Molecular and Epigenetic Mechanisms of MLL in Human Leukemogenesis

Abstract
:1. Introduction
2. MLL in Normal Hematopoiesis
3. Activity of the Wild Type MLL Protein Complex

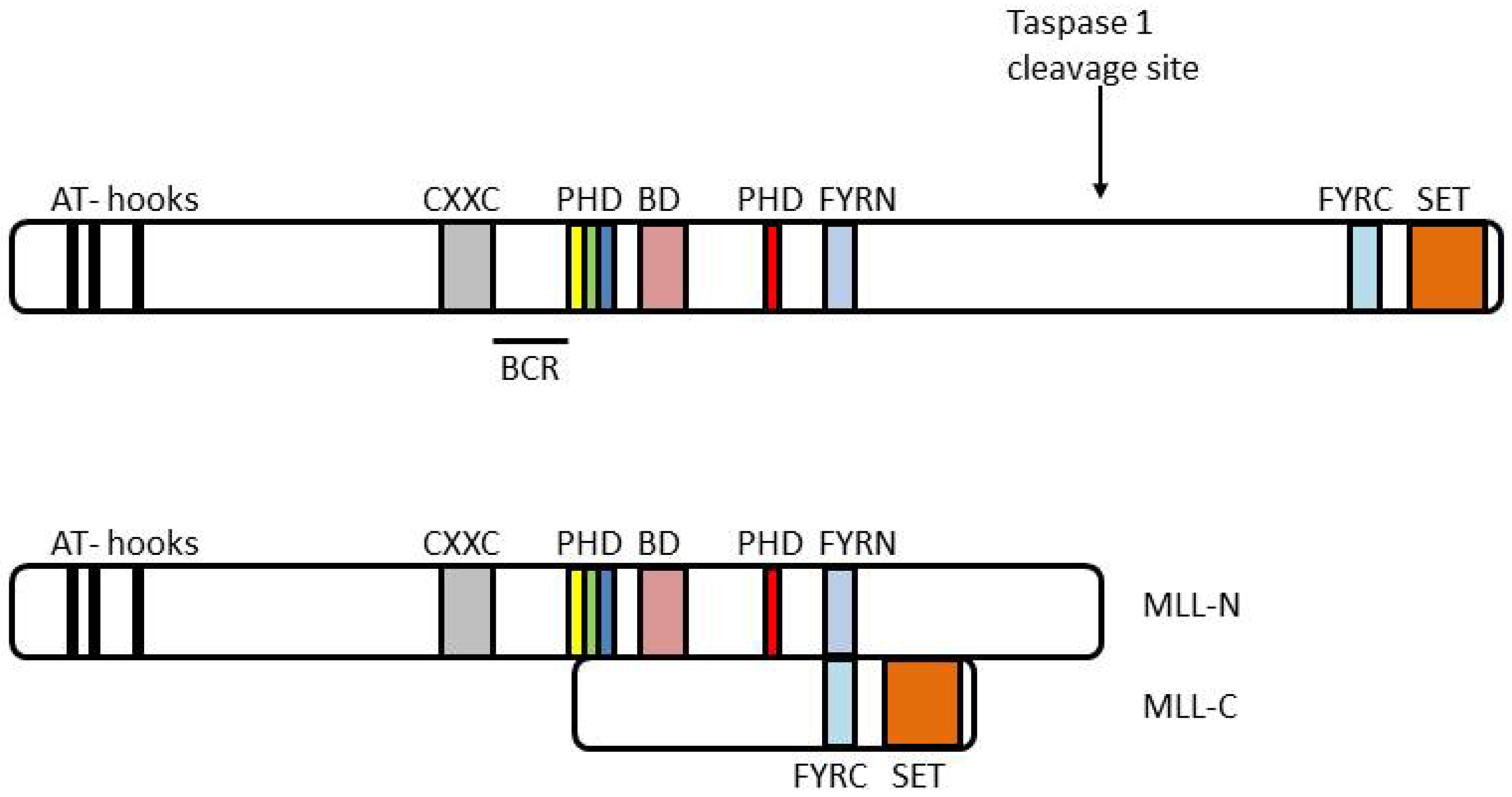{kind=link}
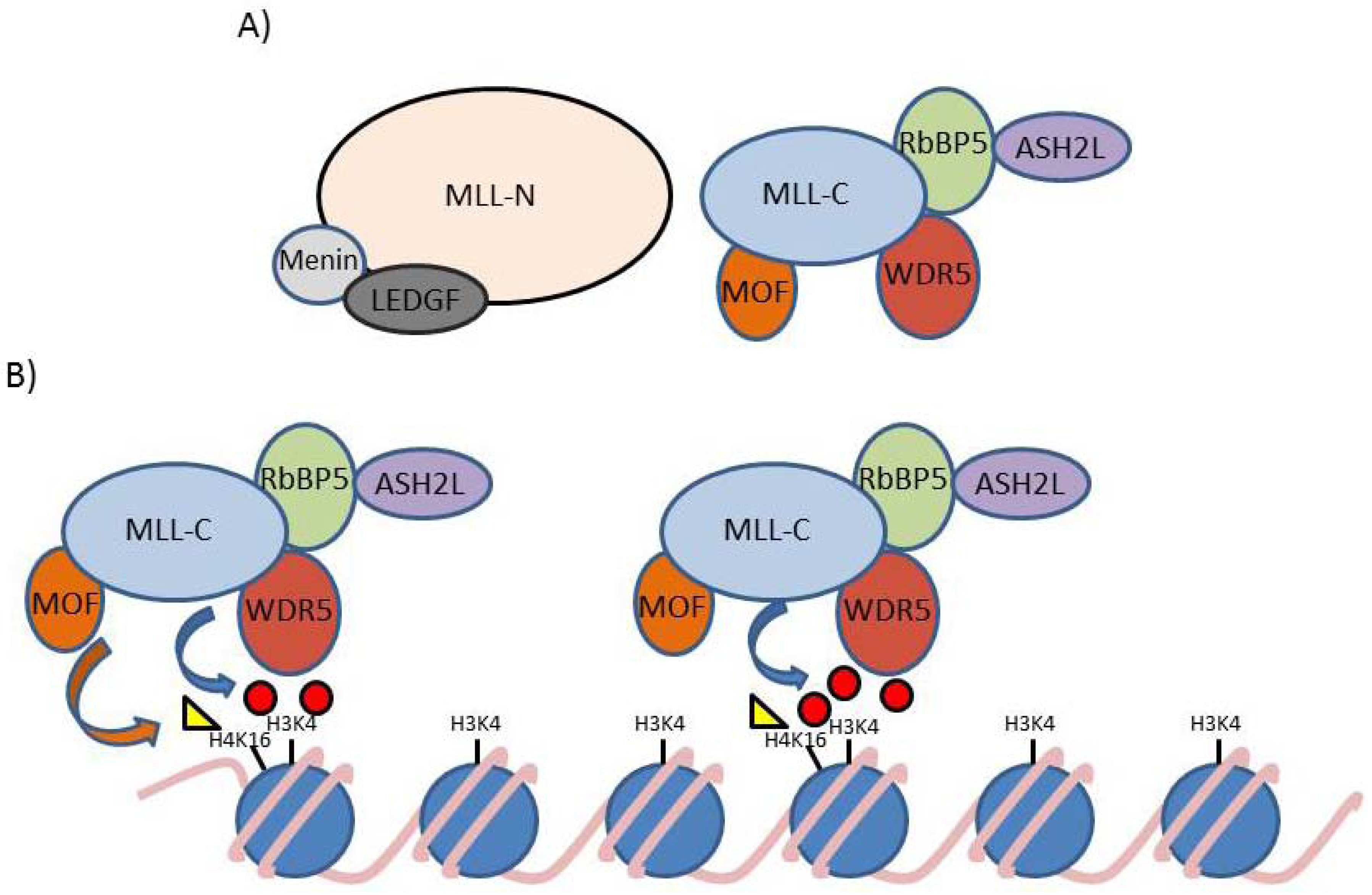{kind=link}
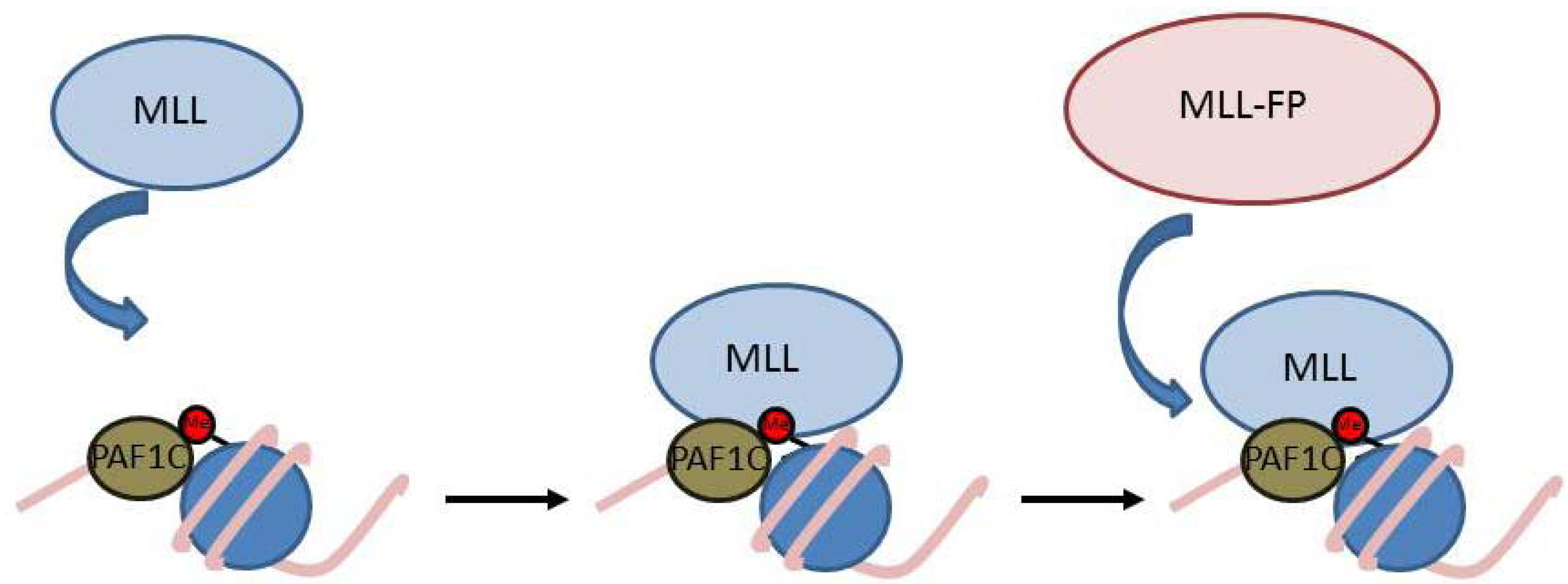{kind=link}
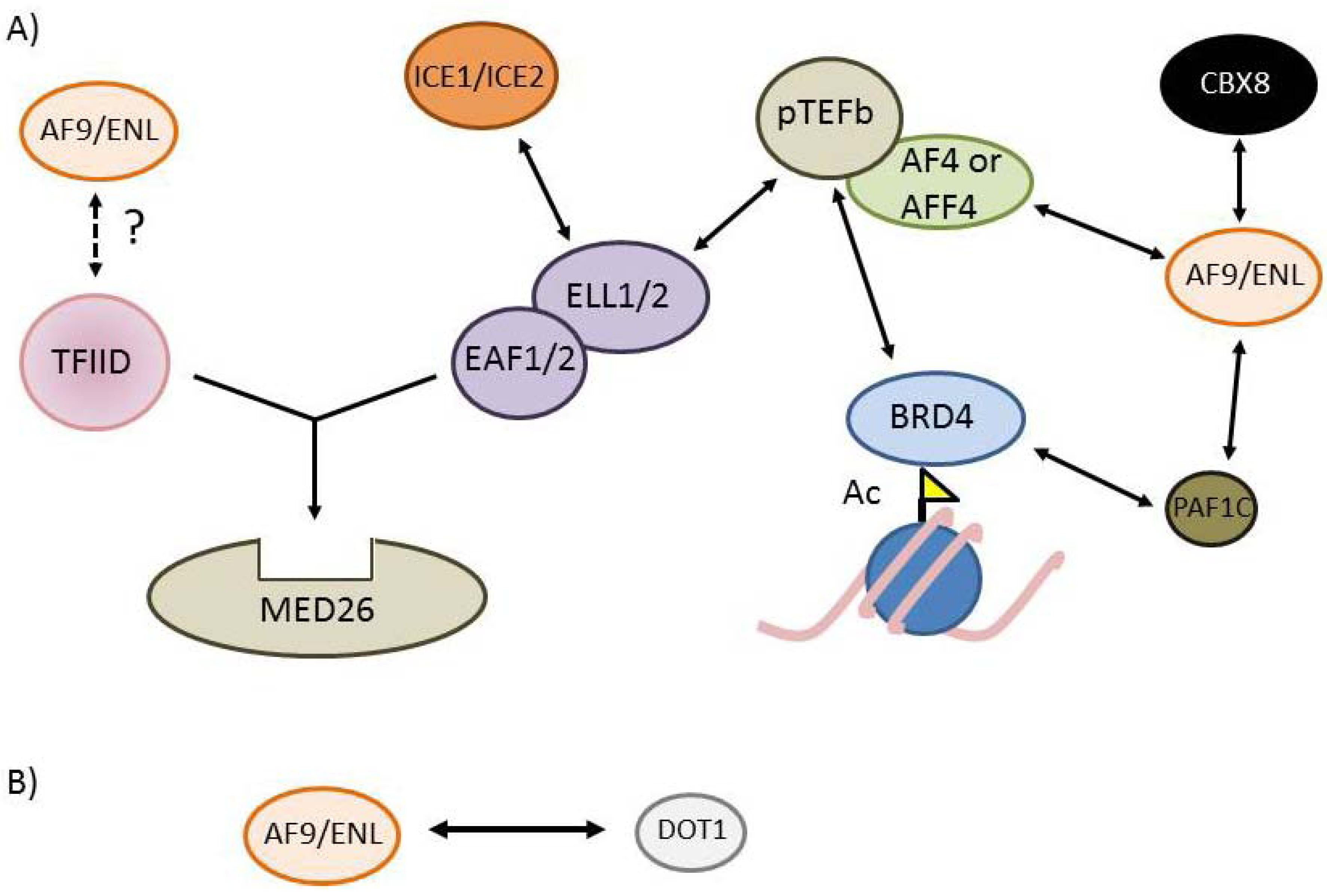{kind=link}
{kind=link}
| NCBI Name | Aliases (commonly used aliases in bold) | Human Chromosome Position | Important References |
|---|---|---|---|
| MLL | MLL1, HRX, TRX1, ALL-1, CXXC7, HTRX1, MLL1A, MLL/GAS7, TET1-MLL, KMT2A | 11q23 | [39,40,41,60] |
| MLL2 | ALR, KMS, MLL4, AAD10, KABUK1, TNRC21, CAGL114, KMT2B, KMT2D | 12q13 | [61] Note: this is the MLL that is mutated in Kabuki syndrome [62], sometimes MLL4 (below) is mistakenly referenced |
| MLL3 | HALR, KMT2C | 7q36.1 | [63,64] |
| MLL4 | MLL2, HRX2, TRX2, WBP7, KMT2D | 19q13.1 | Originally called MLL2 in [65,66], renamed MLL4 in [63] but still commonly referred to as MLL2 in [51,54,67,68,69] |
| MLL5 | HDCMC04P, KMT2E | 7q22.1 | [70] |
| SETD1A | SET1A, SET1, KMT2F | 16p11.2 | [71] |
| SETD1B | SET1B, KMT2G | 12q24.31 | [72] |
| NCBI Name | Aliases (commonly used aliases in bold) | Human Chromosome Position | MLL interaction site | Evidence for a direct interaction with MLL | Structural data supporting interaction |
|---|---|---|---|---|---|
| MEN1 | Menin, MEAI, SCG2 | 11q13 | N terminus | [86] | [87,88,89,90] |
| PSIP1 | p52, p75, PAIP, DFS70, LEDGF, PSIP2 | 9p22.3 | N terminus | [91] | [90] |
| PAF1 | 19q13.1 | CXXC region | [69,78] | No data | |
| CTR9 | SH2BP1, TSBP, p150, p150TSP | 11p15.3 | CXXC region | [78] | No data |
| BMI-1 | RNF51 | 10p11.23 | CXXC region | [83] | No data |
| ELAC2 | ELC2, HPC2 | 17p11.2 | CXXC region | [83] | No data |
| CTBP1 | CtBP | 4p16 | CXXC region | [83] | No data |
| HDAC1 | HD1, RPD3, RPD3L1 | 1p34 | CXXC region | [83] | No data |
| PPIE | CYP-33, CYP33 | 1p32 | PHD finger 3 | [79] | [82] |
| ASB2 | 14q31-q32 | PHD fingers | [85] | No data | |
| HCFC1 | CFF, HCF-1, HCF1, HFC1, VCAF | Xq28 | adjacent to BD | [92] | No data |
| HCFC2 | HCF-2, HCF2 | 12q23.3 | adjacent to BD | [92] | No data |
| CREBBP | CBP, RSTS, KAT3A | 16p13.3 | MLL-C | [93] | No data |
| KAT8 | MOF, hMOF, MYST1 | 16p11.2 | MLL-C | [94] | No data |
| WDR5 | SWD3 | 9q34 | SET domain | [95,96] | [97,98,99,100,101,102,103,104,105,106] |
| RBBP5 | RbBP5, SWD1 | 1q32 | SET domain | [95,96,107] | [104,105,108] |
| ASH2L * | ASH2, ASH2L1, ASH2L2, Bre2 | 8p11.2 | SET domain | [95,96,107] | [105,108,109] |
| DPY30 * | DPY-30, Saf19 | 2p22.3 | SET domain | [96,110] | [111] |
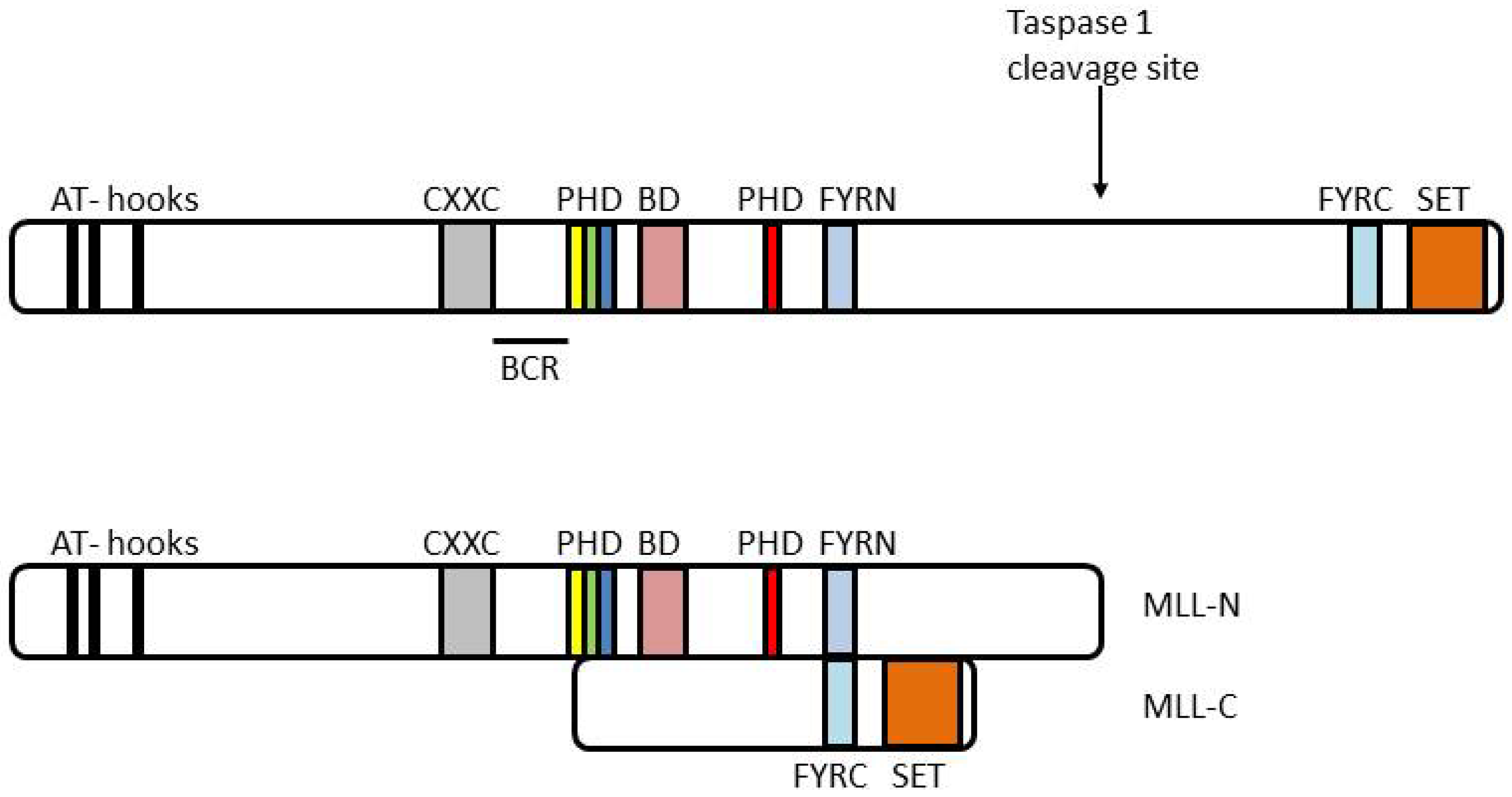
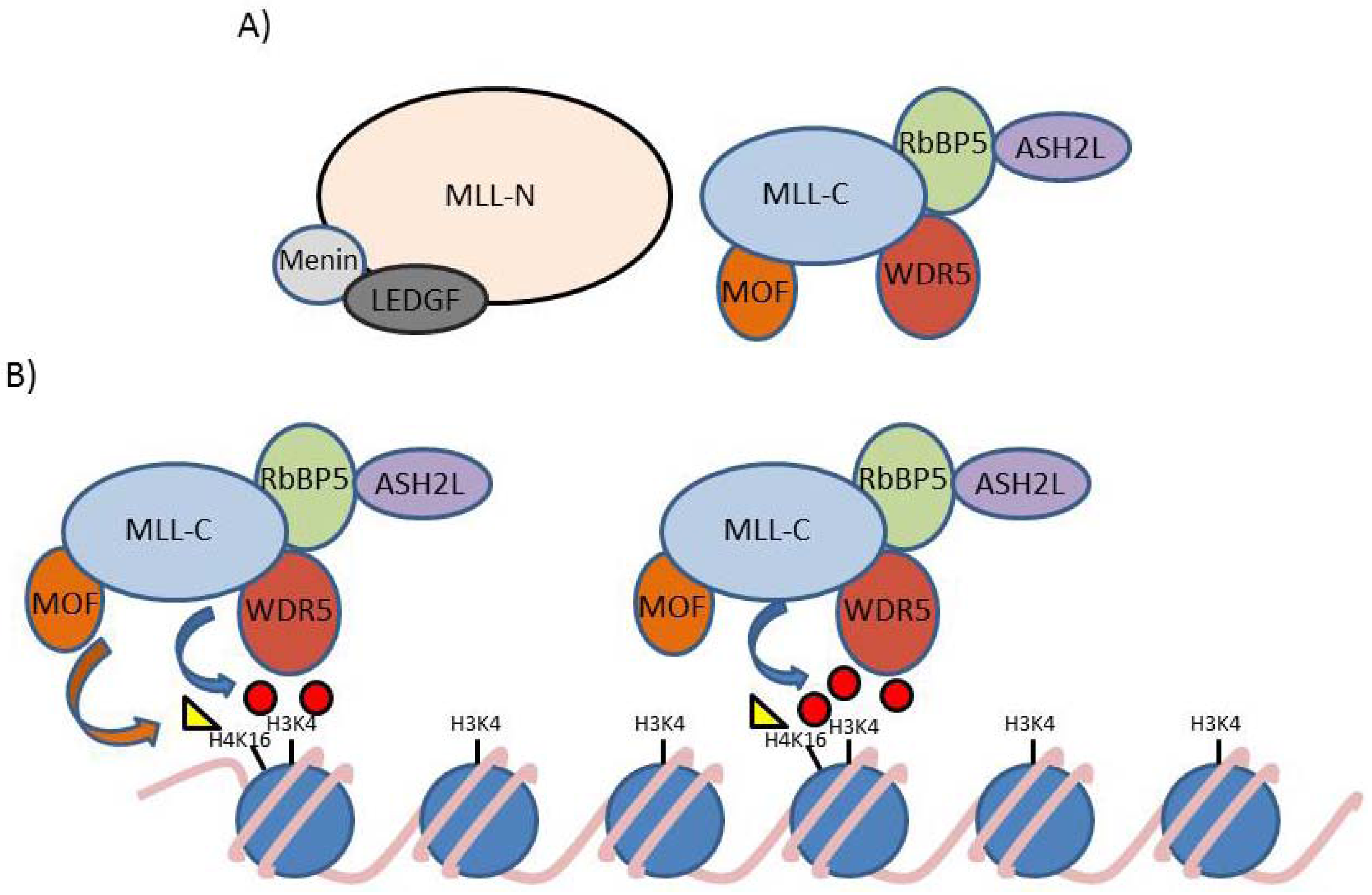
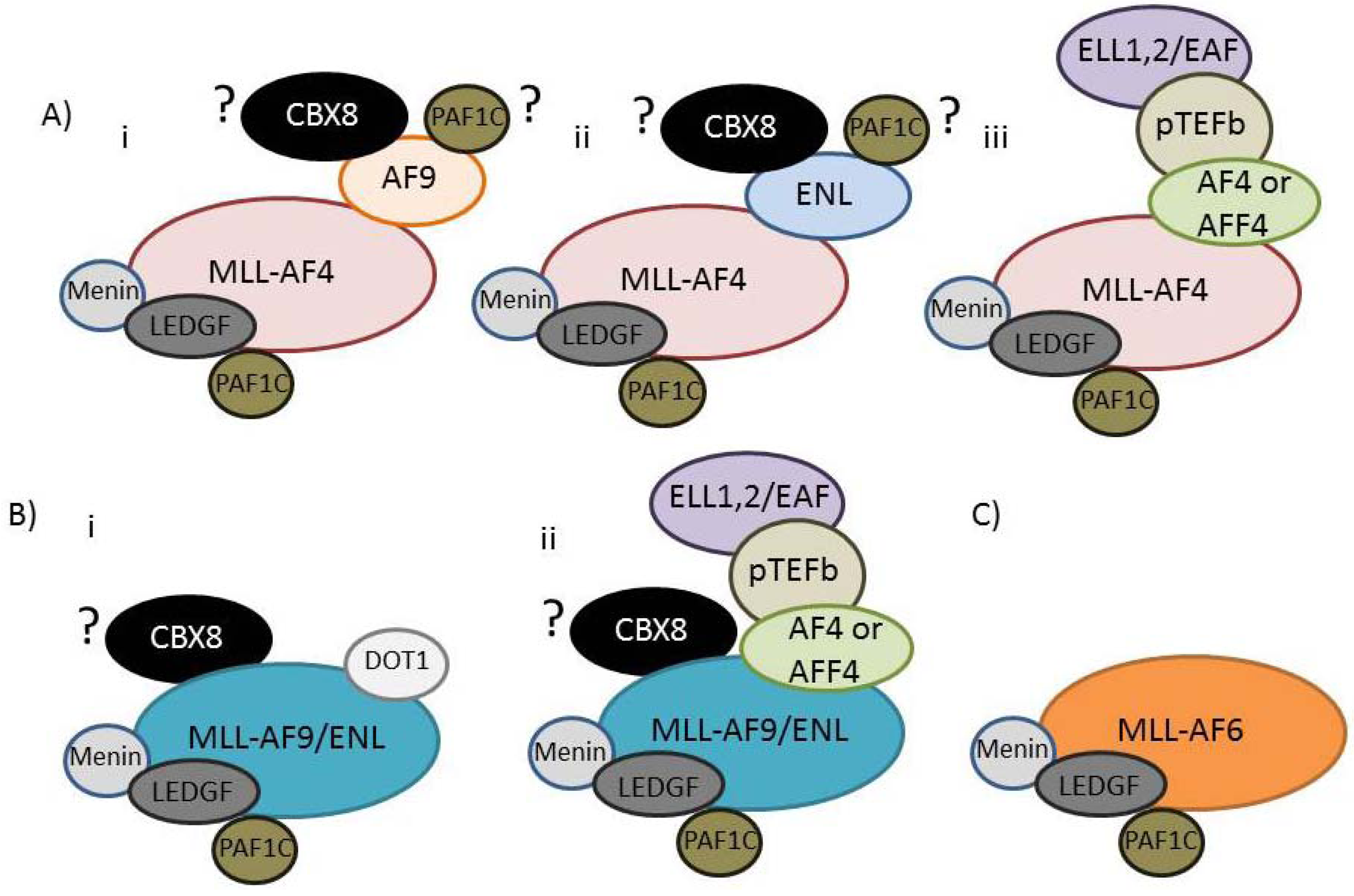
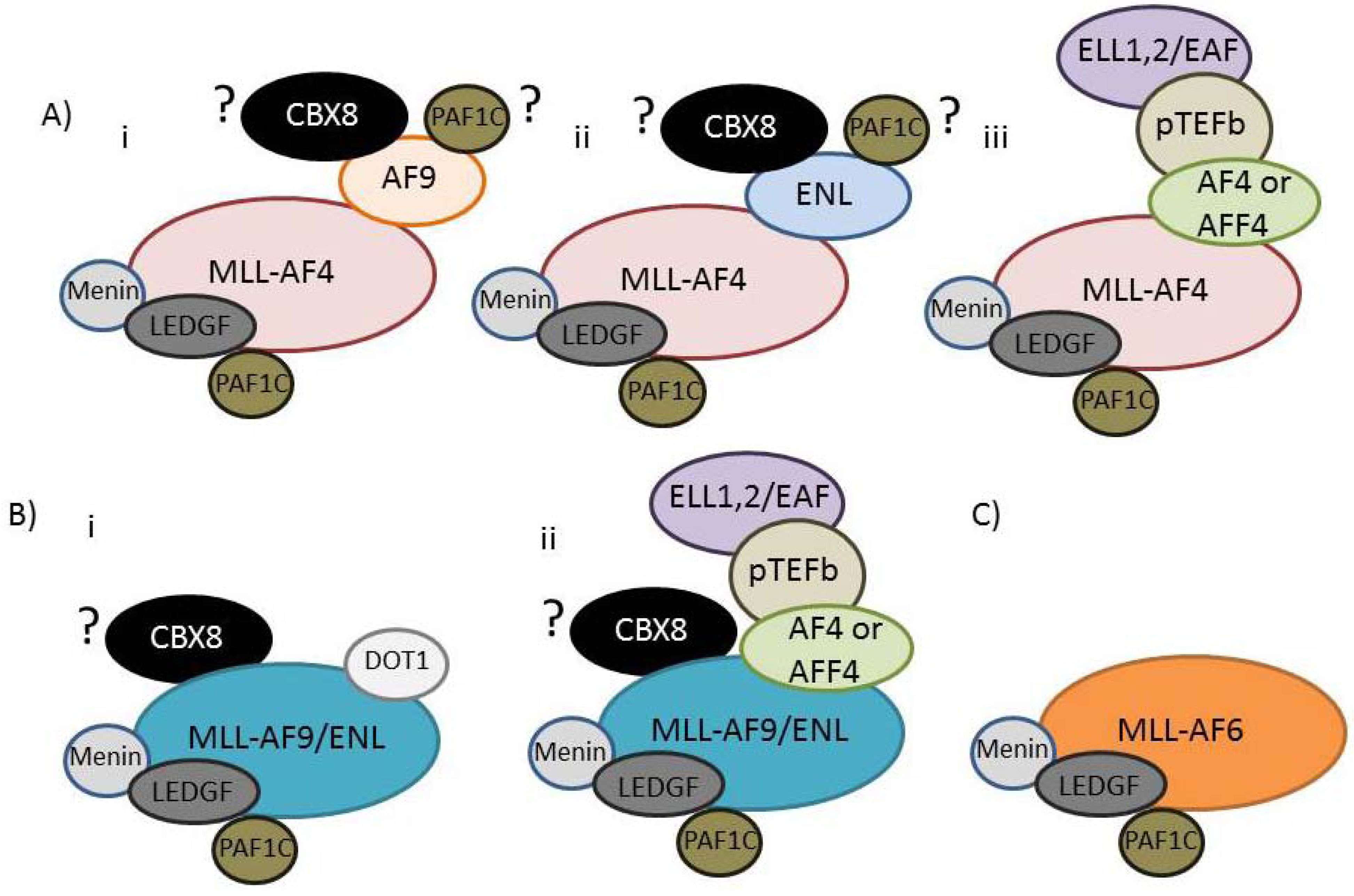
4. MLL and Leukemia
- (1)
- Which key downstream gene targets are essential for MLL-FP mediated leukemogenesis?
- (2)
- How do different MLL-FPs control epigenetic gene regulation on a molecular level?
5. Recruitment of MLL and MLL-FPs to Gene Targets

6. Important MLL-FP Regulatory Targets
6.1. Gene Targets
6.2. MicroRNA Targets
7. The MLL-FP Interactome

| NCBI Name | Aliases (commonly used aliases in bold) | Human Chromosome Position | Established Direct interactions (A question mark indicates a presumed but not fully established interaction) |
|---|---|---|---|
| MLLT3 | AF9, YEATS3 | 9p22 | DOT1L, AF4, AFF4, CBX8, PAF1, CYCT1 |
| MLLT1 | ENL, LTG19, YEATS1 | 19p13.3 | DOT1L, AF4, AFF4, CBX8, PAF1, CYCT1? |
