Synthetic Genetic Targeting of Genome Instability in Cancer

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Discussion
2.1. Therapeutically Exploiting the Molecular Origins of Genome Instability
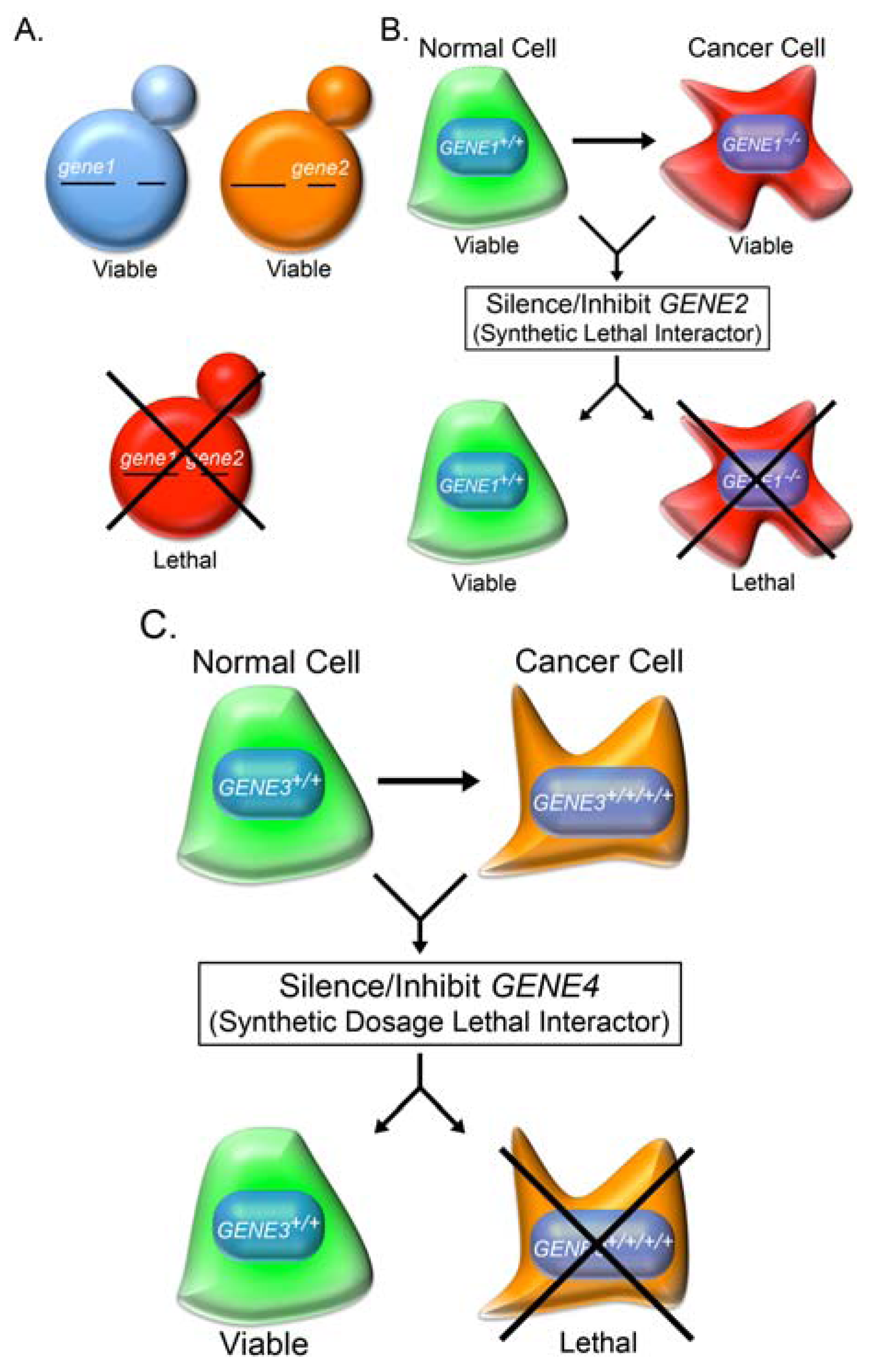
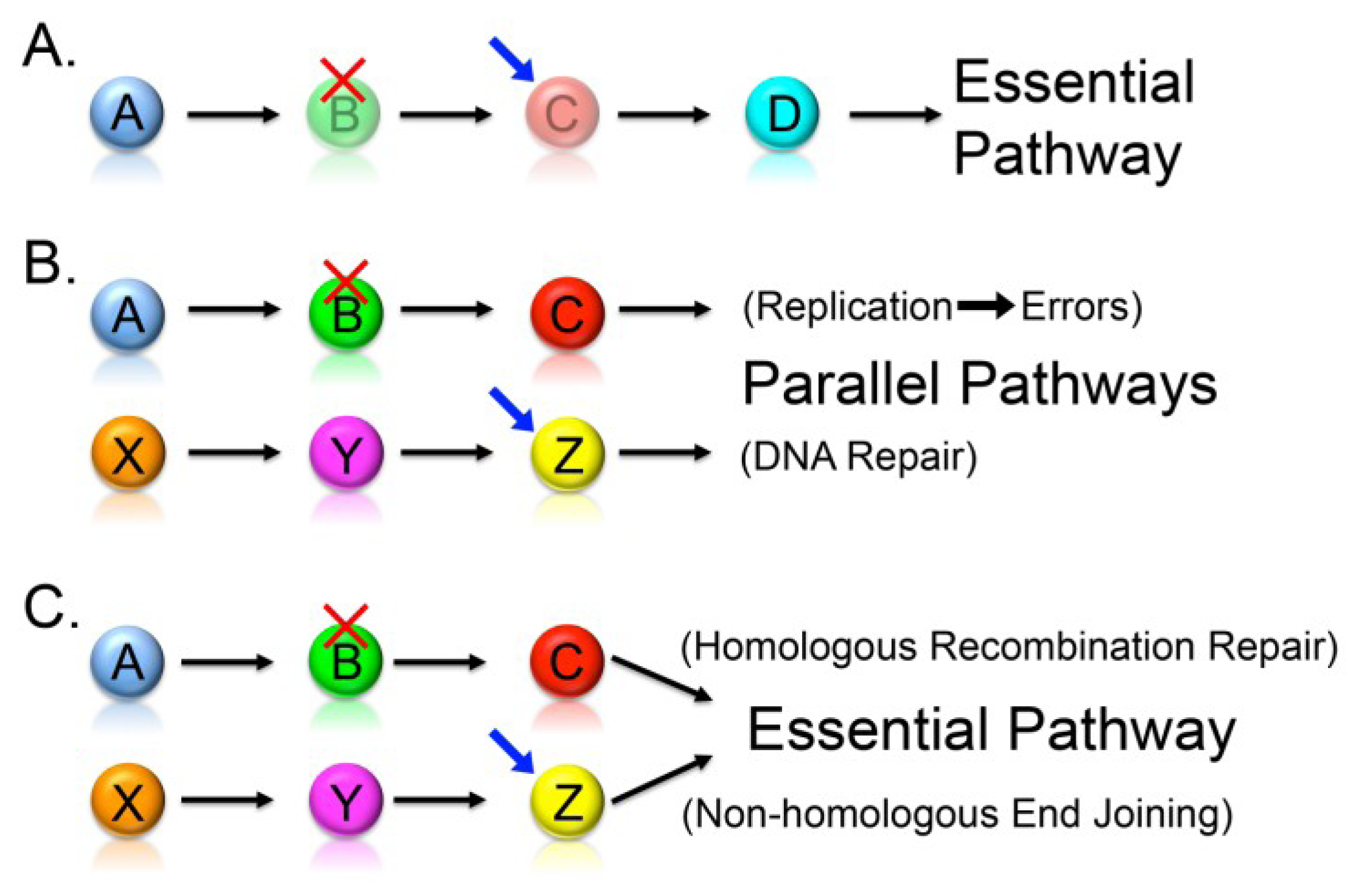
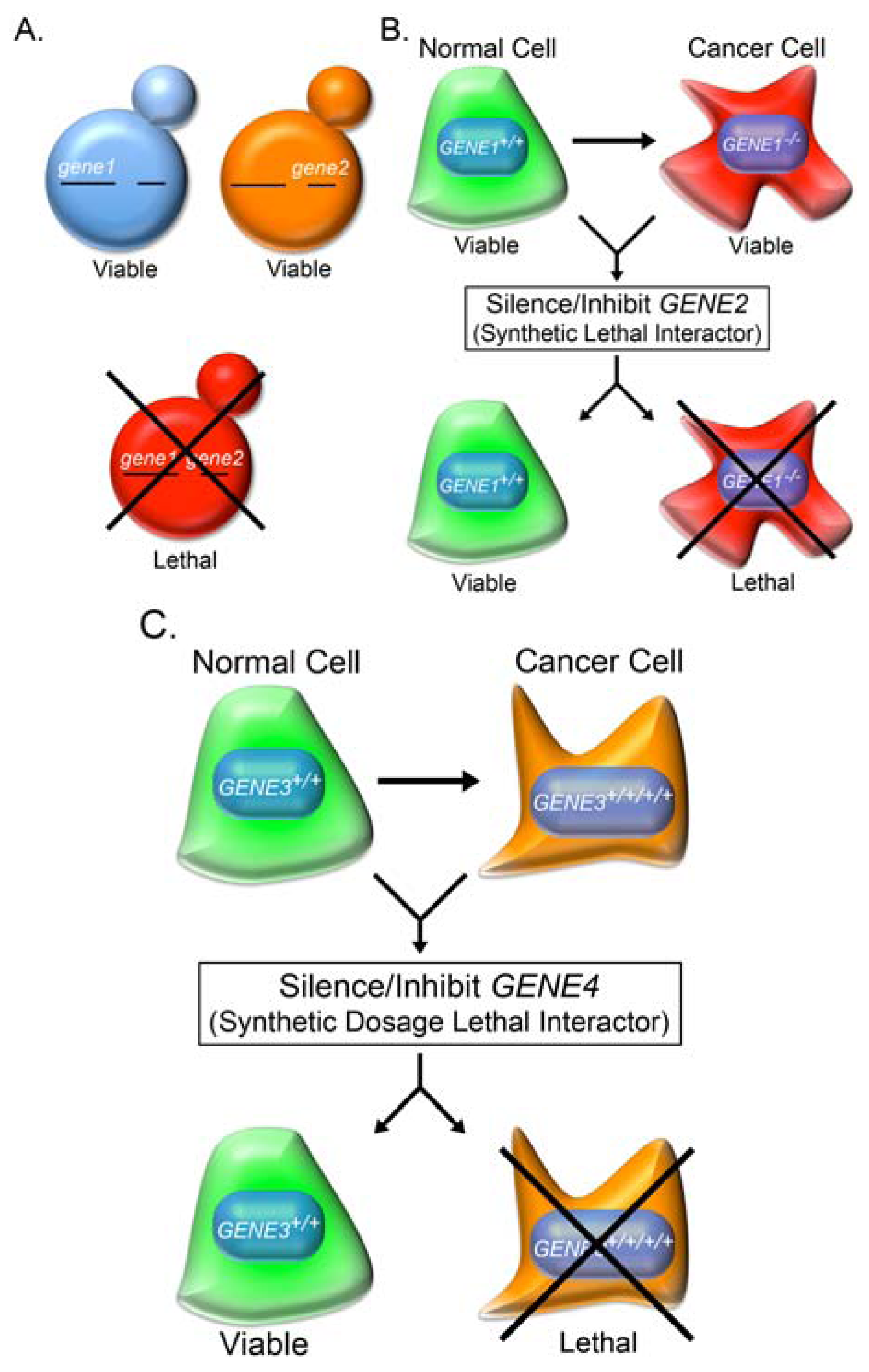
2.2. Synthetic Lethality
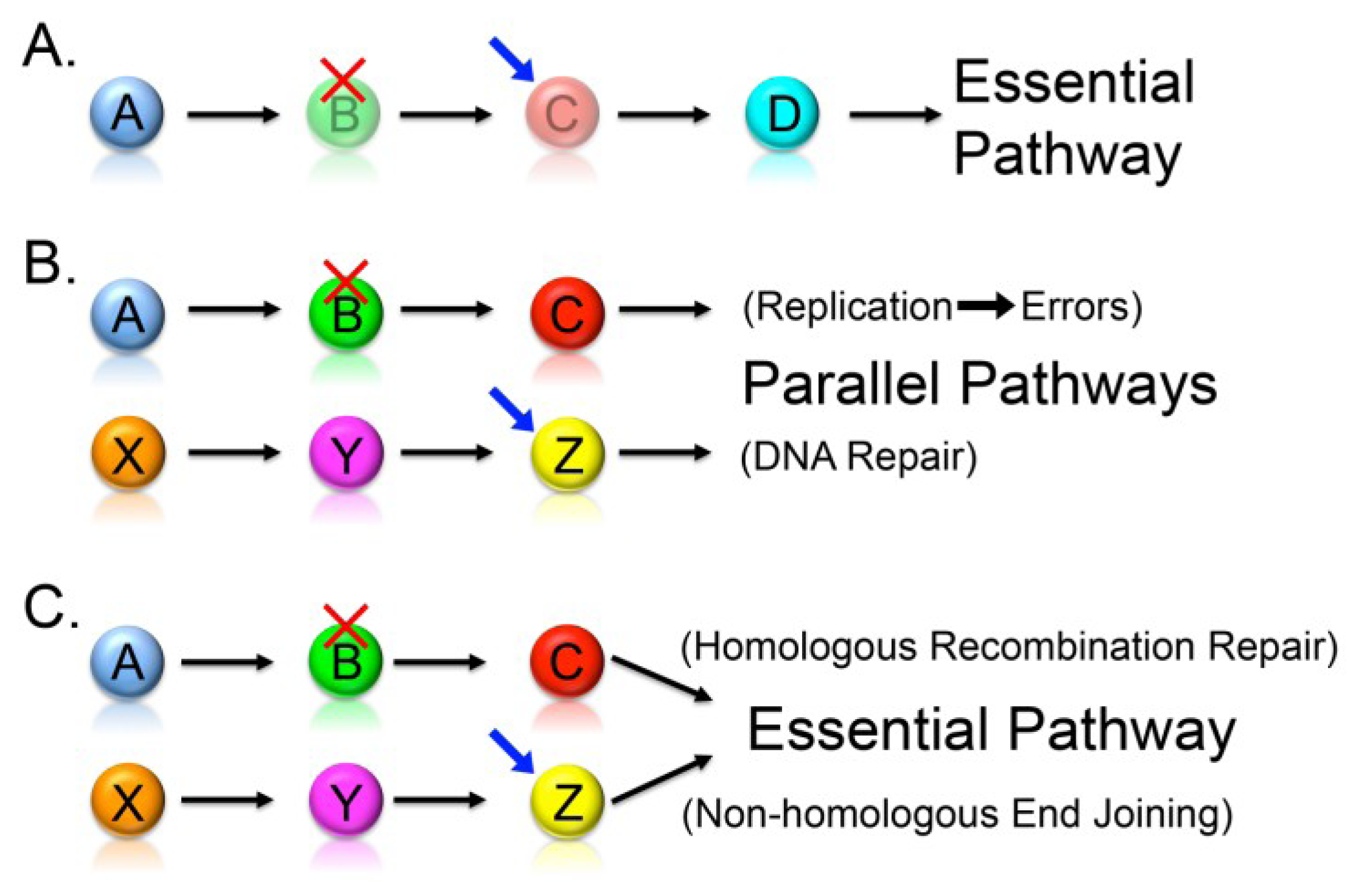
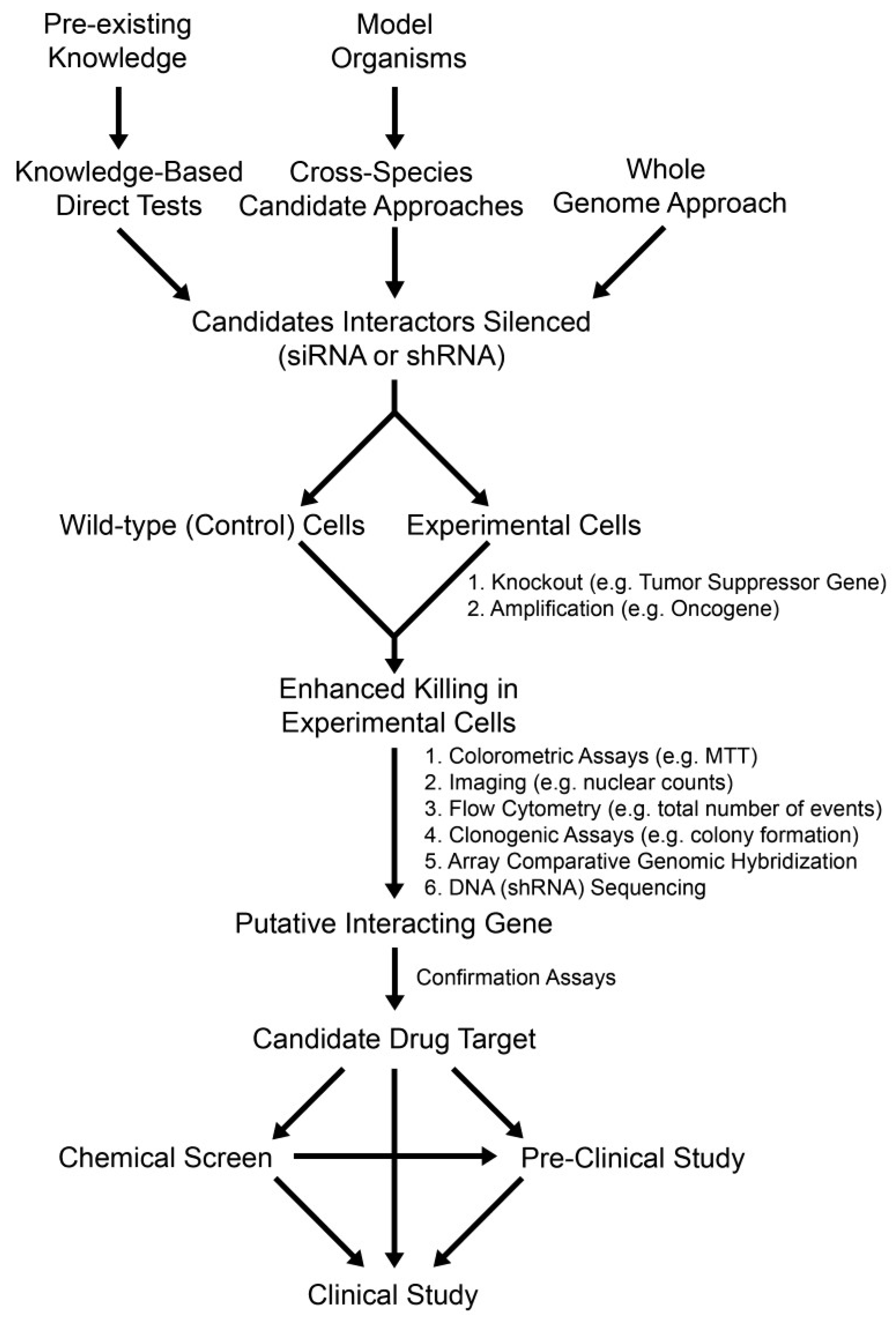
2.3. Knowledge-Based Direct Tests to Identify Synthetic Lethal Interactors
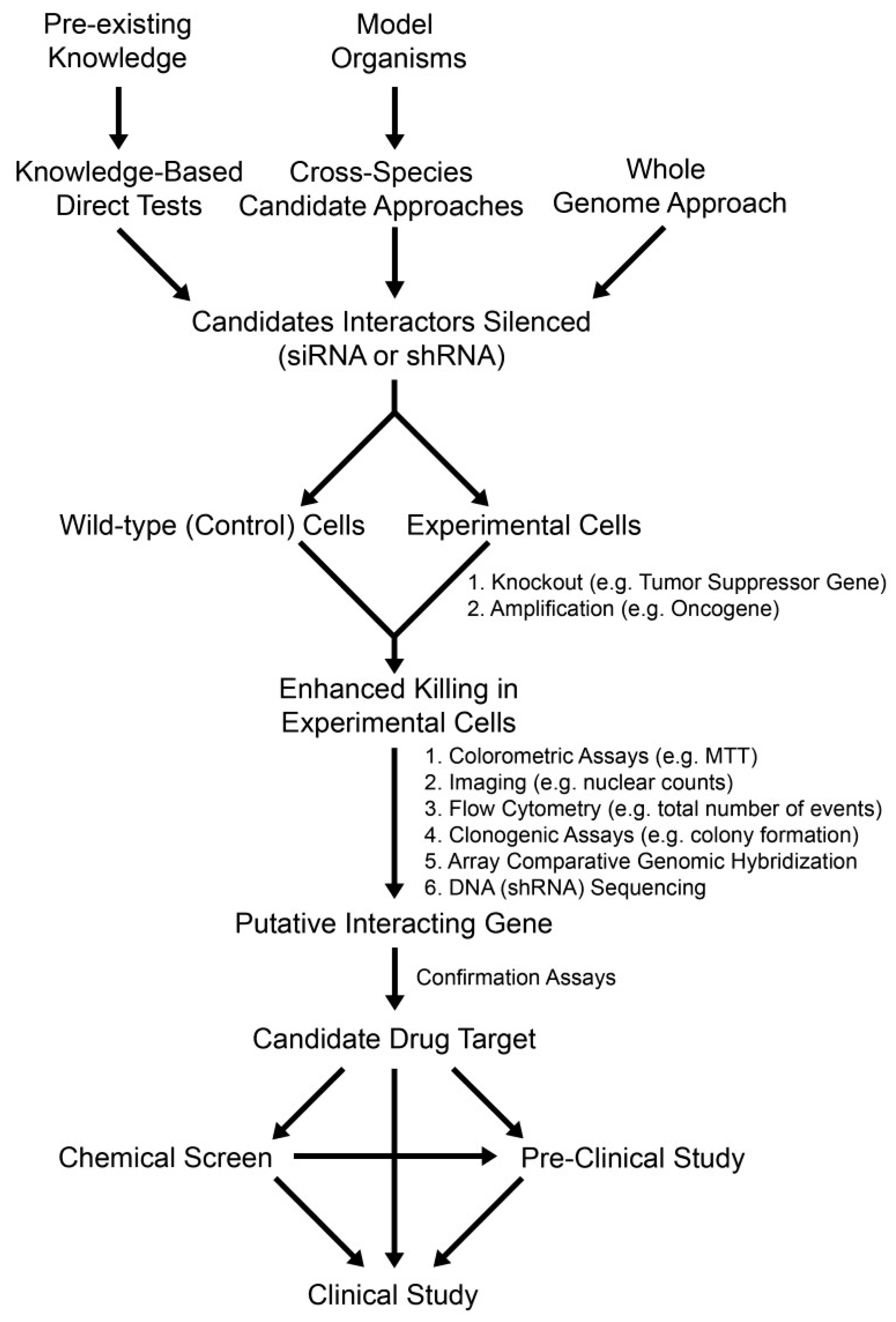
2.4. Cross-Species Candidate Gene Approaches to Uncover Conserved Synthetic Lethal Interactors
2.5. Whole Genome RNAi-Based Screens to Identify Synthetic Lethal Interactors
2.6. Synthetic Dosage Lethality
3. Conclusions—Synthetic Genetic Approaches in a Personalized Medicine Era
Acknowledgments
References
- Ferlay, J.S.H.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Globocan 2008 v2.0, Cancer Incidence And Mortality Worldwide: Iarc Cancerbase No. 10. International Agency for Research on Cancer: Lyon, France, 2010. [Google Scholar]
