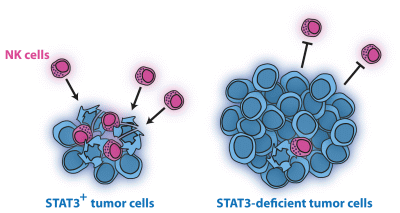
Loss of STAT3 in Lymphoma Relaxes NK Cell-Mediated Tumor Surveillance


 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
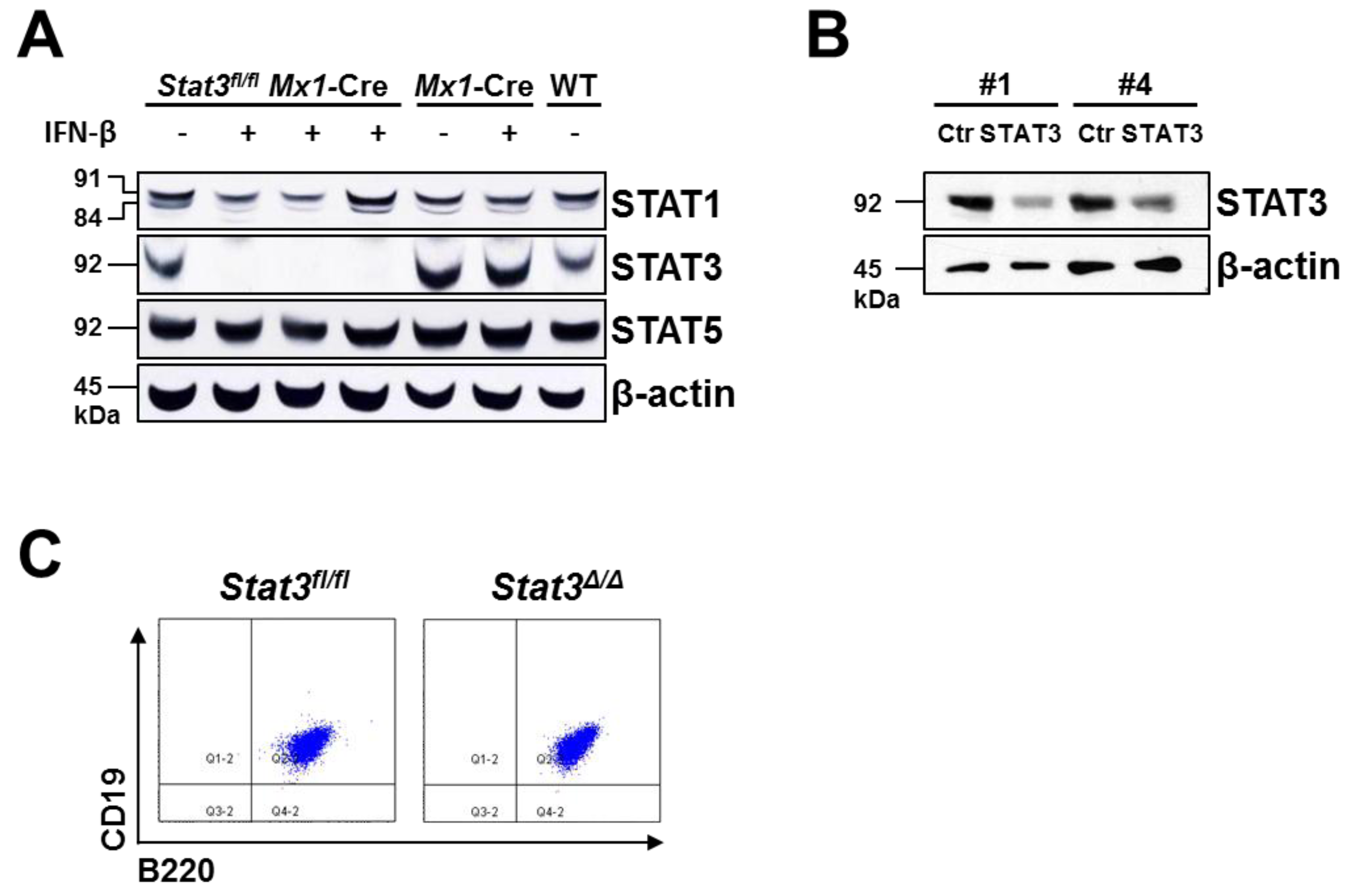
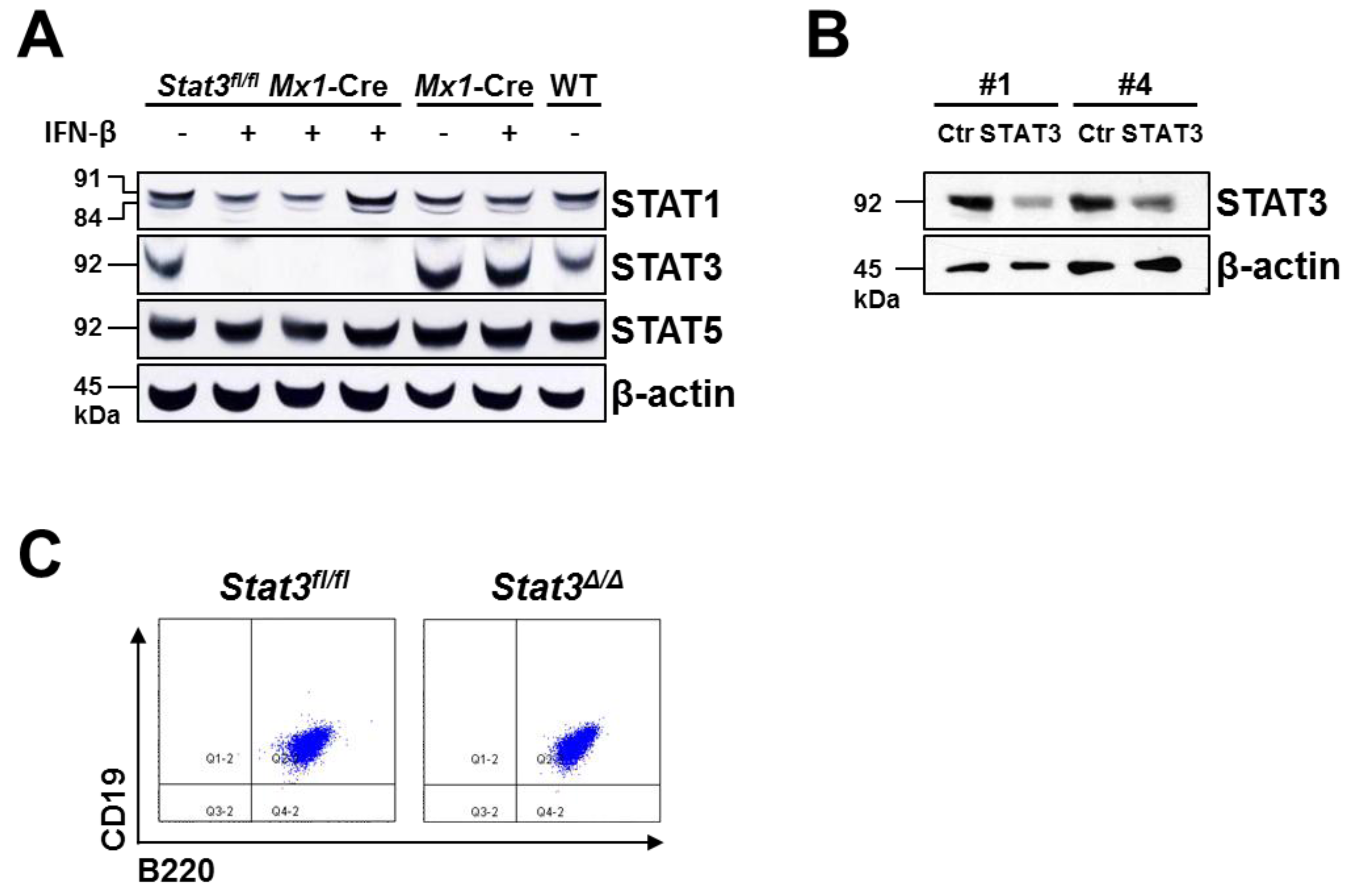
2.1. Generation of STAT3-Deficient BCR/ABL+ Pro-B Cell Lines
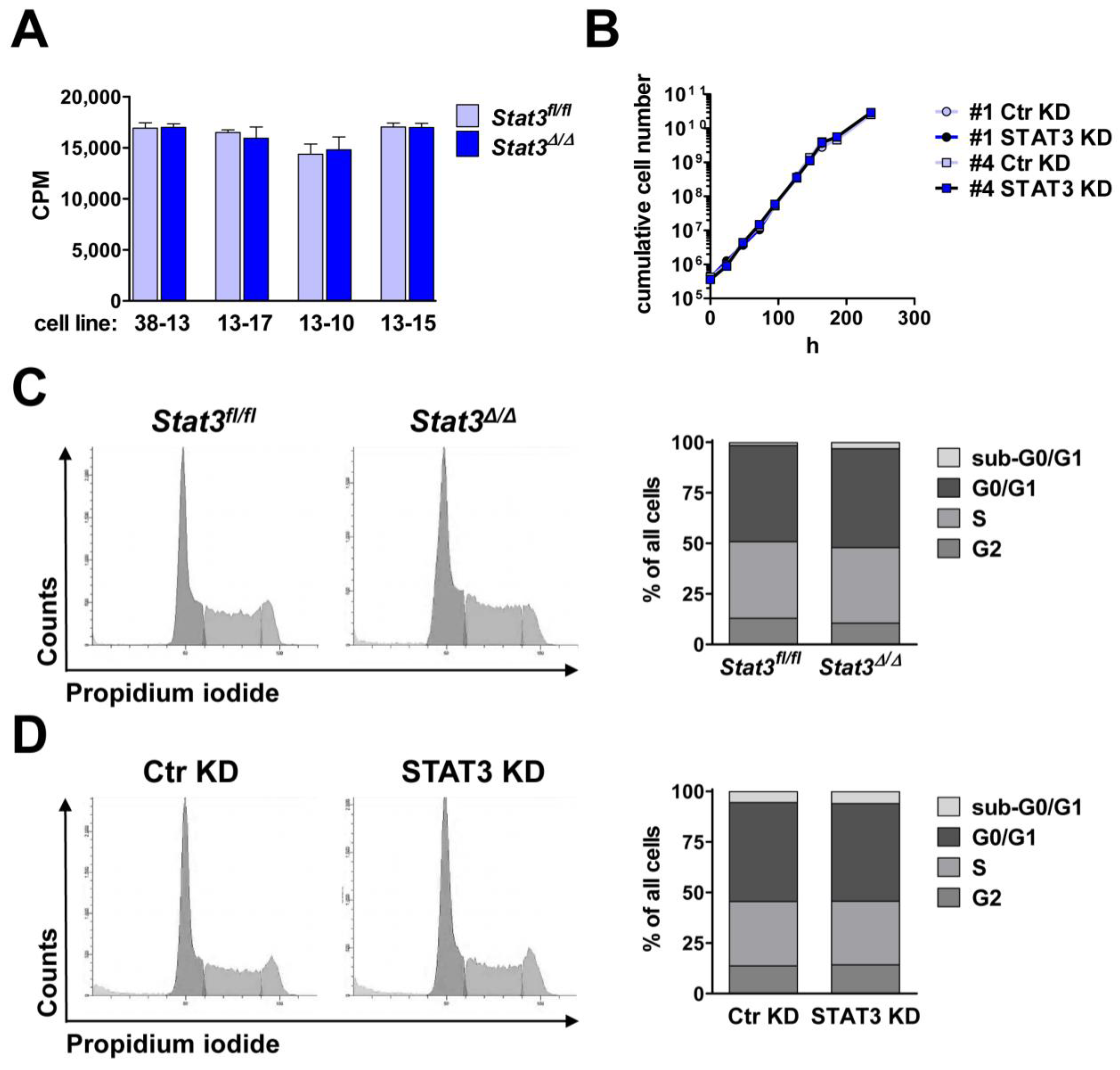
2.2. In Vitro Proliferation of BCR/ABL+ Cells Expressing or Lacking STAT3
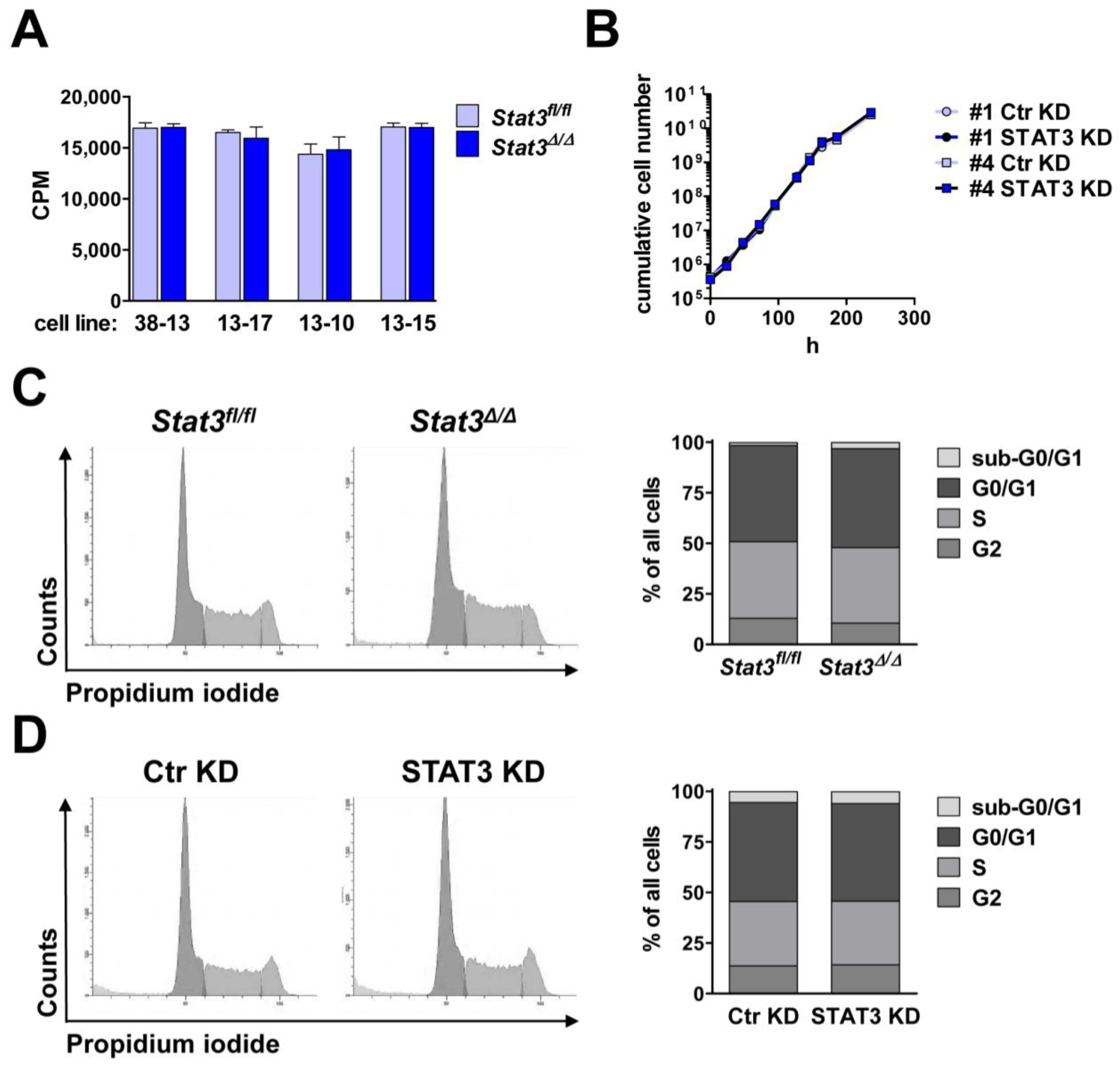
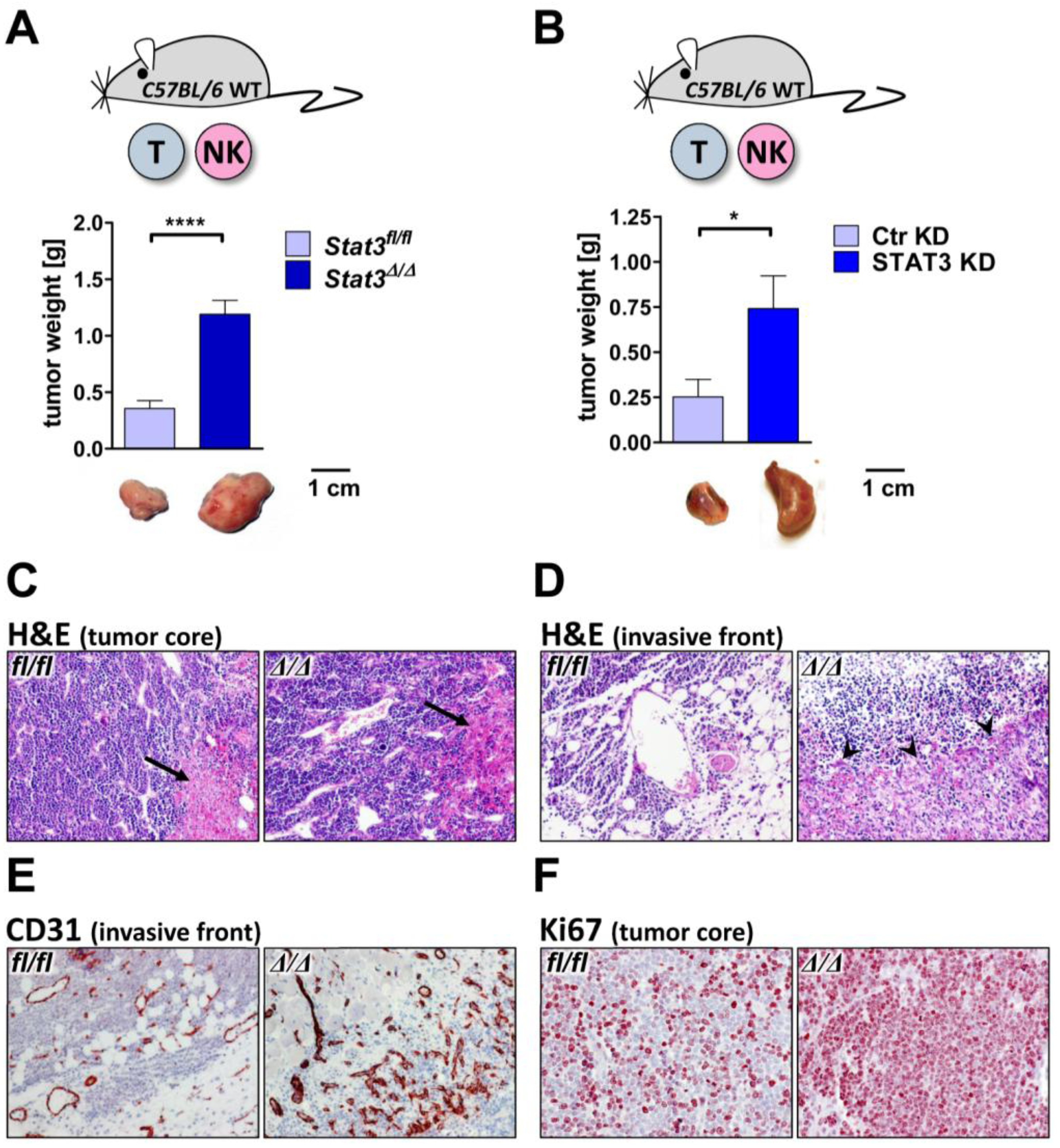
2.3. Lymphoma Development in Vivo
2.4. Cytokine Profile in STAT3-Deficient Lymphomas


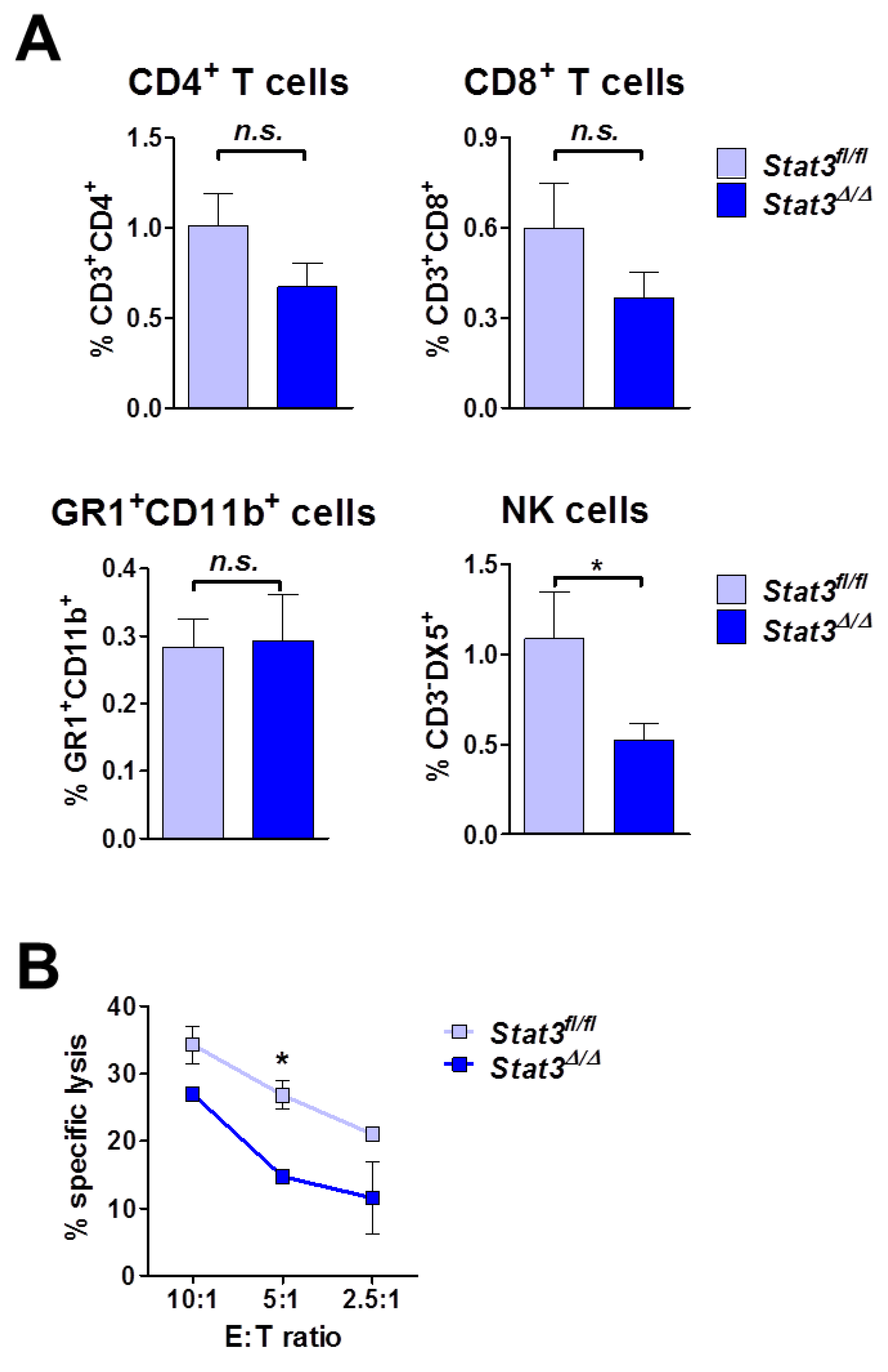
2.5. Lymphoma-Infiltrating Leukocytes

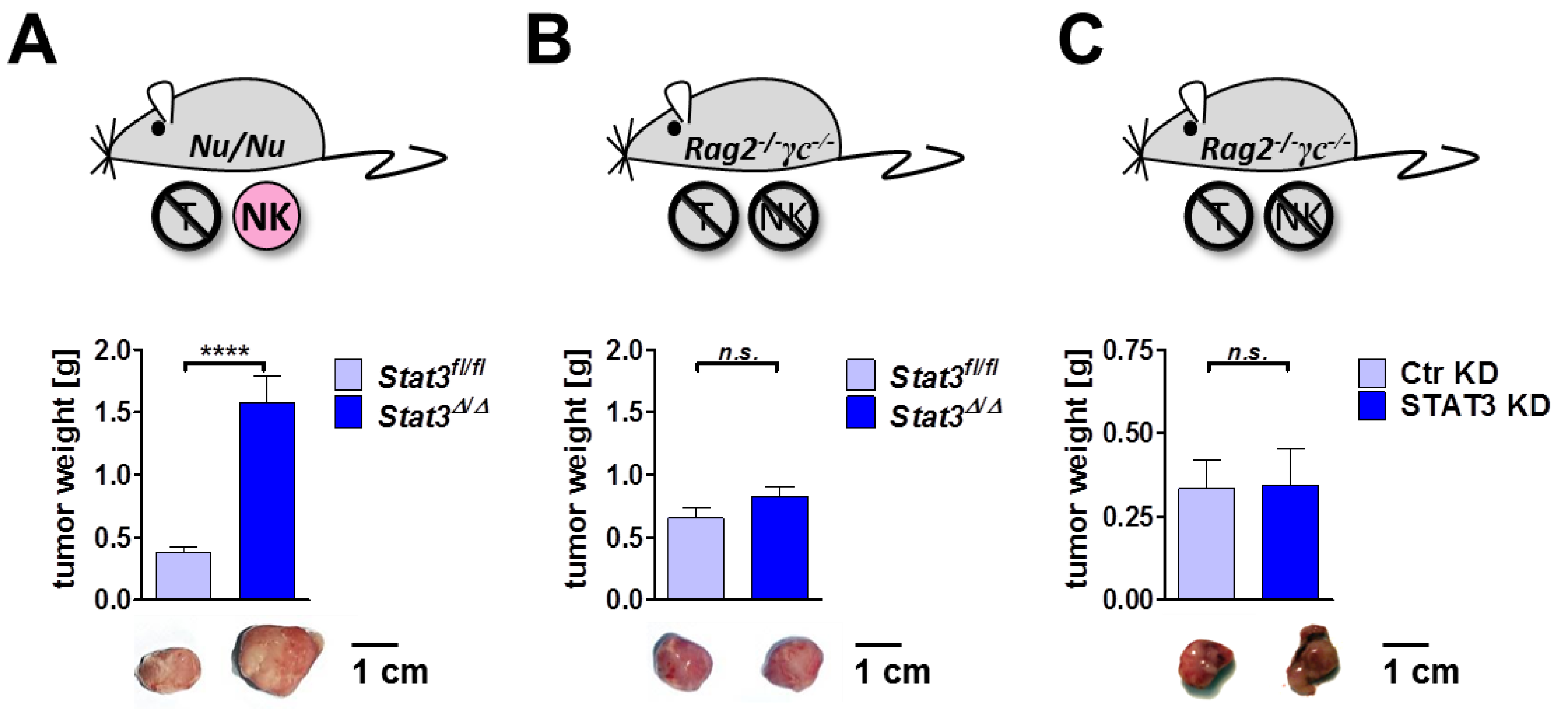
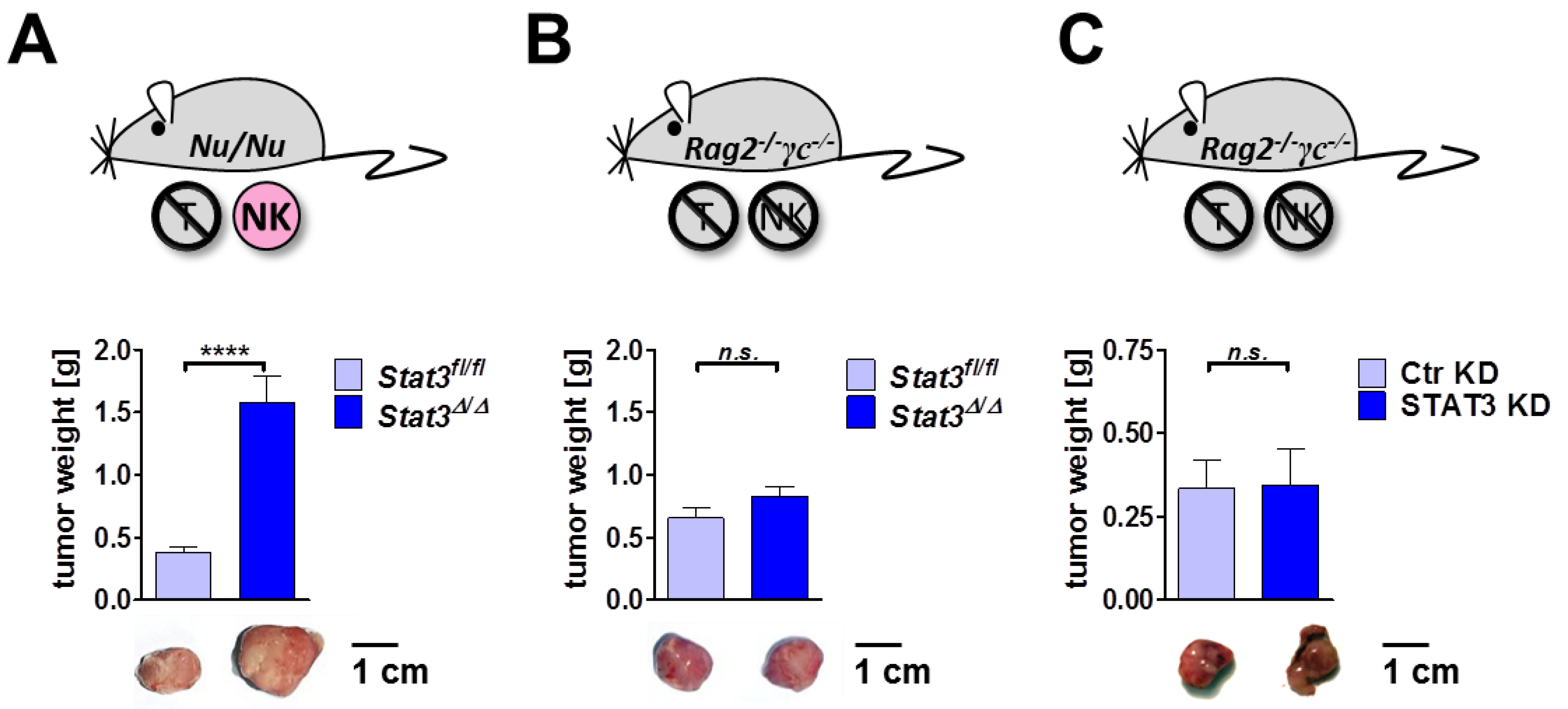
2.6. Lymphoma Development in Immune-Compromised Recipients
3. Experimental
3.1. Mice
3.2. Generation of BCR/ABL+ Cell Lines, in Vitro Deletion of Endogenous Stat3 and shRNA-Mediated Knockdown
3.3. Protein Extracts and Western Blotting
3.4. Transplantation of Tumor Cells
3.5. Immunohistochemistry
3.6. Flow Cytometry
3.7. [3H]-Thymidine Incorporation
3.8. Real-Time PCR Analysis
3.9. In Vitro NK Cell Cytotoxicity Assay
4. Conclusions
Acknowledgments
Author contributions
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Weinstein, I.B. Cancer. Addiction to oncogenes—The Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef]
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef]
- Bowman, T.; Garcia, R.; Turkson, J.; Jove, R. STATs in oncogenesis. Oncogene 2000, 19, 2474–2488. [Google Scholar] [CrossRef]
- Yu, H.; Jove, R. The STATs of cancer—New molecular targets come of age. Nat. Rev. Cancer 2004, 4, 97–105. [Google Scholar] [CrossRef]
- Ling, X.; Arlinghaus, R.B. Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. 2005, 65, 2532–2536. [Google Scholar] [CrossRef]