| ELL | C19orf17, ELL1, MEN, PPP1R68 | 19p13.1 | EAF1, EAF2,ICE1, ICE2, CYCT1 |
| ELL2 | 5q15 | EAF1, EAF2,ICE1, ICE2, CYCT1 | |
| EAF1 | 3p25.1 | ELL, ELL2, MED26, CYCT1 | |
| EAF2 | BM-040, BM040, TRAITS, U19 | 3q13.33 | ELL, ELL2, MED26, CYCT1 |
| CCNT1(pTEFb) | CCNT, CYCT1, HIVE1,Cyclin T1 | 12q13.11 | CDK9, ELL, ELL2, AF4, AFF4, AF9 |
| CDK9(pTEFb) | RP11-228B15.5, C-2k, CDC2L4, CTK1, PITALRE, TAK | 9q34.1 | CYCT1 |
| AFF1 | AF4, MLLT2, PBM1 | 4q21 | AF9, ENL, CYCT1 |
| AFF2 | FMR2, FMR2P, FRAXE, MRX2, OX19 | Xq28 | N/A |
| AFF3 | LAF4, MLLT2-like | 2q11.2-q12 | N/A |
| AFF4 | HSPC092, AF5q31, MCEF | 5q31 | AF9, ENL, CYCT1 |
| DOT1L | DOT1, KMT4 | 19p13.3 | ENL, AF9 |
| CBX8 | PC3, RC1 | 17q25.3 | ENL, AF9 |
| BRD4 | CAP, HUNK1, HUNKI, MCAP | 19p13.1 | CYCT1, PAF1 complex? |
| PAF1 | 19q13.1 | ENL, AF9, BRD4? |
8. A Unifying Molecular Model for the Six Common MLL-FPs?
9. Epigenetic and Transcriptional Mechanisms of the MLL-FP Interactome
10. Therapeutic Inhibitors of MLL-FP Leukemias
11. Conclusions
Acknowledgments
References
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Berdasco, M.; Esteller, M. Aberrant epigenetic landscape in cancer: How cellular identity goes awry. Dev. Cell 2010, 19, 698–711. [Google Scholar] [CrossRef]
- Quintas-Cardama, A.; Santos, F.P.; Garcia-Manero, G. Histone deacetylase inhibitors for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Leukemia 2011, 25, 226–235. [Google Scholar] [CrossRef]
- Hock, H. A complex polycomb issue: The two faces of EZH2 in cancer. Genes Dev. 2012, 26, 751–755. [Google Scholar] [CrossRef]
- Akalin, A.; Garrett-Bakelman, F.E.; Kormaksson, M.; Busuttil, J.; Zhang, L.; Khrebtukova, I.; Milne, T.A.; Huang, Y.; Biswas, D.; Hess, J.L.; et al. Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLoS Genet. 2012, 8, e1002781. [Google Scholar]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Skrabanek, L.; Li, Y.; Jiemjit, A.; Fandy, T.E.; Paietta, E.; Fernandez, H.; Tallman, M.S.; Greally, J.M.; Carraway, H.; et al. Mds and secondary aml display unique patterns and abundance of aberrant DNA methylation. Blood 2009, 114, 3448–3458. [Google Scholar] [CrossRef]
- Figueroa, M.E.; Wouters, B.J.; Skrabanek, L.; Glass, J.; Li, Y.; Erpelinck-Verschueren, C.A.; Langerak, A.W.; Lowenberg, B.; Fazzari, M.; Greally, J.M.; et al. Genome-wide epigenetic analysis delineates a biologically distinct immature acute leukemia with myeloid/T-lymphoid features. Blood 2009, 113, 2795–2804. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Turcan, S.; Rohle, D.; Goenka, A.; Walsh, L.A.; Fang, F.; Yilmaz, E.; Campos, C.; Fabius, A.W.; Lu, C.; Ward, P.S.; et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 2012, 483, 479–483. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Feng, Z.; Lemieux, M.E.; Faber, J.; Vempati, S.; Sinha, A.U.; Xia, X.; Jesneck, J.; Bracken, A.P.; Silverman, L.B.; et al. H3k79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 2008, 14, 355–368. [Google Scholar] [CrossRef]
- Milne, T.A.; Martin, M.E.; Brock, H.W.; Slany, R.K.; Hess, J.L. Leukemogenic MLL fusion proteins bind across a broad region of the Hox a9 locus, promoting transcription and multiple histone modifications. Cancer Res. 2005, 65, 11367–11374. [Google Scholar] [CrossRef]
- Garber, K. Genetic discoveries propagate new epigenetic drugs. J. Natl. Cancer Inst. 2012, 104, 174–176. [Google Scholar] [CrossRef]