- Cahill, D.P.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Genetic instability and darwinian selection in tumours. Trends Cell Biol. 1999, 9, M57–M60. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar] [CrossRef]
- Rajagopalan, H.; Jallepalli, P.V.; Rago, C.; Velculescu, V.E.; Kinzler, K.W.; Vogelstein, B.; Lengauer, C. Inactivation of hcdc4 can cause chromosomal instability. Nature 2004, 428, 77–81. [Google Scholar] [CrossRef]
- Issa, J.P. Cpg island methylator phenotype in cancer. Nat. Rev. Cancer 2004, 4, 988–993. [Google Scholar] [CrossRef]
- Boyer, J.C.; Umar, A.; Risinger, J.I.; Lipford, J.R.; Kane, M.; Yin, S.; Barrett, J.C.; Kolodner, R.D.; Kunkel, T.A. Microsatellite instability, mismatch repair deficiency, and genetic defects in human cancer cell lines. Cancer Res. 1995, 55, 6063–6070. [Google Scholar]
- Gazzoli, I.; Loda, M.; Garber, J.; Syngal, S.; Kolodner, R.D. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the mlh1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res. 2002, 62, 3925–3928. [Google Scholar]
- Loeb, L.A. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991, 51, 3075–3079. [Google Scholar]
- Loeb, L.A. A mutator phenotype in cancer. Cancer Res. 2001, 61, 3230–3239. [Google Scholar]
- Loeb, L.A.; Bielas, J.H.; Beckman, R.A. Cancers exhibit a mutator phenotype: Clinical implications. Cancer Res. 2008, 68, 3551–3557. [Google Scholar] [CrossRef]
- Wahlberg, S.S.; Schmeits, J.; Thomas, G.; Loda, M.; Garber, J.; Syngal, S.; Kolodner, R.D.; Fox, E. Evaluation of microsatellite instability and immunohistochemistry for the prediction of germ-line msh2 and mlh1 mutations in hereditary nonpolyposis colon cancer families. Cancer Res. 2002, 62, 3485–3492. [Google Scholar]
- Bae, J.M.; Kim, J.H.; Kang, G.H. Epigenetic alterations in colorectal cancer: The CpG island methylator phenotype. Histol. Histopathol. 2013, 28, 585–595. [Google Scholar]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Lengauer, C.; Kinzler, K.W.; Vogelstein, B. Genetic instability in colorectal cancers. Nature 1997, 386, 623–627. [Google Scholar] [CrossRef]
- Beheshti, B.; Park, P.C.; Sweet, J.M.; Trachtenberg, J.; Jewett, M.A.; Squire, J.A. Evidence of chromosomal instability in prostate cancer determined by spectral karyotyping (sky) and interphase fish analysis. Neoplasia 2001, 3, 62–69. [Google Scholar]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef]
- Mai, S.; Hanley-Hyde, J.; Rainey, G.J.; Kuschak, T.I.; Paul, J.T.; Littlewood, T.D.; Mischak, H.; Stevens, L.M.; Henderson, D.W.; Mushinski, J.F. Chromosomal and extrachromosomal instability of the cyclin d2 gene is induced by myc overexpression. Neoplasia 1999, 1, 241–252. [Google Scholar]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef]
- Barretina, J.; Taylor, B.S.; Banerji, S.; Ramos, A.H.; Lagos-Quintana, M.; Decarolis, P.L.; Shah, K.; Socci, N.D.; Weir, B.A.; Ho, A.; et al. Subtype-specific genomic alterations define new targets for soft-tissue sarcoma therapy. Nat. Genet. 2010, 42, 715–721. [Google Scholar] [Green Version]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615.[Green Version]
- Parsons, D.W.; Li, M.; Zhang, X.; Jones, S.; Leary, R.J.; Lin, J.C.; Boca, S.M.; Carter, H.; Samayoa, J.; Bettegowda, C.; et al. The genetic landscape of the childhood cancer medulloblastoma. Science 2011, 331, 435–439. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70.