- Bar-Natan, M.; Nelson, E.A.; Xiang, M.; Frank, D.A. STAT signaling in the pathogenesis and treatment of myeloid malignancies. JAK-STAT 2012, 1, 55–64. [Google Scholar] [CrossRef]
- Hoelbl, A.; Schuster, C.; Kovacic, B.; Zhu, B.; Wickre, M.; Hoelzl, M.A.; Fajmann, S.; Grebien, F.; Warsch, W.; Stengl, G.; et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol. Med. 2010, 2, 98–110. [Google Scholar] [CrossRef]
- Bromberg, J. Stat proteins and oncogenesis. J. Clin. Investig. 2002, 109, 1139–1142. [Google Scholar]
- Buettner, R.; Mora, L.B.; Jove, R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Cancer Res. 2002, 8, 945–954. [Google Scholar]
- Azare, J.; Leslie, K.; Al-Ahmadie, H.; Gerald, W.; Weinreb, P.H.; Violette, S.M.; Bromberg, J. Constitutively activated Stat3 induces tumorigenesis and enhances cell motility of prostate epithelial cells through integrin beta 6. Mol. Cell Biol. 2007, 27, 4444–4453. [Google Scholar] [CrossRef]
- Abdulghani, J.; Gu, L.; Dagvadorj, A.; Lutz, J.; Leiby, B.; Bonuccelli, G.; Lisanti, M.P.; Zellweger, T.; Alanen, K.; Mirtti, T.; et al. Stat3 promotes metastatic progression of prostate cancer. Am. J. Pathol. 2008, 172, 1717–1728. [Google Scholar] [CrossRef]
- Barbieri, I.; Pensa, S.; Pannellini, T.; Quaglino, E.; Maritano, D.; Demaria, M.; Voster, A.; Turkson, J.; Cavallo, F.; Watson, C.J.; et al. Constitutively active Stat3 enhances neu-mediated migration and metastasis in mammary tumors via upregulation of Cten. Cancer Res. 2010, 70, 2558–2567. [Google Scholar] [CrossRef]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 supports Ras-dependent oncogenic transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef]
- Zhang, Q.; Raje, V.; Yakovlev, V.A.; Yacoub, A.; Szczepanek, K.; Meier, J.; Derecka, M.; Chen, Q.; Hu, Y.; Sisler, J.; et al. Mitochondrial localized Stat3 promotes breast cancer growth via phosphorylation of serine 727. J. Biol. Chem. 2013, 288, 31280–31288. [Google Scholar] [CrossRef]