- Kwa, F.A.; Balcerczyk, A.; Licciardi, P.; El-Osta, A.; Karagiannis, T.C. Chromatin modifying agents—The cutting edge of anticancer therapy. Drug Discov. Today 2011, 16, 543–547. [Google Scholar] [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Van Holde, K.E.; Allen, J.R.; Tatchell, K.; Weischet, W.O.; Lohr, D. DNA-histone interactions in nucleosomes. Biophys. J. 1980, 32, 271–282. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Milne, T.A.; Dou, Y.; Martin, M.E.; Brock, H.W.; Roeder, R.G.; Hess, J.L. MLL associates specifically with a subset of transcriptionally active target genes. Proc. Natl. Acad. Sci. USA 2005, 102, 14765–14770. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Baker, L.A.; Allis, C.D.; Wang, G.G. PHD fingers in human diseases: Disorders arising from misinterpreting epigenetic marks. Mutat Res. 2008, 647, 3–12. [Google Scholar] [CrossRef]
- Heintzman, N.D.; Hon, G.C.; Hawkins, R.D.; Kheradpour, P.; Stark, A.; Harp, L.F.; Ye, Z.; Lee, L.K.; Stuart, R.K.; Ching, C.W.; et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 2009, 459, 108–112. [Google Scholar] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann, S.A.; Flynn, R.A.; Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Rahl, P.B.; Lin, C.Y.; Seila, A.C.; Flynn, R.A.; McCuine, S.; Burge, C.B.; Sharp, P.A.; Young, R.A. MYC regulates transcriptional pause release. Cell 2010, 141, 432–445. [Google Scholar] [CrossRef] [Green Version]
- Steger, D.J.; Lefterova, M.I.; Ying, L.; Stonestrom, A.J.; Schupp, M.; Zhuo, D.; Vakoc, A.L.; Kim, J.E.; Chen, J.; Lazar, M.A.; et al. DOT1L/KMT4 recruitment and H3K79 methylation are ubiquitously coupled with gene transcription in mammalian cells. Mol. Cell. Biol. 2008, 28, 2825–2839. [Google Scholar] [CrossRef]
- Jude, C.D.; Climer, L.; Xu, D.; Artinger, E.; Fisher, J.K.; Ernst, P. Unique and independent roles for mll in adult hematopoietic stem cells and progenitors. Cell Stem Cell 2007, 1, 324–337. [Google Scholar] [CrossRef]
- Lim, D.A.; Huang, Y.C.; Swigut, T.; Mirick, A.L.; Garcia-Verdugo, J.M.; Wysocka, J.; Ernst, P.; Alvarez-Buylla, A. Chromatin remodelling factor MLL1 is essential for neurogenesis from postnatal neural stem cells. Nature 2009, 458, 529–533. [Google Scholar] [CrossRef]
- McMahon, K.A.; Hiew, S.Y.; Hadjur, S.; Veiga-Fernandes, H.; Menzel, U.; Price, A.J.; Kioussis, D.; Williams, O.; Brady, H.J. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell 2007, 1, 338–345. [Google Scholar] [CrossRef]
- Hess, J.L. MLL: A histone methyltransferase disrupted in leukemia. Trends Mol. Med. 2004, 10, 500–507. [Google Scholar] [CrossRef]
- Milne, T.A.; Briggs, S.D.; Brock, H.W.; Martin, M.E.; Gibbs, D.; Allis, C.D.; Hess, J.L. MLL targets set domain methyltransferase activity to hox gene promoters. Mol. Cell 2002, 10, 1107–1117. [Google Scholar] [CrossRef]
- Nakamura, T.; Mori, T.; Tada, S.; Krajewski, W.; Rozovskaia, T.; Wassell, R.; Dubois, G.; Mazo, A.; Croce, C.M.; Canaani, E. All-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol. Cell 2002, 10, 1119–1128. [Google Scholar] [CrossRef]
- Marschalek, R. Mixed lineage leukemia: Roles in human malignancies and potential therapy. FEBS J. 2010, 277, 1822–1831. [Google Scholar] [CrossRef]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Rusch, M.; Wu, G.; Easton, J.; Parker, M.; Raimondi, S.; Holmfeldt, L.; et al. Whole genome sequence analysis of 22 MLL rearranged infant acute lymphoblastic leukemias reveals remarkably few somatic mutations: A report from the St. Jude Children’s Research Hospital in Washington University Pediatric Cancer Genome Project. In 53rd ASH Annual Meeting and Exposition, San Diego, CA, USA, 10-13 December 2011.
