While the use of Gli inhibitors have been promising, the greatest challenge that remains is the lack of understanding molecular mechanisms surrounding Gli mediated transcription, especially in the context of other active oncogenic pathways. This setback has made Hh inhibition in TIBD especially challenging. Tumors that metastasize to and establish in bone represent a subset of the primary tumor with differential expression of tumorigenic signaling pathways. In the case of breast, prostate, and lung cancer, Hh signaling is known to contribute to tumor progression and a stem-cell like phenotype, but is not found to be a driving mutation. While work from our group demonstrates the importance of Gli on bone destruction in breast to bone metastasis, additional research is required to understand mechanisms in other tumor types. However, based on the increasing number of studies involving Gli inhibition to target Hh signaling, the field is primed to move forward in identifying novel Gli inhibitors as well as elucidating mechanisms controlling dysregulated Hh signaling in cancer progression.
5. Conclusions
Due to its role in tumor progression, metastasis, and treatment failure in many different tumor types, Hh signaling remains an appealing target for the development of therapeutics. Despite its promise in pre-clinical and early clinical studies, Smo inhibition alone is no longer considered an effective treatment for the inhibition of cancer growth, due to patient relapse and resistance. Because of this, combination therapies are currently being tested to evaluate efficacy in several different tumor types [180,181,182,183,184,185,186,187]. Additionally, several pre-clinical compounds targeting downstream of Smo and specifically Gli show great potential, but have proven challenging. Important remaining questions in the field are two-pronged. The first seeks to identify Gli inhibitors that are superior to Smo inhibitors, but are feasible as therapeutics in Hh driven cancers. The second is focused on identifying pragmatic applications of Gli inhibitors in preclinical models of tumorigenesis to prevent progression. This second point is especially important, as information concerning drug mechanisms, pharmacokinetic profiles as well as short-term/long-term side effects is still largely unknown in many tumor types. As additional novel inhibitors are discovered and designed, a better understanding of how Hh mechanisms change in response to inhibition will be required to help refine existing therapies to show more clinical success.
Acknowledgments
Work from the authors’ laboratory was supported by grants from the Department of Veterans Affairs (1I01BX001957) and the NCI (1R01CA163499). We apologize to all colleagues whose work has not been cited due to space limitations.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Petrova, R.; Joyner, A.L. Roles for hedgehog signaling in adult organ homeostasis and repair. Development 2014, 141, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernandez-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Hatton, B.A.; Villavicencio, E.H.; Khanna, P.C.; Friedman, S.D.; Ditzler, S.; Pullar, B.; Robison, K.; White, K.F.; Tunkey, C.; et al. Hedgehog pathway inhibitor saridegib (ipi-926) increases lifespan in a mouse medulloblastoma model. Proc. Natl. Acad. Sci. USA 2012, 109, 7859–7864. [Google Scholar] [CrossRef] [PubMed]
- Gruber, W.; Frischauf, A.M.; Aberger, F. An old friend with new skills: Imiquimod as novel inhibitor of hedgehog signaling in basal cell carcinoma. Oncoscience 2014, 1, 567–573. [Google Scholar] [PubMed]
- Nusslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Fuccillo, M.; Joyner, A.L.; Fishell, G. Morphogen to mitogen: The multiple roles of hedgehog signalling in vertebrate neural development. Nat. Rev. Neurosci. 2006, 7, 772–783. [Google Scholar] [CrossRef] [PubMed]
- Dessaud, E.; McMahon, A.P.; Briscoe, J. Pattern formation in the vertebrate neural tube: A sonic hedgehog morphogen-regulated transcriptional network. Development 2008, 135, 2489–2503. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.T.; He, W.; Hao, C.; Ketova, T.; Pan, F.C.; Wright, C.C.; Litingtung, Y.; Chiang, C. The purkinje neuron acts as a central regulator of spatially and functionally distinct cerebellar precursors. Dev. Cell 2013, 27, 278–292. [Google Scholar] [CrossRef] [PubMed]
- Kenney, A.M.; Cole, M.D.; Rowitch, D.H. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 2003, 130, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Fernandez-Barrena, M.G.; Ortiz-Ruiz, M.C.; Almada, L.L.; Hu, C.; Elsawa, S.F.; Mills, L.D.; Romecin, P.A.; Gulaid, K.H.; Moser, C.D.; et al. Activation of the transcription factor gli1 by wnt signaling underlies the role of sulfatase 2 as a regulator of tissue regeneration. J. Biol. Chem. 2013, 288, 21389–21398. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Andre, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of gli2 and gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Hojo, H.; Ohba, S.; Taniguchi, K.; Shirai, M.; Yano, F.; Saito, T.; Ikeda, T.; Nakajima, K.; Komiyama, Y.; Nakagata, N.; et al. Hedgehog-gli activators direct osteo-chondrogenic function of bone morphogenetic protein toward osteogenesis in the perichondrium. J. Biol. Chem. 2013, 288, 9924–9932. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Liu, C.; Zhao, Y. Decoding ci: From partial degradation to inhibition. Dev. Growth Differ. 2015, 57, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; Placzek, M. Orchestrating ontogenesis: Variations on a theme by sonic hedgehog. Nat. Rev. Genet. 2006, 7, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Alman, B.A. The role of hedgehog signalling in skeletal health and disease. Nat. Rev. Rheumatol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Platt, K.A.; Censullo, P.; Ruiz i Altaba, A. Gli1 is a target of sonic hedgehog that induces ventral neural tube development. Development 1997, 124, 2537–2552. [Google Scholar] [PubMed]
