Do Non-Genomically Encoded Fusion Transcripts Cause Recurrent Chromosomal Translocations?

Abstract
:1. Introduction

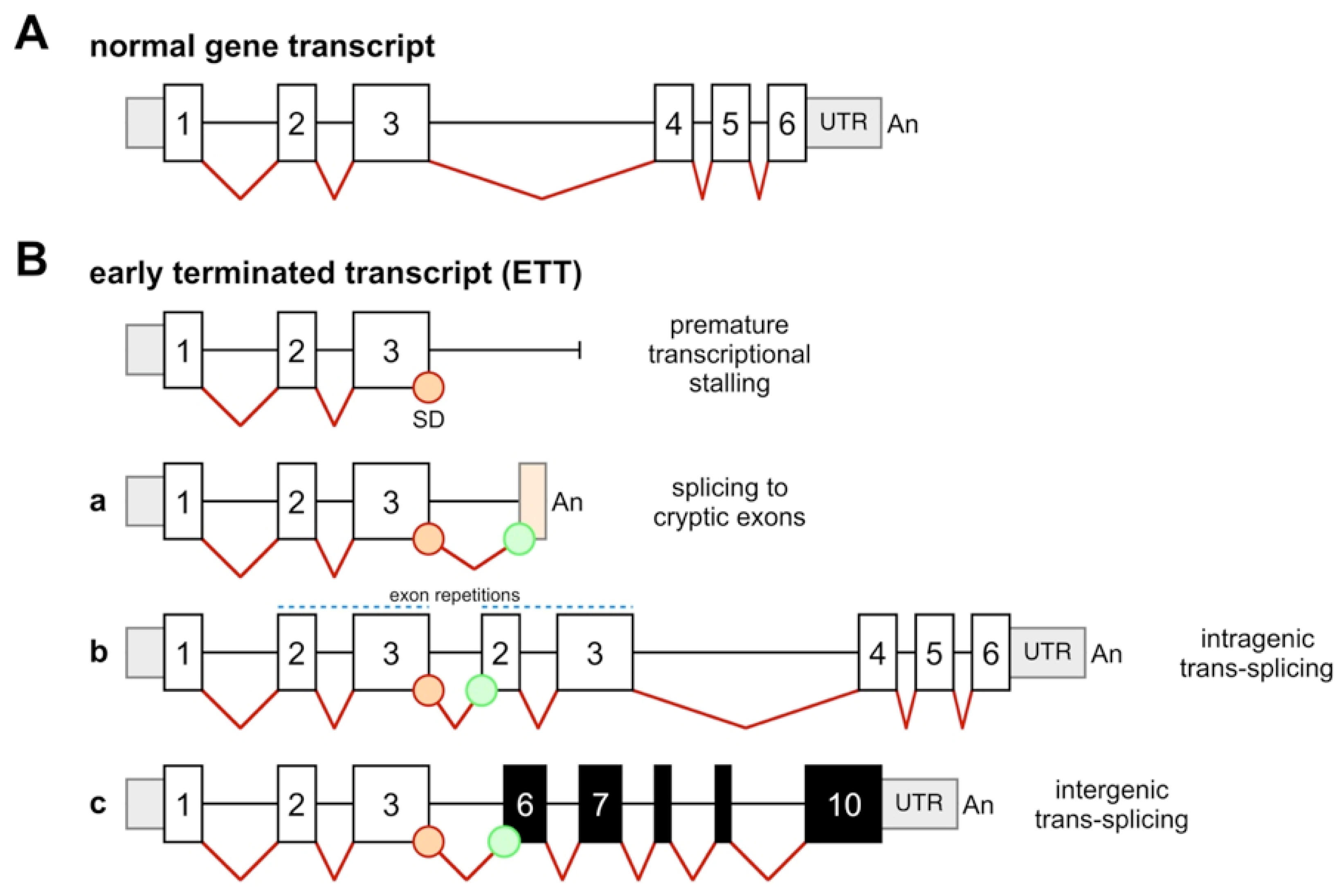
2. Results and Discussion
2.1. Choosing Additional Genes for Determining EETs and NGEFTs
2.2. Identification of Early-Terminated Transcripts in Breakpoint Cluster Regions of Frequently Rearranging Genes
2.3. Abundance of ETTs
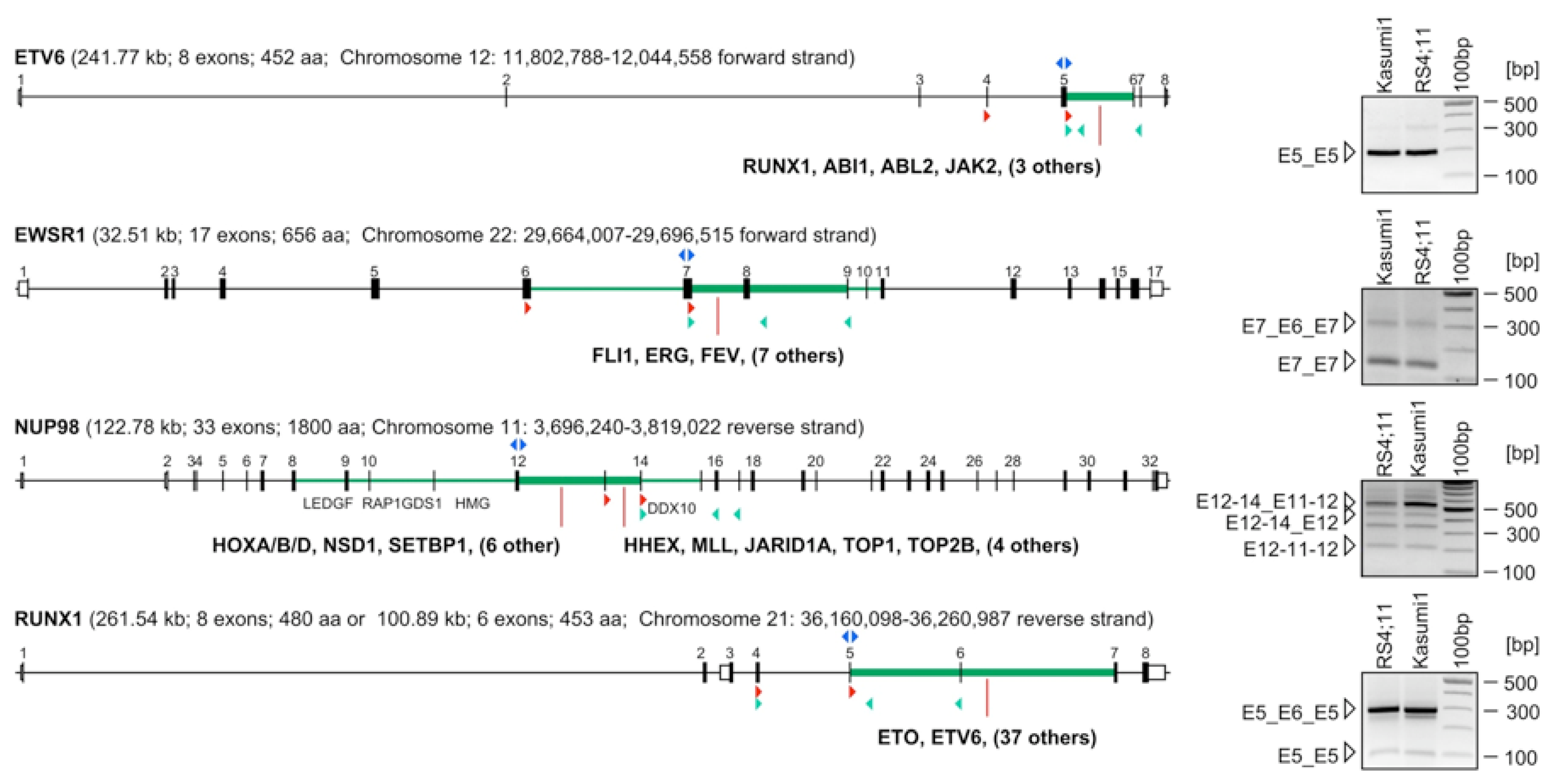
3. Experimental Section
3.1. Cell Lines and Patient Material
3.2. Identification of Exon Repetitions and Fusion Transcripts
4. Conclusions
Appendix

{kind=link}
{kind=link}
{kind=link}
| (A) ETV6—3'RACE experiments |
| cryptic exon within intron 5 |
| (tag|)agacggggtttcaccatgttagccaggatggtcttgatctcctgacctcgcgatccgcctgcgtcggcctcctaaagtgctaggattacaggcgtgagccaccgcgcctggccggtactgtcccttttttatttaaatcatgtatttctagatttttctctggccccaagtgcaccaccagtgttgtctcagtttcactgttataaatcatttacatcagggatgttgaatccagaaaataaaatggcaaacaaaaatga(an) |
| cryptic poly-A site intron 4: …ttcttgcctgaggtttagacaaatccaggaagtttaattgttaacttgataagctggtc(an) |
| cryptic poly-A site intron 5: …acatcagggatgttgaatccagaaaataaaatggcaaacaaaaa(an) |
| cryptic poly-A site intron 5: …agcgaaaacatttaaaataaaaggtttatgagcatgc(an) |
| cryptic poly-A site intron 5: …cattgagcacctactgtctgccagagtaataaagcgctgtagaagcaaagca(an) |
| cryptic poly-A site intron 6: …gaaggaaataattaaagatgcatcttgccatttgttcctga(an) |
| cryptic poly-A site intron 7: …ttatgtctgtcctgtaataaagccaaaactaaatgaaca(an) |
| ETV6 fusion mRNA: ETV6 exon 5::MGST3 exon 6 (in-frame fusion): |
| CACGCCATGCCCATTGGGAGAATAGCAG::AACCCAGCAAGCGTAGTCGAGGAGCCCT |
| [microsomal glutathione S-transferase 3 (MGST3) is located on chromosome 1q24, consists of 6 exons and encodes 152 amino acids] |
| (B) EWSR1—3'-RACE experiments |
| cryptic exon within intron 8 |
| (cag|)gttacaaagtgagagccttgtatacacttcaatacttaaaaagtacccgtactcagtactcagccggcagcataatgaaaagtgggactagacacggtgtccatatggagaggaaaaatatacatagaatatttttaacaaaatgtattcattgtataaatggaatccttctgtaactttggtaactgcatacttgttgtttggtaatgaaccagaggaggtataatactctagaattgtgtaacattaaagtgtaaacttttgtgtttaaa(an) |
| cryptic exon within intron 8 |
| (cag|)gttacaaagtgagagccttgtatacacttcaatacttaaaaagtacccgtactcagtactcagccggcagcataatgaaaagtgggactagacacggtgtccatatggagaggaaaaatatacatagaatatttttaacaaaatgtattcattgta(an) |