- Kim, D.J.; Angel, J.M.; Sano, S.; DiGiovanni, J. Constitutive activation and targeted disruption of signal transducer and activator of transcription 3 (Stat3) in mouse epidermis reveal its critical role in UVB-induced skin carcinogenesis. Oncogene 2009, 28, 950–960. [Google Scholar] [CrossRef]
- Weber-Nordt, R.M.; Egen, C.; Wehinger, J.; Ludwig, W.; Gouilleux-Gruart, V.; Mertelsmann, R.; Finke, J. Constitutive activation of STAT proteins in primary lymphoid and myeloid leukemia cells and in Epstein-Barr virus (EBV)-related lymphoma cell lines. Blood 1996, 88, 809–816. [Google Scholar]
- Lin, T.S.; Mahajan, S.; Frank, D.A. STAT signaling in the pathogenesis and treatment of leukemias. Oncogene 2000, 19, 2496–2504. [Google Scholar] [CrossRef]
- Benekli, M.; Baer, M.R.; Baumann, H.; Wetzler, M. Signal transducer and activator of transcription proteins in leukemias. Blood 2003, 101, 2940–2954. [Google Scholar] [CrossRef]
- Benekli, M.; Baumann, H.; Wetzler, M. Targeting signal transducer and activator of transcription signaling pathway in leukemias. J. Clin. Oncol. 2009, 27, 4422–4432. [Google Scholar] [CrossRef]
- Ecker, A.; Simma, O.; Hoelbl, A.; Kenner, L.; Beug, H.; Moriggl, R.; Sexl, V. The dark and the bright side of Stat3: Proto-oncogene and tumor-suppressor. Front. Biosci. 2009, 14, 2944–2958. [Google Scholar]
- Lee, J.; Kim, J.C.K.; Lee, S.-E.; Quinley, C.; Kim, H.; Herdman, S.; Corr, M.; Raz, E. Signal transducer and activator of transcription 3 (STAT3) protein suppresses adenoma-to-carcinoma transition in Apcmin/+ mice via regulation of Snail-1 (SNAI) protein stability. J. Biol. Chem. 2012, 287, 18182–18189. [Google Scholar]
- De la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; Depinho, R.A.; Bonni, A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef]
