Leptin’s Pro-Angiogenic Signature in Breast Cancer

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Angiogenesis and Tumor Angiogenesis
3. Leptin and OB-R Expression and Signaling
4. Regulation of Leptin Expression
5. Leptin’s Impact on Tumor Angiogenesis
5.1. Leptin Regulation of VEGF and VEGFR2
5.2. Leptin Regulation of IL-1
5.3. NILCO: Notch, IL-1, Leptin Crosstalk Outcome
5.4. Leptin Regulation of MMPs and Integrins
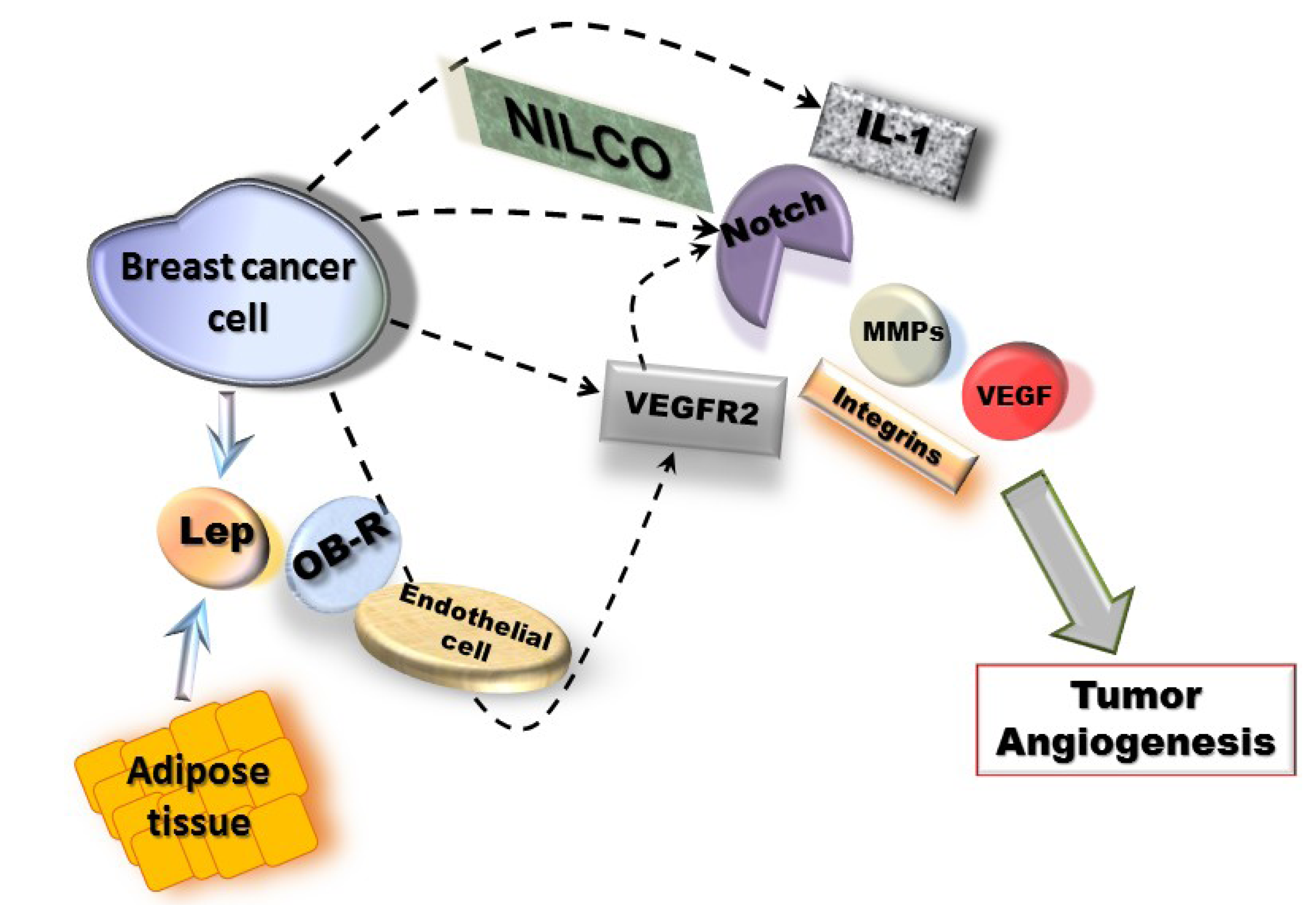
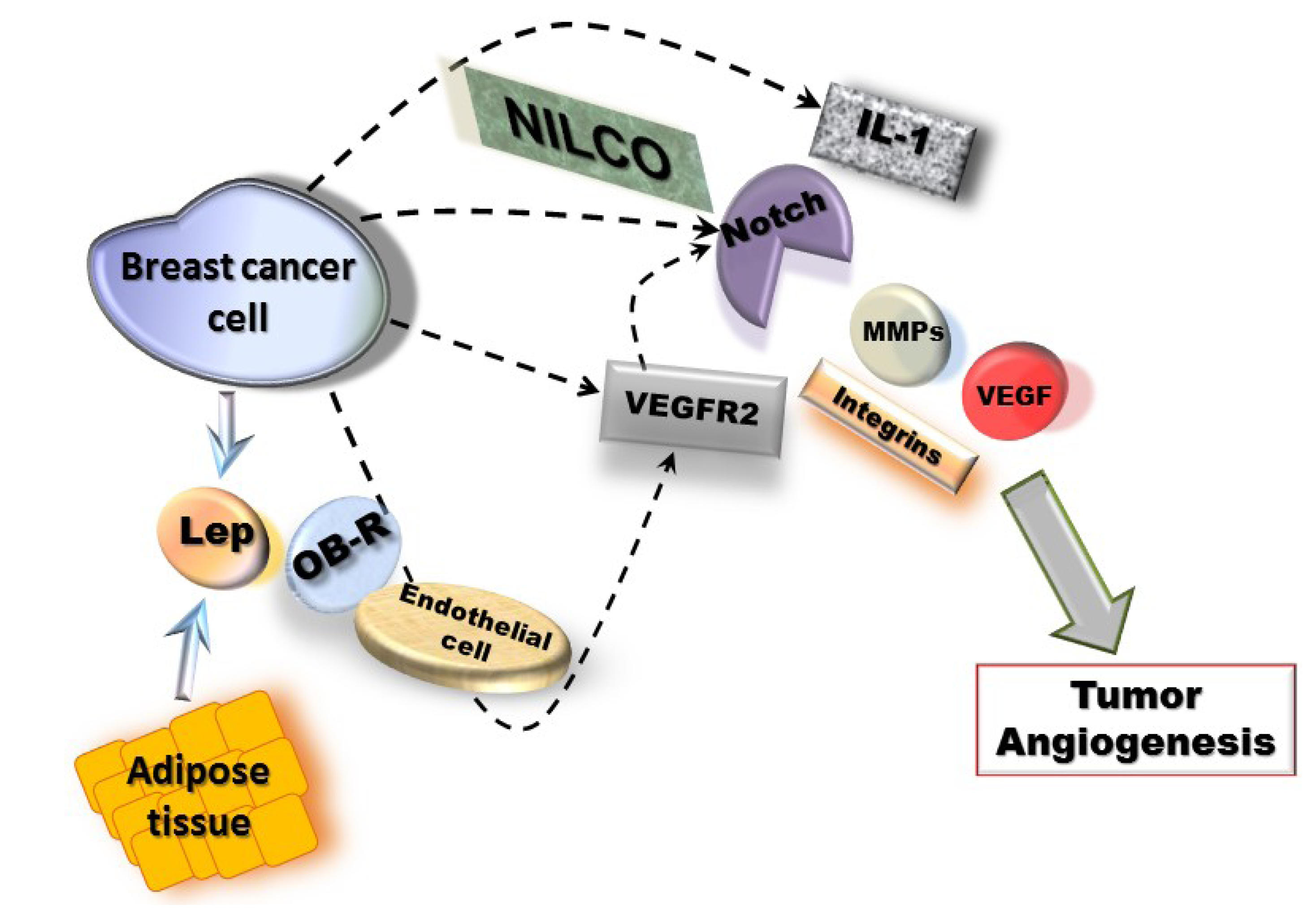
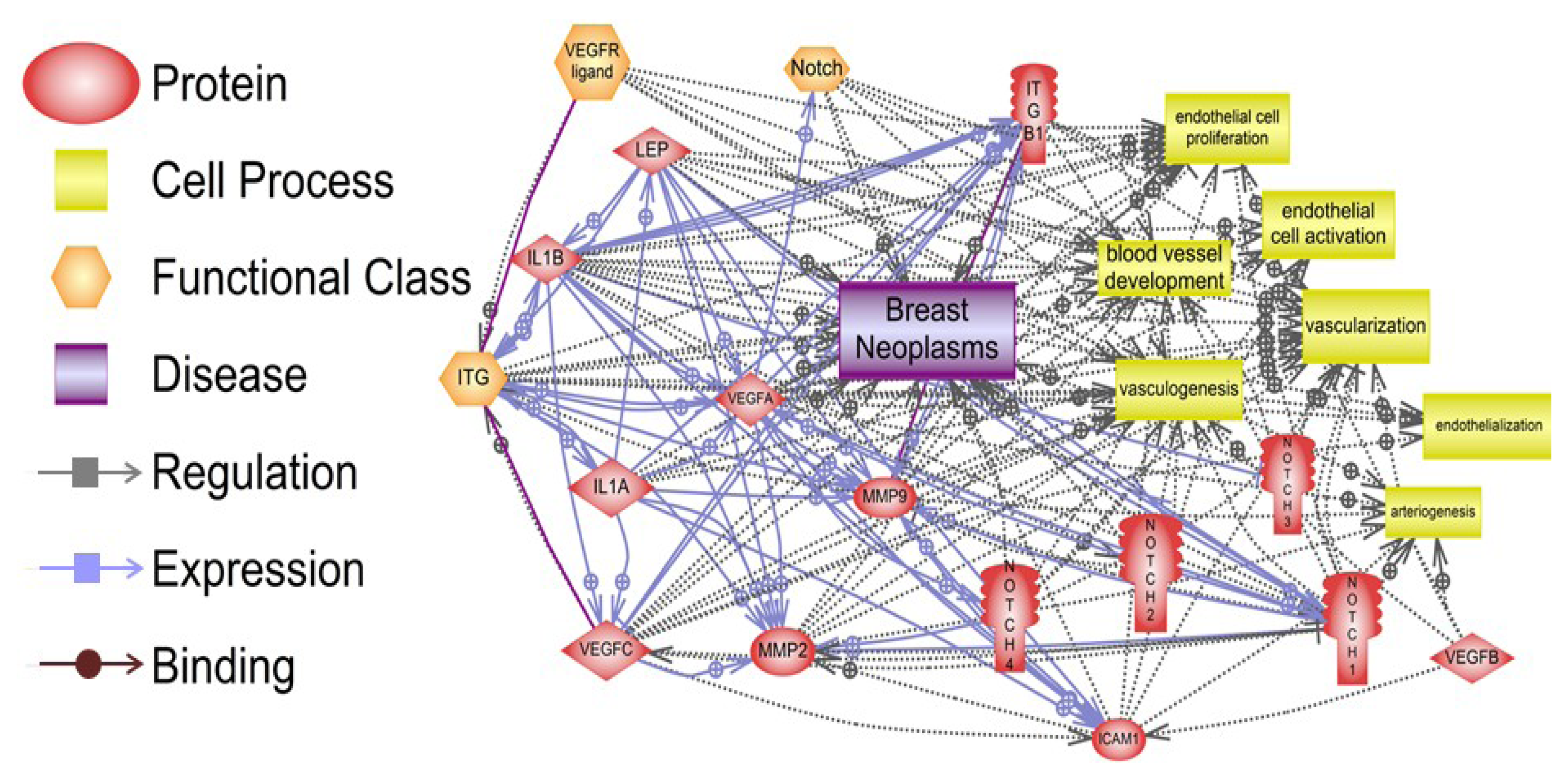
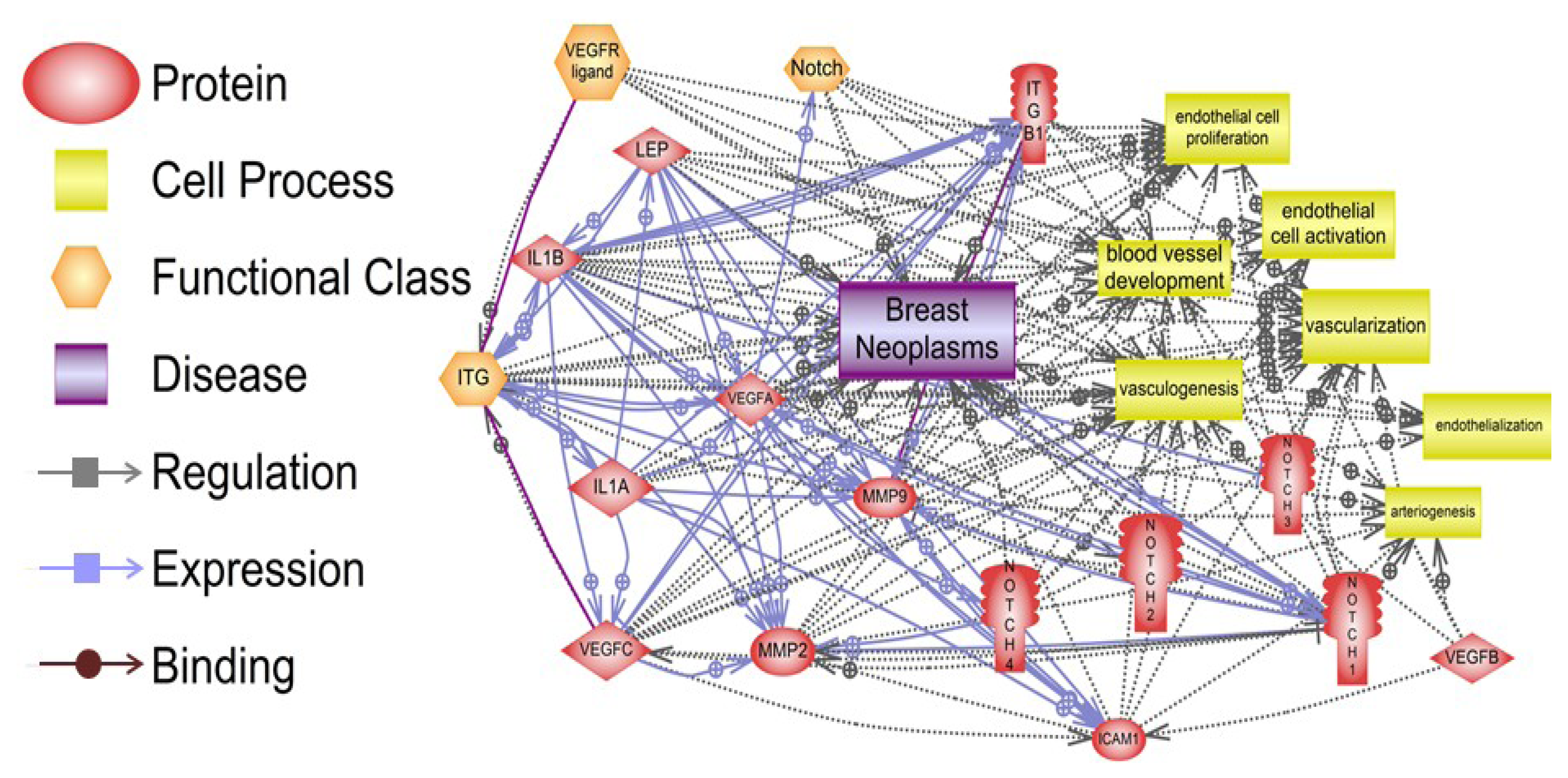
6. Pathway Studio 9 Analysis of Leptin Pro-Angiogenic Networks in Breast Cancer
7. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Basen-Engquist, K.; Chang, M. Obesity and cancer risk: Recent review and evidence. Curr. Oncol. Rep. 2011, 13, 71–76. [Google Scholar] [CrossRef]
- Pischon, T.; Nöthlings, U.; Boeing, H. Obesity and cancer. Proc. Nutr. Soc. 2008, 67, 128–145. [Google Scholar] [CrossRef]
- Vucenik, I.; Stains, J.P. Obesity and cancer risk: Evidence, mechanisms, and recommendations. Ann. NY Acad. Sci. 2012, 1271, 37–43. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest. 2007, 117, 175–184. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Deyoung, S.M.; Bodzin, J.L.; Saltiel, A.R. Increased inflammatory properties of adipose tissue macrophages recruited during diet-induced obesity. Diabetes 2007, 56, 16–23. [Google Scholar] [CrossRef]
- Clemons, M.; Goss, P. Estrogen and the risk of breast cancer. N. Engl. J. Med. 2001, 344, 276–285. [Google Scholar] [CrossRef]
- Bernstein, L.; Ross, R.K. Endogenous hormones and breast cancer risk. Epidemiol. Rev. 1993, 15, 48–65. [Google Scholar]
- Johnston, S.R. New strategies in estrogen receptor-positive breast cancer. Clin. Cancer Res. 2010, 16, 1979–1987. [Google Scholar] [CrossRef]
- Rose, D.P.; Vona-Davis, L. Interaction between menopausal status and obesity in affecting breast cancer risk. Maturitas 2010, 66, 33–38. [Google Scholar] [CrossRef]
- Gabriely, I.; Ma, X.H.; Yang, X.M.; Rossetti, L.; Barzilai, N. Leptin resistance during aging is independent of fat mass. Diabetes 2002, 51, 1016–1021. [Google Scholar] [CrossRef]
- Guo, S.; Liu, M.; Wang, G.; Torroella-Kouri, M.; Gonzalez-Perez, R.R. Oncogenic role and therapeutic target of leptin signaling in breast cancer and cancer stem cells. Biochim. Biophys. Acta 2012, 1825, 207–222. [Google Scholar]
- Jarde, T.; Caldefie-Chezet, F.; Damez, M.; Mishellany, F.; Penault-Llorca, F.; Guillot, J.; Vasson, M.P. Leptin and leptin receptor involvement in cancer development: A study on human primary breast carcinoma. Oncol. Rep. 2008, 19, 905–911. [Google Scholar]
- Henson, M.C.; Castracane, V.D. Leptin: Roles and regulation in primate pregnancy. Semin. Reprod. Med. 2002, 20, 113–122. [Google Scholar] [CrossRef]
- Chen, X.; Zha, X.; Chen, W.; Zhu, T.; Qiu, J.; Roe, O.D.; Li, J.; Wang, Z.; Yin, Y. Leptin attenuates the anti-estrogen effect of tamoxifen in breast cancer. Biomed. Pharmacother. 2013, 67, 22–30. [Google Scholar] [CrossRef]
- Catalano, S.; Mauro, L.; Marsico, S.; Giordano, C.; Rizza, P.; Rago, V.; Montanaro, D.; Maggiolini, M.; Panno, M.L.; Ando, S. Leptin induces, via ERK1/ERK2 signal, functional activation of estrogen receptor alpha in MCF-7 cells. J. Biol. Chem. 2004, 279, 19908–19915. [Google Scholar] [CrossRef]
- Catalano, S.; Marsico, S.; Giordano, C.; Mauro, L.; Rizza, P.; Panno, M.L.; Ando, S. Leptin enhances, via AP-1, expression of aromatase in the MCF-7 cell line. J. Biol. Chem. 2003, 278, 28668–28676. [Google Scholar]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef]