- Bardini, M.; Galbiati, M.; Lettieri, A.; Bungaro, S.; Gorletta, T.A.; Biondi, A.; Cazzaniga, G. Implementation of array based whole-genome high-resolution technologies confirms the absence of secondary copy-number alterations in MLL-AF4-positive infant all patients. Leukemia 2011, 25, 175–178. [Google Scholar] [CrossRef]
- Bardini, M.; Spinelli, R.; Bungaro, S.; Mangano, E.; Corral, L.; Cifola, I.; Fazio, G.; Giordan, M.; Basso, G.; de Rossi, G.; et al. DNA copy-number abnormalities do not occur in infant ALL with t(4;11)/MLL-AF4. Leukemia 2010, 24, 169–176. [Google Scholar]
- Djabali, M.; Selleri, L.; Parry, P.; Bower, M.; Young, B.D.; Evans, G.A. A trithorax-like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat. Genet. 1992, 2, 113–118. [Google Scholar] [CrossRef]
- Gu, Y.; Nakamura, T.; Alder, H.; Prasad, R.; Canaani, O.; Cimino, G.; Croce, C.M.; Canaani, E. The t(4;11) chromosome translocation of human acute leukemias fuses the ALL-1 gene, related to drosophila trithorax, to the AF-4 gene. Cell 1992, 71, 701–708. [Google Scholar] [CrossRef]
- Tkachuk, D.C.; Kohler, S.; Cleary, M.L. Involvement of a homolog of drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 1992, 71, 691–700. [Google Scholar] [CrossRef]
- Hanson, R.D.; Hess, J.L.; Yu, B.D.; Ernst, P.; van Lohuizen, M.; Berns, A.; van der Lugt, N.M.; Shashikant, C.S.; Ruddle, F.H.; Seto, M.; et al. Mammalian trithorax and polycomb-group homologues are antagonistic regulators of homeotic development. Proc. Natl. Acad. Sci. USA 1999, 96, 14372–14377. [Google Scholar]
- Yu, B.D.; Hanson, R.D.; Hess, J.L.; Horning, S.E.; Korsmeyer, S.J. Mll, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 10632–10636. [Google Scholar] [CrossRef]
- Yu, B.D.; Hess, J.L.; Horning, S.E.; Brown, G.A.; Korsmeyer, S.J. Altered hox expression and segmental identity in MLL-mutant mice. Nature 1995, 378, 505–508. [Google Scholar] [CrossRef]
- Krumlauf, R. Hox genes in vertebrate development. Cell 1994, 78, 191–201. [Google Scholar] [CrossRef]
- Beck, S.; Faradji, F.; Brock, H.; Peronnet, F. Maintenance of Hox gene expression patterns. Adv. Exp. Med. Biol. 2010, 689, 41–62. [Google Scholar]
- Terranova, R.; Agherbi, H.; Boned, A.; Meresse, S.; Djabali, M. Histone and DNA methylation defects at Hox genes in mice expressing a set domain-truncated form of mll. Proc. Natl. Acad. Sci. USA 2006, 103, 6629–6634. [Google Scholar] [CrossRef]
- Ernst, P.; Mabon, M.; Davidson, A.J.; Zon, L.I.; Korsmeyer, S.J. An MLL-dependent hox program drives hematopoietic progenitor expansion. Curr. Biol. 2004, 14, 2063–2069. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Allis, C.D.; Wysocka, J. Methylation of lysine 4 on histone H3: Intricacy of writing and reading a single epigenetic mark. Mol. Cell 2007, 25, 15–30. [Google Scholar]
- Goo, Y.H.; Sohn, Y.C.; Kim, D.H.; Kim, S.W.; Kang, M.J.; Jung, D.J.; Kwak, E.; Barlev, N.A.; Berger, S.L.; Chow, V.T.; et al. Activating signal cointegrator 2 belongs to a novel steady-state complex that contains a subset of trithorax group proteins. Mol. Cell. Biol. 2003, 23, 140–149. [Google Scholar]
- Glaser, S.; Schaft, J.; Lubitz, S.; Vintersten, K.; van der Hoeven, F.; Tufteland, K.R.; Aasland, R.; Anastassiadis, K.; Ang, S.L.; Stewart, A.F. Multiple epigenetic maintenance factors implicated by the loss of MLL2 in mouse development. Development 2006, 133, 1423–1432. [Google Scholar]
- Lee, S.; Lee, D.K.; Dou, Y.; Lee, J.; Lee, B.; Kwak, E.; Kong, Y.Y.; Lee, S.K.; Roeder, R.G.; Lee, J.W. Coactivator as a target gene specificity determinant for histone H3 lysine 4 methyltransferases. Proc. Natl. Acad. Sci. USA 2006, 103, 15392–15397. [Google Scholar]
- Issaeva, I.; Zonis, Y.; Rozovskaia, T.; Orlovsky, K.; Croce, C.M.; Nakamura, T.; Mazo, A.; Eisenbach, L.; Canaani, E. Knockdown of ALR (MLL2) reveals alr target genes and leads to alterations in cell adhesion and growth. Mol. Cell. Biol. 2007, 27, 1889–1903. [Google Scholar]
- Glaser, S.; Lubitz, S.; Loveland, K.L.; Ohbo, K.; Robb, L.; Schwenk, F.; Seibler, J.; Roellig, D.; Kranz, A.; Anastassiadis, K.; et al. The histone 3 lysine 4 methyltransferase, MLL2, is only required briefly in development and spermatogenesis. Epigenetics Chromatin 2009, 2, 5. [Google Scholar] [CrossRef]
- Wang, P.; Lin, C.; Smith, E.R.; Guo, H.; Sanderson, B.W.; Wu, M.; Gogol, M.; Alexander, T.; Seidel, C.; Wiedemann, L.M.; et al. Global analysis of H3K4 methylation defines MLL family member targets and points to a role for MLL1-mediated H3K4 methylation in the regulation of transcriptional initiation by rna polymerase II. Mol. Cell. Biol. 2009, 29, 6074–6085. [Google Scholar]
- Heuser, M.; Yap, D.B.; Leung, M.; de Algara, T.R.; Tafech, A.; McKinney, S.; Dixon, J.; Thresher, R.; Colledge, B.; Carlton, M.; et al. Loss of MLL5 results in pleiotropic hematopoietic defects, reduced neutrophil immune function, and extreme sensitivity to DNA demethylation. Blood 2009, 113, 1432–1443. [Google Scholar]
- Sebastian, S.; Sreenivas, P.; Sambasivan, R.; Cheedipudi, S.; Kandalla, P.; Pavlath, G.K.; Dhawan, J. MLL5, a trithorax homolog, indirectly regulates H3K4 methylation, represses cyclin a2 expression, and promotes myogenic differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 4719–4724. [Google Scholar]
- Yap, D.B.; Walker, D.C.; Prentice, L.M.; McKinney, S.; Turashvili, G.; Mooslehner-Allen, K.; de Algara, T.R.; Fee, J.; de Tassigny, X.; Colledge, W.H.; et al. MLL5 is required for normal spermatogenesis. PLoS One 2011, 6, e27127. [Google Scholar]
- Zhang, Y.; Wong, J.; Klinger, M.; Tran, M.T.; Shannon, K.M.; Killeen, N. MLL5 contributes to hematopoietic stem cell fitness and homeostasis. Blood 2009, 113, 1455–1463. [Google Scholar]
- Cimino, G.; Lo Coco, F.; Biondi, A.; Elia, L.; Luciano, A.; Croce, C.M.; Masera, G.; Mandelli, F.; Canaani, E. ALL-1 gene at chromosome 11q23 is consistently altered in acute leukemia of early infancy. Blood 1993, 82, 544–546. [Google Scholar]
- Prasad, R.; Zhadanov, A.B.; Sedkov, Y.; Bullrich, F.; Druck, T.; Rallapalli, R.; Yano, T.; Alder, H.; Croce, C.M.; Huebner, K.; et al. Structure and expression pattern of human ALR, a novel gene with strong homology to ALL-1 involved in acute leukemia and to drosophila trithorax. Oncogene 1997, 15, 549–560. [Google Scholar]
- Ng, S.B.; Bigham, A.W.; Buckingham, K.J.; Hannibal, M.C.; McMillin, M.J.; Gildersleeve, H.I.; Beck, A.E.; Tabor, H.K.; Cooper, G.M.; Mefford, H.C.; et al. Exome sequencing identifies MLL2 mutations as a cause of kabuki syndrome. Nat. Genet. 2010, 42, 790–793. [Google Scholar]
- Ruault, M.; Brun, M.E.; Ventura, M.; Roizes, G.; de Sario, A. MLL3, a new human member of the TRX/MLL gene family, maps to 7q36, a chromosome region frequently deleted in myeloid leukaemia. Gene 2002, 284, 73–81. [Google Scholar] [CrossRef]
- Tan, Y.C.; Chow, V.T. Novel human HALR (MLL3) gene encodes a protein homologous to ALR and to ALL-1 involved in leukemia, and maps to chromosome 7q36 associated with leukemia and developmental defects. Cancer Detect. Prev. 2001, 25, 454–469. [Google Scholar]
- FitzGerald, K.T.; Diaz, M.O. Mll2: A new mammalian member of the TRX/MLL family of genes. Genomics 1999, 59, 187–192. [Google Scholar] [CrossRef]
- Huntsman, D.G.; Chin, S.F.; Muleris, M.; Batley, S.J.; Collins, V.P.; Wiedemann, L.M.; Aparicio, S.; Caldas, C. MLL2, the second human homolog of the drosophila trithorax gene, maps to 19q13.1 and is amplified in solid tumor cell lines. Oncogene 1999, 18, 7975–7984. [Google Scholar]
- Bach, C.; Mueller, D.; Buhl, S.; Garcia-Cuellar, M.P.; Slany, R.K. Alterations of the CxxC domain preclude oncogenic activation of mixed-lineage leukemia 2. Oncogene 2009, 28, 815–823. [Google Scholar]
- Hughes, C.M.; Rozenblatt-Rosen, O.; Milne, T.A.; Copeland, T.D.; Levine, S.S.; Lee, J.C.; Hayes, D.N.; Shanmugam, K.S.; Bhattacharjee, A.; Biondi, C.A.; et al. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol. Cell 2004, 13, 587–597. [Google Scholar]
- Milne, T.A.; Kim, J.; Wang, G.G.; Stadler, S.C.; Basrur, V.; Whitcomb, S.J.; Wang, Z.; Ruthenburg, A.J.; Elenitoba-Johnson, K.S.; Roeder, R.G.; et al. Multiple interactions recruit MLL1 and mll1 fusion proteins to the hoxa9 locus in leukemogenesis. Mol. Cell 2010, 38, 853–863. [Google Scholar]
- Emerling, B.M.; Bonifas, J.; Kratz, C.P.; Donovan, S.; Taylor, B.R.; Green, E.D.; Le Beau, M.M.; Shannon, K.M. MLL5, a homolog of drosophila trithorax located within a segment of chromosome band 7q22 implicated in myeloid leukemia. Oncogene 2002, 21, 4849–4854. [Google Scholar] [CrossRef]
- Wysocka, J.; Myers, M.P.; Laherty, C.D.; Eisenman, R.N.; Herr, W. Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor Hcf-1. Genes Dev. 2003, 17, 896–911. [Google Scholar] [CrossRef]
- Lee, J.H.; Tate, C.M.; You, J.S.; Skalnik, D.G. Identification and characterization of the human Set1b histone H3-Lys4 methyltransferase complex. J. Biol. Chem. 2007, 282, 13419–13428. [Google Scholar] [CrossRef]
- Zeleznik-Le, N.J.; Harden, A.M.; Rowley, J.D. 11q23 translocations split the “at-hook” cruciform DNA-binding region and the transcriptional repression domain from the activation domain of the mixed-lineage leukemia (MLL) gene. Proc. Natl. Acad. Sci. USA 1994, 91, 10610–10614. [Google Scholar]
- Allen, M.D.; Grummitt, C.G.; Hilcenko, C.; Min, S.Y.; Tonkin, L.M.; Johnson, C.M.; Freund, S.M.; Bycroft, M.; Warren, A.J. Solution structure of the nonmethyl-CpG-binding CXXC domain of the leukaemia-associated MLL histone methyltransferase. EMBO J. 2006, 25, 4503–4512. [Google Scholar]
- Birke, M.; Schreiner, S.; Garcia-Cuellar, M.P.; Mahr, K.; Titgemeyer, F.; Slany, R.K. The MT domain of the proto-oncoprotein MLL binds to CpG-containing DNA and discriminates against methylation. Nucleic Acids Res. 2002, 30, 958–965. [Google Scholar] [CrossRef]
- Cierpicki, T.; Risner, L.E.; Grembecka, J.; Lukasik, S.M.; Popovic, R.; Omonkowska, M.; Shultis, D.D.; Zeleznik-Le, N.J.; Bushweller, J.H. Structure of the MLL CXXC domain-DNA complex and its functional role in MLL-AF9 leukemia. Nat. Struct Mol. Biol. 2010, 17, 62–68. [Google Scholar]
- Xu, C.; Bian, C.; Lam, R.; Dong, A.; Min, J. The structural basis for selective binding of non-methylated CpG islands by the CFP1 CXXC domain. Nat. Commun. 2011, 2, 227. [Google Scholar] [CrossRef]
- Muntean, A.G.; Tan, J.; Sitwala, K.; Huang, Y.; Bronstein, J.; Connelly, J.A.; Basrur, V.; Elenitoba-Johnson, K.S.; Hess, J.L. The PAF complex synergizes with MLL fusion proteins at Hox Loci to promote leukemogenesis. Cancer Cell 2010, 17, 609–621. [Google Scholar] [CrossRef]
- Fair, K.; Anderson, M.; Bulanova, E.; Mi, H.; Tropschug, M.; Diaz, M.O. Protein interactions of the MLL PHD fingers modulate MLL target gene regulation in human cells. Mol. Cell. Biol. 2001, 21, 3589–3597. [Google Scholar] [CrossRef]
- Hom, R.A.; Chang, P.Y.; Roy, S.; Musselman, C.A.; Glass, K.C.; Selezneva, A.I.; Gozani, O.; Ismagilov, R.F.; Cleary, M.L.; Kutateladze, T.G. Molecular mechanism of MLL PHD3 and RNA recognition by the Cyp33 RRM domain. J. Mol. Biol. 2010, 400, 145–154. [Google Scholar] [CrossRef]
- Park, S.; Osmers, U.; Raman, G.; Schwantes, R.H.; Diaz, M.O.; Bushweller, J.H. The PHD3 domain of MLL acts as a Cyp33-regulated switch between MLL-mediated activation and repression. Biochemistry 2010, 49, 6576–6586. [Google Scholar]
- Wang, Z.; Song, J.; Milne, T.A.; Wang, G.G.; Li, H.; Allis, C.D.; Patel, D.J. Pro isomerization in MLL1 PHD3-bromo cassette connects H3K4me readout to Cyp33 and HDAC-mediated repression. Cell 2010, 141, 1183–1194. [Google Scholar] [CrossRef]