- Musteanu, M.; Blaas, L.; Mair, M.; Schlederer, M.; Bilban, M.; Tauber, S.; Esterbauer, H.; Mueller, M.; Casanova, E.; Kenner, L.; et al. Stat3 is a negative regulator of intestinal tumor progression in Apc(Min) mice. Gastroenterology 2010, 138, 1003–1011.e5. [Google Scholar] [CrossRef]
- Mair, M.; Zollner, G.; Schneller, D.; Musteanu, M.; Fickert, P.; Gumhold, J.; Schuster, C.; Fuchsbichler, A.; Bilban, M.; Tauber, S.; et al. Signal transducer and activator of transcription 3 protects from liver injury and fibrosis in a mouse model of sclerosing cholangitis. Gastroenterology 2010, 138, 2499–2508. [Google Scholar] [CrossRef]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef]
- Stoiber, D.; Kovacic, B.; Schuster, C.; Schellack, C.; Karaghiosoff, M.; Kreibich, R.; Weisz, E.; Artwohl, M.; Kleine, O.C.; Müller, M.; et al. TYK2 is a key regulator of the surveillance of B lymphoid tumors. J. Clin. Investig. 2004, 114, 1650–1658. [Google Scholar]
- Kovacic, B.; Stoiber, D.; Moriggl, R.; Weisz, E.; Ott, R.G.; Kreibich, R.; Levy, D.E.; Beug, H.; Freissmuth, M.; Sexl, V. STAT1 acts as a tumor promoter for leukemia development. Cancer Cell 2006, 10, 77–87. [Google Scholar]
- Baron, F.; Turhan, A.G.; Giron-Michel, J.; Azzarone, B.; Bentires-Alj, M.; Bours, V.; Bourhis, J.H.; Chouaib, S.; Caignard, A. Leukemic target susceptibility to natural killer cytotoxicity: Relationship with BCR-ABL expression. Blood 2002, 99, 2107–2113. [Google Scholar] [CrossRef]
- Maghazachi, A.A. Role of chemokines in the biology of natural killer cells. Curr. Top. Microbiol. Immunol. 2010, 341, 37–58. [Google Scholar]
- Kärre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R.; Karre, K. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678. [Google Scholar] [CrossRef]
