dNTP Supply Gene Expression Patterns after P53 Loss

Abstract
:1. Introduction
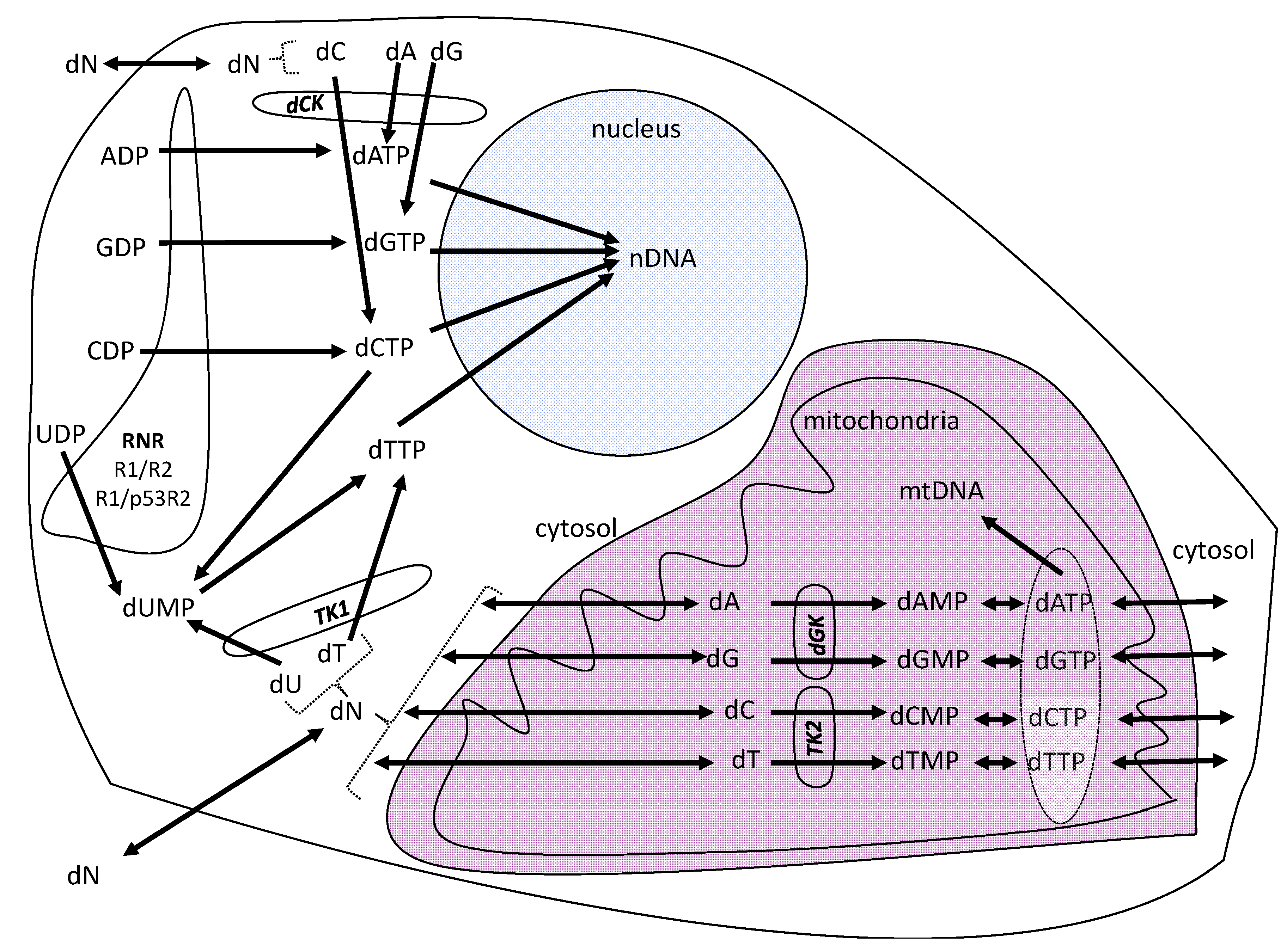
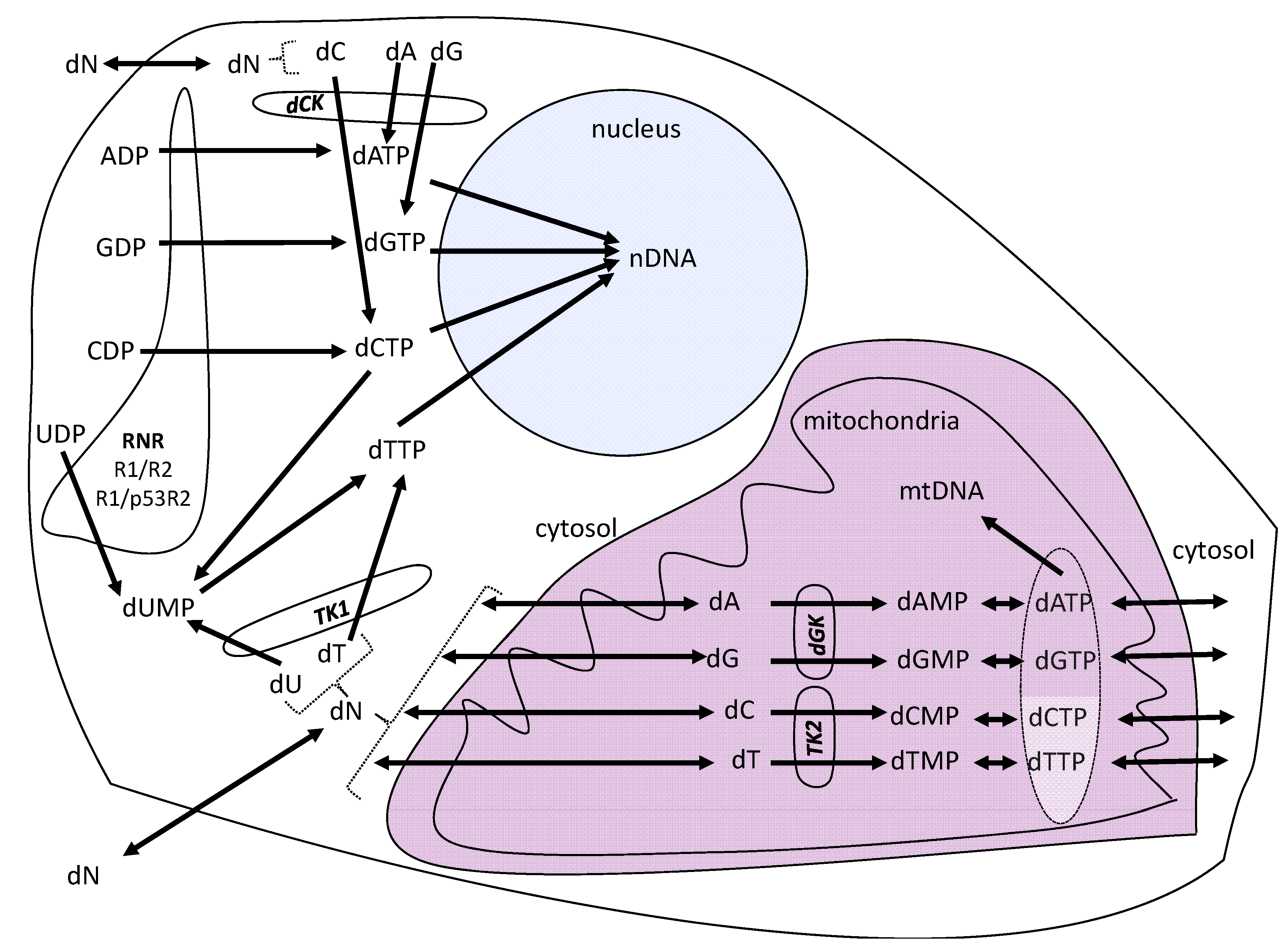
2. Results and Discussion
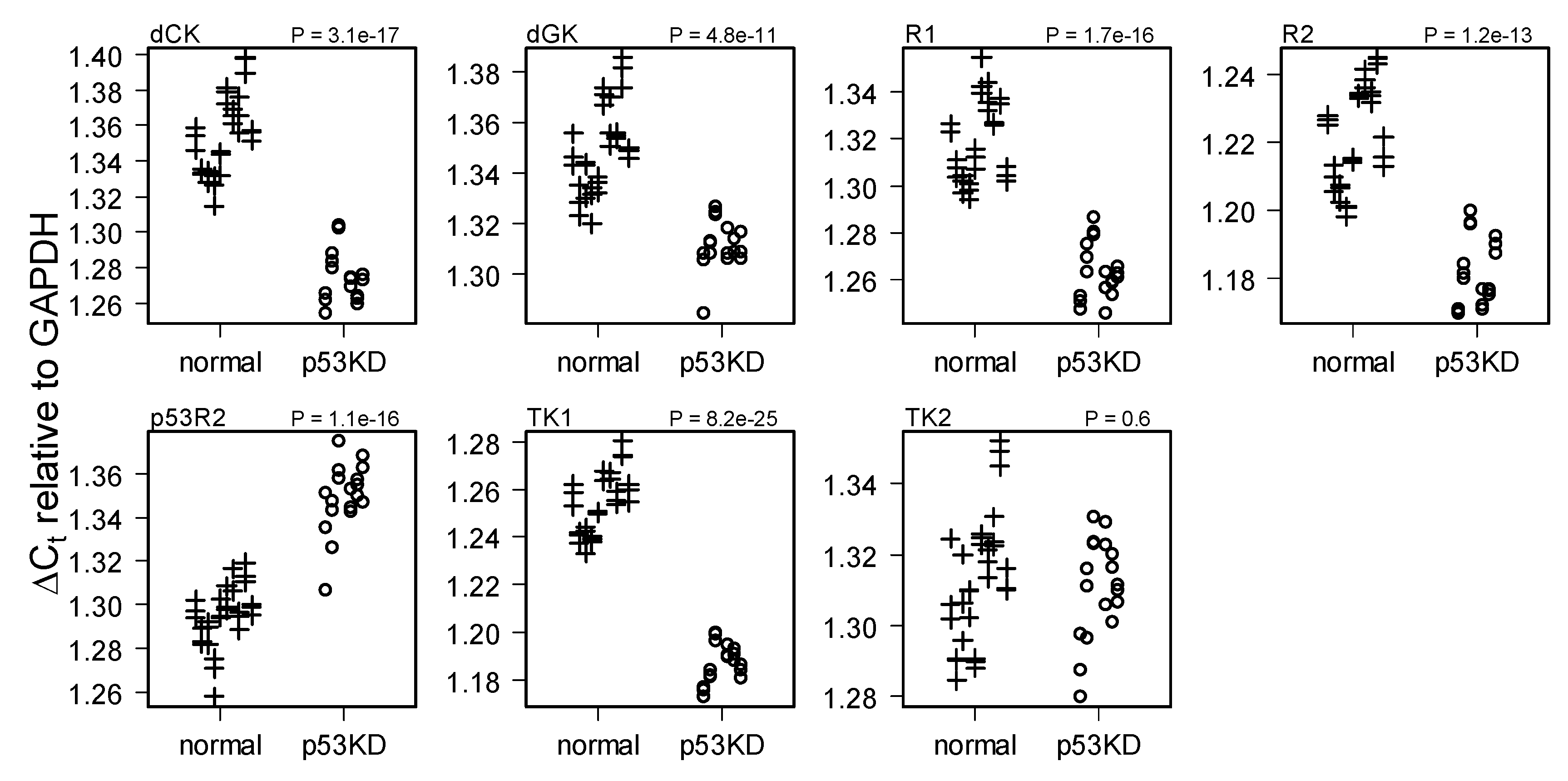
2.1. Comparison of HMEC with and without p53
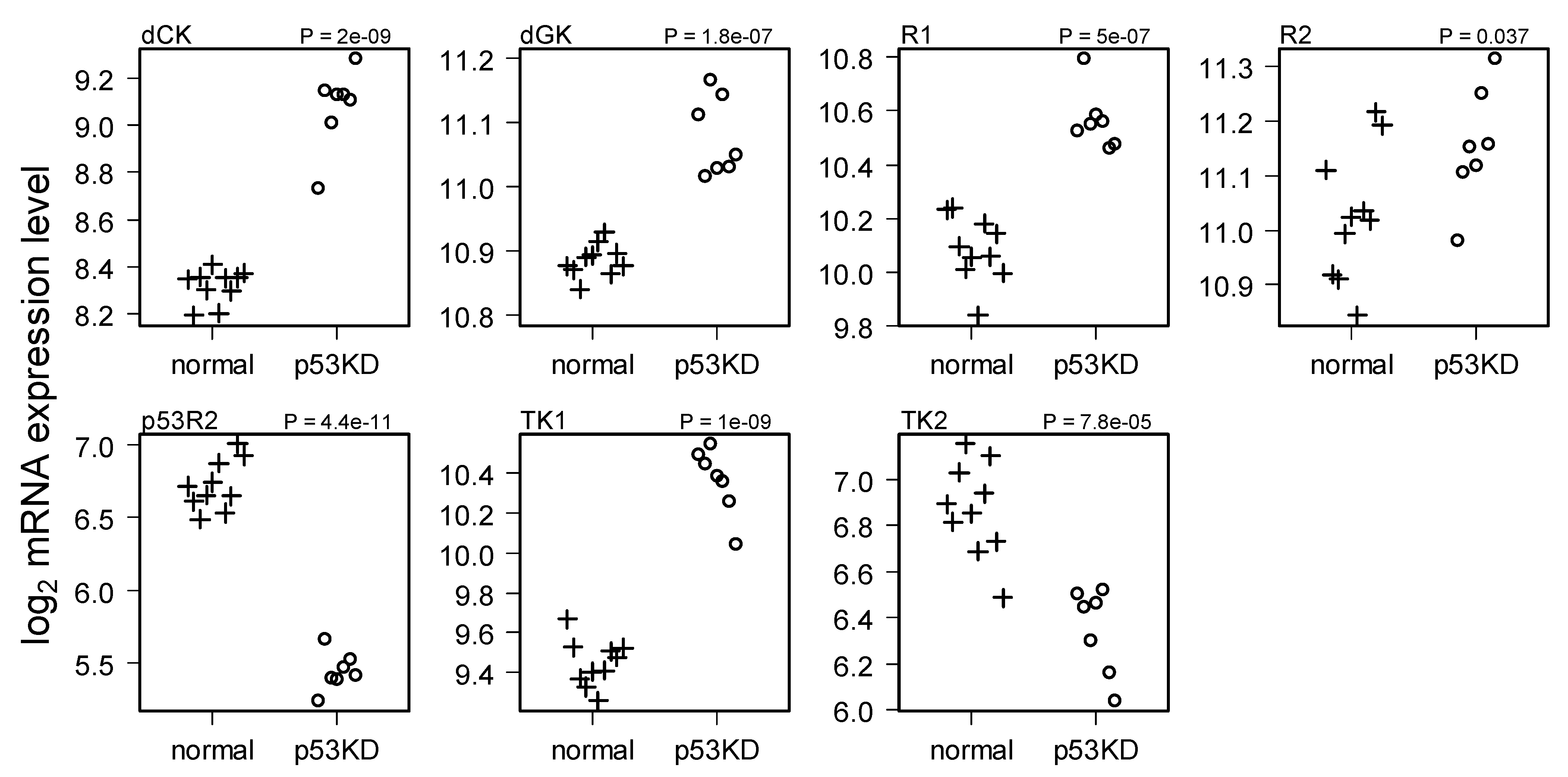
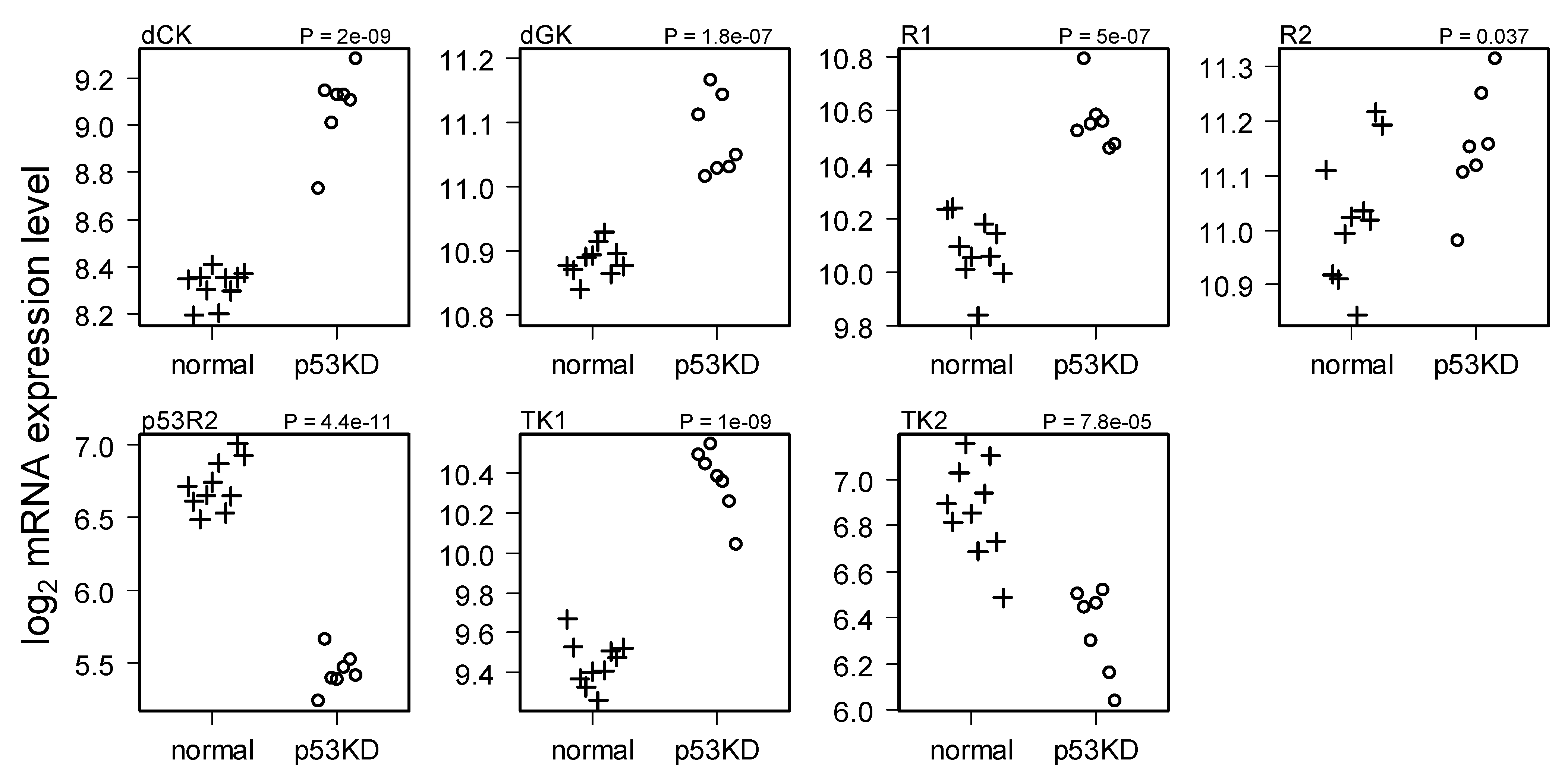
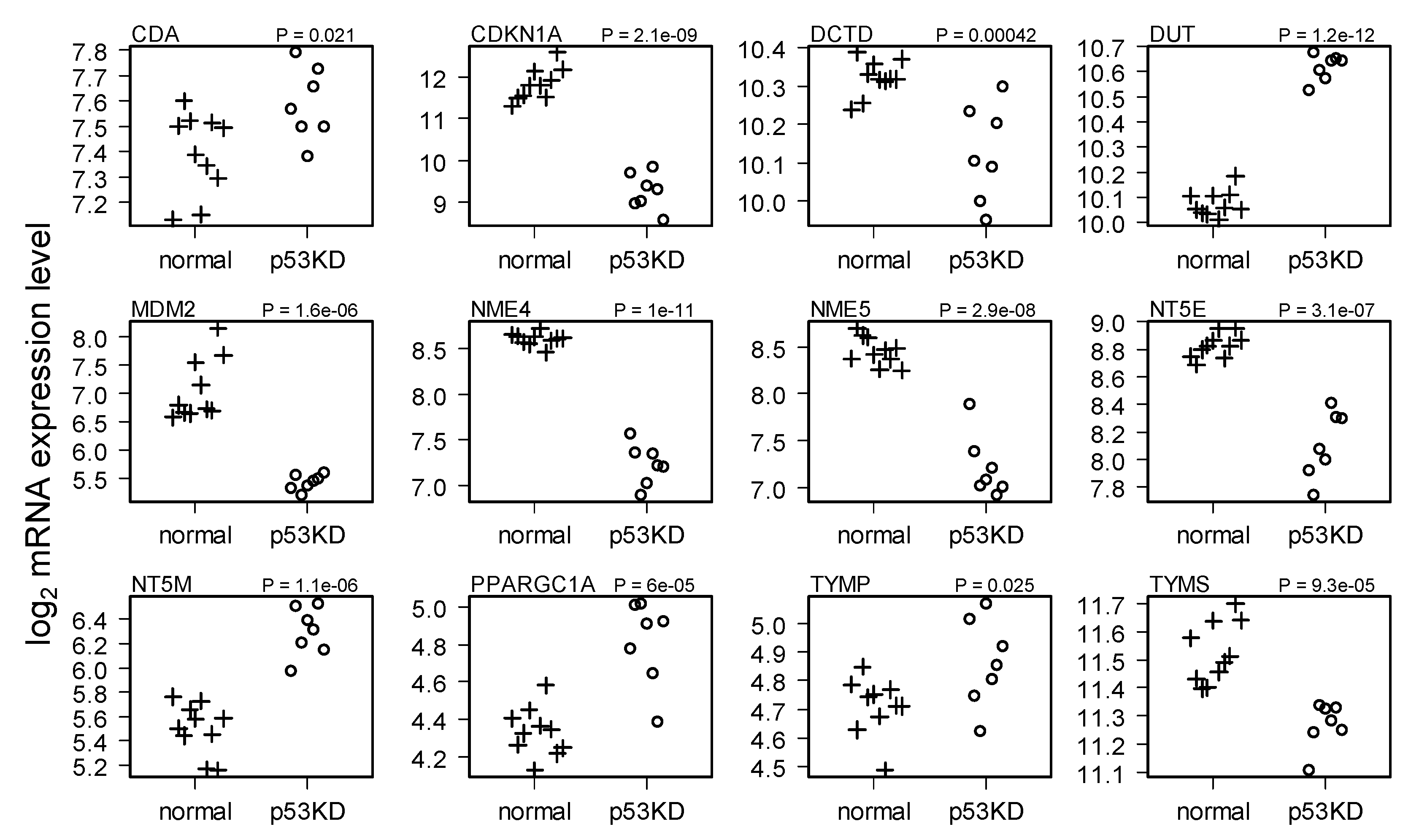
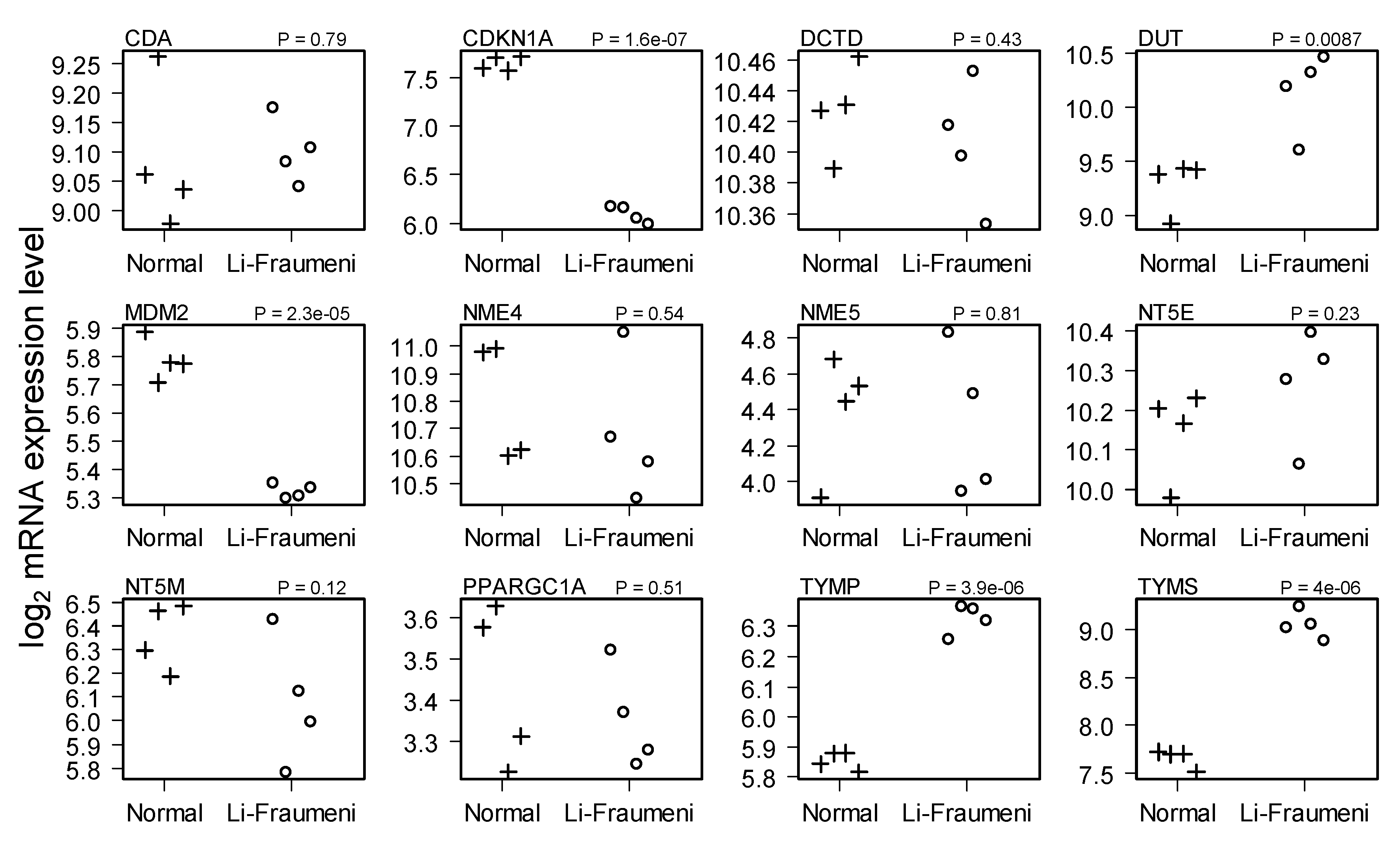
2.2. Comparison of Normal and Li-Fraumeni Gene Expression

{kind=link}
{kind=link}
{kind=link}
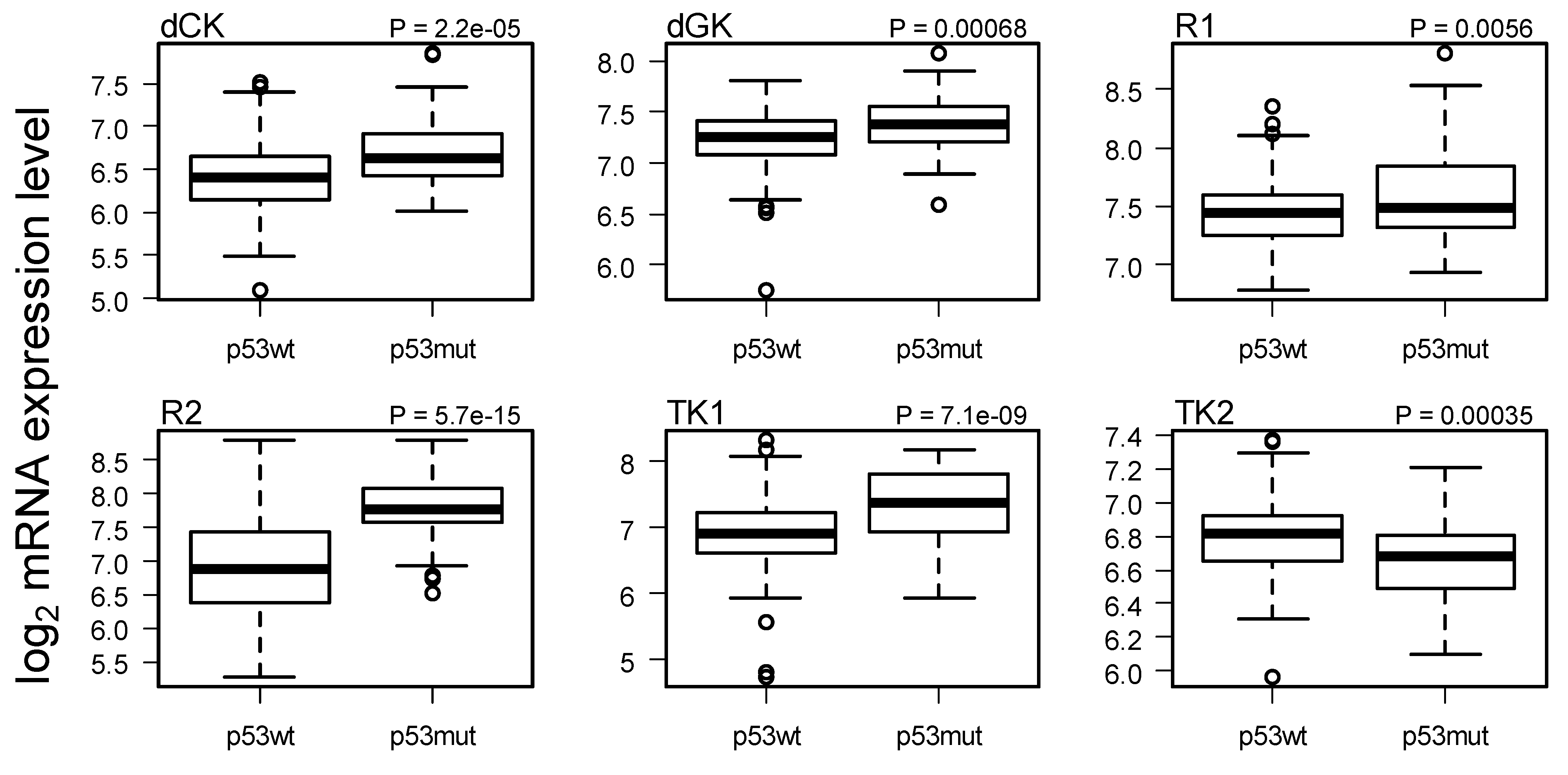{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
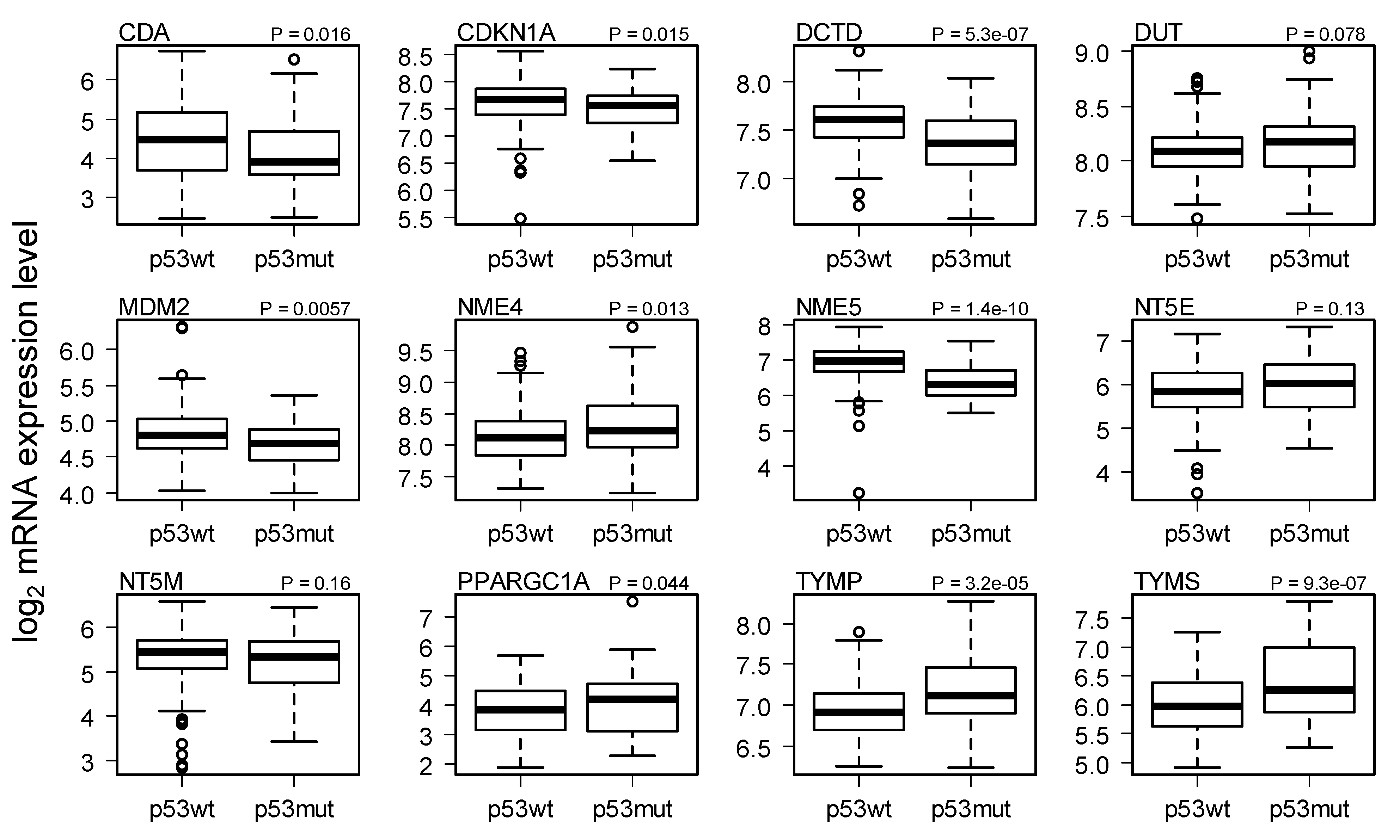
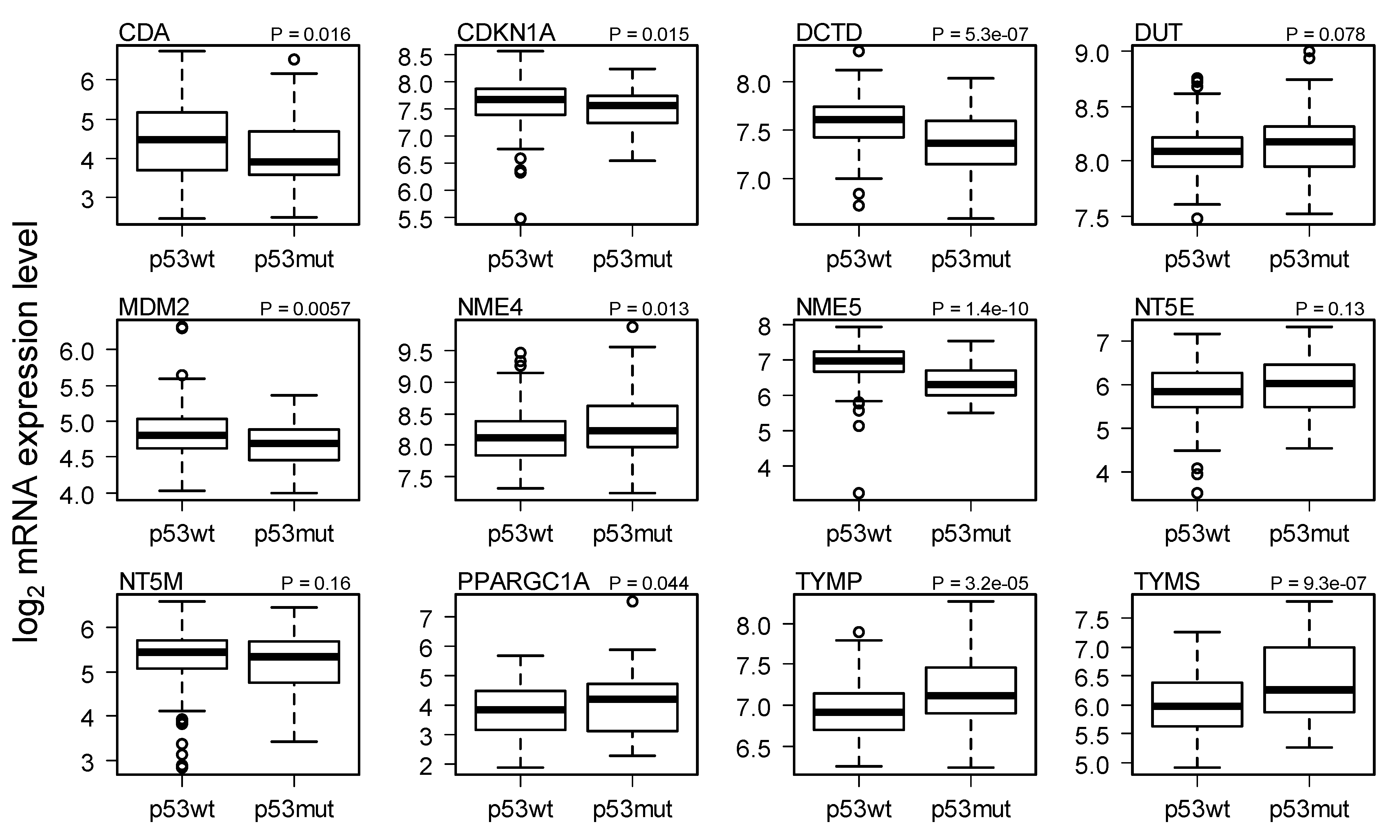
2.3. Comparison of Gene Expression in Breast Cancer with and without p53 Mutated
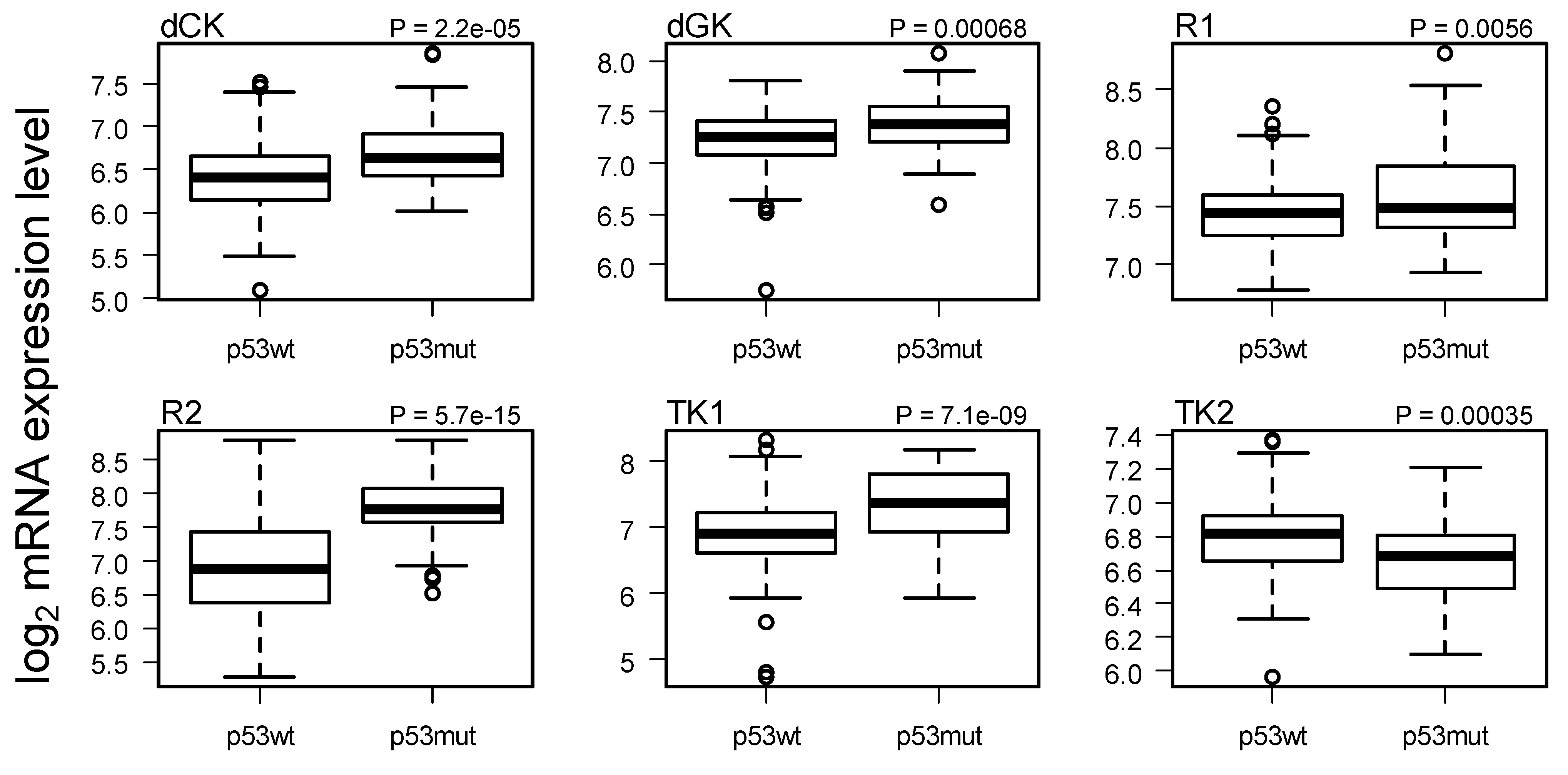
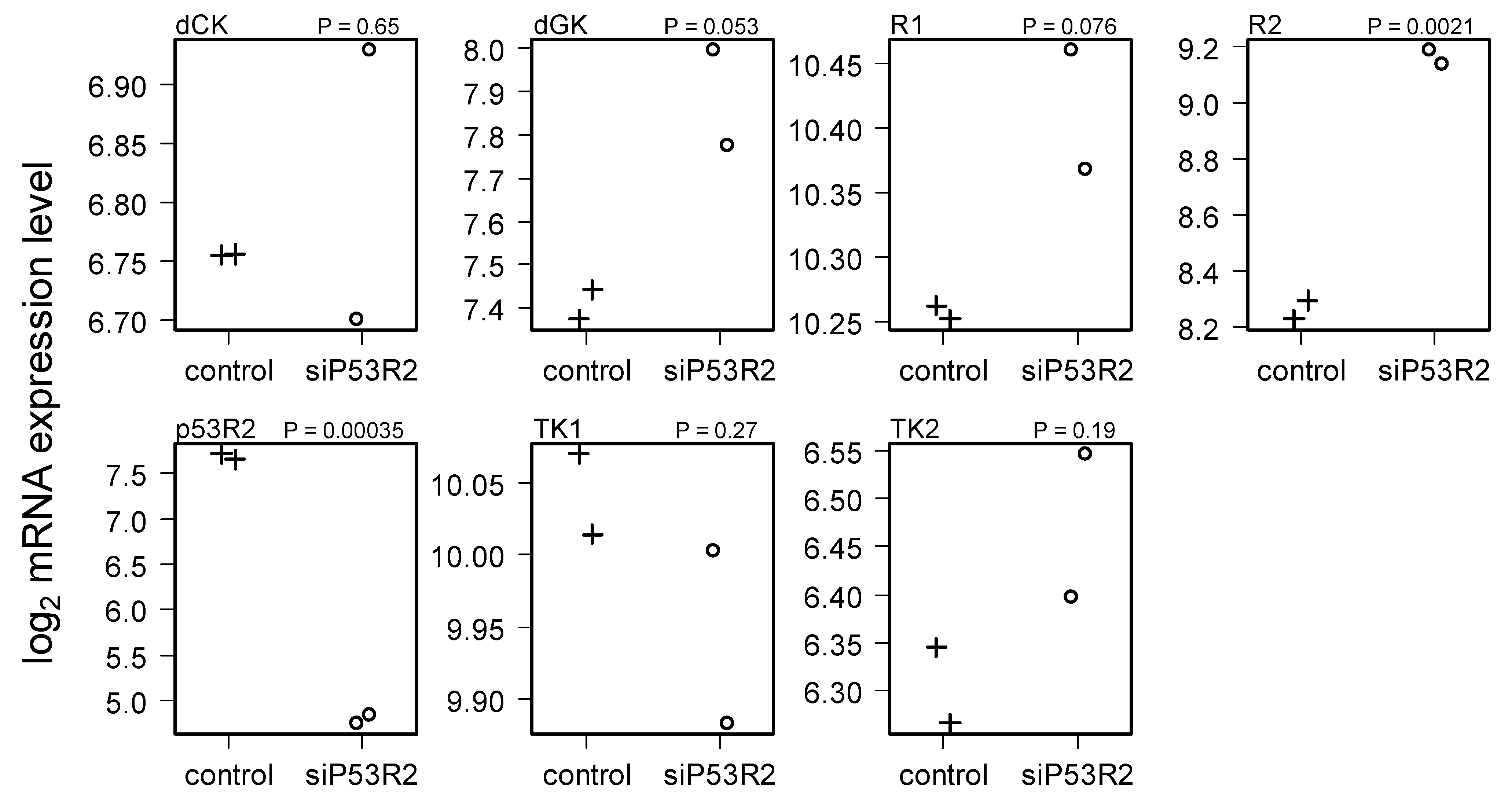
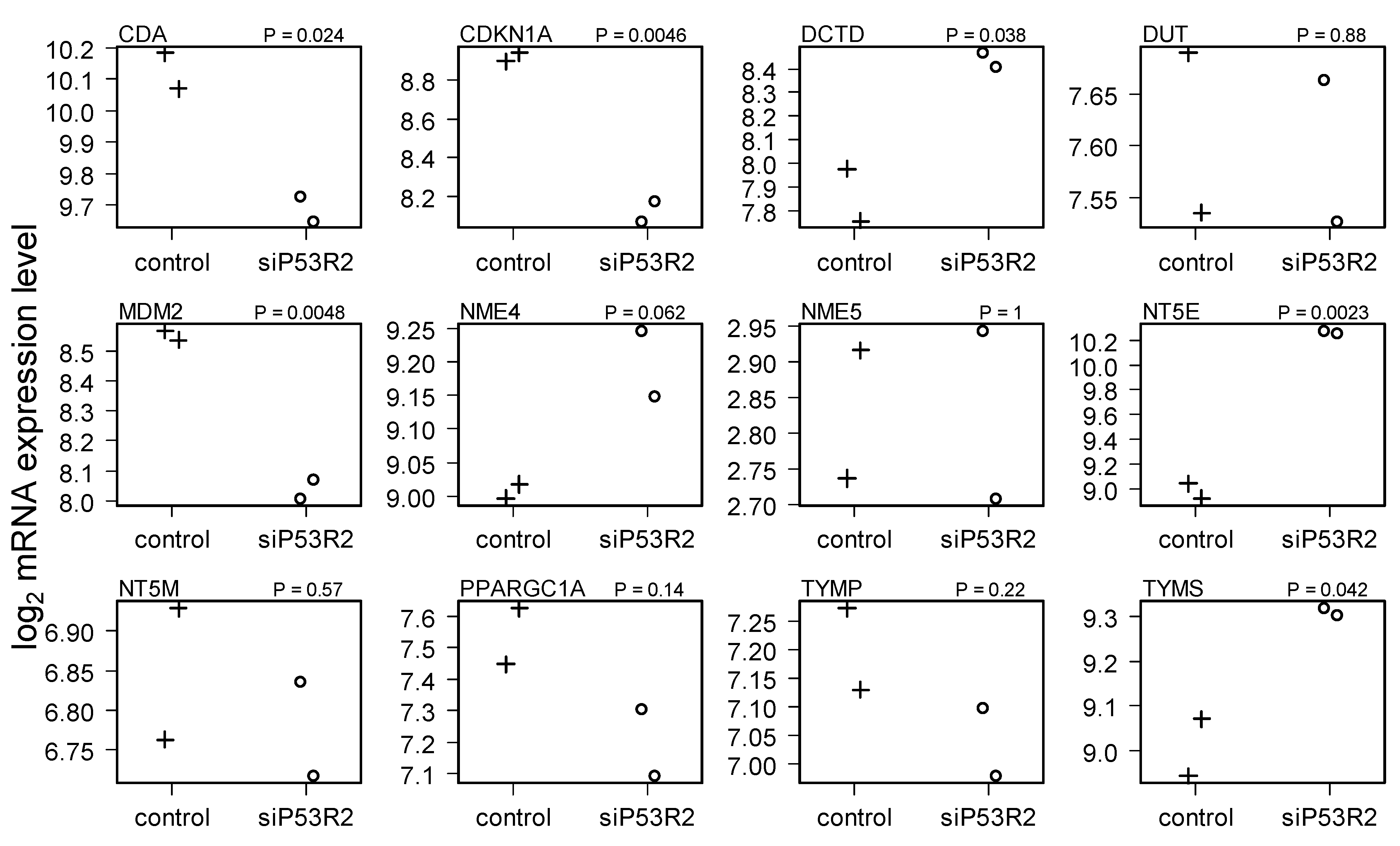
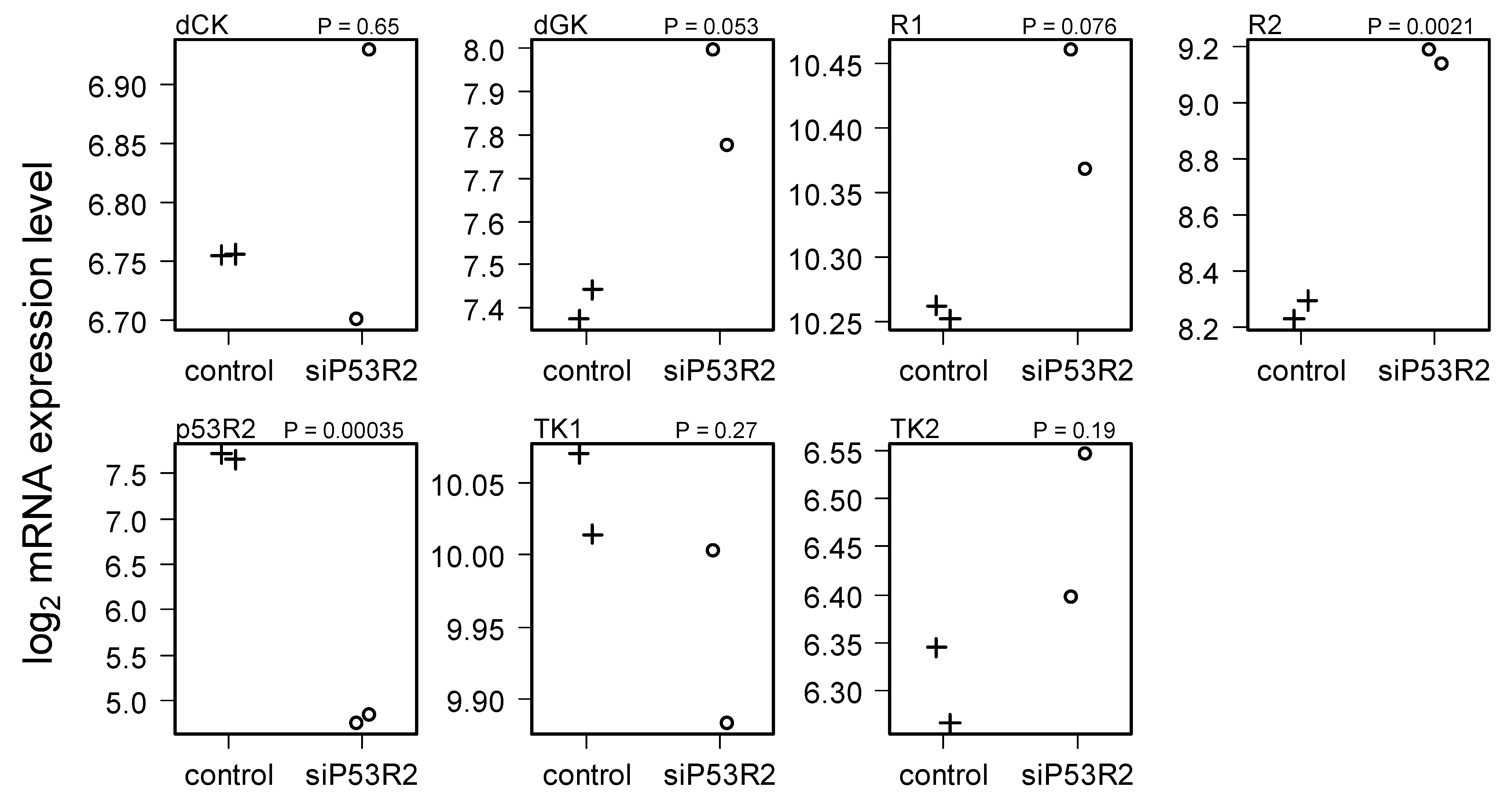
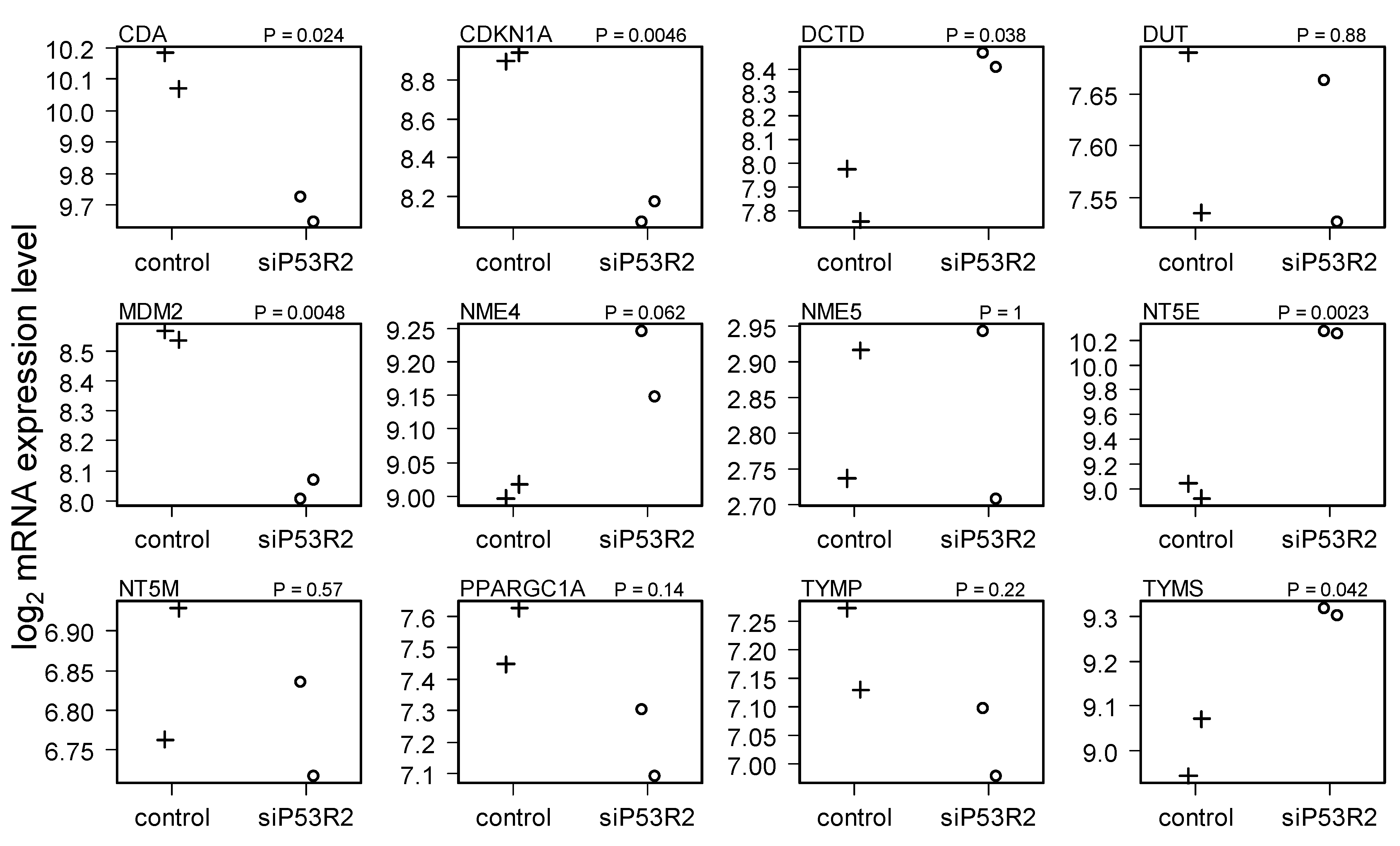
2.4. Gene Expression in Cells Treated with siP53R2
