Histologic and Genetic Advances in Refining the Diagnosis of “Undifferentiated Pleomorphic Sarcoma”

Abstract
:1. Introduction
2. Sarcoma
3. MFH, Morphologic Reassessment, Immunohistochemistry and Ultra-Structural Re-Evaluation

{kind=link}
| Tumor | Number of patients | |
|---|---|---|
| Myxofibrosarcoma | 29 | |
| High grade | 23 | |
| Intermediate grade | 3 | |
| Low grade | 3 | |
| Myogenic sarcoma | 30 | |
| High grade leiomyosarcoma | 20 | |
| Pleomorphic rhabdomyosarcoma | 1 | |
| High grade myogenic sarcoma | 9 | |
| NOS | ||
| Liposarcoma | 4 | |
| Pleomorphic liposarcoma | 3 | |
| Low grade liposarcoma | 1 | |
| Malignant peripheral nerve sheath tumor | 2 | |
| Low grade | 1 | |
| High grade | 1 | |
| Soft tissue osteosarcoma | 3 | |
| Malignant mesenchymoma | 2 | |
| Myofibroblastic sarcoma | 11 | |
| High grade sarcoma without specific line of differentiation | 2 | |
| Possibly high grade myofibroblastic sarcoma | 9 | |
| Dermatofibrosarcoma protuberans with fibrosarcomatous change | 1 | |
| Low grade fibromyxoid sarcoma | 1 | |
| High-grade sarcoma with specific line of differ. | 16 | |
| Pleomorphic | 12 | |
| Spindle-cell | 2 | |
| Spindle cell/myxoid | 1 | |
| Pleomorphic/myxoid | 1 | |
| Sarcomatoid carcinoma | 1 | |
| Total of all tumors | 100 | |
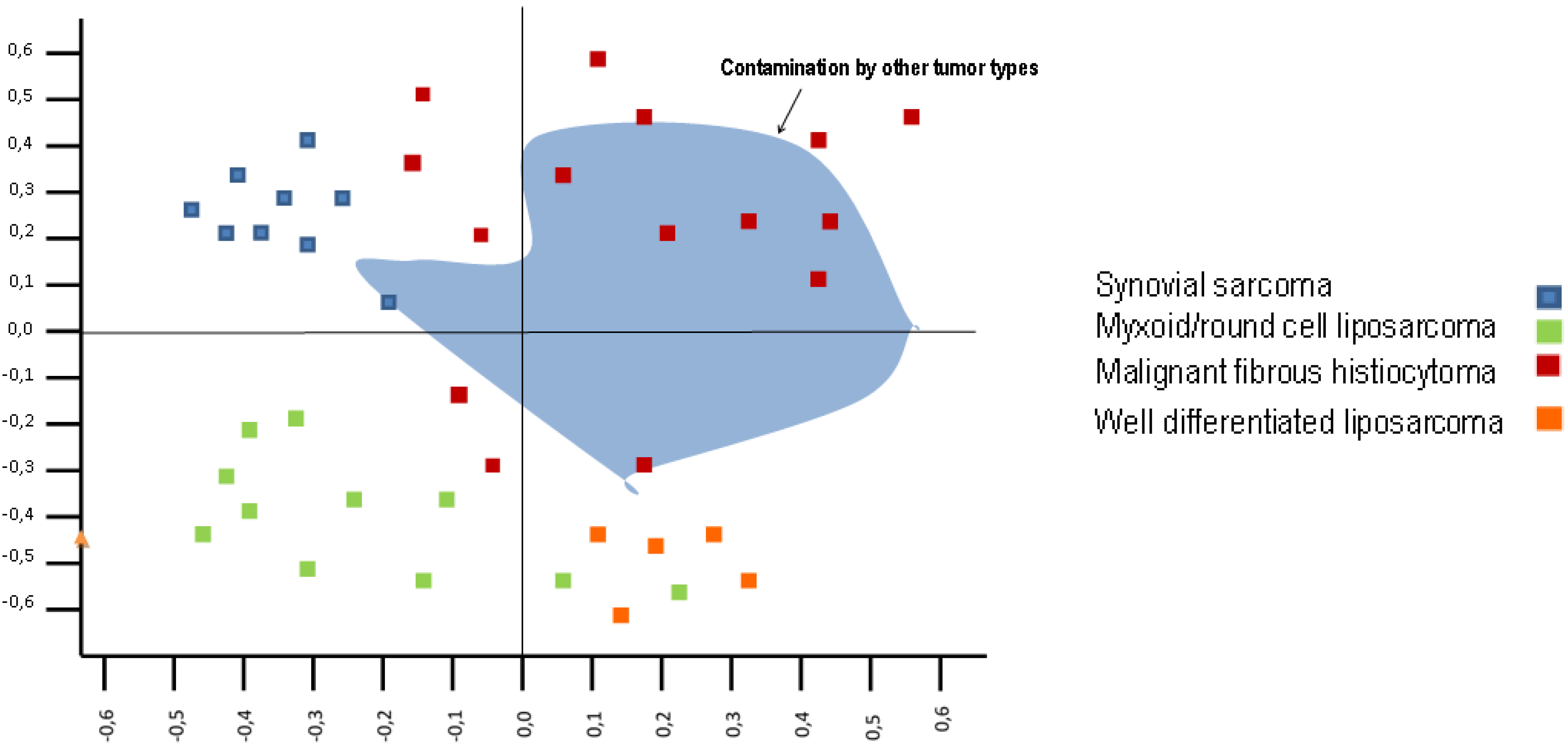
4. Sarcoma Series and Gene Expression Profiles
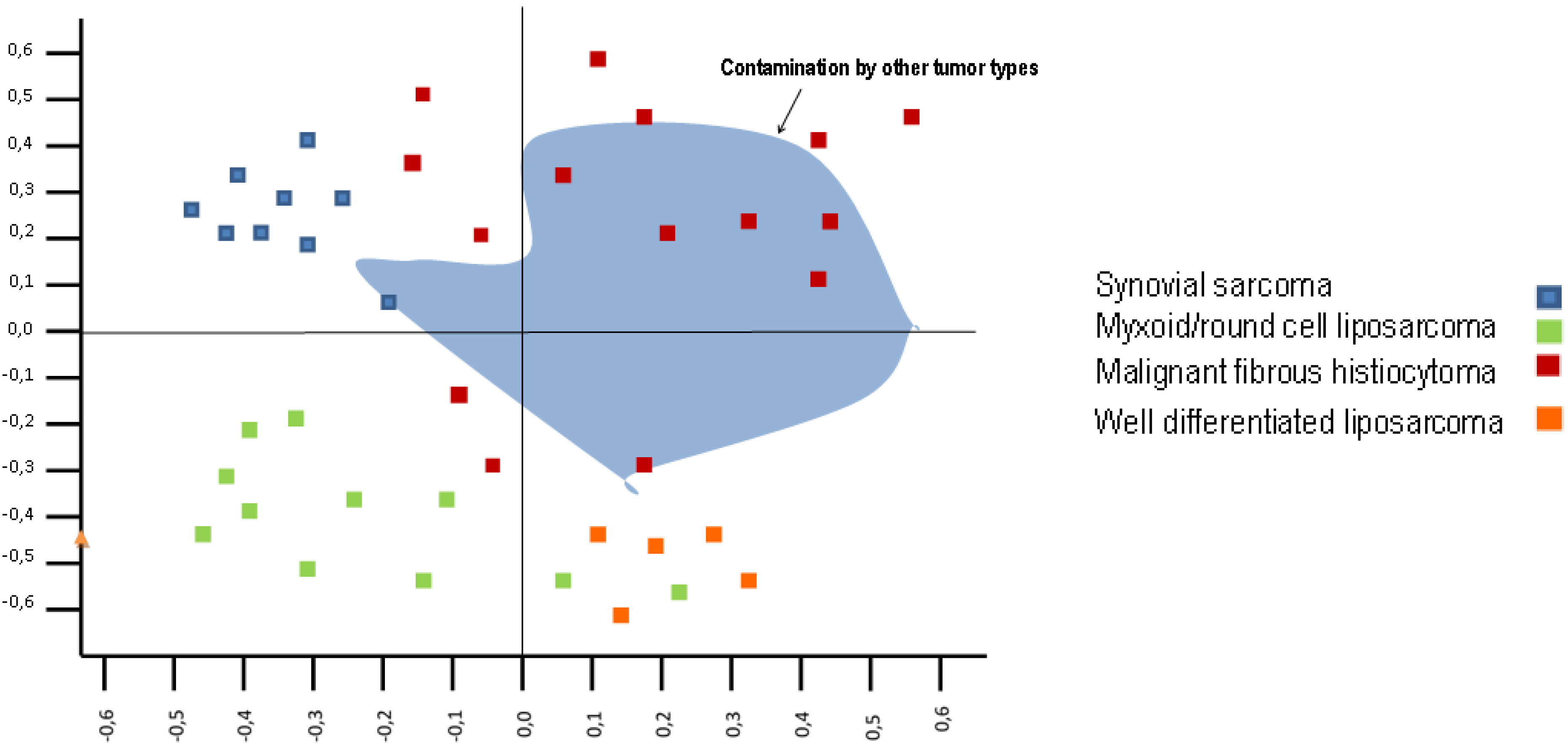
5. Undifferentiated Pleomorphic Sarcoma and Gene Expression
6. Undifferentiated Pleomorphic Sarcoma and Tumor Location
| Histologic subtype | Frequencies |
|---|---|
| MFH | 7–30% |
| Poorly differentiated sarcomas (MFH, fibrosarcoma, malignant hemangiopericytoma, unclassified sarcoma) | 16–50% |
| Liposarcoma | 20–40% |
| Leiomyosarcoma | 10–30% |
7. New Advances in the Diagnosis of Carcinomas: A New Era That May Influence UPS Diagnostic Research
8. The Importance of Finding True Lineage Differentiation in Undifferentiated Pleomorphic Sarcoma
9. Modelling Sarcoma Using Mesenchymal Stem Cells
10. Analysis of Multiple Sarcoma Expression Databases
| Distinct differentiation states | Selected differentially expressed genes |
|---|---|
| Smooth muscle differentiation genes | Myosin light chain kinase |
| Calponin 1 | |
| Smooth muscle actin γ2 | |
| Alpha 2 macroglobulin | |