- Xia, Z.B.; Anderson, M.; Diaz, M.O.; Zeleznik-Le, N.J. MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc. Natl. Acad. Sci. USA 2003, 100, 8342–8347. [Google Scholar]
- Chang, P.Y.; Hom, R.A.; Musselman, C.A.; Zhu, L.; Kuo, A.; Gozani, O.; Kutateladze, T.G.; Cleary, M.L. Binding of the MLL PHD3 finger to histone H3K4me3 is required for Mll-dependent gene transcription. J. Mol. Biol. 2010, 400, 137–144. [Google Scholar] [CrossRef]
- Wang, J.; Muntean, A.G.; Hess, J.L. ECSASB2 mediates MLL degradation during hematopoietic differentiation. Blood 2012, 119, 1151–1161. [Google Scholar] [CrossRef]
- Yokoyama, A.; Somervaille, T.C.; Smith, K.S.; Rozenblatt-Rosen, O.; Meyerson, M.; Cleary, M.L. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 2005, 123, 207–218. [Google Scholar] [CrossRef]
- Grembecka, J.; Belcher, A.M.; Hartley, T.; Cierpicki, T. Molecular basis of the mixed lineage leukemia-menin interaction: Implications for targeting mixed lineage leukemias. J. Biol. Chem. 2010, 285, 40690–40698. [Google Scholar] [CrossRef]
- Grembecka, J.; He, S.; Shi, A.; Purohit, T.; Muntean, A.G.; Sorenson, R.J.; Showalter, H.D.; Murai, M.J.; Belcher, A.M.; Hartley, T.; et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat. Chem. Biol. 2012, 8, 277–284. [Google Scholar]
- Murai, M.J.; Chruszcz, M.; Reddy, G.; Grembecka, J.; Cierpicki, T. Crystal structure of menin reveals binding site for mixed lineage leukemia (MLL) protein. J. Biol. Chem. 2011, 286, 31742–31748. [Google Scholar]
- Huang, J.; Gurung, B.; Wan, B.; Matkar, S.; Veniaminova, N.A.; Wan, K.; Merchant, J.L.; Hua, X.; Lei, M. The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 2012, 482, 542–546. [Google Scholar] [CrossRef]
- Yokoyama, A.; Cleary, M.L. Menin critically links MLL proteins with LEDGF on cancer-associated target genes. Cancer Cell 2008, 14, 36–46. [Google Scholar] [CrossRef]
- Yokoyama, A.; Wang, Z.; Wysocka, J.; Sanyal, M.; Aufiero, D.J.; Kitabayashi, I.; Herr, W.; Cleary, M.L. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate hox gene expression. Mol. Cell. Biol. 2004, 24, 5639–5649. [Google Scholar] [CrossRef]
- Ernst, P.; Wang, J.; Huang, M.; Goodman, R.H.; Korsmeyer, S.J. MLL and CREB bind cooperatively to the nuclear coactivator CREB-binding protein. Mol. Cell. Biol. 2001, 21, 2249–2258. [Google Scholar] [CrossRef]
- Dou, Y.; Milne, T.A.; Tackett, A.J.; Smith, E.R.; Fukuda, A.; Wysocka, J.; Allis, C.D.; Chait, B.T.; Hess, J.L.; Roeder, R.G. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 2005, 121, 873–885. [Google Scholar] [CrossRef]
- Dou, Y.; Milne, T.A.; Ruthenburg, A.J.; Lee, S.; Lee, J.W.; Verdine, G.L.; Allis, C.D.; Roeder, R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006, 13, 713–719. [Google Scholar] [CrossRef]
- Patel, A.; Dharmarajan, V.; Vought, V.E.; Cosgrove, M.S. On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J. Biol. Chem. 2009, 284, 24242–24256. [Google Scholar]
- Han, Z.; Guo, L.; Wang, H.; Shen, Y.; Deng, X.W.; Chai, J. Structural basis for the specific recognition of methylated histone H3 lysine 4 by the WD-40 protein WDR5. Mol. Cell 2006, 22, 137–144. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Wang, W.; Graybosch, D.M.; Li, H.; Allis, C.D.; Patel, D.J.; Verdine, G.L. Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat. Struct. Mol. Biol. 2006, 13, 704–712. [Google Scholar] [CrossRef]
- Schuetz, A.; Allali-Hassani, A.; Martin, F.; Loppnau, P.; Vedadi, M.; Bochkarev, A.; Plotnikov, A.N.; Arrowsmith, C.H.; Min, J. Structural basis for molecular recognition and presentation of histone H3 by WDR5. EMBO J. 2006, 25, 4245–4252. [Google Scholar]
- Patel, A.; Dharmarajan, V.; Cosgrove, M.S. Structure of WDR5 bound to mixed lineage leukemia protein-1 peptide. J. Biol. Chem. 2008, 283, 32158–32161. [Google Scholar]
- Patel, A.; Vought, V.E.; Dharmarajan, V.; Cosgrove, M.S. A conserved arginine-containing motif crucial for the assembly and enzymatic activity of the mixed lineage leukemia protein-1 core complex. J. Biol. Chem. 2008, 283, 32162–32175. [Google Scholar] [CrossRef]
- Song, J.J.; Kingston, R.E. WDR5 interacts with mixed lineage leukemia (MLL) protein via the histone H3-binding pocket. J. Biol. Chem. 2008, 283, 35258–35264. [Google Scholar] [CrossRef]
- Karatas, H.; Townsend, E.C.; Bernard, D.; Dou, Y.; Wang, S. Analysis of the binding of mixed lineage leukemia 1 (Mll1) and histone 3 peptides to WD repeat domain 5 (WDR5) for the design of inhibitors of the MLL1-WDR5 interaction. J. Med. Chem. 2010, 53, 5179–5185. [Google Scholar] [CrossRef]
- Odho, Z.; Southall, S.M.; Wilson, J.R. Characterization of a novel WDR5-binding site that recruits RBBP5 through a conserved motif to enhance methylation of histone H3 lysine 4 by mixed lineage leukemia protein-1. J. Biol. Chem. 2010, 285, 32967–32976. [Google Scholar]
- Avdic, V.; Zhang, P.; Lanouette, S.; Groulx, A.; Tremblay, V.; Brunzelle, J.; Couture, J.F. Structural and biochemical insights into MLL1 core complex assembly. Structure 2011, 19, 101–108. [Google Scholar] [CrossRef]
- Takahashi, Y.H.; Westfield, G.H.; Oleskie, A.N.; Trievel, R.C.; Shilatifard, A.; Skiniotis, G. Structural analysis of the core compass family of histone H3K4 methylases from yeast to human. Proc. Natl. Acad. Sci. USA 2011, 108, 20526–20531. [Google Scholar]
- Cao, F.; Chen, Y.; Cierpicki, T.; Liu, Y.; Basrur, V.; Lei, M.; Dou, Y. An Ash2l/RbBP5 heterodimer stimulates the MLL1 methyltransferase activity through coordinated substrate interactions with the MLL1 set domain. PLoS One 2010, 5, e14102. [Google Scholar]
- Southall, S.M.; Wong, P.S.; Odho, Z.; Roe, S.M.; Wilson, J.R. Structural basis for the requirement of additional factors for MLL1 set domain activity and recognition of epigenetic marks. Mol. Cell 2009, 33, 181–191. [Google Scholar] [CrossRef]
- Steward, M.M.; Lee, J.S.; O’Donovan, A.; Wyatt, M.; Bernstein, B.E.; Shilatifard, A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat. Struct. Mol. Biol. 2006, 13, 852–854. [Google Scholar] [CrossRef]
- Patel, A.; Vought, V.E.; Dharmarajan, V.; Cosgrove, M.S. A novel non-set domain multi-subunit methyltransferase required for sequential nucleosomal histone H3 methylation by the mixed lineage leukemia protein-1 (MLL1) core complex. J. Biol. Chem. 2011, 286, 3359–3369. [Google Scholar] [CrossRef]
- Wang, X.; Lou, Z.; Dong, X.; Yang, W.; Peng, Y.; Yin, B.; Gong, Y.; Yuan, J.; Zhou, W.; Bartlam, M.; et al. Crystal structure of the C-terminal domain of human DPY-30-like protein: A component of the histone methyltransferase complex. J. Mol. Biol. 2009, 390, 530–537. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Ernst, P.; Erdjument-Bromage, H.; Tempst, P.; Korsmeyer, S.J. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol. Cell. Biol. 2003, 23, 186–194. [Google Scholar] [CrossRef]
- Yokoyama, A.; Kitabayashi, I.; Ayton, P.M.; Cleary, M.L.; Ohki, M. Leukemia proto-oncoprotein MLL is proteolytically processed into 2 fragments with opposite transcriptional properties. Blood 2002, 100, 3710–3718. [Google Scholar] [CrossRef]
- Wysocka, J.; Swigut, T.; Milne, T.A.; Dou, Y.; Zhang, X.; Burlingame, A.L.; Roeder, R.G.; Brivanlou, A.H.; Allis, C.D. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 2005, 121, 859–872. [Google Scholar] [CrossRef]
- Milne, T.A.; Hughes, C.M.; Lloyd, R.; Yang, Z.; Rozenblatt-Rosen, O.; Dou, Y.; Schnepp, R.W.; Krankel, C.; Livolsi, V.A.; Gibbs, D.; et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. USA 2005, 102, 749–754. [Google Scholar]
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Collins, F.S.; Emmert-Buck, M.R.; Debelenko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A.; et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276, 404–407. [Google Scholar]
- Caslini, C.; Yang, Z.; El-Osta, M.; Milne, T.A.; Slany, R.K.; Hess, J.L. Interaction of mll amino terminal sequences with menin is required for transformation. Cancer Res. 2007, 67, 7275–7283. [Google Scholar]
- Onodera, A.; Yamashita, M.; Endo, Y.; Kuwahara, M.; Tofukuji, S.; Hosokawa, H.; Kanai, A.; Suzuki, Y.; Nakayama, T. STAT6-mediated displacement of polycomb by trithorax complex establishes long-term maintenance of GATA3 expression in t helper type 2 cells. J. Exp. Med. 2010, 207, 2493–2506. [Google Scholar] [CrossRef]
- Thiel, A.T.; Blessington, P.; Zou, T.; Feather, D.; Wu, X.; Yan, J.; Zhang, H.; Liu, Z.; Ernst, P.; Koretzky, G.A.; et al. MLL-AF9-induced leukemogenesis requires coexpression of the wild-type MLL allele. Cancer Cell 2010, 17, 148–159. [Google Scholar] [CrossRef]
- Schoch, C.; Schnittger, S.; Klaus, M.; Kern, W.; Hiddemann, W.; Haferlach, T. AML with 11q23/MLL abnormalities as defined by the who classification: Incidence, partner chromosomes, fab subtype, age distribution, and prognostic impact in an unselected series of 1897 cytogenetically analyzed aml cases. Blood 2003, 102, 2395–2402. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Armstrong, S.A. MLL translocations, histone modifications and leukaemia stem-cell development. Nat. Rev. Cancer 2007, 7, 823–833. [Google Scholar] [CrossRef]
- Muntean, A.G.; Hess, J.L. The pathogenesis of mixed-lineage leukemia. Annu. Rev. Pathol. 2012, 7, 283–301. [Google Scholar] [CrossRef]
- Dorrance, A.M.; Liu, S.; Yuan, W.; Becknell, B.; Arnoczky, K.J.; Guimond, M.; Strout, M.P.; Feng, L.; Nakamura, T.; Yu, L.; et al. MLL partial tandem duplication induces aberrant hox expression in vivo via specific epigenetic alterations. J. Clin. Invest. 2006, 116, 2707–2716. [Google Scholar] [CrossRef]
- Betz, B.L.; Hess, J.L. Acute myeloid leukemia diagnosis in the 21st century. Arch. Pathol. Lab. Med. 2010, 134, 1427–1433. [Google Scholar]
- Zorko, N.A.; Bernot, K.M.; Whitman, S.P.; Siebenaler, R.F.; Ahmed, E.H.; Marcucci, G.G.; Yanes, D.A.; McConnell, K.K.; Mao, C.; Kalu, C.; et al. MLL-partial tandem duplication and Flt3-internal tandem duplication in a double knock-in mouse recapitulates features of counterpart human acute myeloid leukemias. Blood 2012, 120, 1130–1136. [Google Scholar] [CrossRef]
- Hu, Z.; Li, X.M.; Jorgensen, M.L.; Slayton, W.B. MLL/AF-4 leukemic cells recruit new blood vessels but do not incorporate into capillaries in culture or in a NOD/SCID xenograft model. Leukemia 2009, 23, 990–993. [Google Scholar] [CrossRef]
- Henderson, M.J.; Choi, S.; Beesley, A.H.; Baker, D.L.; Wright, D.; Papa, R.A.; Murch, A.; Campbell, L.J.; Lock, R.B.; Norris, M.D.; et al. A xenograft model of infant leukaemia reveals a complex MLL translocation. Br. J. Haematol. 2008, 140, 716–719. [Google Scholar] [CrossRef]
- Rodriguez-Perales, S.; Cano, F.; Lobato, M.N.; Rabbitts, T.H. MLL gene fusions in human leukaemias: In vivo modelling to recapitulate these primary tumourigenic events. Int. J. Hematol. 2008, 87, 3–9. [Google Scholar] [CrossRef]
- Corral, J.; Lavenir, I.; Impey, H.; Warren, A.J.; Forster, A.; Larson, T.A.; Bell, S.; McKenzie, A.N.; King, G.; Rabbitts, T.H. An MLL-AF9 fusion gene made by homologous recombination causes acute leukemia in chimeric mice: A method to create fusion oncogenes. Cell 1996, 85, 853–861. [Google Scholar] [CrossRef]
- Somervaille, T.C.; Cleary, M.L. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006, 10, 257–268. [Google Scholar] [CrossRef]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. The wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science 2010, 327, 1650–1653. [Google Scholar] [CrossRef]