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337.
- Cancer Genome Atlas Research Network; Hammerman, P.S.; Hayes, D.N.; Wilkerson, M.D.; Schultz, N.; Bose, R.; Chu, A.; Collisson, E.A.; Cope, L.; Creighton, C.J.; et al. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The origin and evolution of mutations in acute myeloid leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef]
- Choi, C.M.; Seo, K.W.; Jang, S.J.; Oh, Y.M.; Shim, T.S.; Kim, W.S.; Lee, D.S.; Lee, S.D. Chromosomal instability is a risk factor for poor prognosis of adenocarcinoma of the lung: Fluorescence in situ hybridization analysis of paraffin-embedded tissue from korean patients. Lung Cancer 2009, 64, 66–70. [Google Scholar] [CrossRef]
- Heilig, C.E.; Loffler, H.; Mahlknecht, U.; Janssen, J.W.; Ho, A.D.; Jauch, A.; Kramer, A. Chromosomal instability correlates with poor outcome in patients with myelodysplastic syndromes irrespectively of the cytogenetic risk group. J. Cell. Mol. Med. 2010, 14, 895–902. [Google Scholar] [CrossRef]
- Murayama-Hosokawa, S.; Oda, K.; Nakagawa, S.; Ishikawa, S.; Yamamoto, S.; Shoji, K.; Ikeda, Y.; Uehara, Y.; Fukayama, M.; McCormick, F.; et al. Genome-wide single-nucleotide polymorphism arrays in endometrial carcinomas associate extensive chromosomal instability with poor prognosis and unveil frequent chromosomal imbalances involved in the pi3-kinase pathway. Oncogene 2010, 29, 1897–1908. [Google Scholar] [CrossRef]
- Nakamura, H.; Saji, H.; Idiris, A.; Kawasaki, N.; Hosaka, M.; Ogata, A.; Saijo, T.; Kato, H. Chromosomal instability detected by fluorescence in situ hybridization in surgical specimens of non-small cell lung cancer is associated with poor survival. Clin. Cancer Res. 2003, 9, 2294–2299. [Google Scholar]
- Sato, H.; Uzawa, N.; Takahashi, K.; Myo, K.; Ohyama, Y.; Amagasa, T. Prognostic utility of chromosomal instability detected by fluorescence in situ hybridization in fine-needle aspirates from oral squamous cell carcinomas. BMC Cancer 2010, 10, 182. [Google Scholar]
- Suehiro, Y.; Okada, T.; Okada, T.; Anno, K.; Okayama, N.; Ueno, K.; Hiura, M.; Nakamura, M.; Kondo, T.; Oga, A.; et al. Aneuploidy predicts outcome in patients with endometrial carcinoma and is related to lack of cdh13 hypermethylation. Clin. Cancer Res. 2008, 14, 3354–3361. [Google Scholar] [CrossRef]
- Tilly, H.; Rossi, A.; Stamatoullas, A.; Lenormand, B.; Bigorgne, C.; Kunlin, A.; Monconduit, M.; Bastard, C. Prognostic value of chromosomal abnormalities in follicular lymphoma. Blood 1994, 84, 1043–1049. [Google Scholar]
- Walther, A.; Houlston, R.; Tomlinson, I. Association between chromosomal instability and prognosis in colorectal cancer: A meta-analysis. Gut 2008, 57, 941–950. [Google Scholar] [CrossRef]
- Yoo, J.W.; Seo, K.W.; Jang, S.J.; Oh, Y.M.; Shim, T.S.; Kim, W.S.; Lee, D.S.; Lee, S.D.; Choi, C.M. The relationship between the presence of chromosomal instability and prognosis of squamous cell carcinoma of the lung: Fluorescence in situ hybridization analysis of paraffin-embedded tissue from 47 korean patients. J. Korean Med. Sci. 2010, 25, 863–867. [Google Scholar] [CrossRef]
- Kinzler, K.W.; Vogelstein, B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature 1997. [Google Scholar] [CrossRef]
- Farnebo, M.; Bykov, V.J.; Wiman, K.G. The p53 tumor suppressor: A master regulator of diverse cellular processes and therapeutic target in cancer. Biochem. Biophys. Res. Commun. 2010, 396, 85–89. [Google Scholar] [CrossRef]
- Chinnam, M.; Goodrich, D.W. Rb1, development, and cancer. Curr. Top. Dev. Biol. 2011, 94, 129–169. [Google Scholar] [CrossRef]
- Vermeij, J.; Teugels, E.; Bourgain, C.; Xiangming, J.; in ’t Veld, P.; Ghislain, V.; Neyns, B.; de Greve, J. Genomic activation of the EGFR and HER2-neu genes in a significant proportion of invasive epithelial ovarian cancers. BMC Cancer 2008, 8, 3. [Google Scholar] [CrossRef]
- Baselga, J.; Swain, S.M. Novel anticancer targets: Revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 2009, 9, 463–475. [Google Scholar] [CrossRef]
- Tsuda, H.; Hirohashi, S.; Hirota, T.; Shimosato, Y. Alterations in copy number of c-erbb-2 and c-myc proto-oncogenes in advanced stage of human breast cancer. Acta Pathol. Jpn. 1991, 41, 19–23. [Google Scholar]
- Delgado, M.D.; Albajar, M.; Gomez-Casares, M.T.; Batlle, A.; Leon, J. Myc oncogene in myeloid neoplasias. Clin. Transl. Oncol. 2013, 15, 87–94. [Google Scholar] [CrossRef]
- Leon, J.; Ferrandiz, N.; Acosta, J.C.; Delgado, M.D. Inhibition of cell differentiation: A critical mechanism for myc-mediated carcinogenesis? Cell Cycle 2009, 8, 1148–1157. [Google Scholar]
- Liebermann, D.A.; Hoffman, B. Differentiation primary response genes and proto-oncogenes as positive and negative regulators of terminal hematopoietic cell differentiation. Stem Cells 1994, 12, 352–369. [Google Scholar] [CrossRef]
- Johnson, L.; Mercer, K.; Greenbaum, D.; Bronson, R.T.; Crowley, D.; Tuveson, D.A.; Jacks, T. Somatic activation of the k-ras oncogene causes early onset lung cancer in mice. Nature 2001, 410, 1111–1116. [Google Scholar] [CrossRef]