| EWSR1 fusion mRNA: EWSR1 exon 7::PRPF6 exon 20 (out-of-frame fusion): |
| CAACAGAGCAGCAGCTACGGGCAGCAGA::GCTGTTTTGGAGTCAGCGGAAGATCACCA |
| [pre-mRNA processing factor 6 (PRP6) is located on 20q13.3, consists of 21 exons and encodes 941 amino acids] |
| (C) NUP98—3'-RACE experiments |
| cryptic poly-A site exon 16: TGCTGGAAATAAACACAGCAACAGCAA(an) |
| additional exon repetition: E14_E13-14-15-16 (out-of-frame; poly-adenylated within exon 16) |
| (D) RUNX1—3'-RACE experiments |
| cryptic exon within intron 6 |
| (cag|)atggggtttcagcatgttggccaggctcgtctcgaactcctgaccacaggt---gctacaaaaggatttcttttaaccaa(an) |
| cryptic poly-A site intron 5: ccccgtaaagccaaaaagttgcattgtcaatactctcgctactcaaa(an) |
| cryptic poly-A site intron 5: tactcagggaccatgtctcagattccttgggttcaac(an) |
Acknowledgements
References
- Reichel, M.; Gillert, E.; Nilson, I.; Siegler, G.; Greil, J.; Fey, G.H.; Marschalek, R. Fine structure of translocation breakpoints in leukemic blasts with chromosomal translocation t(4;11): The DNA damage-repair model of translocation. Oncogene 1998, 17, 3035–3044. [Google Scholar]
- Marcucci, G.; Strout, M.P.; Bloomfield, C.D.; Caligiuri, M.A. Detection of unique ALL1 (MLL) fusion transcripts in normal human bone marrow and blood: Distinct origin of normal versus leukemic ALL1 fusion transcripts. Cancer Res. 1998, 58, 790–793. [Google Scholar]
- Uckun, F.M.; Herman-Hatten, K.; Crotty, M.L.; Sensel, M.G.; Sather, H.N.; Tuel-Ahlgren, L.; Sarquis, M.B.; Bostrom, B.; Nachman, J.B.; Steinherz, P.G.; et al. Clinical significance of MLL-AF4 fusion transcript expression in the absence of a cytogenetically detectable t(4;11)(q21;q23) chromosomal translocation. Blood 1998, 92, 810–821. [Google Scholar]
- Biernaux, C.; Loos, M.; Sels, A.; Huez, G.; Stryckmans, P. Detection of major bcr-abl gene expression at a very low level in blood cells of some healthy individuals. Blood 1995, 86, 3118–3122. [Google Scholar]
- Bose, S.; Deininger, M.; Gora-Tybor, J.; Goldman, J.M.; Melo, J.V. The presence of typical and atypical BCR-ABL fusion genes in leukocytes of normal individuals: Biologic significance and implications for the assessment of minimal residual disease. Blood 1998, 92, 3362–3367. [Google Scholar]