- Kumar, A.R.; Hudson, W.A.; Chen, W.; Nishiuchi, R.; Yao, Q.; Kersey, J.H. Hoxa9 influences the phenotype but not the incidence of MLL-AF9 fusion gene leukemia. Blood 2004, 103, 1823–1828. [Google Scholar] [CrossRef]
- Chen, W.; Kumar, A.R.; Hudson, W.A.; Li, Q.; Wu, B.; Staggs, R.A.; Lund, E.A.; Sam, T.N.; Kersey, J.H. Malignant transformation initiated by MLL-AF9: Gene dosage and critical target cells. Cancer Cell 2008, 13, 432–440. [Google Scholar] [CrossRef]
- So, C.W.; Cleary, M.L. MLL-AFX requires the transcriptional effector domains of AFX to transform myeloid progenitors and transdominantly interfere with forkhead protein function. Mol. Cell. Biol. 2002, 22, 6542–6552. [Google Scholar] [CrossRef]
- So, C.W.; Cleary, M.L. Common mechanism for oncogenic activation of mll by forkhead family proteins. Blood 2003, 101, 633–639. [Google Scholar] [CrossRef]
- So, C.W.; Karsunky, H.; Passegue, E.; Cozzio, A.; Weissman, I.L.; Cleary, M.L. MLL-GAS7 transforms multipotent hematopoietic progenitors and induces mixed lineage leukemias in mice. Cancer Cell 2003, 3, 161–171. [Google Scholar] [CrossRef]
- So, C.W.; Karsunky, H.; Wong, P.; Weissman, I.L.; Cleary, M.L. Leukemic transformation of hematopoietic progenitors by MLL-GAS7 in the absence of Hoxa7 or Hoxa9. Blood 2004, 103, 3192–3199. [Google Scholar] [CrossRef]
- So, C.W.; Lin, M.; Ayton, P.M.; Chen, E.H.; Cleary, M.L. Dimerization contributes to oncogenic activation of MLL chimeras in acute leukemias. Cancer Cell 2003, 4, 99–110. [Google Scholar] [CrossRef]
- Yeung, J.; Esposito, M.T.; Gandillet, A.; Zeisig, B.B.; Griessinger, E.; Bonnet, D.; So, C.W. Beta-catenin mediates the establishment and drug resistance of MLL leukemic stem cells. Cancer Cell 2010, 18, 606–618. [Google Scholar] [CrossRef]
- Balgobind, B.V.; Raimondi, S.C.; Harbott, J.; Zimmermann, M.; Alonzo, T.A.; Auvrignon, A.; Beverloo, H.B.; Chang, M.; Creutzig, U.; Dworzak, M.N.; et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: Results of an international retrospective study. Blood 2009, 114, 2489–2496. [Google Scholar]
- Pigazzi, M.; Masetti, R.; Bresolin, S.; Beghin, A.; di Meglio, A.; Gelain, S.; Trentin, L.; Baron, E.; Giordan, M.; Zangrando, A.; et al. MLL partner genes drive distinct gene expression profiles and genomic alterations in pediatric acute myeloid leukemia: An AIEOP study. Leukemia 2011, 25, 560–563. [Google Scholar] [CrossRef]
- Pui, C.H.; Carroll, W.L.; Meshinchi, S.; Arceci, R.J. Biology, risk stratification, and therapy of pediatric acute leukemias: An update. J. Clin. Oncol. 2011, 29, 551–565. [Google Scholar] [CrossRef]
- Dobson, C.L.; Warren, A.J.; Pannell, R.; Forster, A.; Rabbitts, T.H. Tumorigenesis in mice with a fusion of the leukaemia oncogene mll and the bacterial lacz gene. EMBO J. 2000, 19, 843–851. [Google Scholar] [CrossRef]
- Martin, M.E.; Milne, T.A.; Bloyer, S.; Galoian, K.; Shen, W.; Gibbs, D.; Brock, H.W.; Slany, R.; Hess, J.L. Dimerization of MLL fusion proteins immortalizes hematopoietic cells. Cancer Cell 2003, 4, 197–207. [Google Scholar] [CrossRef]
- Meyer, C.; Kowarz, E.; Hofmann, J.; Renneville, A.; Zuna, J.; Trka, J.; Ben Abdelali, R.; Macintyre, E.; de Braekeleer, E.; de Braekeleer, M.; et al. New insights to the mll recombinome of acute leukemias. Leukemia 2009, 23, 1490–1499. [Google Scholar] [CrossRef]
- Bursen, A.; Schwabe, K.; Ruster, B.; Henschler, R.; Ruthardt, M.; Dingermann, T.; Marschalek, R. The AF4.MLL fusion protein is capable of inducing all in mice without requirement of MLL.AF4. Blood 2010, 115, 3570–3579. [Google Scholar]
- Benedikt, A.; Baltruschat, S.; Scholz, B.; Bursen, A.; Arrey, T.N.; Meyer, B.; Varagnolo, L.; Muller, A.M.; Karas, M.; Dingermann, T.; et al. The leukemogenic AF4-MLL fusion protein causes P-TEFB kinase activation and altered epigenetic signatures. Leukemia 2011, 25, 135–144. [Google Scholar] [CrossRef]
- Thomas, M.; Gessner, A.; Vornlocher, H.P.; Hadwiger, P.; Greil, J.; Heidenreich, O. Targeting MLL-AF4 with short interfering rnas inhibits clonogenicity and engraftment of t(4;11)-positive human leukemic cells. Blood 2005, 106, 3559–3566. [Google Scholar] [CrossRef]
- Kowarz, E.; Burmeister, T.; Lo Nigro, L.; Jansen, M.W.; Delabesse, E.; Klingebiel, T.; Dingermann, T.; Meyer, C.; Marschalek, R. Complex MLL rearrangements in t(4;11) leukemia patients with absent AF4.MLL fusion allele. Leukemia 2007, 21, 1232–1238. [Google Scholar] [CrossRef]
- Greaves, M.F.; Maia, A.T.; Wiemels, J.L.; Ford, A.M. Leukemia in twins: Lessons in natural history. Blood 2003, 102, 2321–2333. [Google Scholar] [CrossRef]
- Liu, H.; Takeda, S.; Kumar, R.; Westergard, T.D.; Brown, E.J.; Pandita, T.K.; Cheng, E.H.; Hsieh, J.J. Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature 2010, 467, 343–346. [Google Scholar]
- Takacova, S.; Slany, R.; Bartkova, J.; Stranecky, V.; Dolezel, P.; Luzna, P.; Bartek, J.; Divoky, V. DNA damage response and inflammatory signaling limit the MLL-ENL-induced leukemogenesis in vivo. Cancer Cell 2012, 21, 517–531. [Google Scholar] [CrossRef]
- Collins, E.C.; Pannell, R.; Simpson, E.M.; Forster, A.; Rabbitts, T.H. Inter-chromosomal recombination of MLL and AF9 genes mediated by cre-loxP in mouse development. EMBO Rep. 2000, 1, 127–132. [Google Scholar] [CrossRef]
- Forster, A.; Pannell, R.; Drynan, L.F.; McCormack, M.; Collins, E.C.; Daser, A.; Rabbitts, T.H. Engineering de novo reciprocal chromosomal translocations associated with MLL to replicate primary events of human cancer. Cancer Cell 2003, 3, 449–458. [Google Scholar] [CrossRef]
- Drynan, L.F.; Pannell, R.; Forster, A.; Chan, N.M.; Cano, F.; Daser, A.; Rabbitts, T.H. MLL fusions generated by Cre-loxP-mediated de novo translocations can induce lineage reassignment in tumorigenesis. EMBO J. 2005, 24, 3136–3146. [Google Scholar] [CrossRef]
- Metzler, M.; Forster, A.; Pannell, R.; Arends, M.J.; Daser, A.; Lobato, M.N.; Rabbitts, T.H. A conditional model of MLL-AF4 B-cell tumourigenesis using invertor technology. Oncogene 2006, 25, 3093–3103. [Google Scholar] [CrossRef]
- Cano, F.; Drynan, L.F.; Pannell, R.; Rabbitts, T.H. Leukaemia lineage specification caused by cell-specific MLL-ENL translocations. Oncogene 2008, 27, 1945–1950. [Google Scholar] [CrossRef]
- Ernst, P.; Fisher, J.K.; Avery, W.; Wade, S.; Foy, D.; Korsmeyer, S.J. Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev. Cell 2004, 6, 437–443. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Li, H.; Patel, D.J.; Allis, C.D. Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell Biol. 2007, 8, 983–994. [Google Scholar] [CrossRef]
- Bartke, T.; Vermeulen, M.; Xhemalce, B.; Robson, S.C.; Mann, M.; Kouzarides, T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 2010, 143, 470–484. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Li, H.; Milne, T.A.; Dewell, S.; McGinty, R.K.; Yuen, M.; Ueberheide, B.; Dou, Y.; Muir, T.W.; Patel, D.J.; et al. Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell 2011, 145, 692–706. [Google Scholar] [CrossRef]
- Kim, J.; Guermah, M.; Roeder, R.G. The human PAF1 complex acts in chromatin transcription elongation both independently and cooperatively with SII/TFIIS. Cell 2010, 140, 491–503. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Staunton, J.E.; Silverman, L.B.; Pieters, R.; den Boer, M.L.; Minden, M.D.; Sallan, S.E.; Lander, E.S.; Golub, T.R.; Korsmeyer, S.J. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 2002, 30, 41–47. [Google Scholar] [CrossRef]
- Ferrando, A.A.; Armstrong, S.A.; Neuberg, D.S.; Sallan, S.E.; Silverman, L.B.; Korsmeyer, S.J.; Look, A.T. Gene expression signatures in MLL-rearranged t-lineage and b-precursor acute leukemias: Dominance of HOX dysregulation. Blood 2003, 102, 262–268. [Google Scholar] [CrossRef]
- Rozovskaia, T.; Feinstein, E.; Mor, O.; Foa, R.; Blechman, J.; Nakamura, T.; Croce, C.M.; Cimino, G.; Canaani, E. Upregulation of Meis1 and HoxA9 in acute lymphocytic leukemias with the t(4:11) abnormality. Oncogene 2001, 20, 874–878. [Google Scholar] [CrossRef]
- Yeoh, E.J.; Ross, M.E.; Shurtleff, S.A.; Williams, W.K.; Patel, D.; Mahfouz, R.; Behm, F.G.; Raimondi, S.C.; Relling, M.V.; Patel, A.; et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell 2002, 1, 133–143. [Google Scholar] [CrossRef]
- Kroon, E.; Krosl, J.; Thorsteinsdottir, U.; Baban, S.; Buchberg, A.M.; Sauvageau, G. Hoxa9 transforms primary bone marrow cells through specific collaboration with Meis1a but not Pbx1b. EMBO J. 1998, 17, 3714–3725. [Google Scholar] [CrossRef]
- Thorsteinsdottir, U.; Kroon, E.; Jerome, L.; Blasi, F.; Sauvageau, G. Defining roles for HOX and MEIS1 genes in induction of acute myeloid leukemia. Mol. Cell. Biol. 2001, 21, 224–234. [Google Scholar] [CrossRef]
- Zeisig, B.B.; Milne, T.; Garcia-Cuellar, M.P.; Schreiner, S.; Martin, M.E.; Fuchs, U.; Borkhardt, A.; Chanda, S.K.; Walker, J.; Soden, R.; et al. Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol. Cell. Biol. 2004, 24, 617–628. [Google Scholar] [CrossRef]
- Ayton, P.M.; Cleary, M.L. Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9. Genes Dev. 2003, 17, 2298–2307. [Google Scholar] [CrossRef]
- Faber, J.; Krivtsov, A.V.; Stubbs, M.C.; Wright, R.; Davis, T.N.; van den Heuvel-Eibrink, M.; Zwaan, C.M.; Kung, A.L.; Armstrong, S.A. Hoxa9 is required for survival in human MLL-rearranged acute leukemias. Blood 2009, 113, 2375–2385. [Google Scholar] [CrossRef]
- Orlovsky, K.; Kalinkovich, A.; Rozovskaia, T.; Shezen, E.; Itkin, T.; Alder, H.; Ozer, H.G.; Carramusa, L.; Avigdor, A.; Volinia, S.; et al. Down-regulation of homeobox genes Meis1 and hoxa in MLL-rearranged acute leukemia impairs engraftment and reduces proliferation. Proc. Natl. Acad. Sci. USA 2011, 108, 7956–7961. [Google Scholar]
- Ross, M.E.; Mahfouz, R.; Onciu, M.; Liu, H.C.; Zhou, X.; Song, G.; Shurtleff, S.A.; Pounds, S.; Cheng, C.; Ma, J.; et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood 2004, 104, 3679–3687. [Google Scholar] [CrossRef]
- Stam, R.W.; Schneider, P.; Hagelstein, J.A.; van der Linden, M.H.; Stumpel, D.J.; de Menezes, R.X.; de Lorenzo, P.; Valsecchi, M.G.; Pieters, R. Gene expression profiling-based dissection of MLL translocated and MLL germline acute lymphoblastic leukemia in infants. Blood 2010, 115, 2835–2844. [Google Scholar]
- Trentin, L.; Giordan, M.; Dingermann, T.; Basso, G.; Te Kronnie, G.; Marschalek, R. Two independent gene signatures in pediatric t(4;11) acute lymphoblastic leukemia patients. Eur. J. Haematol. 2009, 83, 406–419. [Google Scholar] [CrossRef]
- Kumar, A.R.; Li, Q.; Hudson, W.A.; Chen, W.; Sam, T.; Yao, Q.; Lund, E.A.; Wu, B.; Kowal, B.J.; Kersey, J.H. A role for Meis1 in MLL-fusion gene leukemia. Blood 2009, 113, 1756–1758. [Google Scholar] [CrossRef]
- Wang, Q.F.; Wu, G.; Mi, S.; He, F.; Wu, J.; Dong, J.; Luo, R.T.; Mattison, R.; Kaberlein, J.J.; Prabhakar, S.; et al. MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood 2011, 117, 6895–6905. [Google Scholar] [CrossRef]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006, 442, 818–822. [Google Scholar]
- Schwieger, M.; Schuler, A.; Forster, M.; Engelmann, A.; Arnold, M.A.; Delwel, R.; Valk, P.J.; Lohler, J.; Slany, R.K.; Olson, E.N.; et al. Homing and invasiveness of MLL/ENL leukemic cells is regulated by MEF2C. Blood 2009, 114, 2476–2488. [Google Scholar]
- Kuipers, J.E.; Coenen, E.A.; Balgobind, B.V.; Stary, J.; Baruchel, A.; de Haas, V.; de Bont, E.S.; Reinhardt, D.; Kaspers, G.J.; Cloos, J.; et al. High IGSF4 expression in pediatric M5 acute myeloid leukemia with t(9;11)(p22;q23). Blood 2011, 117, 928–935. [Google Scholar]