- Wang, B.; Fallon, J.F.; Beachy, P.A. Hedgehog-regulated processing of gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 2000, 100, 423–434. [Google Scholar] [CrossRef]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic hedgehog signaling regulates gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3365–3377. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Li, X.U.; Zhou, C.; Wen, Y.; Shen, Y.; Zhou, L.; Li, J. Effects and mechanisms of blocking the hedgehog signaling pathway in human gastric cancer cells. Oncol. Lett. 2015, 9, 1997–2002. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Mei, L.; Pan, L.; Xiong, W.; Zhu, H.; Ruan, H.; Zou, C.; Tang, L.; Iguchi, T.; Wu, X. Hedgehog signaling through gli1 and gli2 is required for epithelial-mesenchymal transition in human trophoblasts. Biochim. Biophys. Acta 2015, 1850, 1438–1448. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Fan, Q.; Zhang, X.; Xie, J. A role for transcription factor stat3 signaling in oncogene smoothened-driven carcinogenesis. J. Biol. Chem. 2012, 287, 38356–38366. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.H.; Jung, D.E.; Park, Y.N.; Song, S.Y.; Park, S.W. Aberrant hedgehog ligands induce progressive pancreatic fibrosis by paracrine activation of myofibroblasts and ductular cells in transgenic zebrafish. PLoS ONE 2011, 6, e27941. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting wnt, notch, and hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Kicheva, A.; Cohen, M.; Briscoe, J. Developmental pattern formation: Insights from physics and biology. Science 2012, 338, 210–212. [Google Scholar] [CrossRef] [PubMed]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Kindblom, J.M.; Nilsson, O.; Hurme, T.; Ohlsson, C.; Savendahl, L. Expression and localization of indian hedgehog (Ihh) and parathyroid hormone related protein (PTHrP) in the human growth plate during pubertal development. J. Endocrinol. 2002, 174, R1–R6. [Google Scholar] [CrossRef] [PubMed]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Brauer, P.R.; Xiao, L.; McGuire, M.H.; Yee, J.A. Expression of parathyroid hormone-related peptide (PTHrP) and its receptor (PTH1R) during the histogenesis of cartilage and bone in the chicken mandibular process. J. Anat. 2002, 201, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Karperien, M.; Lanser, P.; de Laat, S.W.; Boonstra, J.; Defize, L.H. Parathyroid hormone related peptide mRNA expression during murine postimplantation development: Evidence for involvement in multiple differentiation processes. Int. J. Dev. Biol. 1996, 40, 599–608. [Google Scholar] [PubMed]
- Long, F.; Linsenmayer, T.F. Regulation of growth region cartilage proliferation and differentiation by perichondrium. Development 1998, 125, 1067–1073. [Google Scholar] [PubMed]
- Chung, U.I.; Schipani, E.; McMahon, A.P.; Kronenberg, H.M. Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J. Clin. Investig. 2001, 107, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Long, F.; Chung, U.I.; Ohba, S.; McMahon, J.; Kronenberg, H.M.; McMahon, A.P. Ihh signaling is directly required for the osteoblast lineage in the endochondral skeleton. Development 2004, 131, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Joeng, K.S.; Long, F. The Gli2 transcriptional activator is a crucial effector for Ihh signaling in osteoblast development and cartilage vascularization. Development 2009, 136, 4177–4185. [Google Scholar] [CrossRef] [PubMed]
- Abzhanov, A.; Rodda, S.J.; McMahon, A.P.; Tabin, C.J. Regulation of skeletogenic differentiation in cranial dermal bone. Development 2007, 134, 3133–3144. [Google Scholar] [CrossRef] [PubMed]
- Lenton, K.; James, A.W.; Manu, A.; Brugmann, S.A.; Birker, D.; Nelson, E.R.; Leucht, P.; Helms, J.A.; Longaker, M.T. Indian hedgehog positively regulates calvarial ossification and modulates bone morphogenetic protein signaling. Genesis 2011, 49, 784–796. [Google Scholar] [CrossRef] [PubMed]
- Rice, D.P.; Connor, E.C.; Veltmaat, J.M.; Lana-Elola, E.; Veistinen, L.; Tanimoto, Y.; Bellusci, S.; Rice, R. Gli3xt-j/xt-j mice exhibit lambdoid suture craniosynostosis which results from altered osteoprogenitor proliferation and differentiation. Hum. Mol. Genet. 2010, 19, 3457–3467. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.; Seelow, D.; Jehee, F.S.; Perlyn, C.A.; Alonso, L.G.; Bueno, D.F.; Donnai, D.; Josifova, D.; Mathijssen, I.M.; Morton, J.E.; et al. Rab23 mutations in carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am. J. Hum. Genet. 2007, 80, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Raggatt, L.J.; Partridge, N.C. Cellular and molecular mechanisms of bone remodeling. J. Biol. Chem. 2010, 285, 25103–25108. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Takahashi, N.; Martin, T.J. Modulation of osteoclast differentiation. Endocr. Rev. 1992, 13, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Tyrovola, J.B. The “mechanostat theory” of frost and the OPG/Rankl/RANK system. J. Cell. Biochem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S.; Kawaguchi, H.; Kugimiya, F.; Ogasawara, T.; Kawamura, N.; Saito, T.; Ikeda, T.; Fujii, K.; Miyajima, T.; Kuramochi, A.; et al. Patched1 haploinsufficiency increases adult bone mass and modulates Gli3 repressor activity. Dev. Cell 2008, 14, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.K.; Bi, Y.; Wan, C.; Chuang, P.T.; Clemens, T.; Young, M.; Yang, Y. Hedgehog signaling in mature osteoblasts regulates bone formation and resorption by controlling PTHrP and RANKL expression. Dev. Cell 2008, 14, 674–688. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Akiyama, H.; Shigeno, C.; Iyama, K.; Matsuoka, H.; Nakamura, T. Hedgehog signaling molecules in bone marrow cells at the initial stage of fracture repair. Biochem. Biophys. Res. Commun. 1999, 262, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Miyaji, T.; Nakase, T.; Iwasaki, M.; Kuriyama, K.; Tamai, N.; Higuchi, C.; Myoui, A.; Tomita, T.; Yoshikawa, H. Expression and distribution of transcripts for sonic hedgehog in the early phase of fracture repair. Histochem. Cell Biol. 2003, 119, 233–237. [Google Scholar] [PubMed]
- Horikiri, Y.; Shimo, T.; Kurio, N.; Okui, T.; Matsumoto, K.; Iwamoto, M.; Sasaki, A. Sonic hedgehog regulates osteoblast function by focal adhesion kinase signaling in the process of fracture healing. PLoS ONE 2013, 8, e76785. [Google Scholar] [CrossRef] [PubMed]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Christiansen, J.; Wicking, C.; Zaphiropoulos, P.G.; Chidambaram, A.; Gerrard, B.; Vorechovsky, I.; Bale, A.E.; Toftgard, R.; Dean, M.; et al. A mammalian patched homolog is expressed in target tissues of sonic hedgehog and maps to a region associated with developmental abnormalities. J. Biol. Chem. 1996, 271, 12125–12128. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Gailani, M.R.; Stahle-Backdahl, M.; Leffell, D.J.; Glynn, M.; Zaphiropoulos, P.G.; Pressman, C.; Unden, A.B.; Dean, M.; Brash, D.E.; Bale, A.E.; et al. The role of the human homologue of drosophila patched in sporadic basal cell carcinomas. Nat. Genet. 1996, 14, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Bonifas, J.M.; Pennypacker, S.; Chuang, P.T.; McMahon, A.P.; Williams, M.; Rosenthal, A.; de Sauvage, F.J.; Epstein, E.H., Jr. Activation of expression of hedgehog target genes in basal cell carcinomas. J. Investig. Dermatol. 2001, 116, 739–742. [Google Scholar] [CrossRef] [PubMed]
- Gorlin, R.J. Nevoid basal-cell carcinoma syndrome. Medicine (Baltimore) 1987, 66, 98–113. [Google Scholar] [CrossRef] [PubMed]
- Muller, E., II; Hudgins, L. 9q22.3 Microdeletion. In GeneReviews®; Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Dolan, C.R., Fong, C.T., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Muller, E.A.; Aradhya, S.; Atkin, J.F.; Carmany, E.P.; Elliott, A.M.; Chudley, A.E.; Clark, R.D.; Everman, D.B.; Garner, S.; Hall, B.D.; et al. Microdeletion 9q22.3 syndrome includes metopic craniosynostosis, hydrocephalus, macrosomia, and developmental delay. Am. J. Med. Genet. A 2012, 158A, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wojnowski, L.; Zimmer, A.M.; Hall, J.; Miller, G.; Zimmer, A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of gorlin syndrome. Nat. Med. 1998, 4, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Nitzki, F.; Becker, M.; Frommhold, A.; Schulz-Schaeffer, W.; Hahn, H. Patched knockout mouse models of basal cell carcinoma. J. Skin Cancer 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, A.; Levesque, M.P.; Dummer, R.; Kabashima, K. Hedgehog signaling in basal cell carcinoma. J. Dermatol. Sci. 2015, 78, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Marzuka, A.G.; Book, S.E. Basal cell carcinoma: Pathogenesis, epidemiology, clinical features, diagnosis, histopathology, and management. Yale J. Biol. Med. 2015, 88, 167–179. [Google Scholar] [PubMed]
- Gorlin, R.J. Nevoid basal cell carcinoma syndrome. Dermatol. Clin. 1995, 13, 113–125. [Google Scholar] [PubMed]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Kohler, B.; Schonicke, A.; Scharwachter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, T.; Waha, A.; Koch, A.; Kraus, J.; Albrecht, S.; Tonn, J.; Sorensen, N.; Berthold, F.; Henk, B.; Schmandt, N.; et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of drosophila patched. Cancer Res. 1997, 57, 2085–2088. [Google Scholar] [PubMed]
- Raffel, C.; Jenkins, R.B.; Frederick, L.; Hebrink, D.; Alderete, B.; Fults, D.W.; James, C.D. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997, 57, 842–845. [Google Scholar] [PubMed]