| Cysteine and glycine-rich protein 1 | |
| Chromosome 9 ORF 3 | |
| Peripheral nerve differentiation genes | Jagged 1 |
| Glycoprotein M6B | |
| Olfactomedin 1 | |
| Nerve growth factor beta | |
| Peripheral myelin protein 22 | |
| Fibroblast differentiation genes | Kallikrein-related peptide 7 |
| Lumican | |
| Cadherin 1 | |
| Complement factor H | |
| Ficolin | |
| Fibroblast growth factor receptor 3 | |
| Adipocyte differentiation genes | Fatty acid binding protein 4 |
| PPAR gamma | |
| IGF-1 | |
| Palmdelphin | |
| Acyl-CoA synthetase long-chain members 1 and 5 |
| Distinct differentiation states | Selected differentially expressed genes |
|---|---|
| Synovial sarcoma genes | Synovial sarcoma, X Breakpoint 1 |
| Cyclin D1 | |
| BCL2 | |
| Vitronectin | |
| Cell adhesion molecule 1 | |
| RIMS binding protein 2 | |
| Malignant nerve sheath genes | Synuclein alpha |
| Synaptotagmin 17 | |
| Transducin beta like 2 | |
| Intercellular adhesion molecule 3 | |
| S100 calcium binding protein A3 | |
| Dual specific phosphatase 4 |
11. Undifferentiated Pleomorphic Sarcoma the Past and the Future
12. Conclusions
References
- Crozat, A.; Aman, P.; Mandahl, N.; Ron, D. Fusion of CHOP to a novel RNA-binding protein in human myxoid liposarcoma. Nature 1993, 363, 640–644. [Google Scholar] [CrossRef]
- Clark, J.; Rocques, P.J.; Crew, A.J.; Gill, S.; Shipley, J.; Chan, A.M.; Gusterson, B.A. Cooper CSIdentification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat. Genet. 1994, 7, 502–508. [Google Scholar]
- Quesada, J.; Amato, R. The molecular biology of soft-tissue sarcomas and current trends in therapy. Sarcoma 2012, 2012, 849456. [Google Scholar]
- Dunham, M.A.; Neumann, A.A.; Fasching, C.L.; Reddel, R.R. Telomere maintenance by recombination in human cells. Nat. Genet. 2000, 26, 447–450. [Google Scholar]
- Ulaner, G.A.; Hoffman, A.R.; Otero, J.; Huang, H.Y.; Zhao, Z.; Mazumdar, M.; Gorlick, R.; Meyers, P.; Healey, J.H.; Ladanyi, M. Divergent patterns of telomere maintenance mechanisms among human sarcomas: Sharply contrasting prevalence of the alternative lengthening of telomeres mechanism in Ewing’s sarcomas and osteosarcomas. Genes Chromosomes Cancer 2004, 41, 155–162. [Google Scholar] [CrossRef]