3. Experimental Section
3.1. Cell Lines and Culture Conditions
3.2. Viral Vectors and Infection
3.3. RNA Isolation
3.4. Data Analysis
4. Conclusions
Acknowledgments
References
- Vousden, K.H.; Prives, C. P53 and prognosis: New insights and further complexity. Cell 2005, 120, 7–10. [Google Scholar]
- Nakano, K.; Balint, E.; Ashcroft, M.; Vousden, K.H. A ribonucleotide reductase gene is a transcriptional target of p53 and p73. Oncogene 2000, 19, 4283–4289. [Google Scholar] [CrossRef]
- Heinemann, V.; Xu, Y.Z.; Chubb, S.; Sen, A.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Inhibition of ribonucleotide reduction in CCRF-CEM cells by 2',2'-difluorodeoxycytidine. Mol. Pharmacol. 1990, 38, 567–572. [Google Scholar]
- Jones, P.A.; Taylor, S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell 1980, 20, 85–93. [Google Scholar] [CrossRef]
- Ng, K.P.; Ebrahem, Q.; Negrotto, S.; Mahfouz, R.Z.; Link, K.A.; Hu, Z.; Gu, X.; Advani, A.; Kalaycio, M.; Sobecks, R.; et al. p53 independent epigenetic-differentiation treatment in xenotransplant models of acute myeloid leukemia. Leukemia 2011, 25, 1739–1750. [Google Scholar] [CrossRef]