- Eguchi-Ishimae, M.; Eguchi, M.; Ishii, E.; Miyazaki, S.; Ueda, K.; Kamada, N.; Mizutani, S. Breakage and fusion of the TEL (ETV6) gene in immature B lymphocytes induced by apoptogenic signals. Blood 2001, 97, 737–743. [Google Scholar] [CrossRef]
- Quina, A.S.; Gameiro, P.; Sá da Costa, M.; Telhada, M.; Parreira, L. PML-RARA fusion transcripts in irradiated and normal hematopoietic cells. Genes Chrom. Cancer 2000, 29, 266–275. [Google Scholar] [CrossRef]
- Beylot-Barry, M.; Groppi, A.; Vergier, B.; Pulford, K.; Merlio, J.P. Characterization of t(2;5) reciprocal transcripts and genomic breakpoints in CD30+ cutaneous lymphoproliferations. Blood 1998, 91, 4668–4676. [Google Scholar]
- Maes, B.; Vanhentenrijk, V.; Wlodarska, I.; Cools, J.; Peeters, B.; Marynen, P.; de Wolf-Peeters, C. The NPM-ALK and the ATIC-ALK fusion genes can be detected in non-neoplastic cells. Am. J. Pathol. 2001, 158, 2185–2193. [Google Scholar] [CrossRef]
- Mori, H.; Colman, S.M.; Xiao, Z.; Ford, A.M.; Healy, L.E.; Donaldson, C.; Hows, J.M.; Navarrete, C.; Greaves, M. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc. Natl. Acad. Sci. USA 2002, 99, 8242–8247. [Google Scholar]
- Kowarz, E.; Merkens, J.; Karas, M.; Dingermann, T.; Marschalek, R. Premature transcript termination, trans-splicing and DNA repair: A vicious path to cancer. Am. J. Blood Res. 2011, 1, 1–12. [Google Scholar]
- Nilson, I.; Löchner, K.; Siegler, G.; Greil, J.; Beck, J.D.; Fey, G.H.; Marschalek, R. Exon/intron structure of the human ALL-1 (MLL) gene involved in translocations to chromosomal region 11q23 and acute leukaemias. Br. J. Haematol. 1996, 93, 966–972. [Google Scholar]
- Scharf, S.; Zech, J.; Bursen, A.; Schraets, D.; Oliver, P.L.; Kliem, S.; Pfitzner, E.; Gillert, E.; Dingermann, T.; Marschalek, R. Transcription linked to recombination: A gene-internal promoter coincides with the recombination hot spot II of the human MLL gene. Oncogene 2007, 26, 1361–1371. [Google Scholar] [CrossRef]
- Li, H.; Wang, J.; Mor, G.; Sklar, J. A neoplastic gene fusion mimics trans-splicing of RNAs in normal human cells. Science 2008, 321, 1357–1361. [Google Scholar]
- Li, H.; Wang, J.; Ma, X.; Sklar, J. Gene fusions and RNA trans-splicing in normal and neoplastic human cells. Cell Cycle 2009, 8, 218–222. [Google Scholar] [CrossRef]