- Raulet, D.H.; Guerra, N. Oncogenic stress sensed by the immune system: Role of natural killer cell receptors. Nat. Rev. Immunol. 2009, 9, 568–580. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Lee, H.; Pal, S.K.; Reckamp, K.; Figlin, R.A.; Yu, H. STAT3: A target to enhance antitumor immune response. Curr. Top. Microbiol. Immunol. 2011, 344, 41–59. [Google Scholar]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef]
- Kortylewski, M.; Yu, H. Role of Stat3 in suppressing anti-tumor immunity. Curr. Opin. Immunol. 2008, 20, 228–233. [Google Scholar] [CrossRef]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mule, J.; Kerr, W.G.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef]
- Alonzi, T.; Maritano, D.; Gorgoni, B.; Rizzuto, G.; Libert, C.; Poli, V. Essential role of STAT3 in the control of the acute-phase response as revealed by inducible gene inactivation [correction of activation] in the liver. Mol. Cell Biol. 2001, 21, 1621–1632. [Google Scholar] [CrossRef]
- Levy, D.E.; Lee, C. What does Stat3 do? J. Clin. Investig. 2002, 109, 1143–1148. [Google Scholar]
- Fagard, R.; Metelev, V.; Souissi, I.; Baran-Marszak, F. STAT3 inhibitors for cancer therapy: Have all roads been explored? JAK-STAT 2013, 2, e22882. [Google Scholar] [CrossRef]
- Ilaria, R.L., Jr.; van Etten, R.A. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J. Biol. Chem. 1996, 271, 31704–31710. [Google Scholar] [CrossRef]
- Shuai, K.; Halpern, J.; ten Hoeve, J.; Rao, X.; Sawyers, C.L. Constitutive activation of STAT5 by the BCR-ABL oncogene in chronic myelogenous leukemia. Oncogene 1996, 13, 247–254. [Google Scholar]
- Steelman, L.S.; Pohnert, S.C.; Shelton, J.G.; Franklin, R.A.; Bertrand, F.E.; McCubrey, J.A. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 2004, 18, 189–218. [Google Scholar] [CrossRef]