- Shi, J.Y.; Shi, Z.Z.; Zhang, S.J.; Zhu, Y.M.; Gu, B.W.; Li, G.; Bai, X.T.; Gao, X.D.; Hu, J.; Jin, W.; et al. Association between single nucleotide polymorphisms in deoxycytidine kinase and treatment response among acute myeloid leukaemia patients. Pharmacogenetics 2004, 14, 759–768. [Google Scholar] [CrossRef]
- Marechal, R.; Mackey, J.R.; Lai, R.; Demetter, P.; Peeters, M.; Polus, M.; Cass, C.E.; Salmon, I.; Deviere, J.; van Laethem, J.L. Deoxycitidine kinase is associated with prolonged survival after adjuvant gemcitabine for resected pancreatic adenocarcinoma. Cancer 2010, 116, 5200–5206. [Google Scholar] [CrossRef]
- Shao, J.; Zhou, B.; Chu, B.; Yen, Y. Ribonucleotide reductase inhibitors and future drug design. Curr. Cancer Drug Targets 2006, 6, 409–431. [Google Scholar]
- Barrett, T.; Troup, D.B.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; et al. NCBI GEO: Archive for functional genomics data sets—10 years on. Nucleic Acids Res. 2011, 39, D1005–D1010. [Google Scholar]