- Jain, R.K.; Booth, M.F. What brings pericytes to tumor vessels? J. Clin. Invest. 2003, 112, 1134–1136. [Google Scholar]
- Clapp, C.; Thebault, S.; Jeziorski, M.C.; Martínez de la Escalera, G. Peptide hormone regulation of angiogenesis. Physiol. Rev. 2009, 89, 1177–1215. [Google Scholar] [CrossRef]
- Hausman, G.J.; Richardson, R.L. Adipose tissue angiogenesis. J. Anim. Sci. 2004, 82, 925–934. [Google Scholar]
- Mu, H.; Ohashi, R.; Yan, S.; Chai, H.; Yang, H.; Lin, P.; Yao, Q.; Chen, C. Adipokine resistinp romotes in vitro angiogenesis of human endothelial cells. Cardiovasc. Res. 2006, 70, 146–157. [Google Scholar] [CrossRef]
- Rehman, J.; Traktuev, D.; Li, J.; Merfeld-Clauss, S.; Temm-Grove, C.J.; Bovenkerk, J.E.; Pell, C.L.; Johnstone, B.H.; Considine, R.V.; March, K.L. Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation 2004, 109, 1292–1298. [Google Scholar] [CrossRef]
- Shigematsu, S.; Yamauchi, K.; Nakajima, K.; Iijima, S.; Aizawa, T.; Hashizume, K. IGF-1 regulates migration and angiogenesis of human endothelial cells. Endocr. J. 1999, 46, S59–S62. [Google Scholar] [CrossRef]
- Tsigkos, S.; Koutsilieris, M.; Papapetropoulos, A. Angiopoietins in angiogenesis and beyond. Expert Opin. Investig. Drugs 2003, 12, 933–941. [Google Scholar] [CrossRef]
- Bråkenhielm, E.; Cao, R.; Gao, B.; Angelin, B.; Cannon, B.; Parini, P.; Cao, Y. Angiogenesis inhibitor, TNP-470, prevents diet-induced and genetic obesity in mice. Circ. Res. 2004, 94, 1579–1588. [Google Scholar] [CrossRef]
- Bouloumié, A.; Drexler, H.C.; Lafontan, M.; Busse, R. Leptin, the product of ob gene, promotes angiogenesis. Circ. Res. 1998, 83, 1059–1066. [Google Scholar] [CrossRef]
- Sierra-Honigmann, M.R.; Nath, A.K.; Murakami, C.; García-Cardeña, G.; Papapetropoulos, A.; Sessa, W.C.; Madge, L.A.; Schechner, J.S.; Schwabb, M.B.; Polverini, P.J.; et al. Biological action of leptin as an angiogenic factor. Science 1998, 281, 1683–1686. [Google Scholar] [CrossRef]
- Bornstein, S.R.; Abu-Asab, M.; Glasow, A.; Päth, G.; Hauner, H.; Tsokos, M.; Chrousos, G.P.; Scherbaum, W.A. Immunohistochemical and ultrastructural localization of leptin and eptin receptor in human white adipose tissue and differentiating human adipose cells in primary culture. Diabetes 2000, 49, 532–538. [Google Scholar] [CrossRef]
- González, R.R.; Simon, C.; Caballero-Campo, P.; Norman, R.; Chardonnens, D.; Devoto, L.; Bischof, P. Leptin and reproduction. Hum. Reprod. Update 2000, 6, 290–300. [Google Scholar] [CrossRef]
- Newman, G.; Gonzalez-Perez, R. Leptin-cytokine crosstalk in breast cancer. Mol. Cell. Endocrinol. 2013. [Google Scholar] [CrossRef]
- Mendelsohn, M.E.; Karas, R.H. The protective effects of estrogen on the cardiovascular system. N. Engl. J. Med. 1999, 340, 1801–1811. [Google Scholar] [CrossRef]
- Welboren, W.J.; Stunnenberg, H.G.; Sweep, F.C.; Span, P.N. Identifying estrogen receptor target genes. Mol. Oncol. 2007, 1, 138–143. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, R.R.; Leavis, P. Leptin upregulates beta3-integrin expression and interleukin-1beta, upregulates leptin and leptin receptor expression in human endometrial epithelial cell cultures. Endocrine 2001, 16, 21–28. [Google Scholar]
- Heida, N.M.; Leifheit-Nestler, M.; Schroeter, M.R.; Muller, J.P.; Cheng, I.F.; Henkel, S.; Limbourg, A.; Limbourg, F.P.; Alves, F.; Quigley, J.P.; et al. Leptin enhances the potency of circulating angiogenic cells via Src kinase and integrin (alpha)vbeta5: Implications for angiogenesis in human obesity. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 200–206. [Google Scholar] [CrossRef]