- Scheel, C.; Schaefer, K.L.; Jauch, A.; Keller, M.; Wai, D.; Brinkschmidt, C.; van Valen, F.; Boecker, W.; Dockhorn-Dworniczak, B.; Poremba, C. Alternative lengthening of telomeres is associated with chromosomal instability in osteosarcomas. Oncogene 2001, 20, 3835–3844. [Google Scholar]
- Nielsen, T.O.; West, R.B. Translating gene expression into clinical care: Sarcomas as a paradigm. J. Clin. Oncol. 2010, 28, 1796–1805. [Google Scholar] [CrossRef]
- Fletcher, C.D. Pleomorphic malignant fibrous histiocytoma: Fact or fiction? A critical reappraisal based on 159 tumors diagnosed as pleomorphic sarcoma. Am. J. Surg. Pathol. 1992, 16, 213–228. [Google Scholar] [CrossRef]
- Fletcher, C.D.; Gustafson, P.; Rydholm, A.; Willén, H.; Akerman, M. Clinicopathologic re-evaluation of 100 malignant fibrous histiocytomas: Prognostic relevance of subclassification. J. Clin. Oncol. 2001, 19, 3045–3050. [Google Scholar]
- Mentzel, T.; Calonje, E.; Wadden, C.; Camplejohn, R.S.; Beham, A.; Smith, M.A.; Fletcher, C.D. Myxofibrosarcoma. Clinicopathologic analysis of 75 cases with emphasis on the low-grade variant. Am. J. Surg. Pathol. 1996, 20, 391–405. [Google Scholar]
- McCormick, D.; Mentzel, T.; Beham, A.; Fletcher, C.D. Dedifferentiated liposarcoma. Clinicopathologic analysis of 32 cases suggesting a better prognostic subgroup among pleomorphic sarcomas. Am. J. Surg. Pathol. 1994, 18, 1213–1223. [Google Scholar] [CrossRef]
- Henricks, W.H.; Chu, Y.C.; Goldblum, J.R.; Weiss, S.W. Dedifferentiated liposarcoma: A clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am. J. Surg. Pathol. 1997, 21, 271–281. [Google Scholar] [CrossRef]
- Merck, C.; Angervall, L.; Kindblom, L.G.; Odén, A. Myxofibrosarcoma. A malignant soft tissue tumor of fibroblastic-histiocytic origin. A clinicopathologic and prognostic study of 110 cases using multivariate analysis. Acta Pathol. Microbiol. Immunol. Scand. Suppl. 1983, 282, 1–40. [Google Scholar]
- Nielsen, T.O.; West, R.B.; Linn, S.C.; Alter, O.; Knowling, M.A.; O’Connell, J.X.; Zhu, S.; Fero, M.; Sherlock, G.; Pollack, J.R.; et al. Molecular characterisation of soft tissue tumours: A gene expression study. Lancet 2002, 359, 1301–1307. [Google Scholar]