- Spiekermann, K.; Pau, M.; Schwab, R.; Schmieja, K.; Franzrahe, S.; Hiddemann, W. Constitutive activation of STAT3 and STAT5 is induced by leukemic fusion proteins with protein tyrosine kinase activity and is sufficient for transformation of hematopoietic precursor cells. Exp. Hematol. 2002, 30, 262–271. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Kunnumakkara, A.B.; Harikumar, K.B.; Gupta, S.R.; Tharakan, S.T.; Koca, C.; Dey, S.; Sung, B. Signal transducer and activator of transcription-3, inflammation, and cancer: How intimate is the relationship? Ann. NY Acad. Sci. 2009, 1171, 59–76. [Google Scholar] [CrossRef]
- Bromberg, J.F.; Wrzeszczynska, M.H.; Devgan, G.; Zhao, Y.; Pestell, R.G.; Albanese, C.; Darnell, J.E. Stat3 as an oncogene. Cell 1999, 98, 295–303. [Google Scholar] [CrossRef]
- Levy, D.E.; Inghirami, G. STAT3: A multifaceted oncogene. Proc. Natl. Acad. Sci. USA 2006, 103, 10151–10152. [Google Scholar] [CrossRef]
- Ishida, Y.; Kondo, T.; Takayasu, T.; Iwakura, Y.; Mukaida, N. The essential involvement of cross-talk between IFN-gamma and TGF-beta in the skin wound-healing process. J. Immunol. 2004, 172, 1848–1855. [Google Scholar]
- Shinozaki, M.; Okada, Y.; Kitano, A.; Ikeda, K.; Saika, S.; Shinozaki, M. Impaired cutaneous wound healing with excess granulation tissue formation in TNFalpha-null mice. Arch. Dermatol. Res. 2009, 301, 531–537. [Google Scholar] [CrossRef]
- Schall, T.J.; Bacon, K.; Toy, K.J.; Goeddel, D.V. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature 1990, 347, 669–671. [Google Scholar] [CrossRef]
- Allavena, P.; Germano, G.; Marchesi, F.; Mantovani, A. Chemokines in cancer related inflammation. Exp. Cell Res. 2011, 317, 664–673. [Google Scholar] [CrossRef]
- Taub, D.D.; Sayers, T.J.; Carter, C.R.; Ortaldo, J.R. Alpha and beta chemokines induce NK cell migration and enhance NK-mediated cytolysis. J. Immunol. 1995, 155, 3877–3888. [Google Scholar]