- Aguirre, A.J.; Bardeesy, N.; Sinha, M.; Lopez, L.; Tuveson, D.A.; Horner, J.; Redston, M.S.; DePinho, R.A. Activated kras and ink4a/arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003, 17, 3112–3126. [Google Scholar] [CrossRef]
- Haigis, K.M.; Kendall, K.R.; Wang, Y.; Cheung, A.; Haigis, M.C.; Glickman, J.N.; Niwa-Kawakita, M.; Sweet-Cordero, A.; Sebolt-Leopold, J.; Shannon, K.M.; et al. Differential effects of oncogenic k-ras and n-ras on proliferation, differentiation and tumor progression in the colon. Nat. Genet. 2008, 40, 600–608. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Dobzhansky, T. Genetics of natural populations. XIII. Recombination and variability in populations of drosophila pseudoobscura. Genetics 1946, 31, 269–290. [Google Scholar]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar] [CrossRef]
- McManus, K.J.; Barrett, I.J.; Nouhi, Y.; Hieter, P. Specific synthetic lethal killing of rad54b-deficient human colorectal cancer cells by fen1 silencing. Proc. Natl. Acad. Sci. USA 2009, 106, 3276–3281. [Google Scholar] [CrossRef]
- Van Pel, D.M.; Barrett, I.J.; Shimizu, Y.; Sajesh, B.V.; Guppy, B.J.; Pfeifer, T.; McManus, K.J.; Hieter, P. An evolutionarily conserved synthetic lethal interaction network identifies fen1 as a broad-spectrum target for anticancer therapeutic development. PLoS Genet. 2013, 9, e1003254. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Szankasi, P.; Roberts, C.J.; Murray, A.W.; Friend, S.H. Integrating genetic approaches into the discovery of anticancer drugs. Science 1997, 278, 1064–1068. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. Choosing anticancer drug targets in the postgenomic era. J. Clin. Invest. 1999, 104, 1503–1506. [Google Scholar] [CrossRef]
- Friend, S.H.; Oliff, A. Emerging uses for genomic information in drug discovery. N. Engl. J. Med. 1998, 338, 125–126. [Google Scholar] [CrossRef]
- Roy, R.; Chun, J.; Powell, S.N. Brca1 and brca2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2012, 12, 68–78. [Google Scholar]
- El-Khamisy, S.F.; Masutani, M.; Suzuki, H.; Caldecott, K.W. A requirement for parp-1 for the assembly or stability of xrcc1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003, 31, 5526–5533. [Google Scholar] [CrossRef]
- Okano, S.; Lan, L.; Caldecott, K.W.; Mori, T.; Yasui, A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell. Biol. 2003, 23, 3974–3981. [Google Scholar]
- Strom, C.E.; Johansson, F.; Uhlen, M.; Szigyarto, C.A.; Erixon, K.; Helleday, T. Poly (adp-ribose) polymerase (parp) is not involved in base excision repair but parp inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011, 39, 3166–3175. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(adp-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Kraus, W.L.; Hottiger, M.O. PARP-1 and gene regulation: Progress and puzzles. Mol. Aspects Med. 2013. [Google Scholar] [CrossRef]
- Kuzminov, A. Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc. Natl. Acad. Sci. USA 2001, 98, 8241–8246. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of brca2-deficient tumours with inhibitors of poly(adp-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in brca mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. Parp inhibition: Parp1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar]
- Papeo, G.; Casale, E.; Montagnoli, A.; Cirla, A. Parp inhibitors in cancer therapy: An update. Expert Opin. Ther. Pat. 2013, 23, 503–514. [Google Scholar] [CrossRef]
- Glendenning, J.; Tutt, A. Parp inhibitors—Current status and the walk towards early breast cancer. Breast 2011, 20, S12–S19. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(adp-ribose) polymerase in tumors from brca mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Orlando, L.; Schiavone, P.; Fedele, P.; Calvani, N.; Nacci, A.; Cinefra, M.; D’Amico, M.; Mazzoni, E.; Marino, A.; Sponziello, F.; et al. Poly (adp-ribose) polymerase (parp): Rationale, preclinical and clinical evidences of its inhibition as breast cancer treatment. Expert Opin. Ther. Targets 2012, 16, S83–S89. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(adp-ribose) polymerase inhibitor olaparib in patients with brca1 or brca2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Fogelman, D.R.; Wolff, R.A.; Kopetz, S.; Javle, M.; Bradley, C.; Mok, I.; Cabanillas, F.; Abbruzzese, J.L. Evidence for the efficacy of iniparib, a parp-1 inhibitor, in brca2-associated pancreatic cancer. Anticancer Res. 2011, 31, 1417–1420. [Google Scholar]
- O’Shaughnessy, J.; Osborne, C.; Pippen, J.E.; Yoffe, M.; Patt, D.; Rocha, C.; Koo, I.C.; Sherman, B.M.; Bradley, C. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. N. Engl. J. Med. 2011, 364, 205–214. [Google Scholar] [CrossRef]
- Patel, A.G.; de Lorenzo, S.B.; Flatten, K.S.; Poirier, G.G.; Kaufmann, S.H. Failure of iniparib to inhibit poly(adp-ribose) polymerase in vitro. Clin. Cancer Res. 2012, 18, 1655–1662. [Google Scholar] [CrossRef]
