Emerging Biomarkers in Glioblastoma

Abstract
:1. Introduction

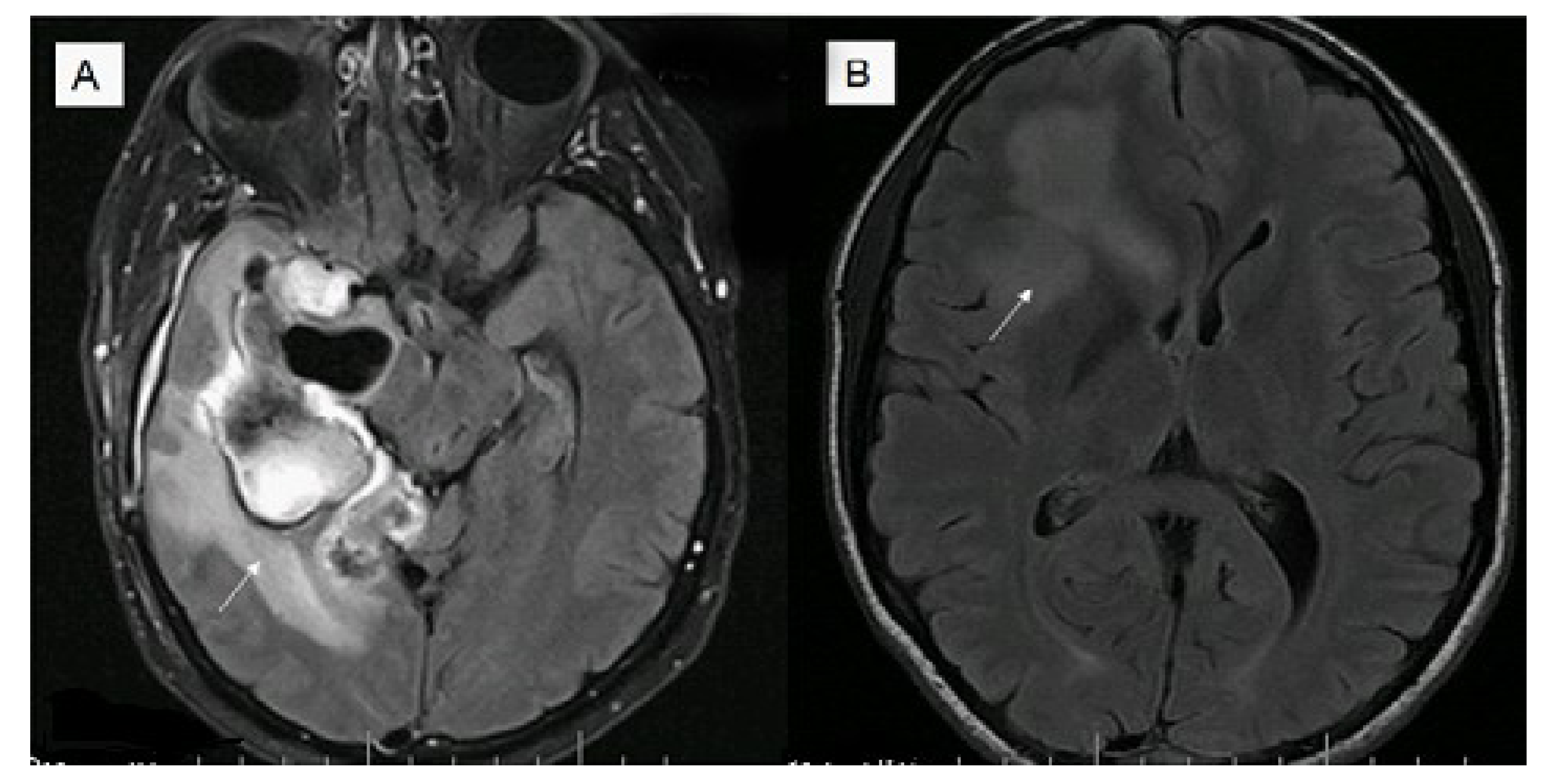{kind=link}
| Verhaak classification [4] | Phillips classification [5] | Jiao classification [6] |
|---|---|---|
| Classical—High EGFR, TP53, longest survival of subgroups in response to aggressive treatment. | Proliferative—Enriched for neural stem cell markers, PTEN loss, EGFR amplified or normal, Akt (protein kinase B) cell signaling pathway activation, shorter survival than proneural subgroup. | I-X glioma—GBM like; multiple molecular subgroups, distinct from IDH1/ATRX/TP53 (I-A glioma) and IDH/CIC/FUBP1 (I-CF glioma) tumors—prognosis approximately 1 year. |
| Proneural—TP53 mutated, IDH1 gene mutated, PDGFRA mutated, patients significantly younger. | Proneural—PTEN intact, EGFR normal, Notch activation, longer survival than proliferative and mesenchymal subgroup. | |
| Mesenchymal—NF1 mutated, TP53 mutated, PTEN mutated. | Mesenchymal—Enriched for neural stem cell markers, PTEN loss, EGFR amplified or normal, Akt cell signaling pathway activation, shorter survival than proneural subgroup. | |
| Neural—mutations in many of same genes as the other 3 subgroups. Oldest patients on average. |
| Molecular/metabolic alteration | Possible biomarker status |
|---|---|
| O(6)-methlyguanine-DNA-methyltransferase (MGMT) promoter methylation | Prognostic, predictive [1] |
| Loss of heterozygosity chromosome 1p 19q | No prognostic significance [8] |
| Loss of heterozygosity 10q | Prognostic [9] |
| Isocitrate dehydrogenase (IDH) mutational status | Prognostic [10] |
| Epidermal growth factor receptor (EGFR) | Prognostic [11] |
| Epidermal growth factor, latrophilin, and 7 transmembrane domain-containing protein 1 on chromosome 1 (ELTD1) | Diagnostic, potentially prognostic [12] |
| Vascular endothelial growth factor (VEGF) | Potentially prognostic [13] |
| Tumor suppressor protein p53 | Diagnostic [14] |
| Phosphatase and tensin homolog (PTEN) | Prognostic, possibly predictive [13] |
| p16INK4a gene | Inconsistent findings [13] |
| Cytochrome c oxidase (CcO) | Potentially prognostic [15] |
| Phospholipid metabolites | Potentially predictive [16] |
| Telomerase messenger expression (hTERT messenger ribonucleic acid [mRNA]) | Potentially diagnostic [17], prognostic [18] |
| microRNAs (miRNAs) | Diagnostic, prognostic [19] |
| Cancer stem cell markers | Potentially prognostic [20,21] |