- Wolter, M.; Reifenberger, J.; Sommer, C.; Ruzicka, T.; Reifenberger, G. Mutations in the human homologue of the drosophila segment polarity gene patched (PTCH) in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1997, 57, 2581–2585. [Google Scholar] [PubMed]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Northcott, P.A.; Hielscher, T.; Dubuc, A.; Mack, S.; Shih, D.; Remke, M.; Al-Halabi, H.; Albrecht, S.; Jabado, N.; Eberhart, C.G.; et al. Pediatric and adult sonic hedgehog medulloblastomas are clinically and molecularly distinct. Acta Neuropathol. 2011, 122, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Kinzler, K.W.; Bigner, S.H.; Bigner, D.D.; Trent, J.M.; Law, M.L.; O’Brien, S.J.; Wong, A.J.; Vogelstein, B. Identification of an amplified, highly expressed gene in a human glioma. Science 1987, 236, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Hui, A.B.; Lo, K.W.; Yin, X.L.; Poon, W.S.; Ng, H.K. Detection of multiple gene amplifications in glioblastoma multiforme using array-based comparative genomic hybridization. Lab. Investig. 2001, 81, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Hamoudi, R.A. Molecular and cytogenetic analysis of glioblastoma multiforme. Cancer Genet. Cytogenet. 2000, 122, 87–92. [Google Scholar] [CrossRef]
- Frattini, V.; Trifonov, V.; Chan, J.M.; Castano, A.; Lia, M.; Abate, F.; Keir, S.T.; Ji, A.X.; Zoppoli, P.; Niola, F.; et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 2013, 45, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. Hedgehog-gli1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Ehtesham, M.; Sarangi, A.; Valadez, J.G.; Chanthaphaychith, S.; Becher, M.W.; Abel, T.W.; Thompson, R.C.; Cooper, M.K. Ligand-dependent activation of the hedgehog pathway in glioma progenitor cells. Oncogene 2007, 26, 5752–5761. [Google Scholar] [CrossRef] [PubMed]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Liu, X.; Chen, L.; Dou, Z.; Lei, X.; Chang, L.; Cai, J.; Cui, Y.; Yang, D.; Sun, Y.; et al. Targeting the SMO oncogene by miR-326 inhibits glioma biological behaviors and stemness. Neuro Oncol. 2015, 17, 243–253. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, S.A.; Machalek, D.A.; Shearer, R.F.; Millar, E.K.; Nair, R.; Schofield, P.; McLeod, D.; Cooper, C.L.; McNeil, C.M.; McFarland, A.; et al. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer Res. 2011, 71, 4002–4014. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Sheen, I.S.; Jeng, W.J.; Yu, M.C.; Hsiau, H.I.; Chang, F.Y. High expression of sonic hedgehog signaling pathway genes indicates a risk of recurrence of breast carcinoma. Onco Targets Ther. 2013, 7, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Ten Haaf, A.; Bektas, N.; von Serenyi, S.; Losen, I.; Arweiler, E.C.; Hartmann, A.; Knuchel, R.; Dahl, E. Expression of the glioma-associated oncogene homolog (gli) 1 in human breast cancer is associated with unfavourable overall survival. BMC Cancer 2009, 9, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, T.; Li, C.; Zhang, X.; Chi, S.; He, N.; Chen, K.; McCormick, F.; Gatalica, Z.; Xie, J. Activation of the hedgehog pathway in advanced prostate cancer. Mol. Cancer 2004, 3, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.; Hernandez, A.M.; Stecca, B.; Kahler, A.J.; DeGueme, A.M.; Barrett, A.; Beyna, M.; Datta, M.W.; Datta, S.; Ruiz i Altaba, A. Inhibition of prostate cancer proliferation by interference with SONIC HEDGEHOG-GLI1 signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 12561–12566. [Google Scholar] [CrossRef] [PubMed]
- Riobo, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P., Jr. Phosphoinositide 3-kinase and AKT are essential for sonic hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef] [PubMed]
- Riobo, N.A.; Haines, G.M.; Emerson, C.P., Jr. Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res. 2006, 66, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.; Schnidar, H.; Neill, G.W.; Hanneder, M.; Klingler, S.; Blaas, L.; Schmid, C.; Hauser-Kronberger, C.; Regl, G.; Philpott, M.P.; et al. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol. Cell. Biol. 2006, 26, 6283–6298. [Google Scholar] [CrossRef] [PubMed]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/Gli in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Li, J.; Gao, T.; Xie, J.; Evers, B.M. Protein kinase Cdelta negatively regulates hedgehog signaling by inhibition of Gli1 activity. J. Biol. Chem. 2009, 284, 2150–2158. [Google Scholar] [CrossRef] [PubMed]
- Zwerner, J.P.; Joo, J.; Warner, K.L.; Christensen, L.; Hu-Lieskovan, S.; Triche, T.J.; May, W.A. The EWS/FLI1 oncogenic transcription factor deregulates gli1. Oncogene 2008, 27, 3282–3291. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Khan, A.A.; Shimokawa, T.; Zhan, J.; Stromblad, S.; Fang, W.; Zhang, H. A feedback regulation between kindlin-2 and GLI1 in prostate cancer cells. FEBS Lett. 2013, 587, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz, I.A.A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Wang, Y.; Fan, C.; Gao, P.; Wang, X.; Wei, G.; Wei, J. Estrogen promotes stemness and invasiveness of ER-positive breast cancer cells through Gli1 activation. Mol. Cancer 2014, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, B.; Lu, Y.; Teng, K.Y.; Nuovo, G.; Li, X.; Shapiro, C.L.; Majumder, S. Hedgehog signaling is a novel therapeutic target in tamoxifen-resistant breast cancer aberrantly activated by PI3K/AKT pathway. Cancer Res. 2012, 72, 5048–5059. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Frolova, N.; Sadlonova, A.; Novak, Z.; Steg, A.; Page, G.P.; Welch, D.R.; Lobo-Ruppert, S.M.; Ruppert, J.M.; Johnson, M.R.; et al. Hedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol. Ther. 2006, 5, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.G.; Pannell, L.K.; Singh, S.; Samant, R.S.; Shevde, L.A. Increased vascularity and spontaneous metastasis of breast cancer by hedgehog signaling mediated upregulation of cyr61. Oncogene 2012, 31, 3370–3380. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Pepicelli, C.V.; Dibble, C.C.; Catbagan, W.; Zarycki, J.L.; Laciak, R.; Gipp, J.; Shaw, A.; Lamm, M.L.; Munoz, A.; et al. Hedgehog signaling promotes prostate xenograft tumor growth. Endocrinology 2004, 145, 3961–3970. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, S.E.; Furic, L.; Buchanan, G.; Larsson, O.; Pedersen, J.; Frydenberg, M.; Risbridger, G.P.; Taylor, R.A. Hedgehog signaling is active in human prostate cancer stroma and regulates proliferation and differentiation of adjacent epithelium. Prostate 2013, 73, 1810–1823. [Google Scholar] [CrossRef] [PubMed]