- Ming, W.J.; Bersani, L.; Mantovani, A. Tumor necrosis factor is chemotactic for monocytes and polymorphonuclear leukocytes. J. Immunol. 1987, 138, 1469–1474. [Google Scholar]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef]
- Orr, M.T.; Lanier, L.L. Natural killer cell education and tolerance. Cell 2010, 142, 847–856. [Google Scholar] [CrossRef]
- Koch, J.; Steinle, A.; Watzl, C.; Mandelboim, O. Activating natural cytotoxicity receptors of natural killer cells in cancer and infection. Trends Immunol. 2013, 34, 182–191. [Google Scholar]
- Kovanen, P.E.; Leonard, W.J. Cytokines and immunodeficiency diseases: Critical roles of the gamma(c)-dependent cytokines interleukins 2, 4, 7, 9, 15, and 21, and their signaling pathways. Immunol. Rev. 2004, 202, 67–83. [Google Scholar] [CrossRef]
- Putz, E.M.; Gotthardt, D.; Hoermann, G.; Csiszar, A.; Wirth, S.; Berger, A.; Straka, E.; Rigler, D.; Wallner, B.; Jamieson, A.M.; et al. CDK8-mediated STAT1-S727 phosphorylation restrains NK cell cytotoxicity and tumor surveillance. Cell Rep. 2013, 4, 437–444. [Google Scholar] [CrossRef]
- Zebedin, E.; Simma, O.; Schuster, C.; Putz, E.M.; Fajmann, S.; Warsch, W.; Eckelhart, E.; Stoiber, D.; Weisz, E.; Schmid, J.A.; et al. Leukemic challenge unmasks a requirement for PI3Kdelta in NK cell-mediated tumor surveillance. Blood 2008, 112, 4655–4664. [Google Scholar] [CrossRef]
- Yu, H.; Kortylewski, M.; Pardoll, D. Crosstalk between cancer and immune cells: Role of STAT3 in the tumour microenvironment. Nat. Rev. Immunol. 2007, 7, 41–51. [Google Scholar] [CrossRef]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar]
- Kühn, R.; Schwenk, F.; Aguet, M.; Rajewsky, K. Inducible gene targeting in mice. Science 1995, 269, 1427–1429. [Google Scholar] [CrossRef]