2. Molecular and Metabolic Alterations in GBM and Their Potential Biomarker Status
2.1. MGMT and DNA Methylation
2.2. Loss of Heterozygosity (LOH) of Chromosomes 1p and 19q
2.3. Loss of Heterozygosity 10q
2.4. IDH
2.5. EGFR
2.6. Epidermal Growth Factor, Latrophilin, and 7 Transmembrane Domain-Containing Protein 1 on Chromosome 1 (ELTD1)
2.7. Vascular Endothelial Growth Factor (VEGF)
2.8. p53
2.9. PTEN
2.10. p16INK4a
2.11. Cytochrome c Oxidase
2.12. Phospholipid Metabolites
2.13. Telomerase Messenger Expression (hTERT Messenger Ribonucleic Acid [mRNA])
3. MicroRNAs (miRNAs)
4. Cancer Stem Cells in GBM
5. Imaging Modalities and Their Potential Biomarker Status
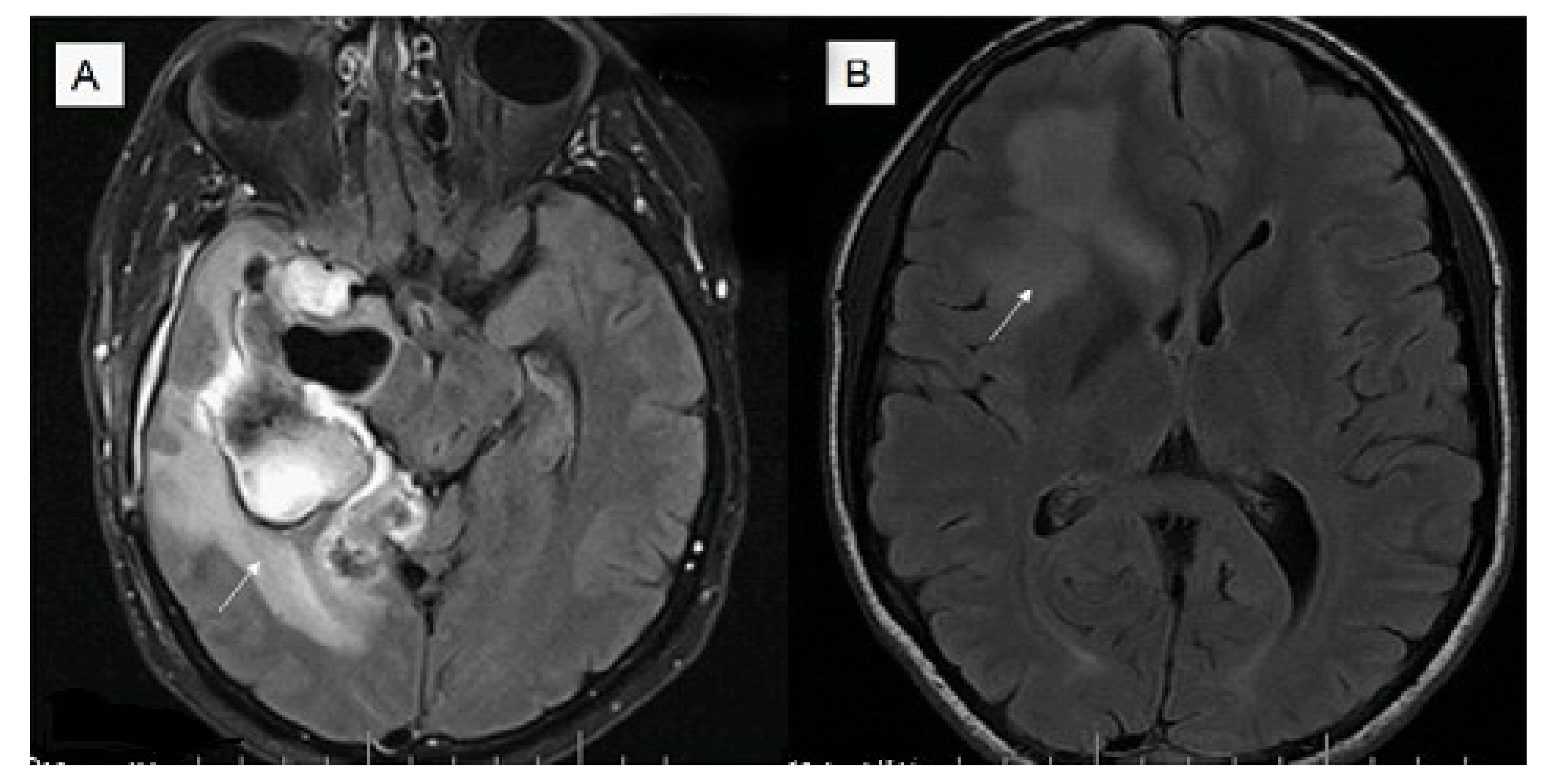
6. Conclusions
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomized phase III study: 5-Year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Polley, M.Y.; Lamborn, K.R.; Chang, S.M.; Butowski, N.; Clarke, J.L.; Prados, M. Conditional probability of survival in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 2011, 29, 4175–4180. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Jiao, Y.; Killela, P.J.; Reitman, Z.J.; Rasheed, A.B.; Heaphy, C.M.; de Wilde, R.F.; Rodriguez, F.J.; Rosemberg, S.; Oba-Shinjo, S.M.; Marie, S.K.N.; et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 2012, 3, 709–722. [Google Scholar]
- Di Stefano, A.L.; Enciso-Mora, V.; Marie, Y.; Desestret, V.; Labussiere, M.; Boisselier, B.; Mokhtari, K.; Idbaih, A.; Hoang-Xuan, K.; Delattre, J.Y.; et al. Association between glioma susceptibility loci and tumour pathology defines specific molecular etiologies. Neurooncology 2013, 15, 542–547. [Google Scholar]
- Hegi, M.E.; Janzer, R.C.; Lambiv, W.L.; Gorlia, T.; Kouwenhoven, M.C.M.; Hartmann, C.; von Deimling, A.; Martinet, D.; Schmutz, N.B.; Diserens, A.C.; et al. Presence of an oligodendroglioma-like component in newly diagnosed glioblastoma identifies a pathogenetically heterogenous subgroup and lacks prognostic value: Central pathology review of the EORTC_26981/NCIC_CE.3 trial. Acta Neuropathol. 2012, 123, 841–852. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.C.; Antweiler, S.; Urban, N.; Mueller, W.; Kuklik, A.; Meyer-Puttlitz, B.; Wiestler, O.D.; Louis, D.N.; Fimmers, R.; von Deimling, A. Impact of genotype and morphology on the prognosis of glioblastoma. J. Neuropathol. Exp. Neurol. 2002, 61, 321–328. [Google Scholar]