- Liu, X.; Shi, Y.; Maag, D.X.; Palma, J.P.; Patterson, M.J.; Ellis, P.A.; Surber, B.W.; Ready, D.B.; Soni, N.B.; Ladror, U.S.; et al. Iniparib nonselectively modifies cysteine-containing proteins in tumor cells and is not a bona fide parp inhibitor. Clin. Cancer Res. 2012, 18, 510–523. [Google Scholar] [CrossRef]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Curtin, N.J. DNA repair dysregulation from cancer driver to therapeutic target. Nat. Rev. Cancer 2012, 12, 801–817. [Google Scholar] [CrossRef]
- Fiedler, D.; Braberg, H.; Mehta, M.; Chechik, G.; Cagney, G.; Mukherjee, P.; Silva, A.C.; Shales, M.; Collins, S.R.; van Wageningen, S.; et al. Functional organization of the s. Cerevisiae phosphorylation network. Cell 2009, 136, 952–963. [Google Scholar] [CrossRef]
- Lin, Y.Y.; Qi, Y.; Lu, J.Y.; Pan, X.; Yuan, D.S.; Zhao, Y.; Bader, J.S.; Boeke, J.D. A comprehensive synthetic genetic interaction network governing yeast histone acetylation and deacetylation. Genes Dev. 2008, 22, 2062–2074. [Google Scholar] [CrossRef]
- Montpetit, B.; Thorne, K.; Barrett, I.; Andrews, K.; Jadusingh, R.; Hieter, P.; Measday, V. Genome-wide synthetic lethal screens identify an interaction between the nuclear envelope protein, apq12p, and the kinetochore in saccharomyces cerevisiae. Genetics 2005, 171, 489–501. [Google Scholar] [CrossRef]
- Zhao, R.; Davey, M.; Hsu, Y.C.; Kaplanek, P.; Tong, A.; Parsons, A.B.; Krogan, N.; Cagney, G.; Mai, D.; Greenblatt, J.; et al. Navigating the chaperone network: An integrative map of physical and genetic interactions mediated by the hsp90 chaperone. Cell 2005, 120, 715–727. [Google Scholar] [CrossRef]
- Lehner, B.; Crombie, C.; Tischler, J.; Fortunato, A.; Fraser, A.G. Systematic mapping of genetic interactions in caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat. Genet. 2006, 38, 896–903. [Google Scholar]
- Byrne, A.B.; Weirauch, M.T.; Wong, V.; Koeva, M.; Dixon, S.J.; Stuart, J.M.; Roy, P.J. A global analysis of genetic interactions in caenorhabditis elegans. J. Biol. 2007, 6, 8. [Google Scholar] [CrossRef]
- Tarailo, M.; Tarailo, S.; Rose, A.M. Synthetic lethal interactions identify phenotypic “interologs” of the spindle assembly checkpoint components. Genetics 2007, 177, 2525–2530. [Google Scholar]
- McLellan, J.; O’Neil, N.; Tarailo, S.; Stoepel, J.; Bryan, J.; Rose, A.; Hieter, P. Synthetic lethal genetic interactions that decrease somatic cell proliferation in caenorhabditis elegans identify the alternative rfc ctf18 as a candidate cancer drug target. Mol. Biol. Cell 2009, 20, 5306–5313. [Google Scholar] [CrossRef]
- Dixon, S.J.; Fedyshyn, Y.; Koh, J.L.; Prasad, T.S.; Chahwan, C.; Chua, G.; Toufighi, K.; Baryshnikova, A.; Hayles, J.; Hoe, K.L.; et al. Significant conservation of synthetic lethal genetic interaction networks between distantly related eukaryotes. Proc. Natl. Acad. Sci. USA 2008, 105, 16653–16658. [Google Scholar] [CrossRef]
- Miyagawa, K.; Tsuruga, T.; Kinomura, A.; Usui, K.; Katsura, M.; Tashiro, S.; Mishima, H.; Tanaka, K. A role for rad54b in homologous recombination in human cells. EMBO J. 2002, 21, 175–180. [Google Scholar]
- Hiramoto, T.; Nakanishi, T.; Sumiyoshi, T.; Fukuda, T.; Matsuura, S.; Tauchi, H.; Komatsu, K.; Shibasaki, Y.; Inui, H.; Watatani, M.; et al. Mutations of a novel human rad54 homologue, rad54b, in primary cancer. Oncogene 1999, 18, 3422–3426. [Google Scholar] [CrossRef]
- Breitkreutz, B.J.; Stark, C.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Livstone, M.; Oughtred, R.; Lackner, D.H.; Bahler, J.; Wood, V.; et al. The biogrid interaction database: 2008 update. Nucleic Acids Res. 2008, 36, D637–D640. [Google Scholar]
- Huang, C.H.; Konagaya, A.; Lanza, V.; Sloot, P.M. Introduction to the special section on biogrid: Biomedical computations on the grid. IEEE Trans. Inf. Technol. Biomed. 2008, 12, 133–137. [Google Scholar] [CrossRef]
- Stark, C.; Breitkreutz, B.J.; Chatr-Aryamontri, A.; Boucher, L.; Oughtred, R.; Livstone, M.S.; Nixon, J.; van Auken, K.; Wang, X.; Shi, X.; et al. The biogrid interaction database: 2011 update. Nucleic Acids Res. 2011, 39, D698–D704. [Google Scholar] [CrossRef]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. Biogrid: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar]
- Chatr-Aryamontri, A.; Breitkreutz, B.J.; Heinicke, S.; Boucher, L.; Winter, A.; Stark, C.; Nixon, J.; Ramage, L.; Kolas, N.; O’Donnell, L.; et al. The biogrid interaction database: 2013 update. Nucleic Acids Res. 2013, 41, D816–D823. [Google Scholar] [CrossRef]
- Breitkreutz, B.J.; Stark, C.; Tyers, M. Osprey: A network visualization system. Genome Biol. 2003, 4, R22. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Dompe, N.; Rivers, C.S.; Li, L.; Cordes, S.; Schwickart, M.; Punnoose, E.A.; Amler, L.; Seshagiri, S.; Tang, J.; Modrusan, Z.; et al. A whole-genome rnai screen identifies an 8q22 gene cluster that inhibits death receptor-mediated apoptosis. Proc. Natl. Acad. Sci. USA 2011, 108, E943–E951. [Google Scholar] [CrossRef]
- Schlabach, M.R.; Luo, J.; Solimini, N.L.; Hu, G.; Xu, Q.; Li, M.Z.; Zhao, Z.; Smogorzewska, A.; Sowa, M.E.; Ang, X.L.; et al. Cancer proliferation gene discovery through functional genomics. Science 2008, 319, 620–624. [Google Scholar] [CrossRef]