- Cao, X.; Shores, E.W.; Hu-Li, J.; Anver, M.R.; Kelsall, B.L.; Russell, S.M.; Drago, J.; Noguchi, M.; Grinberg, A.; Bloom, E.T.; et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor gamma chain. Immunity 1995, 2, 223–238. [Google Scholar] [CrossRef]
- Schreiber, E.; Matthias, P.; Müller, M.M.; Schaffner, W. Rapid detection of octamer binding proteins with “mini-extracts”, prepared from a small number of cells. Nucleic Acids Res. 1989, 17, 6419. [Google Scholar] [CrossRef]
- Hoelbl, A.; Kovacic, B.; Kerenyi, M.A.; Simma, O.; Warsch, W.; Cui, Y.; Beug, H.; Hennighausen, L.; Moriggl, R.; Sexl, V. Clarifying the role of Stat5 in lymphoid development and Abelson-induced transformation. Blood 2006, 107, 4898–4906. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Putz, E.M.; Hoelzl, M.A.; Baeck, J.; Bago-Horvath, Z.; Schuster, C.; Reichholf, B.; Kern, D.; Aberger, F.; Sexl, V.; Hoelbl-Kovacic, A. Loss of STAT3 in Lymphoma Relaxes NK Cell-Mediated Tumor Surveillance. Cancers 2014, 6, 193-210. https://doi.org/10.3390/cancers6010193
Putz EM, Hoelzl MA, Baeck J, Bago-Horvath Z, Schuster C, Reichholf B, Kern D, Aberger F, Sexl V, Hoelbl-Kovacic A. Loss of STAT3 in Lymphoma Relaxes NK Cell-Mediated Tumor Surveillance. Cancers. 2014; 6(1):193-210. https://doi.org/10.3390/cancers6010193
Chicago/Turabian StylePutz, Eva Maria, Maria Agnes Hoelzl, Julia Baeck, Zsuzsanna Bago-Horvath, Christian Schuster, Brian Reichholf, Daniela Kern, Fritz Aberger, Veronika Sexl, and Andrea Hoelbl-Kovacic. 2014. "Loss of STAT3 in Lymphoma Relaxes NK Cell-Mediated Tumor Surveillance" Cancers 6, no. 1: 193-210. https://doi.org/10.3390/cancers6010193
APA StylePutz, E. M., Hoelzl, M. A., Baeck, J., Bago-Horvath, Z., Schuster, C., Reichholf, B., Kern, D., Aberger, F., Sexl, V., & Hoelbl-Kovacic, A. (2014). Loss of STAT3 in Lymphoma Relaxes NK Cell-Mediated Tumor Surveillance. Cancers, 6(1), 193-210. https://doi.org/10.3390/cancers6010193