- Shigemura, K.; Huang, W.C.; Li, X.; Zhau, H.E.; Zhu, G.; Gotoh, A.; Fujisawa, M.; Xie, J.; Marshall, F.F.; Chung, L.W. Active sonic hedgehog signaling between androgen independent human prostate cancer cells and normal/benign but not cancer-associated prostate stromal cells. Prostate 2011, 71, 1711–1722. [Google Scholar] [CrossRef] [PubMed]
- Efstathiou, E.; Karlou, M.; Wen, S.; Hoang, A.; Pettaway, C.A.; Pisters, L.L.; Maity, S.; Troncoso, P.; Logothetis, C.J. Integrated hedgehog signaling is induced following castration in human and murine prostate cancers. Prostate 2013, 73, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Azoulay, S.; Terry, S.; Chimingqi, M.; Sirab, N.; Faucon, H.; Gil Diez de Medina, S.; Moutereau, S.; Maille, P.; Soyeux, P.; Abbou, C.; et al. Comparative expression of hedgehog ligands at different stages of prostate carcinoma progression. J. Pathol. 2008, 216, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Tanner, M.; Levine, A.C.; Levina, E.; Ohouo, P.; Buttyan, R. Androgenic regulation of hedgehog signaling pathway components in prostate cancer cells. Cell Cycle 2009, 8, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Feuerstein, M.A.; Levina, E.; Baghel, P.S.; Carkner, R.D.; Tanner, M.J.; Shtutman, M.; Vacherot, F.; Terry, S.; de la Taille, A.; et al. Hedgehog/GLI supports androgen signaling in androgen deprived and androgen independent prostate cancer cells. Mol. Cancer 2010, 9, 89. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Stecca, B. Cooperative integration between HEDGEHOG-GLI signalling and other oncogenic pathways: Implications for cancer therapy. Expert Rev. Mol. Med. 2015, 17, e5. [Google Scholar] [CrossRef] [PubMed]
- Kakonen, S.M.; Mundy, G.R. Mechanisms of osteolytic bone metastases in breast carcinoma. Cancer 2003, 97, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Mavrogenis, A.F.; Angelini, A.; Vottis, C.; Pala, E.; Calabro, T.; Papagelopoulos, P.J.; Ruggieri, P. Modern palliative treatments for metastatic bone disease: Awareness of merits, demerits and guidance. Clin. J. Pain 2015. [Google Scholar] [CrossRef] [PubMed]
- Mundy, G.R. Mechanisms of bone metastasis. Cancer 1997, 80, 1546–1556. [Google Scholar] [CrossRef]
- Chiechi, A.; Waning, D.L.; Stayrook, K.R.; Buijs, J.T.; Guise, T.A.; Mohammad, K.S. Role of TGF-β in breast cancer bone metastases. Adv. Biosci. Biotechnol. 2013, 4, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Page, J.M.; Merkel, A.R.; Ruppender, N.S.; Guo, R.; Dadwal, U.C.; Cannonier, S.A.; Basu, S.; Guelcher, S.A.; Sterling, J.A. Matrix rigidity regulates the transition of tumor cells to a bone-destructive phenotype through integrin beta3 and TGF-beta receptor type II. Biomaterials 2015, 64, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Kamalakar, A.; Bendre, M.S.; Washam, C.L.; Fowler, T.W.; Carver, A.; Dilley, J.D.; Bracey, J.W.; Akel, N.S.; Margulies, A.G.; Skinner, R.A.; et al. Circulating interleukin-8 levels explain breast cancer osteolysis in mice and humans. Bone 2014, 61, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Merkel, A.R.; Page, J.M.; Ruppender, N.S.; Guelcher, S.A.; Sterling, J.A. Wnt signaling induces gene expression of factors associated with bone destruction in lung and breast cancer. Clin. Exp. Metastasis 2014, 31, 945–959. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Sterling, J.A.; Edwards, J.R.; DeGraff, D.J.; Lee, C.; Park, S.I.; Matusik, R.J. Activation of NF-kappa B signaling promotes growth of prostate cancer cells in bone. PLoS ONE 2013, 8, e60983. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Nguyen, M.P.; Padalecki, S.S.; Grubbs, B.G.; Merkel, A.R.; Oyajobi, B.O.; Matrisian, L.M.; Mundy, G.R.; Sterling, J.A. TGF-beta promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer Res. 2011, 71, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Sterling, J.A.; Oyajobi, B.O.; Grubbs, B.; Padalecki, S.S.; Munoz, S.A.; Gupta, A.; Story, B.; Zhao, M.; Mundy, G.R. The hedgehog signaling molecule Gli2 induces parathyroid hormone-related peptide expression and osteolysis in metastatic human breast cancer cells. Cancer Res. 2006, 66, 7548–7553. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, V.I.; Javelaud, D.; Van Kempen, L.C.; Mohammad, K.S.; Dennler, S.; Luciani, F.; Hoek, K.S.; Juarez, P.; Goydos, J.S.; Fournier, P.J.; et al. GLI2-mediated melanoma invasion and metastasis. J. Natl. Cancer Inst. 2010, 102, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Tiet, T.D.; Hopyan, S.; Nadesan, P.; Gokgoz, N.; Poon, R.; Lin, A.C.; Yan, T.; Andrulis, I.L.; Alman, B.A.; Wunder, J.S. Constitutive hedgehog signaling in chondrosarcoma up-regulates tumor cell proliferation. Am. J. Pathol. 