- Herbert, B.S.; Chanoux, R.A.; Liu, Y.; Baenziger, P.H.; Goswami, C.P.; McClintick, J.N.; Edenberg, H.J.; Pennington, R.E.; Lipkin, S.M.; Kopelovich, L. A molecular signature of normal breast epithelial and stromal cells from Li-Fraumeni syndrome mutation carriers. Oncotarget 2010, 1, 405–422. [Google Scholar]
- Miller, L.D.; Smeds, J.; George, J.; Vega, V.B.; Vergara, L.; Ploner, A.; Pawitan, Y.; Hall, P.; Klaar, S.; Liu, E.T.; et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc. Natl. Acad. Sci. USA 2005, 102, 13550–13555. [Google Scholar]
- Zhang, K.; Wu, J.; Wu, X.; Wang, X.; Wang, Y.; Zhou, N.; Kuo, M.L.; Liu, X.; Zhou, B.; Chang, L.; et al. p53R2 inhibits the proliferation of human cancer cells in association with cell-cycle arrest. Mol. Cancer Ther. 2011, 10, 269–278. [Google Scholar] [CrossRef]
- Kunos, C.A.; Waggoner, S.; von Gruenigen, V.; Eldermire, E.; Pink, J.; Dowlati, A.; Kinsella, T.J. Phase I trial of pelvic radiation, weekly cisplatin, and 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP, NSC #663249) for locally advanced cervical cancer. Clin. Cancer Res. 2010, 16, 1298–1306. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003, 31, e15. [Google Scholar]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. affy—Analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- Garbe, J.C.; Bhattacharya, S.; Merchant, B.; Bassett, E.; Swisshelm, K.; Feiler, H.S.; Wyrobek, A.J.; Stampfer, M.R. Molecular distinctions between stasis and telomere attrition senescence barriers shown by long-term culture of normal human mammary epithelial cells. Cancer Res. 2009, 69, 7557–7568. [Google Scholar] [CrossRef]
- Jackson, M.W.; Agarwal, M.K.; Yang, J.; Bruss, P.; Uchiumi, T.; Agarwal, M.L.; Stark, G.R.; Taylor, W.R. p130/p107/p105Rb-dependent transcriptional repression during DNA-damage-induced cell-cycle exit at G2. J. Cell Sci. 2005, 118, 1821–1832. [Google Scholar]