- Hess, J.L.; Bittner, C.B.; Zeisig, D.T.; Bach, C.; Fuchs, U.; Borkhardt, A.; Frampton, J.; Slany, R.K. C-myb is an essential downstream target for homeobox-mediated transformation of hematopoietic cells. Blood 2006, 108, 297–304. [Google Scholar] [CrossRef]
- Zuber, J.; Rappaport, A.R.; Luo, W.; Wang, E.; Chen, C.; Vaseva, A.V.; Shi, J.; Weissmueller, S.; Fellmann, C.; Taylor, M.J.; et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev. 2011, 25, 1628–1640. [Google Scholar] [CrossRef]
- Mak, A.B.; Nixon, A.M.; Moffat, J. The mixed lineage leukemia (MLL) fusion-associated gene AF4 promotes CD133 transcription. Cancer Res. 2012, 72, 1929–1934. [Google Scholar] [CrossRef]
- Kim, J.; Woo, A.J.; Chu, J.; Snow, J.W.; Fujiwara, Y.; Kim, C.G.; Cantor, A.B.; Orkin, S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010, 143, 313–324. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of bet recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef]
- Guenther, M.G.; Lawton, L.N.; Rozovskaia, T.; Frampton, G.M.; Levine, S.S.; Volkert, T.L.; Croce, C.M.; Nakamura, T.; Canaani, E.; Young, R.A. Aberrant chromatin at genes encoding stem cell regulators in human mixed-lineage leukemia. Genes Dev. 2008, 22, 3403–3408. [Google Scholar] [CrossRef]
- Bernt, K.M.; Zhu, N.; Sinha, A.U.; Vempati, S.; Faber, J.; Krivtsov, A.V.; Feng, Z.; Punt, N.; Daigle, A.; Bullinger, L.; et al. MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 2011, 20, 66–78. [Google Scholar] [CrossRef]
- Dick, J.E. Stem cell concepts renew cancer research. Blood 2008, 112, 4793–4807. [Google Scholar] [CrossRef]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef]
- Lawrie, C.H. Microrna expression in lymphoid malignancies: New hope for diagnosis and therapy? J. Cell. Mol. Med. 2008, 12, 1432–1444. [Google Scholar] [CrossRef]
- Mi, S.; Lu, J.; Sun, M.; Li, Z.; Zhang, H.; Neilly, M.B.; Wang, Y.; Qian, Z.; Jin, J.; Zhang, Y.; et al. Microrna expression signatures accurately discriminate acute lymphoblastic leukemia from acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2007, 104, 19971–19976. [Google Scholar]
- Li, Z.; Lu, J.; Sun, M.; Mi, S.; Zhang, H.; Luo, R.T.; Chen, P.; Wang, Y.; Yan, M.; Qian, Z.; et al. Distinct microrna expression profiles in acute myeloid leukemia with common translocations. Proc. Natl. Acad. Sci. USA 2008, 105, 15535–15540. [Google Scholar]
- Schotte, D.; de Menezes, R.X.; Moqadam, F.A.; Khankahdani, L.M.; Lange-Turenhout, E.; Chen, C.; Pieters, R.; den Boer, M.L. Microrna characterize genetic diversity and drug resistance in pediatric acute lymphoblastic leukemia. Haematologica 2011, 96, 703–711. [Google Scholar] [CrossRef]
- Stumpel, D.J.; Schotte, D.; Lange-Turenhout, E.A.; Schneider, P.; Seslija, L.; de Menezes, R.X.; Marquez, V.E.; Pieters, R.; den Boer, M.L.; Stam, R.W. Hypermethylation of specific microrna genes in MLL-rearranged infant acute lymphoblastic leukemia: Major matters at a micro scale. Leukemia 2011, 25, 429–439. [Google Scholar] [CrossRef]
- Garzon, R.; Pichiorri, F.; Palumbo, T.; Visentini, M.; Aqeilan, R.; Cimmino, A.; Wang, H.; Sun, H.; Volinia, S.; Alder, H.; et al. Microrna gene expression during retinoic acid-induced differentiation of human acute promyelocytic leukemia. Oncogene 2007, 26, 4148–4157. [Google Scholar]
- Popovic, R.; Riesbeck, L.E.; Velu, C.S.; Chaubey, A.; Zhang, J.; Achille, N.J.; Erfurth, F.E.; Eaton, K.; Lu, J.; Grimes, H.L.; et al. Regulation of miR-196b by MLL and its overexpression by MLL fusions contributes to immortalization. Blood 2009, 113, 3314–3322. [Google Scholar] [CrossRef]
- Li, Z.; Huang, H.; Chen, P.; He, M.; Li, Y.; Arnovitz, S.; Jiang, X.; He, C.; Hyjek, E.; Zhang, J.; et al. miR-196b Directly targets both HOXA9/MEIS1 oncogenes and FAS tumour suppressor in MLL-rearranged leukaemia. Nat. Commun. 2012, 3, 688. [Google Scholar]
- Mi, S.; Li, Z.; Chen, P.; He, C.; Cao, D.; Elkahloun, A.; Lu, J.; Pelloso, L.A.; Wunderlich, M.; Huang, H.; et al. Aberrant overexpression and function of the miR-17-92 cluster in MLL-rearranged acute leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 3710–3715. [Google Scholar]
- Fontana, L.; Pelosi, E.; Greco, P.; Racanicchi, S.; Testa, U.; Liuzzi, F.; Croce, C.M.; Brunetti, E.; Grignani, F.; Peschle, C. Micrornas 17-5p-20a-106a control monocytopoiesis through AML1 targeting and M-CSF receptor upregulation. Nat. Cell Biol. 2007, 9, 775–787. [Google Scholar] [CrossRef]
- Garzon, R.; Pichiorri, F.; Palumbo, T.; Iuliano, R.; Cimmino, A.; Aqeilan, R.; Volinia, S.; Bhatt, D.; Alder, H.; Marcucci, G.; et al. Microrna fingerprints during human megakaryocytopoiesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5078–5083. [Google Scholar]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of mirna clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef]
- Koralov, S.B.; Muljo, S.A.; Galler, G.R.; Krek, A.; Chakraborty, T.; Kanellopoulou, C.; Jensen, K.; Cobb, B.S.; Merkenschlager, M.; Rajewsky, N.; et al. Dicer ablation affects antibody diversity and cell survival in the b lymphocyte lineage. Cell 2008, 132, 860–874. [Google Scholar] [CrossRef]
- Wong, P.; Iwasaki, M.; Somervaille, T.C.; Ficara, F.; Carico, C.; Arnold, C.; Chen, C.Z.; Cleary, M.L. The miR-17-92 microRNA polycistron regulates mll leukemia stem cell potential by modulating p21 expression. Cancer Res. 2010, 70, 3833–3842. [Google Scholar] [CrossRef]
- Caslini, C.; Alarcon, A.S.; Hess, J.L.; Tanaka, R.; Murti, K.G.; Biondi, A. The amino terminus targets the mixed lineage leukemia (MLL) protein to the nucleolus, nuclear matrix and mitotic chromosomal scaffolds. Leukemia 2000, 14, 1898–1908. [Google Scholar]
- Aguda, B.D.; Kim, Y.; Piper-Hunter, M.G.; Friedman, A.; Marsh, C.B. Microrna regulation of a cancer network: Consequences of the feedback loops involving miR-17-92, E2F, and Myc. Proc. Natl. Acad. Sci. USA 2008, 105, 19678–19683. [Google Scholar]
- Schotte, D.; Chau, J.C.; Sylvester, G.; Liu, G.; Chen, C.; van der Velden, V.H.; Broekhuis, M.J.; Peters, T.C.; Pieters, R.; den Boer, M.L. Identification of new microrna genes and aberrant microrna profiles in childhood acute lymphoblastic leukemia. Leukemia 2009, 23, 313–322. [Google Scholar] [CrossRef]
- Letai, A.; Sorcinelli, M.D.; Beard, C.; Korsmeyer, S.J. Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell 2004, 6, 241–249. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef]
- Moriya, K.; Suzuki, M.; Watanabe, Y.; Takahashi, T.; Aoki, Y.; Uchiyama, T.; Kumaki, S.; Sasahara, Y.; Minegishi, M.; Kure, S.; et al. Development of a multi-step leukemogenesis model of MLL-rearranged leukemia using humanized mice. PLoS One 2012, 7, e37892. [Google Scholar]
- Tamai, H.; Miyake, K.; Takatori, M.; Miyake, N.; Yamaguchi, H.; Dan, K.; Shimada, T.; Inokuchi, K. Activated k-ras protein accelerates human MLL/AF4-induced leukemo-lymphomogenicity in a transgenic mouse model. Leukemia 2011, 25, 888–891. [Google Scholar] [CrossRef]
- Ono, R.; Kumagai, H.; Nakajima, H.; Hishiya, A.; Taki, T.; Horikawa, K.; Takatsu, K.; Satoh, T.; Hayashi, Y.; Kitamura, T.; et al. Mixed-lineage-leukemia (MLL) fusion protein collaborates with Ras to induce acute leukemia through aberrant Hox expression and Raf activation. Leukemia 2009, 23, 2197–2209. [Google Scholar] [CrossRef]
- Burmeister, T.; Marschalek, R.; Schneider, B.; Meyer, C.; Gokbuget, N.; Schwartz, S.; Hoelzer, D.; Thiel, E. Monitoring minimal residual disease by quantification of genomic chromosomal breakpoint sequences in acute leukemias with MLL aberrations. Leukemia 2006, 20, 451–457. [Google Scholar]
- Langer, T.; Metzler, M.; Reinhardt, D.; Viehmann, S.; Borkhardt, A.; Reichel, M.; Stanulla, M.; Schrappe, M.; Creutzig, U.; Ritter, J.; et al. Analysis of t(9;11) chromosomal breakpoint sequences in childhood acute leukemia: Almost identical mll breakpoints in therapy-related aml after treatment without etoposides. Genes Chromosomes Cancer 2003, 36, 393–401. [Google Scholar] [CrossRef]
- Erfurth, F.; Hemenway, C.S.; de Erkenez, A.C.; Domer, P.H. MLL fusion partners AF4 and AF9 interact at subnuclear foci. Leukemia 2004, 18, 92–102. [Google Scholar] [CrossRef]
- Palermo, C.M.; Bennett, C.A.; Winters, A.C.; Hemenway, C.S. The AF4-mimetic peptide, PFWT, induces necrotic cell death in MV4-11 leukemia cells. Leuk. Res. 2008, 32, 633–642. [Google Scholar] [CrossRef]
- Srinivasan, R.S.; Nesbit, J.B.; Marrero, L.; Erfurth, F.; LaRussa, V.F.; Hemenway, C.S. The synthetic peptide pfwt disrupts AF4-AF9 protein complexes and induces apoptosis in t(4;11) leukemia cells. Leukemia 2004, 18, 1364–1372. [Google Scholar] [CrossRef]
- Zeisig, D.T.; Bittner, C.B.; Zeisig, B.B.; Garcia-Cuellar, M.P.; Hess, J.L.; Slany, R.K. The eleven-nineteen-leukemia protein ENL connects nuclear MLL fusion partners with chromatin. Oncogene 2005, 24, 5525–5532. [Google Scholar]
- Okada, Y.; Feng, Q.; Lin, Y.; Jiang, Q.; Li, Y.; Coffield, V.M.; Su, L.; Xu, G.; Zhang, Y. hDOT1L links histone methylation to leukemogenesis. Cell 2005, 121, 167–178. [Google Scholar] [CrossRef]
- Estable, M.C.; Naghavi, M.H.; Kato, H.; Xiao, H.; Qin, J.; Vahlne, A.; Roeder, R.G. MCEF, the newest member of the AF4 family of transcription factors involved in leukemia, is a positive transcription elongation factor-b-associated protein. J. Biomed. Sci. 2002, 9, 234–245. [Google Scholar] [CrossRef]
- Bitoun, E.; Oliver, P.L.; Davies, K.E. The mixed-lineage leukemia fusion partner AF4 stimulates rna polymerase II transcriptional elongation and mediates coordinated chromatin remodeling. Hum. Mol. Genet. 2007, 16, 92–106. [Google Scholar]
- Mueller, D.; Bach, C.; Zeisig, D.; Garcia-Cuellar, M.P.; Monroe, S.; Sreekumar, A.; Zhou, R.; Nesvizhskii, A.; Chinnaiyan, A.; Hess, J.L.; et al. A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 2007, 110, 4445–4454. [Google Scholar] [CrossRef]
- Mueller, D.; Garcia-Cuellar, M.P.; Bach, C.; Buhl, S.; Maethner, E.; Slany, R.K. Misguided transcriptional elongation causes mixed lineage leukemia. PLoS Biol. 2009, 7, e1000249. [Google Scholar] [CrossRef]
- Biswas, D.; Milne, T.A.; Basrur, V.; Kim, J.; Elenitoba-Johnson, K.S.; Allis, C.D.; Roeder, R.G. Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc. Natl. Acad. Sci. USA 2011, 108, 15751–15756. [Google Scholar]
- Lin, C.; Smith, E.R.; Takahashi, H.; Lai, K.C.; Martin-Brown, S.; Florens, L.; Washburn, M.P.; Conaway, J.W.; Conaway, R.C.; Shilatifard, A. AFF4, a component of the ELL/P-TEFB elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol. Cell 2010, 37, 429–437. [Google Scholar] [CrossRef]
- Yokoyama, A.; Lin, M.; Naresh, A.; Kitabayashi, I.; Cleary, M.L. A higher-order complex containing AF4 and enl family proteins with P-TEFB facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 2010, 17, 198–212. [Google Scholar] [CrossRef]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 2010, 38, 439–451. [Google Scholar] [CrossRef]
- Monroe, S.C.; Jo, S.Y.; Sanders, D.S.; Basrur, V.; Elenitoba-Johnson, K.S.; Slany, R.K.; Hess, J.L. MLL-AF9 and MLL-ENL alter the dynamic association of transcriptional regulators with genes critical for leukemia. Exp. Hematol. 2011, 39, 77–86.e5. [Google Scholar] [CrossRef]
- Lin, C.; Garrett, A.S.; de Kumar, B.; Smith, E.R.; Gogol, M.; Seidel, C.; Krumlauf, R.; Shilatifard, A. Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC). Genes Dev. 2011, 25, 1486–1498. [Google Scholar] [CrossRef]