- Jansen, M.; Yip, S.; Louis, D.N. Molecular pathology in adult neuro-oncology: An update on diagnostic, prognostic and predictive markers. Lancet Neuro. 2010, 9, 717–726. [Google Scholar] [CrossRef]
- Ang, C.; Guiot, M.C.; Ramanakumar, A.V.; Roberge, D.; Kavan, P. Clinical significance of molecular biomarkers in glioblastoma. Can. J. Neurol. Sci. 2010, 37, 625–630. [Google Scholar]
- Towner, R.A.; Jensen, R.L.; Colman, H.; Vaillant, B.; Smith, N.; Casteel, R.; Saunders, D.; Gillespie, D.L.; Silasi-Mansat, R.; Lupu, F.; et al. ELTD1, a potential new biomarker for gliomas. Neurosurgery 2013, 72, 77–90. [Google Scholar] [CrossRef]
- Bell, E.H.; Hadziahmetovic, M.; Chakravarti, A. Evolvement of molecular biomarkers in targeted therapy of malignant gliomas. In Brain Tumors—Current and Emerging Therapeutic Strategies, 1st ed.; Abujamra, A.L., Ed.; InTech Europe: Rijeka, Croatia, 2011; pp. 117–142. [Google Scholar]
- Tabatabai, G.; Stupp, R.; van den Bent, M.J.; Hegi, M.E.; Tonn, J.C.; Wick, W.; Weller, M. Molecular diagnostics of gliomas: The clinical perspective. Acta Neuropathol. 2010, 120, 585–592. [Google Scholar] [CrossRef]
- Kadenbach, B.; Huttemann, M.; Arnold, S.; Lee, I.; Bender, E. Mitochondrial energy metabolism is regulated via nuclear-coded subunits of cytochrome c oxidase. Free Radic. Biol. Med. 2000, 29, 211–221. [Google Scholar] [CrossRef]
- Hattingen, E.; Bahr, O.; Rieger, J.; Blasel, S.; Steinbach, J.; Pilatus, U. Phospholipid metabolites in recurrent glioblastoma: In vivo markers detect different tumor phenotypes before and under antiangiogenic therapy. PLoS One 2013, 8, e56439. [Google Scholar]
- Shervington, A.; Patel, R.; Lu, C.; Cruickshanks, N.; Lea, R.; Roberts, G.; Dawson, T.; Shervington, L. Telomerase subunits expression variation between biopsy samples and cell lines derived from malignant glioma. Brain Res. 2007, 1134, 45–52. [Google Scholar]
- Boldrini, L.; Pistolesi, S.; Gisfredi, S.; Ursino, S.; Ali, G.; Pieracci, N.; Basolo, F.; Parenti, G.; Fontanini, G. Telomerase activity and hTERT mRNA expression in glial tumors. Int. J. Oncol. 2006, 28, 1555–1560. [Google Scholar]
- Qiu, S.; Lin, S.; Hu, D.; Feng, Y.; Tan, Y.; Peng, Y. Interactions of miR-323/miR-326/miR-329 and miR-130a/miR-155/miR-210 as prognostic indicators for clinical outcome of glioblastoma patients. J. Transl. Med. 2013, 11, e10. [Google Scholar] [CrossRef]