- Park, H.Y.; Kwon, H.M.; Lim, H.J.; Hong, B.K.; Lee, J.Y.; Park, B.E.; Jang, Y.; Cho, S.Y.; Kim, H.S. Potential role of leptin in angiogenesis: Leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp. Mol. Med. 2001, 33, 95–102. [Google Scholar] [CrossRef]
- Guo, S.; Colbert, L.S.; Fuller, M.; Zhang, Y.; Gonzalez-Perez, R.R. Vascular endothelial growth factor receptor-2 in breast cancer. Biochim. Biophys. Acta 2010, 1806, 108–121. [Google Scholar]
- Joukov, V.; Pajusola, K.; Kaipainen, A.; Chilov, D.; Lahtinen, I.; Kukk, E.; Saksela, O.; Kalkkinen, N.; Alitalo, K. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. EMBO J. 1996, 15, 290–298. [Google Scholar]
- Meyer, M.; Clauss, M.; Lepple-Wienhues, A.; Waltenberger, J.; Augustin, H.G.; Ziche, M.; Lanz, C.; Büttner, M.; Rziha, H.J.; Dehio, C. A novel vascular endothelial growth factor encoded by Orf virus, VEGF-E, mediates angiogenesis via signalling through VEGFR-2 (KDR) but not VEGFR-1 (Flt-1) receptor tyrosine kinases. EMBO J. 1999, 18, 363–374. [Google Scholar] [CrossRef]
- Park, J.E.; Chen, H.H.; Winer, J.; Houck, K.A.; Ferrara, N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to Flt-1 but not to Flk-1/KDR. J. Biol. Chem. 1994, 269, 25646–25654. [Google Scholar]
- Waltenberger, J.; Claesson-Welsh, L.; Siegbahn, A.; Shibuya, M.; Heldin, C.H. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 1994, 269, 26988–26995. [Google Scholar]
- Carmeliet, P. Angiogenesis in life, disease and medicine. Nature 2005, 438, 932–936. [Google Scholar] [CrossRef]
- Yancopoulos, G.D.; Davis, S.; Gale, N.W.; Rudge, J.S.; Wiegand, S.J.; Holash, J. Vascular-specific growth factors and blood vessel formation. Nature 2000, 407, 242–248. [Google Scholar] [CrossRef]
- Iwasaki, H.; Kawamoto, A.; Tjwa, M.; Horii, M.; Hayashi, S.; Oyamada, A.; Matsumoto, T.; Suehiro, S.; Carmeliet, P.; Asahara, T. PLGF repairs myocardial ischemia through mechanisms of angiogenesis, cardioprotection and recruitment of myo-angiogenic competent marrow progenitors. PLoS One 2011, 6, e24872. [Google Scholar] [CrossRef]
- Garonna, E.; Botham, K.M.; Birdsey, G.M.; Randi, A.M.; Gonzalez-Perez, R.R.; Wheeler-Jones, C.P. Vascular endothelial growth factor receptor-2 couples cyclo-oxygenase-2 with pro-angiogenic actions of leptin on human endothelial cells. PLoS One 2011, 6, e18823. [Google Scholar]
- Cao, Y. Angiogenesis modulates adipogenesis and obesity. J. Clin. Invest. 2007, 117, 2362–2368. [Google Scholar] [CrossRef]
- Sainson, R.C.; Johnston, D.A.; Chu, H.C.; Holderfield, M.T.; Nakatsu, M.N.; Crampton, S.P.; Davis, J.; Conn, E.; Hughes, C.C. TNF primes endothelial cells for angiogenic sprouting by inducing a tip cell phenotype. Blood 2008, 111, 4997–5007. [Google Scholar] [CrossRef]
- Schneider, B.P.; Miller, K.D. Angiogenesis of breast cancer. J. Clin. Oncol. 2005, 23, 1782–1790. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Dvorak, H.F.; Brown, L.F.; Detmar, M.; Dvorak, A.M. Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am. J. Pathol. 1995, 146, 1029–1039. [Google Scholar]
- Dvorak, H.F.; Nagy, J.A.; Dvorak, J.T.; Dvorak, A.M. Identification and characterization of the blood vessels of solid tumors that are leaky to circulating macromolecules. Am. J. Pathol. 1988, 133, 95–109. [Google Scholar]
- Jensen, H.M.; Chen, I.; deVault, M.R.; Lewis, A.E. Angiogenesis induced by “normal” human breast tissue: A probable marker for precancer. Science 1982, 218, 293–295. [Google Scholar]