2006, 168, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Bovee, J.V.; van den Broek, L.J.; Cleton-Jansen, A.M.; Hogendoorn, P.C. Up-regulation of PTHrP and BCL-2 expression characterizes the progression of osteochondroma towards peripheral chondrosarcoma and is a late event in central chondrosarcoma. Lab. Investig. 2000, 80, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Bovee, J.V.; Jadnanansing, N.A.; Taminiau, A.H.; Hogendoorn, P.C. Expression of cartilage growth plate signalling molecules in chondroblastoma. J. Pathol. 2004, 202, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Pateder, D.B.; Gish, M.W.; O’Keefe, R.J.; Hicks, D.G.; Teot, L.A.; Rosier, R.N. Parathyroid hormone-related peptide expression in cartilaginous tumors. Clin. Orthop. Relat. Res. 2002, 403, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Kunisada, T.; Moseley, J.M.; Slavin, J.L.; Martin, T.J.; Choong, P.F. Co-expression of parathyroid hormone-related protein (PTHrP) and PTH/PTHrP receptor in cartilaginous tumours: A marker for malignancy? Pathology 2002, 34, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Chang, C.J.; Lin, C.P.; Chang, S.Y.; Chu, P.Y.; Tai, S.K.; Li, W.Y.; Chao, K.S.; Chen, Y.J. Expression of hedgehog signaling molecules as a prognostic indicator of oral squamous cell carcinoma. Head Neck 2012, 34, 1556–1561. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Wang, L.; Zuo, H.; Zhang, Z.; Chen, W.; Mao, L.; Zhang, P. HH/GLI signalling as a new therapeutic target for patients with oral squamous cell carcinoma. Oral Oncol. 2011, 47, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J.; Brown, J.S.; Woolgar, J.A.; Lowe, D.; Rogers, S.N.; Vaughan, E.D. The influence of the pattern of mandibular invasion on recurrence and survival in oral squamous cell carcinoma. Head Neck 2004, 26, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Lowe, D.; Kalavrezos, N.; D’Souza, J.; Magennis, P.; Woolgar, J. Patterns of invasion and routes of tumor entry into the mandible by oral squamous cell carcinoma. Head Neck 2002, 24, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Honami, T.; Shimo, T.; Okui, T.; Kurio, N.; Hassan, N.M.; Iwamoto, M.; Sasaki, A. Sonic hedgehog signaling promotes growth of oral squamous cell carcinoma cells associated with bone destruction. Oral Oncol. 2012, 48, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the hedgehog signaling pathway in cancer: Beyond smoothened. Oncotarget 2015, 6, 13899–13913. [Google Scholar] [PubMed]
- Di Magno, L.; Coni, S.; Di Marcotullio, L.; Canettieri, G. Digging a hole under hedgehog: Downstream inhibition as an emerging anticancer strategy. Biochim. Biophys. Acta 2015, 1856, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Infante, P.; Alfonsi, R.; Botta, B.; Mori, M.; di Marcotullio, L. Targeting GLI factors to inhibit the hedgehog pathway. Trends Pharmacol. Sci. 2015, 36, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of hedgehog signaling by direct binding of cyclopamine to smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Zhou, H.; Liu, Y.; Liu, Z.; Liu, J.; Tang, J.; Li, J.; Zhang, J.; Sheng, W.; Zhao, Y.; et al. Hyaluronic acid functional amphipathic and redox-responsive polymer particles for the co-delivery of doxorubicin and cyclopamine to eradicate breast cancer cells and cancer stem cells. Nanoscale 2015, 7, 8607–8618. [Google Scholar] [CrossRef] [PubMed]
- Chitkara, D.; Singh, S.; Kumar, V.; Danquah, M.; Behrman, S.W.; Kumar, N.; Mahato, R.I. Micellar delivery of cyclopamine and gefitinib for treating pancreatic cancer. Mol. Pharmcol. 2012, 9, 2350–2357. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, R.J.; Hutson, P.R.; Hannam, P.W.; Nydza, R.J.; Washington, I.M.; Moore, R.W.; Girdaukas, G.G.; Peterson, R.E.; Bushman, W. Dose- and route-dependent teratogenicity, toxicity, and pharmacokinetic profiles of the hedgehog signaling antagonist cyclopamine in the mouse. Toxicol. Sci. 2008, 104, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Bisht, S.; Feldmann, G.; Koorstra, J.B.; Mullendore, M.; Alvarez, H.; Karikari, C.; Rudek, M.A.; Lee, C.K.; Maitra, A.; Maitra, A. In vivo characterization of a polymeric nanoparticle platform with potential oral drug delivery capabilities. Mol. Cancer Ther. 2008, 7, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.R.; Lescarbeau, A.; Grogan, M.J.; Tan, E.; Lin, G.; Austad, B.C.; Yu, L.C.; Behnke, M.L.; Nair, S.J.; Hagel, M.; et al. Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). J. Med. Chem. 2009, 52, 4400–4418. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Guicherit, O.M.; Zaharian, B.I.; Xu, Y.; Chai, L.; Wichterle, H.; Kon, C.; Gatchalian, C.; Porter, J.A.; Rubin, L.L.; et al. Identification of a small molecule inhibitor of the hedgehog signaling pathway: Effects on basal cell carcinoma-like lesions. Proc. Natl. Acad. Sci. USA 2003, 100, 4616–4621. [Google Scholar] [CrossRef] [PubMed]
- Romer, J.T.; Kimura, H.; Magdaleno, S.; Sasai, K.; Fuller, C.; Baines, H.; Connelly, M.; Stewart, C.F.; Gould, S.; Rubin, L.L.; et al. Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(+/−)p53(−/−) mice. Cancer Cell 2004, 6, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a commonly used antifungal that inhibits hedgehog pathway activity and cancer growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.L.; Matsui, W. Hedgehog pathway as a drug target: Smoothened inhibitors in development. Onco Targets Ther. 2012, 5, 47–58. [Google Scholar] [CrossRef] [PubMed]