- Metellus, P.; Nanni-Metellus, I.; Delfino, C.; Colin, C.; Tchogandjian, A.; Coulibaly, B.; Fina, F.; Loundou, A.; Barrie, M.; Chinot, O.; et al. Prognostic impact of CD133 mRNA expression in 48 glioblastoma patients treated with concomitant radiochemotherapy: A prospective patient cohort at a single institution. Ann. Surg. Oncol. 2011, 18, 2937–2945. [Google Scholar]
- Yan, X.; Ma, L.; Yi, D.; Yoon, J.G.; Diercks, A.; Flotz, G.; Price, N.D.; Hood, L.E.; Tian, Q. A CD133-related gene expression signature identifies an aggressive glioblatoma subtype with excessive mutations. Proc. Natl. Acad. Sci. USA 2011, 108, 1591–1596. [Google Scholar]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Mason, S.; McDonald, K. MGMT testing for glioma in clinical laboratories: Discordance with methylation analyses prevents the implementation of routine immunohistochemistry. J. Cancer Res. Clin. Oncol. 2012, 138, 1789–1797. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef]
- Shinawi, T.; Hill, V.K.; Krex, D.; Schackert, G.; Gentle, D.; Morris, M.R.; Wei, W.; Cruickshank, G.; Maher, E.R.; Latif, F. DNA methylation profiles of long- and short-term glioblastoma survivors. Epigenetics 2013, 8, 149–156. [Google Scholar] [CrossRef]
- Riemenschneider, M.J.; Jeuken, J.W.; Wesseling, P.; Reifenberger, G. Molecular diagnostics of gliomas: State of the art. Acta Neuropathol. 2010, 120, 567–584. [Google Scholar] [CrossRef]
- Gladson, C.L.; Prayson, R.A.; Liu, W.M. The pathobiology of glioma tumors. Annu. Rev. Pathol. 2010, 5, 33–50. [Google Scholar] [CrossRef]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; di Patre, P.L.; Burkhard, C.; Schuler, D.; Probst-Hensch, N.M.; Maiorka, P.C.; et al. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef]
- Fults, D.; Pedone, C.A.; Thompson, G.E.; Uchiyama, C.M.; Gumpper, K.L.; IIiev, D.; Vinson, V.L.; Tavtigian, S.V.; Perry, W.L., 3rd. Microsatellite deletion mapping on chromosome 10q and mutation analysis of MMAC1, FAS, and MXI1 in human glioblastoma multiforme. Int. J. Oncol. 1998, 12, 905–910. [Google Scholar]
- Wooten, E.C.; Fults, D.; Duggirala, R.; Williams, K.; Kyritsis, A.P.; Bondy, M.L.; Levin, V.A.; O’Connell, P. A study of loss of heterozygosity at 70 loci in anaplastic astrocytoma and glioblastoma multiforme with implications for tumor evolution. Neurooncology 1999, 1, 169–176. [Google Scholar]