- Jaffe, T.; Schwartz, B. Leptin promotes motility and invasiveness in human colon cancer cells by activating multiple signal-transduction pathways. Int. J. Cancer 2008, 123, 2543–2556. [Google Scholar] [CrossRef]
- Ratke, J.; Entschladen, F.; Niggemann, B.; Zänker, K.S.; Lang, K. Leptin stimulates the migration of colon carcinoma cells by multiple signaling pathways. Endocr. Relat. Cancer Cell 2010, 17, 179–189. [Google Scholar] [CrossRef]
- Chen, C.; Chang, Y.C.; Liu, C.L.; Liu, T.P.; Chang, K.J.; Guo, I.C. Leptin induces proliferation and anti-apoptosis in human hepatocarcinoma cells by up-regulating cyclin D1 and down-regulating Bax via a janus kinase 2-linked pathway. Endocr. Relat. Cancer 2007, 14, 513–529. [Google Scholar] [CrossRef]
- Lawrence, J.E.; Cook, N.J.; Rovin, R.A.; Winn, R.J. Leptin promotes glioblastoma. Neurol. Res. Int. 2012. [Google Scholar] [CrossRef]
- Riolfi, M.; Ferla, R.; del Valle, L.; Piña-Oviedo, S.; Scolaro, L.; Micciolo, R.; Guidi, M.; Terrasi, M.; Cetto, G.L.; Surmacz, E. Leptin and its receptor are overexpressed in brain tumors and correlate with the degree of malignancy. Brain Pathol. 2010, 20, 481–489. [Google Scholar] [CrossRef]
- Andò, S.; Catalano, S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat. Rev. Endocrinol. 2011, 8, 263–275. [Google Scholar] [CrossRef]
- Grossmann, M.E.; Cleary, M.P. The balance between leptin and adiponectin in the control of carcinogenesis—Focus on mammary tumorigenesis. Biochimie 2012, 94, 2164–2171. [Google Scholar] [CrossRef]
- Jarde, T.; Caldefie-Chezet, F.; Damez, M.; Mishellany, F.; Perrone, D.; Penault-Llorca, F.; Guillot, J.; Vasson, M.P. Adiponectin and leptin expression in primary ductal breast cancer and in adjacent healthy epithelial and myoepithelial tissue. Histopathology 2008, 53, 484–487. [Google Scholar] [CrossRef]
- Ishikawa, M.; Kitayama, J.; Nagawa, H. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin. Cancer Res. 2004, 10, 4325–4331. [Google Scholar] [CrossRef]
- Colbert, L.; McGlothen, T.; Wilson, K.; Gillespie, C.; Dickson, T.; Guo, S.; Gonzalez-Perez, R.R. Differential expression of nilco reveals pathogenesis of human breast cancer. Cancer Res. 2012. [Google Scholar] [CrossRef]
- Gonzalez, R.R.; Leavis, P.C. A peptide derived from the human leptin molecule is a potent inhibitor of the leptin receptor function in rabbit endometrial cells. Endocrine 2003, 21, 185–195. [Google Scholar] [CrossRef]
- Banks, A.S.; Davis, S.M.; Bates, S.H.; Myers, M.G. Activation of downstream signals by the long form of the leptin receptor. J. Biol. Chem. 2000, 275, 14563–14572. [Google Scholar] [CrossRef]
- Frühbeck, G. Intracellular signalling pathways activated by leptin. Biochem. J. 2006, 393, 7–20. [Google Scholar] [CrossRef]
- Kallen, C.B.; Lazar, M.A. Antidiabetic thiazolidinediones inhibit leptin (OB) gene expression in 3T3-L1 adipocytes. Proc. Natl. Acad. Sci. USA 1996, 93, 5793–5796. [Google Scholar] [CrossRef]
- Fried, S.K.; Ricci, M.R.; Russell, C.D.; Laferrère, B. Regulation of leptin production in humans. J. Nutr. 2000, 130, 3127S–3131S. [Google Scholar]
- Faggioni, R.; Fantuzzi, G.; Fuller, J.; Dinarello, C.A.; Feingold, K.R.; Grunfeld, C. IL-1 beta mediates leptin induction during inflammation. Am. J. Physiol. 1998, 274, R204–R208. [Google Scholar]
- Janik, J.E.; Curti, B.D.; Considine, R.V.; Rager, H.C.; Powers, G.C.; Alvord, W.G.; Smith, J.W.N.; Gause, B.L.; Kopp, W.C. Interleukin 1 alpha increases serum leptin concentrations in humans. J. Clin. Endocrinol. Metab. 1997, 82, 3084–3086. [Google Scholar] [CrossRef]