- De Smaele, E.; Ferretti, E.; Gulino, A. Vismodegib, a small-molecule inhibitor of the hedgehog pathway for the treatment of advanced cancers. Curr. Opin. Investig. Drugs 2010, 11, 707–718. [Google Scholar] [PubMed]
- Jimeno, A.; Weiss, G.J.; Miller, W.H., Jr.; Gettinger, S.; Eigl, B.J.; Chang, A.L.; Dunbar, J.; Devens, S.; Faia, K.; Skliris, G.; et al. Phase I study of the hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin. Cancer Res. 2013, 19, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- D’Amato, C.; Rosa, R.; Marciano, R.; D’Amato, V.; Formisano, L.; Nappi, L.; Raimondo, L.; di Mauro, C.; Servetto, A.; Fulciniti, F.; et al. Inhibition of hedgehog signalling by NVP-LDE225 (Erismodegib) interferes with growth and invasion of human renal cell carcinoma cells. Br. J. Cancer 2014, 111, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Justilien, V.; Fields, A.P. Molecular pathways: Novel approaches for improved therapeutic targeting of hedgehog signaling in cancer stem cells. Clin. Cancer Res. 2015, 21, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.J.; Messersmith, W.A.; Shaik, M.N.; Li, S.; Zheng, X.; McLachlan, K.R.; Cesari, R.; Courtney, R.; Levin, W.J.; El-Khoueiry, A.B. A phase I study of PF-04449913, an oral hedgehog inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Ibuki, N.; Ghaffari, M.; Pandey, M.; Iu, I.; Fazli, L.; Kashiwagi, M.; Tojo, H.; Nakanishi, O.; Gleave, M.E.; Cox, M.E. Tak-441, a novel investigational smoothened antagonist, delays castration-resistant progression in prostate cancer by disrupting paracrine hedgehog signaling. Int. J. Cancer 2013, 133, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Tawbi, H.A.; Thomas, A.L.; Stoller, R.G.; Turtschi, C.P.; Baselga, J.; Sarantopoulos, J.; Mahalingam, D.; Shou, Y.; Moles, M.A.; et al. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor sonidegib (LDE225) in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Kim, J.; Spaunhurst, K.; Montoya, J.; Khodosh, R.; Chandra, K.; Fu, T.; Gilliam, A.; Molgo, M.; Beachy, P.A.; et al. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J. Clin. Oncol. 2014, 32, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N. Engl. J. Med. 2012, 366, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B.; Fehrenbacher, L.; Holloway, R.; Amit, A.; Karlan, B.; Slomovitz, B.; Sabbatini, P.; Fu, L.; Yauch, R.L.; Chang, I.; et al. A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin. Cancer Res. 2012, 18, 6509–6518. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Le Cesne, A.; Bellera, C.; Piperno-Neumann, S.; Duffaud, F.; Penel, N.; Cassier, P.; Domont, J.; Takebe, N.; Kind, M.; et al. GDC-0449 in patients with advanced chondrosarcomas: A French Sarcoma Group/US and French National Cancer Institute single-arm phase II collaborative study. Ann. Oncol. 2013, 24, 2922–2926. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, G.J.; Alicke, B.; Weinmann, L.; Januario, T.; West, K.; Modrusan, Z.; Burdick, D.; Goldsmith, R.; Robarge, K.; Sutherlin, D.; et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011, 71, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Peukert, S.; He, F.; Dai, M.; Zhang, R.; Sun, Y.; Miller-Moslin, K.; McEwan, M.; Lagu, B.; Wang, K.; Yusuff, N.; et al. Discovery of NVP-LEQ506, a second-generation inhibitor of smoothened. ChemMedChem 2013, 8, 1261–1265. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Flaveny, C.A.; Giambelli, C.; Fei, D.L.; Han, L.; Hang, B.I.; Bai, F.; Pei, X.H.; Nose, V.; Burlingame, O.; et al. Repurposing the FDA-approved pinworm drug pyrvinium as a novel chemotherapeutic agent for intestinal polyposis. PLoS ONE 2014, 9, e101969. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed]
- Infante, P.; Mori, M.; Alfonsi, R.; Ghirga, F.; Aiello, F.; Toscano, S.; Ingallina, C.; Siler, M.; Cucchi, D.; Po, A.; et al. GLI1/DNA interaction is a druggable target for hedgehog-dependent tumors. EMBO J. 2015, 34, 200–217. [Google Scholar] [CrossRef] [PubMed]
- Agyeman, A.; Jha, B.K.; Mazumdar, T.; Houghton, J.A. Mode and specificity of binding of the small molecule GANT61 to GLI determines inhibition of GLI-DNA binding. Oncotarget 2014, 5, 4492–4503. [Google Scholar] [PubMed]
- Srivastava, R.K.; Kaylani, S.Z.; Edrees, N.; Li, C.; Talwelkar, S.S.; Xu, J.; Palle, K.; Pressey, J.G.; Athar, M. GLI inhibitor GANT-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting Shh/AKT-mTOR axis. Oncotarget 2014, 5, 12151–12165. [Google Scholar] [PubMed]
- Matsumoto, T.; Tabata, K.; Suzuki, T. The GANT61, a GLI inhibitor, induces caspase-independent apoptosis of SK-N-LO cells. Biol. Pharm. Bull. 2014, 37, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef] [PubMed]
- Wolff, F.; Loipetzberger, A.; Gruber, W.; Esterbauer, H.; Aberger, F.; Frischauf, A.M. Imiquimod directly inhibits hedgehog signalling by stimulating adenosine receptor/protein kinase A-mediated GLI phosphorylation. Oncogene 2013, 32, 5574–5581. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the hedgehog pathway by preventing ciliary accumulation and reducing stability of the GLI2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, T.; Arai, M.A.; Koyano, T.; Kowithayakorn, T.; Ishibashi, M. Naturally occurring small-molecule inhibitors of hedgehog/GLI-mediated transcription. Chembiochem 2008, 9, 1082–1092. [Google Scholar] [CrossRef] [PubMed]