- Silva, J.M.; Marran, K.; Parker, J.S.; Silva, J.; Golding, M.; Schlabach, M.R.; Elledge, S.J.; Hannon, G.J.; Chang, K. Profiling essential genes in human mammary cells by multiplex rnai screening. Science 2008, 319, 617–620. [Google Scholar] [CrossRef]
- Xie, L.; Gazin, C.; Park, S.M.; Zhu, L.J.; Debily, M.A.; Kittler, E.L.; Zapp, M.L.; Lapointe, D.; Gobeil, S.; Virbasius, C.M.; et al. A synthetic interaction screen identifies factors selectively required for proliferation and tert transcription in p53-deficient human cancer cells. PLoS Genet. 2012, 8, e1003151. [Google Scholar] [CrossRef]
- Kroll, E.S.; Hyland, K.M.; Hieter, P.; Li, J.J. Establishing genetic interactions by a synthetic dosage lethality phenotype. Genetics 1996, 143, 95–102. [Google Scholar]
- Yan, H.; Gibson, S.; Tye, B.K. Mcm2 and mcm3, two proteins important for ars activity, are related in structure and function. Genes Dev. 1991, 5, 944–957. [Google Scholar] [CrossRef]
- Li, J.J.; Herskowitz, I. Isolation of orc6, a component of the yeast origin recognition complex by a one-hybrid system. Science 1993, 262, 1870–1874. [Google Scholar]
- Molenaar, J.J.; Ebus, M.E.; Geerts, D.; Koster, J.; Lamers, F.; Valentijn, L.J.; Westerhout, E.M.; Versteeg, R.; Caron, H.N. Inactivation of CDK2 is synthetically lethal to mycn over-expressing cancer cells. Proc. Natl. Acad. Sci. USA 2009, 106, 12968–12973. [Google Scholar] [CrossRef]
- Wartiovaara, K.; Barnabe-Heider, F.; Miller, F.D.; Kaplan, D.R. N-myc promotes survival and induces s-phase entry of postmitotic sympathetic neurons. J. Neurosci. 2002, 22, 815–824. [Google Scholar]
- Knoepfler, P.S.; Cheng, P.F.; Eisenman, R.N. N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev. 2002, 16, 2699–2712. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of n-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar]
- Garson, J.A.; Pemberton, L.F.; Sheppard, P.W.; Varndell, I.M.; Coakham, H.B.; Kemshead, J.T. N-myc gene expression and oncoprotein characterisation in medulloblastoma. Br. J. Cancer 1989, 59, 889–894. [Google Scholar] [CrossRef]
- Nisen, P.D.; Zimmerman, K.A.; Cotter, S.V.; Gilbert, F.; Alt, F.W. Enhanced expression of the n-myc gene in wilms’ tumors. Cancer Res. 1986, 46, 6217–6222. [Google Scholar]
- Schwab, M. Enhanced expression of the cellular oncogene mycn and progression of human neuroblastoma. Adv. Enzyme Regul. 1991, 31, 329–338. [Google Scholar] [CrossRef]
- Schwab, M. Mycn in neuronal tumours. Cancer Lett. 2004, 204, 179–187. [Google Scholar] [CrossRef]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A genome-wide rnai screen identifies multiple synthetic lethal interactions with the ras oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef]
- Villaruz, L.C.; Socinski, M.A.; Cunningham, D.E.; Chiosea, S.I.; Burns, T.F.; Siegfried, J.M.; Dacic, S. The prognostic and predictive value of kras oncogene substitutions in lung adenocarcinoma. Cancer 2013, 119, 2268–2274. [Google Scholar] [CrossRef]
- Zhao, S.; Choi, M.; Overton, J.D.; Bellone, S.; Roque, D.M.; Cocco, E.; Guzzo, F.; English, D.P.; Varughese, J.; Gasparrini, S.; et al. Landscape of somatic single-nucleotide and copy-number mutations in uterine serous carcinoma. Proc. Natl. Acad. Sci. USA 2013, 110, 2916–2921. [Google Scholar] [CrossRef]
- Jenwitheesuk, E.; Horst, J.A.; Rivas, K.L.; van Voorhis, W.C.; Samudrala, R. Novel paradigms for drug discovery: Computational multitarget screening. Trends Pharmacol. Sci. 2008, 29, 62–71. [Google Scholar] [CrossRef]
- Lopez-Vallejo, F.; Caulfield, T.; Martinez-Mayorga, K.; Giulianotti, M.A.; Nefzi, A.; Houghten, R.A.; Medina-Franco, J.L. Integrating virtual screening and combinatorial chemistry for accelerated drug discovery. Comb. Chem. High Throughput Screen. 2011, 14, 475–487. [Google Scholar] [CrossRef]
- Guvench, O.; MacKerell, A.D., Jr. Computational evaluation of protein-small molecule binding. Curr. Opin. Struct. Biol. 2009, 19, 56–61. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [CrossRef]
- Furey, T.S. Chip-seq and beyond: New and improved methodologies to detect and characterize protein-DNA interactions. Nat. Rev. Genet. 2012, 13, 840–852. [Google Scholar] [CrossRef]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef]
- Robertson, A.G.; Bilenky, M.; Tam, A.; Zhao, Y.; Zeng, T.; Thiessen, N.; Cezard, T.; Fejes, A.P.; Wederell, E.D.; Cullum, R.; et al. Genome-wide relationship between histone h3 lysine 4 mono- and tri-methylation and transcription factor binding. Genome Res. 2008, 18, 1906–1917. [Google Scholar] [CrossRef]
- Neff, T.; Armstrong, S.A. Chromatin maps, histone modifications and leukemia. Leukemia 2009, 23, 1243–1251. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sajesh, B.V.; Guppy, B.J.; McManus, K.J. Synthetic Genetic Targeting of Genome Instability in Cancer. Cancers 2013, 5, 739-761. https://doi.org/10.3390/cancers5030739
Sajesh BV, Guppy BJ, McManus KJ. Synthetic Genetic Targeting of Genome Instability in Cancer. Cancers. 2013; 5(3):739-761. https://doi.org/10.3390/cancers5030739
Chicago/Turabian StyleSajesh, Babu V., Brent J. Guppy, and Kirk J. McManus. 2013. "Synthetic Genetic Targeting of Genome Instability in Cancer" Cancers 5, no. 3: 739-761. https://doi.org/10.3390/cancers5030739