- Kakkar, A.; Suri, V.; Jha, P.; Srivastava, A.; Sharma, V.; Pathak, P.; Sharma, M.C.; Sharma, M.S.; Kale, S.S.; Chosdol, K.; et al. Loss of heterozygosity on chromosome 10q in glioblastomas, and its association with other genetic alterations and survival in Indian patients. Neurol. India 2011, 59, 254–261. [Google Scholar] [CrossRef]
- Fischer, I.; Aldape, K. Molecular tools: Biology, prognosis, and therapeutic triage. Neuroimaging Clin. N. Am. 2010, 20, 273–282. [Google Scholar] [CrossRef]
- Simpson, L.; Parsons, R. PTEN: Life as a tumor suppressor. Exp. Cell Res. 2001, 264, 29–41. [Google Scholar] [CrossRef]
- Kim, B.; Myung, J.K.; Seo, J.H.; Park, C.K.; Paek, S.H.; Kim, D.G.; Jung, H.W.; Park, S.H. The clinicopathologic values of the molecules associated with the main pathogenesis of the glioblastoma. J. Neurol. Sci. 2010, 294, 112–118. [Google Scholar] [CrossRef]
- Griguer, C.; Cantor, A.B.; Fathallah-Shaykh, H.M.; Gillespie, G.Y.; Gordon, A.S.; Markert, J.M.; Radovanovic, I.; Clement-Schatio, V.; Shannon, C.N.; Oliva, C.R. Prognostic relevance of Cytochrome c Oxidase in primary Glioblastoma Multiforme. PLoS One 2013, 8, e61035. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011, 39, D152–D157. [Google Scholar] [CrossRef]
- Mizoguchi, M.; Guan, Y.; Yoshimoto, K.; Hata, N.; Amano, T.; Nakamizo, A.; Sasaki, T. Clinical implications of microRNAs in human glioblastoma. Front. Oncol. 2013, 3, e19. [Google Scholar]
- Wu, Z.; Sun, L.; Wang, H.; Yao, J.; Jiang, C.; Xu, W.; Yang, Z. MiR-328 expression is decreased in high-grade gliomas and is associated with worse survival in primary glioblastoma. PLoS One 2012, 7, e47270. [Google Scholar]
- Lakomy, R.; Sana, J.; Hankeova, S.; Fadrus, P.; Kren, L.; Lzicarova, E.; Svoboda, M.; Dolezelova, H.; Smrcka, M.; Vyzula, R.; et al. MiR-195, miR-196b, miR-181c, miR-21 expression levels and o-6-methylguanine-DNA methyltransferase methylation status are associated with clinical outcome in glioblastoma patients. Cancer Sci. 2011, 102, 2186–2190. [Google Scholar] [CrossRef]
- Wang, Q.; Pengcun, L.; Ailin, L.; Jiang, W.; Wang, H.; Wang, J.; Xie, K. Plasma specific miRNAa as predictive biomarkers for diagnosis and prognosis of glioma. J. Exp. Clin. CancerRes. 2012, 31, e97. [Google Scholar] [CrossRef]
- Fox, J.L.; Dews, M.; Minn, A.J.; Thomas-Tikhonenko, A. Targeting of TGFβ signature and its essential component CTGF by miR-18 correlates with improved survival in glioblastoma. RNA 2013, 19, 177–190. [Google Scholar] [CrossRef]