- Brummelkamp, T.R.; Bernards, R.; Agami, R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002, 296, 550–553. [Google Scholar] [CrossRef]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: New York, NY, USA, 2005; pp. 397–420. [Google Scholar]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, Y.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol. 2004, 5, R80. [Google Scholar] [CrossRef] [Green Version]
- Bourdon, A.; Minai, L.; Serre, V.; Jais, J.P.; Sarzi, E.; Aubert, S.; Chretien, D.; de Lonlay, P.; Paquis-Flucklinger, V.; Arakawa, H.; et al. Mutation of RRM2B, encoding p53-controlled ribonucleotide reductase (p53R2), causes severe mitochondrial DNA depletion. Nat. Genet. 2007, 39, 776–780. [Google Scholar]
- Gower, W.R., Jr.; Carr, M.C.; Ives, D.H. Deoxyguanosine kinase. Distinct molecular forms in mitochondria and cytosol. J. Biol. Chem. 1979, 254, 2180–2183. [Google Scholar]
- Colly, L.P.; Richel, D.J.; Arentsen-Honders, M.W.; Kester, M.G.; ter Riet, P.M.; Willemze, R. Increase in Ara-C sensitivity in Ara-C sensitive and -resistant leukemia by stimulation of the salvage and inhibition of the de novo pathway. Ann. Hematol. 1992, 65, 26–32. [Google Scholar] [CrossRef]