- Bosco-Clement, G.; Zhang, F.; Chen, Z.; Zhou, H.M.; Li, H.; Mikami, I.; Hirata, T.; Yagui-Beltran, A.; Lui, N.; Do, H.T.; et al. Targeting GLI transcription activation by small molecule suppresses tumor growth. Oncogene 2014, 33, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Spiller, S.E.; Ditzler, S.H.; Pullar, B.J.; Olson, J.M. Response of preclinical medulloblastoma models to combination therapy with 13-cis retinoic acid and suberoylanilide hydroxamic acid (SAHA). J. Neurooncol. 2008, 87, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Spiller, S.E.; Ravanpay, A.C.; Hahn, A.W.; Olson, J.M. Suberoylanilide hydroxamic acid is effective in preclinical studies of medulloblastoma. J. Neurooncol. 2006, 79, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Dhanyamraju, P.K.; Holz, P.S.; Finkernagel, F.; Fendrich, V.; Lauth, M. Histone deacetylase 6 represents a novel drug target in the oncogenic hedgehog signaling pathway. Mol. Cancer Ther. 2015, 14, 727–739. [Google Scholar] [CrossRef] [PubMed]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of bet bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of hedgehog pathway transcriptional output through bet bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Mechlin, C.W.; Tanner, M.J.; Chen, M.; Buttyan, R.; Levin, R.M.; Mian, B.M. Gli2 expression and human bladder transitional carcinoma cell invasiveness. J. Urol. 2010, 184, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Desch, P.; Asslaber, D.; Kern, D.; Schnidar, H.; Mangelberger, D.; Alinger, B.; Stoecher, M.; Hofbauer, S.W.; Neureiter, D.; Tinhofer, I.; et al. Inhibition of GLI, but not smoothened, induces apoptosis in chronic lymphocytic leukemia cells. Oncogene 2010, 29, 4885–4895. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; Devecchio, J.; Agyeman, A.; Shi, T.; Houghton, J.A. Blocking hedgehog survival signaling at the level of the GLI genes induces DNA damage and extensive cell death in human colon carcinoma cells. Cancer Res. 2011, 71, 5904–5914. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Li, Y.; Li, Z.; Wang, Y.; Wang, P.; Liang, Y. Gli inhibitor GANT61 causes apoptosis in myeloid leukemia cells and acts in synergy with rapamycin. Leuk. Res. 2012, 36, 742–748. [Google Scholar] [CrossRef] [PubMed]
- Keysar, S.B.; Le, P.N.; Anderson, R.T.; Morton, J.J.; Bowles, D.W.; Paylor, J.J.; Vogler, B.W.; Thorburn, J.; Fernandez, P.; Glogowska, M.J.; et al. Hedgehog signaling alters reliance on EGF receptor signaling and mediates anti-EGFR therapeutic resistance in head and neck cancer. Cancer Res. 2013, 73, 3381–3392. [Google Scholar] [CrossRef] [PubMed]
- Chari, N.S.; Romano, R.A.; Koster, M.I.; Jaks, V.; Roop, D.; Flores, E.R.; Teglund, S.; Sinha, S.; Gruber, W.; Aberger, F.; et al. Interaction between the TP63 and SHH pathways is an important determinant of epidermal homeostasis. Cell Death Differ. 2013, 20, 1080–1088. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.T.; Hermann, P.C.; Witthauer, J.; Rubio-Viqueira, B.; Leicht, S.F.; Huber, S.; Ellwart, J.W.; Mustafa, M.; Bartenstein, P.; D’Haese, J.G.; et al. Combined targeted treatment to eliminate tumorigenic cancer stem cells in human pancreatic cancer. Gastroenterology 2009, 137, 1102–1113. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Trabulo, S.M.; Sainz, B., Jr.; Balic, A.; Garcia, E.; Hahn, S.A.; Vandana, M.; Sahoo, S.K.; Tunici, P.; Bakker, A.; et al. Multimodal treatment eliminates cancer stem cells and leads to long-term survival in primary human pancreatic cancer tissue xenografts. PLoS ONE 2013, 8, e66371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domingo-Domenech, J.; Vidal, S.J.; Rodriguez-Bravo, V.; Castillo-Martin, M.; Quinn, S.A.; Rodriguez-Barrueco, R.; Bonal, D.M.; Charytonowicz, E.; Gladoun, N.; de la Iglesia-Vicente, J.; et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell 2012, 22, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Ferruzzi, P.; Mennillo, F.; De Rosa, A.; Giordano, C.; Rossi, M.; Benedetti, G.; Magrini, R.; Pericot Mohr, G.; Miragliotta, V.; Magnoni, L.; et al. In vitro and in vivo characterization of a novel hedgehog signaling antagonist in human glioblastoma cell lines. Int. J. Cancer 2012, 131, E33–E44. [Google Scholar] [CrossRef] [PubMed]
- Markant, S.L.; Esparza, L.A.; Sun, J.; Barton, K.L.; McCoig, L.M.; Grant, G.A.; Crawford, J.R.; Levy, M.L.; Northcott, P.A.; Shih, D.; et al. Targeting sonic hedgehog-associated medulloblastoma through inhibition of Aurora and Polo-like kinases. Cancer Res. 2013, 73, 6310–6322. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, G.; Dhara, S.; Fendrich, V.; Bedja, D.; Beaty, R.; Mullendore, M.; Karikari, C.; Alvarez, H.; Iacobuzio-Donahue, C.; Jimeno, A.; et al. Blockade of hedgehog signaling inhibits pancreatic cancer invasion and metastases: A new paradigm for combination therapy in solid cancers. Cancer Res. 2007, 67, 2187–2196. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cannonier, S.A.; Sterling, J.A. The Role of Hedgehog Signaling in Tumor Induced Bone Disease. Cancers 2015, 7, 1658-1683. https://doi.org/10.3390/cancers7030856
AMA Style
Cannonier SA, Sterling JA. The Role of Hedgehog Signaling in Tumor Induced Bone Disease. Cancers. 2015; 7(3):1658-1683. https://doi.org/10.3390/cancers7030856
Chicago/Turabian StyleCannonier, Shellese A., and Julie A. Sterling. 2015. "The Role of Hedgehog Signaling in Tumor Induced Bone Disease" Cancers 7, no. 3: 1658-1683. https://doi.org/10.3390/cancers7030856