- Nakayama, R.; Nemoto, T.; Takahashi, H.; Ohta, T.; Kawai, A.; Seki, K.; Yoshida, T.; Toyama, Y.; Ichikawa, H.; Hasegawa, T. Gene expression analysis of soft tissue sarcomas: Characterization and reclassification of malignant fibrous histiocytoma. Mod. Pathol. 2007, 20, 749–759. [Google Scholar] [CrossRef]
- Obermann, H.; Samalecos, A.; Osterhoff, C.; Schröder, B.; Heller, R.; Kirchhoff, C. HE6, a two-subunit heptahelical receptor associated with apical membranes of efferent and epididymal duct epithelia. Mol. Reprod. Dev. 2003, 64, 13–26. [Google Scholar] [CrossRef]
- Baird, K.; Davis, S.; Antonescu, C.R.; Harper, U.L.; Walker, R.L.; Chen, Y.; Glatfelter, A.A.; Duray, P.H.; Meltzer, P.S. Gene expression profiling of human sarcomas: Insights into sarcoma biology. Cancer Res. 2005, 65, 9226–9235. [Google Scholar]
- Skubitz, K.M.; Francis, P.; Skubitz, A.P.; Luo, X.; Nilbert, M. Gene expression identifies heterogeneity of metastatic propensity in high-grade soft tissue sarcomas. Cancer 2012, 118, 4235–4243. [Google Scholar] [CrossRef]
- Stoeckle, E.; Coindre, J.M.; Bonvalot, S.; Kantor, G.; Terrier, P.; Bonichon, F.; Nguyen Bui, B. French Federation of Cancer Centers Sarcoma Group. Prognostic factors in retroperitoneal sarcoma: A multivariate analysis of a series of 165 patients of the French Cancer Center Federation Sarcoma Group. Cancer 2001, 92, 359–368. [Google Scholar] [CrossRef]
- Pinson, C.W.; ReMine, S.G.; Fletcher, W.S.; Braasch, J.W. Long-term results with primary retroperitoneal tumors. Arch. Surg. 1989, 124, 1168–1173. [Google Scholar]
- Catton, C.N.; O’Sullivan, B.; Kotwall, C.; Cummings, B.; Hao, Y.; Fornasier, V. Outcome and prognosis in retroperitoneal soft tissue sarcoma. Int. J. Radiat. Oncol. Biol. Phys. 1994, 29, 1005–1010. [Google Scholar] [CrossRef]
- Kilkenny, J.W., 3rd; Bland, K.I.; Copeland, E.M., 3rd. Retroperitoneal sarcoma: The University of Florida experience. J. Am. Coll. Surg. 1996, 182, 329–339. [Google Scholar]
- Karakousis, C.P.; Velez, A.F.; Gerstenbluth, R.; Driscoll, D.L. Resectability and survival in retroperitoneal sarcomas. Ann. Surg. Oncol. 1996, 3, 150–158. [Google Scholar] [CrossRef]
- Lewis, J.J.; Leung, D.; Woodruff, J.M.; Brennan, M.F. Retroperitoneal soft-tissue sarcoma: Analysis of 500 patients treated and followed at a single institution. Ann. Surg. 1998, 228, 355–365. [Google Scholar] [CrossRef]