- He, N.; Chan, C.K.; Sobhian, B.; Chou, S.; Xue, Y.; Liu, M.; Alber, T.; Benkirane, M.; Zhou, Q. Human polymerase-associated factor complex (PAFC) connects the super elongation complex (SEC) to rna polymerase ii on chromatin. Proc. Natl. Acad. Sci. USA 2011, 108, E636–E645. [Google Scholar]
- Smith, E.; Lin, C.; Shilatifard, A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 2011, 25, 661–672. [Google Scholar] [CrossRef]
- Liu, M.; Hsu, J.; Chan, C.; Li, Z.; Zhou, Q. The ubiquitin ligase siah1 controls ELL2 stability and formation of super elongation complexes to modulate gene transcription. Mol. Cell 2012, 46, 325–334. [Google Scholar] [CrossRef]
- Luo, Z.; Lin, C.; Guest, E.; Garrett, A.S.; Mohaghegh, N.; Swanson, S.; Marshall, S.; Florens, L.; Washburn, M.P.; Shilatifard, A. The super elongation complex family of rna polymerase II elongation factors: Gene target specificity and transcriptional output. Mol. Cell. Biol. 2012, 32, 2608–2617. [Google Scholar] [CrossRef]
- Smith, E.R.; Lin, C.; Garrett, A.S.; Thornton, J.; Mohaghegh, N.; Hu, D.; Jackson, J.; Saraf, A.; Swanson, S.K.; Seidel, C.; et al. The little elongation complex regulates small nuclear RNA transcription. Mol. Cell 2011, 44, 954–965. [Google Scholar] [CrossRef]
- Takahashi, H.; Parmely, T.J.; Sato, S.; Tomomori-Sato, C.; Banks, C.A.; Kong, S.E.; Szutorisz, H.; Swanson, S.K.; Martin-Brown, S.; Washburn, M.P.; et al. Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell 2011, 146, 92–104. [Google Scholar] [CrossRef]
- Garcia-Cuellar, M.P.; Zilles, O.; Schreiner, S.A.; Birke, M.; Winkler, T.H.; Slany, R.K. The ENL moiety of the childhood leukemia-associated MLL-ENL oncoprotein recruits human polycomb 3. Oncogene 2001, 20, 411–419. [Google Scholar] [CrossRef]
- Hemenway, C.S.; de Erkenez, A.C.; Gould, G.C. The polycomb protein MPc3 interacts with AF9, an MLL fusion partner in t(9;11)(p22;q23) acute leukemias. Oncogene 2001, 20, 3798–3805. [Google Scholar] [CrossRef]
- Tan, J.; Jones, M.; Koseki, H.; Nakayama, M.; Muntean, A.G.; Maillard, I.; Hess, J.L. CBX8, a polycomb group protein, is essential for MLL-AF9-induced leukemogenesis. Cancer Cell 2011, 20, 563–575. [Google Scholar] [CrossRef]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFB for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFB and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef]
- Liedtke, M.; Ayton, P.M.; Somervaille, T.C.; Smith, K.S.; Cleary, M.L. Self-association mediated by the RAS association 1 domain of AF6 activates the oncogenic potential of MLL-AF6. Blood 2010, 116, 63–70. [Google Scholar] [CrossRef]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef]
- Muse, G.W.; Gilchrist, D.A.; Nechaev, S.; Shah, R.; Parker, J.S.; Grissom, S.F.; Zeitlinger, J.; Adelman, K. RNA polymerase is poised for activation across the genome. Nat. Genet. 2007, 39, 1507–1511. [Google Scholar]
- Nechaev, S.; Fargo, D.C.; dos Santos, G.; Liu, L.; Gao, Y.; Adelman, K. Global analysis of short rnas reveals widespread promoter-proximal stalling and arrest of pol II in drosophila. Science 2010, 327, 335–338. [Google Scholar]
- Zeitlinger, J.; Stark, A.; Kellis, M.; Hong, J.W.; Nechaev, S.; Adelman, K.; Levine, M.; Young, R.A. RNA polymerase stalling at developmental control genes in the drosophila melanogaster embryo. Nat. Genet. 2007, 39, 1512–1516. [Google Scholar] [CrossRef]
- Kininis, M.; Isaacs, G.D.; Core, L.J.; Hah, N.; Kraus, W.L. Postrecruitment regulation of RNA polymerase II directs rapid signaling responses at the promoters of estrogen target genes. Mol. Cell. Biol. 2009, 29, 1123–1133. [Google Scholar] [CrossRef]
- Marshall, N.F.; Price, D.H. Purification of P-TEFB, a transcription factor required for the transition into productive elongation. J. Biol. Chem. 1995, 270, 12335–12338. [Google Scholar]
- Marshall, N.F.; Peng, J.; Xie, Z.; Price, D.H. Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J. Biol. Chem. 1996, 271, 27176–27183. [Google Scholar]
- Yamaguchi, Y.; Takagi, T.; Wada, T.; Yano, K.; Furuya, A.; Sugimoto, S.; Hasegawa, J.; Handa, H. NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 1999, 97, 41–51. [Google Scholar] [CrossRef]
- Wada, T.; Takagi, T.; Yamaguchi, Y.; Watanabe, D.; Handa, H. Evidence that P-TEFB alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J. 1998, 17, 7395–7403. [Google Scholar] [CrossRef]
- Chao, S.H.; Price, D.H. Flavopiridol inactivates P-TEFB and blocks most rna polymerase II transcription in vivo. J. Biol. Chem. 2001, 276, 31793–31799. [Google Scholar] [CrossRef]
- Shilatifard, A.; Duan, D.R.; Haque, D.; Florence, C.; Schubach, W.H.; Conaway, J.W.; Conaway, R.C. ELL2, a new member of an ell family of RNA polymerase II elongation factors. Proc. Natl. Acad. Sci. USA 1997, 94, 3639–3643. [Google Scholar] [CrossRef]
- Shilatifard, A.; Lane, W.S.; Jackson, K.W.; Conaway, R.C.; Conaway, J.W. An RNA polymerase II elongation factor encoded by the human ell gene. Science 1996, 271, 1873–1876. [Google Scholar]
- Shilatifard, A.; Conaway, R.C.; Conaway, J.W. The RNA polymerase II elongation complex. Annu. Rev. Biochem 2003, 72, 693–715. [Google Scholar] [CrossRef]
- Bursen, A.; Moritz, S.; Gaussmann, A.; Dingermann, T.; Marschalek, R. Interaction of AF4 wild-type and AF4.MLL fusion protein with SIAH proteins: Indication for t(4;11) pathobiology? Oncogene 2004, 23, 6237–6249. [Google Scholar] [CrossRef]
- Oliver, P.L.; Bitoun, E.; Clark, J.; Jones, E.L.; Davies, K.E. Mediation of AF4 protein function in the cerebellum by SIAH proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 14901–14906. [Google Scholar]
- Muntean, A.G.; Giannola, D.; Udager, A.M.; Hess, J.L. The PHD fingers of MLL block MLL fusion protein-mediated transformation. Blood 2008, 112, 4690–4693. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Taranova, O.; He, J.; Zhang, Y. DOT1L, the H3K79 methyltransferase, is required for MLL-AF9-mediated leukemogenesis. Blood 2011, 117, 6912–6922. [Google Scholar] [CrossRef]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule dot1l inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef]
- Chang, M.J.; Wu, H.; Achille, N.J.; Reisenauer, M.R.; Chou, C.W.; Zeleznik-Le, N.J.; Hemenway, C.S.; Zhang, W. Histone H3 lysine 79 methyltransferase Dot1 is required for immortalization by mll oncogenes. Cancer Res. 2010, 70, 10234–10242. [Google Scholar]
- Jo, S.Y.; Granowicz, E.M.; Maillard, I.; Thomas, D.; Hess, J.L. Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by mll translocation. Blood 2011, 117, 4759–4768. [Google Scholar]
- Krogan, N.J.; Dover, J.; Wood, A.; Schneider, J.; Heidt, J.; Boateng, M.A.; Dean, K.; Ryan, O.W.; Golshani, A.; Johnston, M.; et al. The Paf1 complex is required for histone H3 methylation by COMPASS and Dot1p: Linking transcriptional elongation to histone methylation. Mol. Cell 2003, 11, 721–729. [Google Scholar] [CrossRef]
- Pradeepa, M.M.; Sutherland, H.G.; Ule, J.; Grimes, G.R.; Bickmore, W.A. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genet. 2012, 8, e1002717. [Google Scholar] [CrossRef]
- Brown-Bryan, T.A.; Leoh, L.S.; Ganapathy, V.; Pacheco, F.J.; Mediavilla-Varela, M.; Filippova, M.; Linkhart, T.A.; Gijsbers, R.; Debyser, Z.; Casiano, C.A. Alternative splicing and caspase-mediated cleavage generate antagonistic variants of the stress oncoprotein LEDGF/P75. Mol. Cancer Res. 2008, 6, 1293–1307. [Google Scholar] [CrossRef]
- Ge, H.; Si, Y.; Roeder, R.G. Isolation of cdnas encoding novel transcription coactivators p52 and p75 reveals an alternate regulatory mechanism of transcriptional activation. EMBO J. 1998, 17, 6723–6729. [Google Scholar] [CrossRef]
- Liedtke, M.; Cleary, M.L. Therapeutic targeting of mll. Blood 2009, 113, 6061–6068. [Google Scholar] [CrossRef]
- Bernt, K.M.; Armstrong, S.A. Targeting epigenetic programs in MLL-rearranged leukemias. Hematology Am. Soc. Hematol. Educ. Program 2011, 354–360. [Google Scholar]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Arrowsmith, C.H.; Bountra, C.; Fish, P.V.; Lee, K.; Schapira, M. Epigenetic protein families: A new frontier for drug discovery. Nat. Rev. Drug Discov. 2012, 11, 384–400. [Google Scholar] [CrossRef]
- Brennan, P.; Filippakopoulos, P.; Knapp, S. The therapeutic potential of acetyl-lysine and methyl-lysine effector domains. Drug Discov. Today Ther. Strateg. 2012, in press. [Google Scholar]
- Deshpande, A.J.; Bradner, J.; Armstrong, S.A. Chromatin modifications as therapeutic targets in MLL-rearranged leukemia. Trends Immunol. 2012, in press. [Google Scholar]
- Yao, Y.; Chen, P.; Diao, J.; Cheng, G.; Deng, L.; Anglin, J.L.; Prasad, B.V.; Song, Y. Selective inhibitors of histone methyltransferase dot1l: Design, synthesis, and crystallographic studies. J. Am Chem. Soc 2011, 133, 16746–16749. [Google Scholar]
- Harris, W.J.; Huang, X.; Lynch, J.T.; Spencer, G.J.; Hitchin, J.R.; Li, Y.; Ciceri, F.; Blaser, J.G.; Greystoke, B.F.; Jordan, A.M.; et al. The histone demethylase KDM1A sustains the oncogenic potential of MLL-AF9 leukemia stem cells. Cancer Cell 2012, 21, 473–487. [Google Scholar] [CrossRef]
- Somervaille, T.C.; Matheny, C.J.; Spencer, G.J.; Iwasaki, M.; Rinn, J.L.; Witten, D.M.; Chang, H.Y.; Shurtleff, S.A.; Downing, J.R.; Cleary, M.L. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 2009, 4, 129–140. [Google Scholar] [CrossRef]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. ET bromodomain inhibition as a therapeutic strategy to target MYC. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar]
- Mertz, J.A.; Conery, A.R.; Bryant, B.M.; Sandy, P.; Balasubramanian, S.; Mele, D.A.; Bergeron, L.; Sims, R.J., 3rd. Targeting myc dependence in cancer by inhibiting bet bromodomains. Proc. Natl. Acad. Sci. USA 2011, 108, 16669–16674. [Google Scholar]
- Bennett, C.A.; Winters, A.C.; Barretto, N.N.; Hemenway, C.S. Molecular targeting of MLL-rearranged leukemia cell lines with the synthetic peptide PFWT synergistically enhances the cytotoxic effect of established chemotherapeutic agents. Leuk. Res. 2009, 33, 937–947. [Google Scholar] [CrossRef]
- Wang, Z.; Smith, K.S.; Murphy, M.; Piloto, O.; Somervaille, T.C.; Cleary, M.L. Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature 2008, 455, 1205–1209. [Google Scholar]
- Armstrong, S.A.; Kung, A.L.; Mabon, M.E.; Silverman, L.B.; Stam, R.W.; Den Boer, M.L.; Pieters, R.; Kersey, J.H.; Sallan, S.E.; Fletcher, J.A.; et al. Inhibition of FLT3 in MLL. Validation of a therapeutic target identified by gene expression based classification. Cancer Cell 2003, 3, 173–183. [Google Scholar] [CrossRef]
- Ayton, P.M.; Chen, E.H.; Cleary, M.L. Binding to nonmethylated cpg DNA is essential for target recognition, transactivation, and myeloid transformation by an mll oncoprotein. Mol. Cell. Biol. 2004, 24, 10470–10478. [Google Scholar] [CrossRef]
- Balkhi, M.Y.; Trivedi, A.K.; Geletu, M.; Christopeit, M.; Bohlander, S.K.; Behre, H.M.; Behre, G. Proteomics of acute myeloid leukaemia: Cytogenetic risk groups differ specifically in their proteome, interactome and post-translational protein modifications. Oncogene 2006, 25, 7041–7058. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ballabio, E.; Milne, T.A. Molecular and Epigenetic Mechanisms of MLL in Human Leukemogenesis. Cancers 2012, 4, 904-944. https://doi.org/10.3390/cancers4030904
Ballabio E, Milne TA. Molecular and Epigenetic Mechanisms of MLL in Human Leukemogenesis. Cancers. 2012; 4(3):904-944. https://doi.org/10.3390/cancers4030904
Chicago/Turabian StyleBallabio, Erica, and Thomas A. Milne. 2012. "Molecular and Epigenetic Mechanisms of MLL in Human Leukemogenesis" Cancers 4, no. 3: 904-944. https://doi.org/10.3390/cancers4030904