- Strissel, P.L.; Strick, R.; Tomek, R.J.; Roe, B.A.; Rowley, J.D.; Zeleznik-Le, N.J. DNA structural properties of AF9 are similar to MLL and could act as recombination hot spots resulting in MLL/AF9 translocations and leukemogenesis. Hum. Mol. Genet. 2000, 9, 1671–1679. [Google Scholar] [CrossRef]
- Hensel, J.P.; Gillert, E.; Fey, G.H.; Marschalek, R. Breakpoints of t(4;11) translocations in the human MLL and AF4 genes in ALL patients are preferentially clustered outside of high-affinity matrix attachment regions. J. Cell Biochem. 2001, 82, 299–309. [Google Scholar] [CrossRef]
- Strick, R.; Zhang, Y.; Emmanuel, N.; Strissel, P.L. Common chromatin structures at breakpoint cluster regions may lead to chromosomal translocations found in chronic and acute leukemias. Hum. Genet. 2006, 119, 479–495. [Google Scholar] [CrossRef]
- Strissel, P.L.; Strick, R.; Rowley, J.D.; Zeleznik-Le, N.J. An in vivo topoisomerase II cleavage site and a DNase I hypersensitive site colocalize near exon 9 in the MLL breakpoint cluster region. Blood 1998, 92, 3793–3803. [Google Scholar]
- Aplan, P.D.; Chervinsky, D.S.; Stanulla, M.; Burhans, W.C. Site-specific DNA cleavage within the MLL breakpoint cluster region induced by topoisomerase II inhibitors. Blood 1996, 87, 2649–2658. [Google Scholar]
- Nasr, F.; Macintyre, E.; Venuat, A.M.; Bayle, C.; Carde, P.; Ribrag, V. Translocation t(4;11)(q21;q23) and MLL gene rearrangement in acute lymphoblastic leukemia secondary to anti topoisomerase II anticancer agents. Leuk. Lymphoma 1997, 25, 399–401. [Google Scholar]
- Megonigal, M.D.; Cheung, N.K.; Rappaport, E.F.; Nowell, P.C.; Wilson, R.B.; Jones, D.H.; Addya, K.; Leonard, D.G.; Kushner, B.H.; Williams, T.M.; et al. Detection of leukemia-associated MLL-GAS7 translocation early during chemotherapy with DNA topoisomerase II inhibitors. Proc. Natl. Acad. Sci. USA 2000, 97, 2814–2819. [Google Scholar]
- Felix, C.A. Leukemias related to treatment with DNA topoisomerase II inhibitors. Med. Pediatr. Oncol. 2001, 36, 525–535. [Google Scholar] [CrossRef]
- Stanulla, M.; Wang, J.; Chervinsky, D.S.; Thandla, S.; Aplan, P.D. DNA cleavage within the MLL breakpoint cluster region is a specific event which occurs as part of higher-order chromatin fragmentation during the initial stages of apoptosis. Mol. Cell Biol. 1997, 17, 4070–4079. [Google Scholar]