- Wong, S.T.; Zhang, X.Q.; Zhuang, J.T.; Chan, H.L.; Li, C.H.; Leung, G.K. MicroRNA-21 inhibition enhances in vitro chemosensitivity of temozolomide-resistant glioblastoma cells. Anticancer Res. 2012, 32, 2835–2841. [Google Scholar]
- Cho, D.Y.; Lin, S.Z.; Yang, W.K.; Hsu, D.M.; Lin, H.L.; Lee, H.C.; Lee, W.Y.; Chiu, S.C. The role of cancer stem cells (CD133(+)) in malignant gliomas. Cell Transplant. 2011, 20, 121–125. [Google Scholar] [CrossRef]
- Melguizo, C.; Prados, J.; Gonzalez, B.; Ortiz, R.; Concha, A.; Alvarez, P.J.; Madeddu, R.; Perazzoli, G.; Oliver, J.A.; Lopez, R.; et al. MGMT promoter methylation status and MGMT and CD133 immunohistochemical expression as prognostic markers in glioblastoma patients treated with temozolomide plus radiotherapy. J. Transl. Med. 2012, 10, e250. [Google Scholar] [CrossRef]
- Beier, D.; Schulz, J.B.; Beier, C.P. Chemoresistance of glioblastoma cancer stem cells-much more complex than expected. Mol. Cancer 2011, 10, e128. [Google Scholar] [CrossRef] [Green Version]
- Chiang, M.F.; Chou, P.Y.; Wang, W.J.; Sze, C.I.; Chang, N.S. Tumor suppressor WWOX and p53 alterations and drug resistance in glioblastomas. Front. Oncol. 2013, 3, e43. [Google Scholar]
- Soda, Y.; Myskiw, C.; Rommel, A.; Verma, I.M. Mechanisms of neovascularization and resistance to anti-angiogenic therapies in glioblastoma multiforme. J. Mol. Med.(Berl) 2013, 91, 439–448. [Google Scholar] [CrossRef]
- Jamal, M.; Rath, B.H.; Tsang, P.S.; Camphausen, K.; Tofilon, P.J. The brain microenvironment preferentially enhances the radioresistance of CD133 (+) glioblastoma stem-like cells. Neoplasia 2012, 14, 150–158. [Google Scholar]
- He, J.; Liu, Y.; Zhu, T.; Zhu, J.; Dimeco, F.; Vescovi, A.L.; Heth, J.A.; Muraszko, K.M.; Fan, X.; Lubman, D.M. CD90 is identified as a candidate marker for cancer stem cells in primary high-grade gliomas using tissue microarrays. Mol. Cell. Proteomics 2012. [Google Scholar] [CrossRef]
- Jain, R.; Poisson, L.; Narang, J.; Gutman, D.; Scarpace, L.; Hwang, S.N.; Holder, C.; Wintermark, M.; Colen, R.R.; Kirby, J.; et al. Genomic mapping and survival prediction in glioblastoma: Molecular subclassification strengthened by hemodynamic imaging biomarkers. Radiology 2013, 267, 212–220. [Google Scholar] [CrossRef]