- Kumar, S.; Kishimoto, H.; Chua, H.L.; Badve, S.; Miller, K.D.; Bigsby, R.M.; Nakshatri, H. Interleukin-1 alpha promotes tumor growth and cachexia in MCF-7 xenograft model of breast cancer. Am. J. Pathol. 2003, 163, 2531–2541. [Google Scholar] [CrossRef]
- Henson, M.C.; Castracane, V.D. Leptin in pregnancy. Biol. Reprod. 2000, 63, 1219–1228. [Google Scholar] [CrossRef]
- Marttunen, M.B.; Andersson, S.; Hietanen, P.; Karonen, S.L.; Koistinen, H.A.; Koivisto, V.A.; Tiitinen, A.; Ylikorkala, O. Antiestrogenic tamoxifen and toremifene increase serum leptin levels in postmenopausal breast cancer patients. Maturitas 2000, 35, 175–179. [Google Scholar] [CrossRef]
- De la Brousse, F.C.; Shan, B.; Chen, J.L. Identification of the promoter of the mouse obese gene. Proc. Natl. Acad. Sci. USA 1996, 93, 4096–4101. [Google Scholar] [CrossRef]
- Hwang, C.S.; Mandrup, S.; MacDougald, O.A.; Geiman, D.E.; Lane, M.D. Transcriptional activation of the mouse obese (ob) gene by CCAAT/enhancer binding protein alpha. Proc. Natl. Acad. Sci. USA 1996, 93, 873–877. [Google Scholar] [CrossRef]
- Kim, J.B.; Sarraf, P.; Wright, M.; Yao, K.M.; Mueller, E.; Solanes, G.; Lowell, B.B.; Spiegelman, B.M. Nutritional and insulin regulation of fatty acid synthetase and leptin gene expression through ADD1/SREBP1. J. Clin. Invest. 1998, 101, 1–9. [Google Scholar] [CrossRef]
- Wrann, C.D.; Eguchi, J.; Bozec, A.; Xu, Z.; Mikkelsen, T.; Gimble, J.; Nave, H.; Wagner, E.F.; Ong, S.E.; Rosen, E.D. FOSL2 promotes leptin gene expression in human and mouse adipocytes. J. Clin. Invest. 2012, 122, 1010–1021. [Google Scholar] [CrossRef]
- Wang, B.; Wood, I.S.; Trayhurn, P. Hypoxia induces leptin gene expression and secretion in human preadipocytes: Differential effects of hypoxia on adipokine expression by preadipocytes. J. Endocrinol. 2008, 198, 127–134. [Google Scholar] [CrossRef]
- Meissner, U.; Spranger, R.; Lehner, M.; Allabauer, I.; Rascher, W.; Dötsch, J. Hypoxia-induced leptin production in human trophoblasts does not protect from apoptosis. Eur. J. Endocrinol. 2005, 153, 455–461. [Google Scholar] [CrossRef]
- Cascio, S.; Bartella, V.; Auriemma, A.; Johannes, G.J.; Russo, A.; Giordano, A.; Surmacz, E. Mechanism of leptin expression in breast cancer cells: Role of hypoxia-inducible factor-1alpha. Oncogene 2008, 27, 540–547. [Google Scholar] [CrossRef]
- Bartella, V.; Cascio, S.; Fiorio, E.; Auriemma, A.; Russo, A.; Surmacz, E. Insulin-dependent leptin expression in breast cancer cells. Cancer Res. 2008, 68, 4919–4927. [Google Scholar] [CrossRef]
- Cascio, S.; Ferla, R.; D’Andrea, A.; Gerbino, A.; Bazan, V.; Surmacz, E.; Russo, A. Expression of angiogenic regulators, VEGF and leptin, is regulated by the EGF/PI3K/STAT3 pathway in colorectal cancer cells. J. Cell. Physiol. 2009, 221, 189–194. [Google Scholar] [CrossRef] [Green Version]
- Marchi, M.; Lisi, S.; Curcio, M.; Barbuti, S.; Piaggi, P.; Ceccarini, G.; Nannipieri, M.; Anselmino, M.; di Salvo, C.; Vitti, P.; et al. Human leptin tissue distribution, but not weight loss-dependent change in expression, is associated with methylation of its promoter. Epigenetics 2011, 6, 1198–1206. [Google Scholar] [CrossRef]
- Cao, R.; Brakenhielm, E.; Wahlestedt, C.; Thyberg, J.; Cao, Y. Leptin induces vascular permeability and synergistically stimulates angiogenesis with FGF-2 and VEGF. Proc. Natl. Acad. Sci. USA 2001, 98, 6390–6395. [Google Scholar]
- Gainsford, T.; Willson, T.A.; Metcalf, D.; Handman, E.; McFarlane, C.; Ng, A.; Nicola, N.A.; Alexander, W.S.; Hilton, D.J. Leptin can induce proliferation, differentiation, and functional activation of hemopoietic cells. Proc. Natl. Acad. Sci. USA 1996, 93, 14564–14568. [Google Scholar] [CrossRef]