- Herman, K.; Gruchała, A.; Niezabitowski, A.; Gliński, B.; Lackowska, B. Prognostic factors in retroperitoneal sarcomas: Ploidy of DNA as a predictor of clinical outcome. J. Surg. Oncol. 1999, 71, 32–35. [Google Scholar] [CrossRef]
- Coindre, J.M.; Mariani, O.; Chibon, F.; Mairal, A.; de Saint Aubain Somerhausen, N.; Favre-Guillevin, E.; Bui, N.B.; Stoeckle, E.; Hostein, I.; Aurias, A. Most malignant fibrous histiocytomas developed in the retroperitoneum are dedifferentiated liposarcomas: A review of 25 cases initially diagnosed as malignant fibrous histiocytoma. Mod. Pathol. 2003, 16, 256–262. [Google Scholar] [CrossRef]
- Pilotti, S.; Della Torre, G.; Lavarino, C.; Sozzi, G.; Minoletti, F.; Vergani, B.; Azzarelli, A.; Rilke, F.; Pierotti, M.A. Molecular abnormalities in liposarcoma: Role of MDM2 and CDK4-containing amplicons at 12q13-22. J. Pathol. 1998, 185, 188–190. [Google Scholar] [CrossRef]
- Horvai, A.E.; Schaefer, J.T.; Nakakura, E.K.; O’Donnell, R.J. Immunostaining for peroxisome proliferator gamma distinguishes dedifferentiated liposarcoma from other retroperitoneal sarcomas. Mod. Pathol. 2008, 21, 517–524. [Google Scholar]
- Haun, J.B.; Castro, C.M.; Wang, R.; Peterson, V.M.; Marinelli, B.S.; Lee, H.; Weissleder, R. Micro-NMR for rapid molecular analysis of human tumor samples. Sci. Transl. Med. 2011, 3, 71ra16. [Google Scholar] [CrossRef]
- Tanas, M.R.; Sboner, A.; Oliveira, A.M.; Erickson-Johnson, M.R.; Hespelt, J.; Hanwright, P.J.; Flanagan, J.; Luo, Y.; Fenwick, K.; Natrajan, R.; et al. Identification of a disease-defining gene fusion in epithelioid hemangioendothelioma. Sci. Transl. Med. 2011, 3, 98ra82. [Google Scholar]
- Rodriguez, R.; Rubio, R.; Menendez, P. Modeling sarcomagenesis using multipotent mesenchymal stem cells. Cell Res. 2012, 22, 62–77. [Google Scholar] [CrossRef]
- Helman, L.J.; Meltzer, P. Mechanisms of sarcoma development. Nat. Rev. Cancer 2003, 3, 685–694. [Google Scholar] [CrossRef]
- Fodde, R.; Brabletz, T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol. 2007, 19, 150–158. [Google Scholar] [CrossRef]
- Matushansky, I.; Hernando, E.; Socci, N.D.; Mills, J.E.; Matos, T.A.; Edgar, M.A.; Singer, S.; Maki, R.G.; Cordon-Cardo, C. Derivation of sarcomas from mesenchymal stem cells via inactivation of the Wnt pathway. J. Clin. Invest. 2007, 117, 3248–3257. [Google Scholar] [CrossRef]