- Bradshaw, P.C.; Samuels, D.C. A computational model of mitochondrial deoxynucleotide metabolism and DNA replication. Am. J. Physiol. Cell Physiol. 2005, 288, C989–C1002. [Google Scholar] [CrossRef]
- Bryson, A.E.; Ho, Y.C. Applied Optimal Control; Hemisphere Publishing Corporation: New York, NY, USA, 1975. [Google Scholar]
- Kailath, T. Linear Systems; Prentice-Hall: Englewood Cliffs, NJ, USA, 1980. [Google Scholar]
- Radivoyevitch, T.; Loparo, K.A.; Jackson, R.C.; Sedwick, W.D. On systems and control approaches to therapeutic gain. BMC Cancer 2006, 6, 104. [Google Scholar] [CrossRef]
- Ainseba, B.; Benosman, C. Optimal control for resistance and suboptimal response in CML. Math. Biosci. 2010, 227, 81–93. [Google Scholar] [CrossRef]
- Padhi, R.; Kothari, M. An optimal dynamic inversion-based neuro-adaptive approach for treatment of chronic myelogenous leukemia. Comput. Methods Programs Biomed. 2007, 87, 208–224. [Google Scholar] [CrossRef]
- Nanda, S.; Moore, H.; Lenhart, S. Optimal control of treatment in a mathematical model of chronic myelogenous leukemia. Math. Biosci. 2007, 210, 143–156. [Google Scholar] [CrossRef]
Supplementary Materials

© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Radivoyevitch, T.; Saunthararajah, Y.; Pink, J.; Ferris, G.; Lent, I.; Jackson, M.; Junk, D.; Kunos, C.A. dNTP Supply Gene Expression Patterns after P53 Loss. Cancers 2012, 4, 1212-1224. https://doi.org/10.3390/cancers4041212
Radivoyevitch T, Saunthararajah Y, Pink J, Ferris G, Lent I, Jackson M, Junk D, Kunos CA. dNTP Supply Gene Expression Patterns after P53 Loss. Cancers. 2012; 4(4):1212-1224. https://doi.org/10.3390/cancers4041212
Chicago/Turabian StyleRadivoyevitch, Tomas, Yogen Saunthararajah, John Pink, Gina Ferris, Ian Lent, Mark Jackson, Damian Junk, and Charles A. Kunos. 2012. "dNTP Supply Gene Expression Patterns after P53 Loss" Cancers 4, no. 4: 1212-1224. https://doi.org/10.3390/cancers4041212