- Ferla, R.; Bonomi, M.; Otvos, L.J.; Surmacz, E. Glioblastoma-derived leptin induces tube formation and growth of endothelial cells: Comparison with VEGF effects. BMC Cancer Cell 2011. [Google Scholar] [CrossRef]
- Glasow, A.; Kiess, W.; Anderegg, U.; Berthold, A.; Bottner, A.; Kratzsch, J. Expression of leptin (OB) and leptin receptor (OB-R) in human fibroblasts: Regulation of leptin secretion by insulin. J. Clin. Endocrinol. Metab. 2001, 86, 4472–4479. [Google Scholar] [CrossRef]
- Guo, S.; Liu, M.; Gonzalez-Perez, R.R. Role of notch and its oncogenic signaling crosstalk in breast cancer. Biochim. Biophys. Acta 2011, 1815, 197–213. [Google Scholar]
- Barone, I.; Catalano, S.; Gelsomino, L.; Marsico, S.; Giordano, C.; Panza, S.; Bonofiglio, D.; Bossi, G.; Covington, K.R.; Fuqua, S.A.; et al. Leptin mediates tumor-stromal interactions that promote the invasive growth of breast cancer cells. Cancer Res. 2012, 72, 1416–1427. [Google Scholar] [CrossRef]
- Gonzalez-Perez, R.R.; Xu, Y.; Guo, S.; Watters, A.; Zhou, W.; Leibovich, S.J. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFkappaB/HIF-1alpha activation. Cell. Signal. 2010, 22, 1350–1362. [Google Scholar] [CrossRef]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Hursting, S.D.; Nunez, N.P.; Varticovski, L.; Vinson, C. The obesity-cancer link: Lessons learned from a fatless mouse. Cancer Res. 2007, 67, 2391–2393. [Google Scholar] [CrossRef]
- Rupnick, M.A.; Panigrahy, D.; Zhang, C.Y.; Dallabrida, S.M.; Lowell, B.B.; Langer, R.; Folkman, M.J. Adipose tissue mass can be regulated through the vasculature. Proc. Natl. Acad. Sci. USA 2002, 99, 10730–10735. [Google Scholar]
- Montague, C.T.; Prins, J.B.; Sanders, L.; Digby, J.E.; O’Rahilly, S. Depot- and sex-specific differences in human leptin mrna expression: Implications for the control of regional fat distribution. Diabetes 1997, 46, 342–347. [Google Scholar]
- Montague, C.T.; Prins, J.B.; Sanders, L.; Zhang, J.; Sewter, C.P.; Digby, J.; Byrne, C.D.; O’Rahilly, S. Depot-related gene expression in human subcutaneous and omental adipocytes. Diabetes 1998, 47, 1384–1391. [Google Scholar] [CrossRef]
- Brandon, E.L.; Gu, J.W.; Cantwell, L.; He, Z.; Wallace, G.; Hall, J.E. Obesity promotes melanoma tumor growth: Role of leptin. Cancer Biol. Ther. 2009, 8, 1871–1879. [Google Scholar] [CrossRef]
- Amjadi, F.; Javanmard, S.H.; Zarkesh-Esfahani, H.; Khazaei, M.; Narimani, M. Leptin promotes melanoma tumor growth in mice related to increasing circulating endothelial progenitor cells numbers and plasma NO production. J. Exp. Clin. Cancer Res. 2011. [Google Scholar] [CrossRef]
- Cleary, M.P.; Juneja, S.C.; Phillips, F.C.; Hu, X.; Grande, J.P.; Maihle, N.J. Leptin receptor-deficient MMTV-TGF-alpha/lepr(db)lepr(db) female mice do not develop oncogene-induced mammary tumors. Exp. Biol. Med. 2004, 229, 182–193. [Google Scholar]
- Cleary, M.P.; Phillips, F.C.; Getzin, S.C.; Jacobson, T.L.; Jacobson, M.K.; Christensen, T.A.; Juneja, S.C.; Grande, J.P.; Maihle, N.J. Genetically obese MMTV-TGF-alpha/lep(ob)lep(ob) female mice do not develop mammary tumors. Breast Cancer Res. Treat. 2003, 77, 205–215. [Google Scholar] [CrossRef]
- Gu, J.W.; Young, E.; Patterson, S.G.; Makey, K.L.; Wells, J.; Huang, M.; Tucker, K.B.; Miele, L. Postmenopausal obesity promotes tumor angiogenesis and breast cancer progression in mice. Cancer Biol. Ther. 2011, 11, 910–917. [Google Scholar] [CrossRef]
- Gillespie, C.; Quarshie, A.; Penichet, M.; Gonzalez-Perez, R.R. Potential role of leptin signaling in DMBA-induced mammary tumors by non-responsive C57BL/6J mice fed a high-fat diet. J. Carcinog. Mutagen. 2012. [Google Scholar] [CrossRef]