- Fodde, R.; Brabletz, T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr. Opin. Cell Biol. 2007, 19, 150–158. [Google Scholar] [CrossRef]
- Matthay, K.K.; Villablanca, J.G.; Seeger, R.C.; Stram, D.O.; Harris, R.E.; Ramsay, N.K.; Swift, P.; Shimada, H.; Black, C.T.; Brodeur, G.M.; et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. N. Engl. J. Med. 1999, 341, 1165–1173. [Google Scholar] [CrossRef]
- Rodriguez, R.; Rubio, R.; Masip, M.; Catalina, P.; Nieto, A.; de la Cueva, T.; Arriero, M.; San Martin, N.; de la Cueva, E.; Balomenos, D.; et al. Loss of p53 induces tumorigenesis in p21-deficient mesenchymal stem cells. Neoplasia 2009, 11, 397–407. [Google Scholar]
- Henderson, S.R.; Guiliano, D.; Presneau, N.; McLean, S.; Frow, R.; Vujovic, S.; Anderson, J.; Sebire, N.; Whelan, J.; Athanasou, N.; et al. A molecular map of mesenchymal tumors. Genome Biol. 2005, 6, R76. [Google Scholar] [CrossRef]
- Detwiller, K.Y.; Fernando, N.T.; Segal, N.H.; Ryeom, S.W.; D’Amore, P.A.; Yoon, S.S. Analysis of hypoxia-related gene expression in sarcomas and effect of hypoxia on RNA interference of vascular endothelial cell growth factor A. Cancer Res. 2005, 65, 5881–5889. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Fountzilas, E.; Goldsmith, J.D.; Bhasin, M.; Pillay, K.; Francoeur, N.; Libermann, T.A.; Gebhardt, M.C.; Spentzos, D. Analysis of multiple sarcoma expression datasets: Implications for classification, oncogenic pathway activation and chemotherapy resistance. PLoS One 2010, 5, e9747. [Google Scholar]
- Yuen, J.S.; Macaulay, V.M. Targeting the type 1 insulin-like growth factor receptor as a treatment for cancer. Expert Opin. Ther. Targets 2008, 12, 589–603. [Google Scholar] [CrossRef]
- Blay, J.Y.; Ray-Coquard, I.; Alberti, L.; Ranchère, D. Targeting other abnormal signaling pathways in sarcoma: EGFR in synovial sarcomas, PPAR-gamma in liposarcomas. Cancer Treat. Res. 2004, 120, 151–167. [Google Scholar] [CrossRef]
- Adriaenssens, E.; Vanhecke, E.; Saule, P.; Mougel, A.; Page, A.; Romon, R.; Nurcombe, V.; Le Bourhis, X.; Hondermarck, H. Nerve growth factor is a potential therapeutic target in breast cancer. Cancer Res. 2008, 68, 346–351. [Google Scholar]