- Betti, C.J.; Villalobos, M.J.; Diaz, M.O.; Vaughan, A.T. Apoptotic triggers initiate translocations within the MLL gene involving the nonhomologous end joining repair system. Cancer Res. 2001, 61, 4550–4555. [Google Scholar]
- Stanulla, M.; Chhalliyil, P.; Wang, J.; Jani-Sait, S.N.; Aplan, P.D. Mechanisms of MLL gene rearrangement: Site-specific DNA cleavage within the breakpoint cluster region is independent of chromosomal context. Hum. Mol. Genet. 2001, 10, 2481–2491. [Google Scholar] [CrossRef]
- Sim, S.P.; Liu, L.F. Nucleolytic cleavage of the mixed lineage leukemia breakpoint cluster region during apoptosis. J. Biol. Chem. 2001, 276, 31590–31595. [Google Scholar] [CrossRef]
- Meyer, C.; Kowarz, E.; Hofmann, J.; Renneville, A.; Zuna, J.; Trka, J.; Ben Abdelali, R.; Macintyre, E.; de Braekeleer, E.; de Braekeleer, M.; et al. New insights to the MLL recombinome of acute leukemias. Leukemia 2009, 23, 1490–1499. [Google Scholar] [CrossRef]
- Storici, F.; Bebenek, K.; Kunkel, T.A.; Gordenin, D.A.; Resnick, M.A. RNA-templated DNA repair. Nature 2007, 447, 338–341. [Google Scholar] [CrossRef]
- Storici, F. RNA-mediated DNA modifications and RNA-templated DNA repair. Curr. Opin. Mol. Ther. 2008, 10, 224–230. [Google Scholar]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; et al. ENCODE Project Consortium. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [CrossRef]
- Gingeras, T.R. Implications of chimaeric non-co-linear transcripts. Nature 2009, 461, 206–211. [Google Scholar] [CrossRef]
- Zaphiropoulos, P.G. Trans-splicing in higher eukaryotes: Implications for cancer development? Front. Genet. 2011, 2, 92. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kowarz, E.; Dingermann, T.; Marschalek, R. Do Non-Genomically Encoded Fusion Transcripts Cause Recurrent Chromosomal Translocations? Cancers 2012, 4, 1036-1049. https://doi.org/10.3390/cancers4041036
Kowarz E, Dingermann T, Marschalek R. Do Non-Genomically Encoded Fusion Transcripts Cause Recurrent Chromosomal Translocations? Cancers. 2012; 4(4):1036-1049. https://doi.org/10.3390/cancers4041036
Chicago/Turabian StyleKowarz, Eric, Theo Dingermann, and Rolf Marschalek. 2012. "Do Non-Genomically Encoded Fusion Transcripts Cause Recurrent Chromosomal Translocations?" Cancers 4, no. 4: 1036-1049. https://doi.org/10.3390/cancers4041036