- Gonzalez, R.R.; Cherfils, S.; Escobar, M.; Yoo, J.H.; Carino, C.; Styer, A.K.; Sullivan, B.T.; Sakamoto, H.; Olawaiye, A.; Serikawa, T.; et al. Leptin signaling promotes the growth of mammary tumors and increases the expression of vascular endothelial growth factor (VEGF) and its receptor type two (VEGF-R2). J. Biol. Chem. 2006, 281, 26320–26328. [Google Scholar] [CrossRef]
- Gonzalez, R.R.; Watters, A.; Xu, Y.; Singh, U.P.; Mann, D.R.; Rueda, B.R.; Penichet, M.L. Leptin-signaling inhibition results in efficient anti-tumor activity in estrogen receptor positive or negative breast cancer. Breast Cancer Res. 2009, 11, R36. [Google Scholar] [CrossRef]
- Carino, C.; Olawaiye, A.B.; Cherfils, S.; Serikawa, T.; Lynch, M.P.; Rueda, B.R.; Gonzalez, R.R. Leptin regulation of proangiogenic molecules in benign and cancerous endometrial cells. Int. J. Cancer 2008, 123, 2782–2790. [Google Scholar] [CrossRef]
- Otvos, L., Jr.; Kovalszky, I.; Riolfi, M.; Ferla, R.; Olah, J.; Sztodola, A.; Nama, K.; Molino, A.; Piubello, Q.; Wade, J.D.; et al. Efficacy of a leptin receptor antagonist peptide in a mouse model of triple-negative breast cancer. Eur. J. Cancer 2011, 47, 1578–1584. [Google Scholar] [CrossRef]
- Birmingham, J.M.; Busik, J.V.; Hansen-Smith, F.M.; Fenton, J.I. Novel mechanism for obesity-induced colon cancer progression. Carcinogenesis 2009, 30, 690–697. [Google Scholar] [CrossRef]
- Fuller, M.; Byrd, M.; Guo, S.; Gonzalez-Perez, R.R. Leptin induces Src/Gbr2/Gab2/STAT3 activation and rac-1 crosstalk to regulate VEGF/VEGFR2 in breast cancer. Cancer Res. 2011. [Google Scholar] [CrossRef]
- Frankenberry, K.A.; Somasundar, P.; McFadden, D.W.; Vona-Davis, L.C. Leptin induces cell migration and the expression of growth factors in human prostate cancer cells. Am. J. Surg. 2004, 188, 560–565. [Google Scholar] [CrossRef]
- Cohen, B.; Barkan, D.; Levy, Y.; Goldberg, I.; Fridman, E.; Kopolovic, J.; Rubinstein, M. Leptin induces angiopoietin-2 expression in adipose tissues. J. Biol. Chem. 2001, 276, 7697–7700. [Google Scholar]
- Zhou, W.; Guo, S.; Gonzalez-Perez, R.R. Leptin pro-angiogenic signature in breast cancer is linked to IL-1 signalling. Br. J. Cancer 2011, 104, 128–137. [Google Scholar] [CrossRef]
- Singer, C.F.; Hudelist, G.; Gschwantler-Kaulich, D.; Fink-Retter, A.; Mueller, R.; Walter, I.; Czerwenka, K.; Kubista, E. Interleukin-1alpha protein secretion in breast cancer is associated with poor differentiation and estrogen receptor alpha negativity. Int. J. Gynecol. Cancer 2006, 16 , 556–559. [Google Scholar]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 is required for tumor invasiveness and angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef]
- Elaraj, D.M.; Weinreich, D.M.; Varghese, S.; Puhlmann, M.; Hewitt, S.M.; Carroll, N.M.; Feldman, E.D.; Turner, E.M.; Alexander, H.R. The role of interleukin 1 in growth and metastasis of human cancer xenografts. Clin. Cancer Res. 2006, 12, 1088–1096. [Google Scholar] [CrossRef]
- Apte, R.N.; Dotan, S.; Elkabets, M.; White, M.R.; Reich, E.; Carmi, Y.; Song, X.; Dvozkin, T.; Krelin, Y.; Voronov, E. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006, 25, 387–408. [Google Scholar] [CrossRef]
- Maedler, K.; Sergeev, P.; Ehses, J.A.; Mathe, Z.; Bosco, D.; Berney, T.; Dayer, J.M.; Reinecke, M.; Halban, P.A.; Donath, M.Y.; et al. Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc. Natl. Acad. Sci. USA 2004, 101, 8138–8143. [Google Scholar] [CrossRef]