- Martínez-Torrecuadrada, J.; Cifuentes, G.; López-Serra, P.; Saenz, P.; Martínez, A.; Casal, J.I. Targeting the extracellular domain of fibroblast growth factor receptor 3 with human single-chain Fv antibodies inhibits bladder carcinoma cell line proliferation. Clin. Cancer Res. 2005, 11, 6280–6290. [Google Scholar] [CrossRef]
- Mito, J.K.; Riedel, R.F.; Dodd, L.; Lahat, G.; Lazar, A.J.; Dodd, R.D.; Stangenberg, L.; Eward, W.C.; Hornicek, F.J.; Yoon, S.S.; et al. Cross species genomic analysis identifies a mouse model as undifferentiated pleomorphic sarcoma/malignant fibrous histiocytoma. PLoS One 2009, 4, e8075. [Google Scholar]
- Kwong, L.N.; Costello, J.C.; Liu, H.; Jiang, S.; Helms, T.L.; Langsdorf, A.E.; Jakubosky, D.; Genovese, G.; Muller, F.L.; Jeong, J.H.; et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat. Med. 2012, 18, 1503–1510. [Google Scholar]
- Honoki, K.; Fujii, H.; Tohma, Y.; Tsujiuchi, T.; Kido, A.; Tsukamoto, S.; Mori, T.; Tanaka, Y. Comparison of gene expression profiling in sarcomas and mesenchymal stem cells identifies tumorigenic pathways in chemically induced rat sarcoma model. ISRN Oncol. 2012, 2012, 909453. [Google Scholar]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar]
- Fellner, C. Vismodegib (erivedge) for advanced Basal cell carcinoma. Pharm. Ther. 2012, 37, 670–682. [Google Scholar]
- Lahat, G.; Zhang, P.; Zhu, Q.S.; Torres, K.; Ghadimi, M.; Smith, K.D.; Wang, W.L.; Lazar, A.J.; Lev, D. The expression of c-Met pathway components in unclassified pleomorphic sarcoma/malignant fibrous histiocytoma (UPS/MFH): A tissue microarray study. Histopathology 2011, 59, 556–561. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kelleher, F.C.; Viterbo, A. Histologic and Genetic Advances in Refining the Diagnosis of “Undifferentiated Pleomorphic Sarcoma”. Cancers 2013, 5, 218-233. https://doi.org/10.3390/cancers5010218
Kelleher FC, Viterbo A. Histologic and Genetic Advances in Refining the Diagnosis of “Undifferentiated Pleomorphic Sarcoma”. Cancers. 2013; 5(1):218-233. https://doi.org/10.3390/cancers5010218
Chicago/Turabian StyleKelleher, Fergal C., and Antonella Viterbo. 2013. "Histologic and Genetic Advances in Refining the Diagnosis of “Undifferentiated Pleomorphic Sarcoma”" Cancers 5, no. 1: 218-233. https://doi.org/10.3390/cancers5010218