- Carrillo, J.A.; Lai, A.; Nghiemphu, P.L.; Kim, H.J.; Phillips, H.S.; Kharbanda, S.; Moftakhar, P.; Lalaezari, S.; Yong, W.; Ellingson, B.M.; et al. Relationship between tumor enhancement, edema, IDH1 mutational status, MGMT promoter methylation, and survival in glioblastoma. AJNR Am. J. Neuroradiol. 2012, 33, 1349–1355. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Hollander, A.; Xu, X.; Benci, J.L.; Davis, J.J.; Dorsey, J.F.; Kao, G. 18F-fluorothymidine-PET imaging of glioblastoma multiforme: Effects of radiation therapy on radiotracer uptake and molecular biomarker patterns. Scientific World J. 2013, 2013, 796029. [Google Scholar]
- Pope, W.B.; Lai, A.; Mehta, R.; Kim, H.J.; Qiao, J.; Young, J.R.; Xue, X.; Goldin, J.; Brown, M.S.; Nghiemphu, P.L.; et al. Apparent diffusion coefficient histogram analysis stratifies progression-free survival in newly diagnosed bevacizumab-treated glioblastoma. AJNR Am. J. Neuroradiol. 2011, 32, 882–889. [Google Scholar] [CrossRef]
- Pope, W.B.; Mirsadraei, L.; Lai, A.; Eskin, A.; Qiao, J.; Kim, H.J.; Ellingson, B.; Nghiemphu, P.L.; Kharbanda, S.; Soriano, R.H.; et al. Differential gene expression in glioblastoma defined by ADC histogram analysis: Relationship to extracellular matrix molecules and survival. AJNR Am. J. Neuroradiol. 2012, 33, 1059–1064. [Google Scholar] [CrossRef]
- Pope, W.B.; Qiao, X.J.; Kim, H.J.; Lai, A.; Nghiemphu, P.; Xue, X.; Elingson, B.M.; Schiff, D.; Aregawi, D.; Cha, S.; et al. Apparent diffusion coefficient histogram analysis stratifies progression-free and overall survival in patients with recurrent GBM treated with bevacizumab: A multi-center study. J. Neurooncol. 2012, 108, 491–498. [Google Scholar] [CrossRef]
- Paldino, M.J.; Desjardins, A.; Friedman, H.S.; Vredenburgh, J.J.; Barboriak, D.P. A change in the apparent diffusion coefficient after treatment with bevacizumab is associated with decreased survival in patients with recurrent glioblastoma multiforme. Br. J. Radiol. 2012, 85, 382–389. [Google Scholar] [CrossRef]
- Gokmen-Polar, Y.; Badve, S. Molecular profiling assays in breast cancer: Are we ready for prime time? Oncology 2012, 26, 350–357. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
McNamara, M.G.; Sahebjam, S.; Mason, W.P. Emerging Biomarkers in Glioblastoma. Cancers 2013, 5, 1103-1119. https://doi.org/10.3390/cancers5031103
McNamara MG, Sahebjam S, Mason WP. Emerging Biomarkers in Glioblastoma. Cancers. 2013; 5(3):1103-1119. https://doi.org/10.3390/cancers5031103
Chicago/Turabian StyleMcNamara, Mairéad G., Solmaz Sahebjam, and Warren P. Mason. 2013. "Emerging Biomarkers in Glioblastoma" Cancers 5, no. 3: 1103-1119. https://doi.org/10.3390/cancers5031103