- Gabay, C.; Dreyer, M.; Pellegrinelli, N.; Chicheportiche, R.; Meier, C.A. Leptin directly induces the secretion of interleukin 1 receptor antagonist in human monocytes. J. Clin. Endocrinol. Metab. 2001, 86, 783–791. [Google Scholar] [CrossRef]
- Guo, S.; Gonzalez-Perez, R.R. Notch, IL-1 and leptin crosstalk outcome (NILCO) is critical for leptin-induced proliferation, migration and VEGF/VEGFR-2 expression in breast cancer. PLoS One 2011, 6, e21467. [Google Scholar]
- Polus, A.G.J.; Piatkowska, E.; Dembinska-Kiec, A. Differences in leptin, VEGF, and bFGF-induced angiogenic differentiation of HUVEC and human umbilical blood CD34+ progenitor cells. Eur. J. Biochem. 2003. Abstract No.: P4.1-67. [Google Scholar]
- Lanier, V.; Gillespie, C.; McGlothen, T.; Dickson, T.; Guo, S.; Gonzalez-Perez, R.R. Leptin induces notch in endothelial cells: Role of VEGFR-2 transactivation. Cancer Res. 2012. [Google Scholar] [CrossRef]
- Eliasz, S.; Liang, S.; Chen, Y.; De Marco, M.A.; Machek, O.; Skucha, S.; Miele, L.; Bocchetta, M. Notch-1 stimulates survival of lung adenocarcinoma cells during hypoxia by activating the IGF-1R pathway. Oncogene 2010, 29, 2488–2498. [Google Scholar] [CrossRef]
- Lemoine, A.Y.; Ledoux, S.; Larger, E. Adipose tissue angiogenesis in obesity. Thromb. Haemost. 2013. [Google Scholar] [CrossRef]
- Schroeter, M.R.; Leifheit, M.; Sudholt, P.; Heida, N.M.; Dellas, C.; Rohm, I.; Alves, F.; Zientkowska, M.; Rafail, S.; Puls, M.; et al. Leptin enhances the recruitment of endothelial progenitor cells into neointimal lesions after vascular injury by promoting integrin-mediated adhesion. Circ. Res. 2008, 103, 536–544. [Google Scholar] [CrossRef]
- Huang, C.Y.; Yu, H.; Lai, T.Y.; Yeh, Y.L.; Su, C.C.; Hsu, H.H.; Tsai, F.J.; Tsai, C.H.; Wu, H.C.; Tang, C.H. Leptin increases motility and integrin up-regulation in human prostate cancer cells. J. Cell Physiol. 2011, 226, 1274–1282. [Google Scholar] [CrossRef]
- Yang, S.N.; Chen, H.T.; Tsou, H.K.; Huang, C.Y.; Yang, W.H.; Su, C.M.; Fong, Y.C.; Tseng, W.P.; Tang, C.H. Leptin enhances cell migration in human chondrosarcoma cells through OBRL leptin receptor. Carcinogenesis 2009, 30, 566–574. [Google Scholar] [CrossRef]
- Yeh, W.L.; Lu, D.Y.; Lee, M.J.; Fu, W.M. Leptin induces migration and invasion of glioma cells through MMP-13 production. Glia 2009, 57, 454–464. [Google Scholar] [CrossRef]
- Dong, Z.; Xu, X.; Du, L.; Yang, Y.; Cheng, H.; Zhang, X.; Li, Z.; Wang, L.; Li, J.; Liu, H.; et al. Leptin-mediated regulation of MT1-MMP localization is KIF1B dependent and enhances gastric cancer cell invasion. Carcinogenesis 2013, 34, 974–983. [Google Scholar] [CrossRef]
- Schneider, B.P.; Siedge, G.W.J. Anti-vascular endothelial growth factor therapy for breast cancer: Can we pick the winners? J. Clin. Oncol. 2011, 29, 2444–2447. [Google Scholar] [CrossRef]
- Pàez-Ribes, M.; Allen, E.; Hudock, J.; Takeda, T.; Okuyama, H.; Viñals, F.; Inoue, M.; Bergers, G.; Hanahan, D.; Casanovas, O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009, 5, 220–231. [Google Scholar]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gonzalez-Perez, R.R.; Lanier, V.; Newman, G. Leptin’s Pro-Angiogenic Signature in Breast Cancer. Cancers 2013, 5, 1140-1162. https://doi.org/10.3390/cancers5031140
Gonzalez-Perez RR, Lanier V, Newman G. Leptin’s Pro-Angiogenic Signature in Breast Cancer. Cancers. 2013; 5(3):1140-1162. https://doi.org/10.3390/cancers5031140
Chicago/Turabian StyleGonzalez-Perez, Ruben Rene, Viola Lanier, and Gale Newman. 2013. "Leptin’s Pro-Angiogenic Signature in Breast Cancer" Cancers 5, no. 3: 1140-1162. https://doi.org/10.3390/cancers5031140