
Translation into Clinical Practice of the G1-G7 Molecular Subgroup Classification of Glioblastoma: Comprehensive Demographic and Molecular Pathway Profiling

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
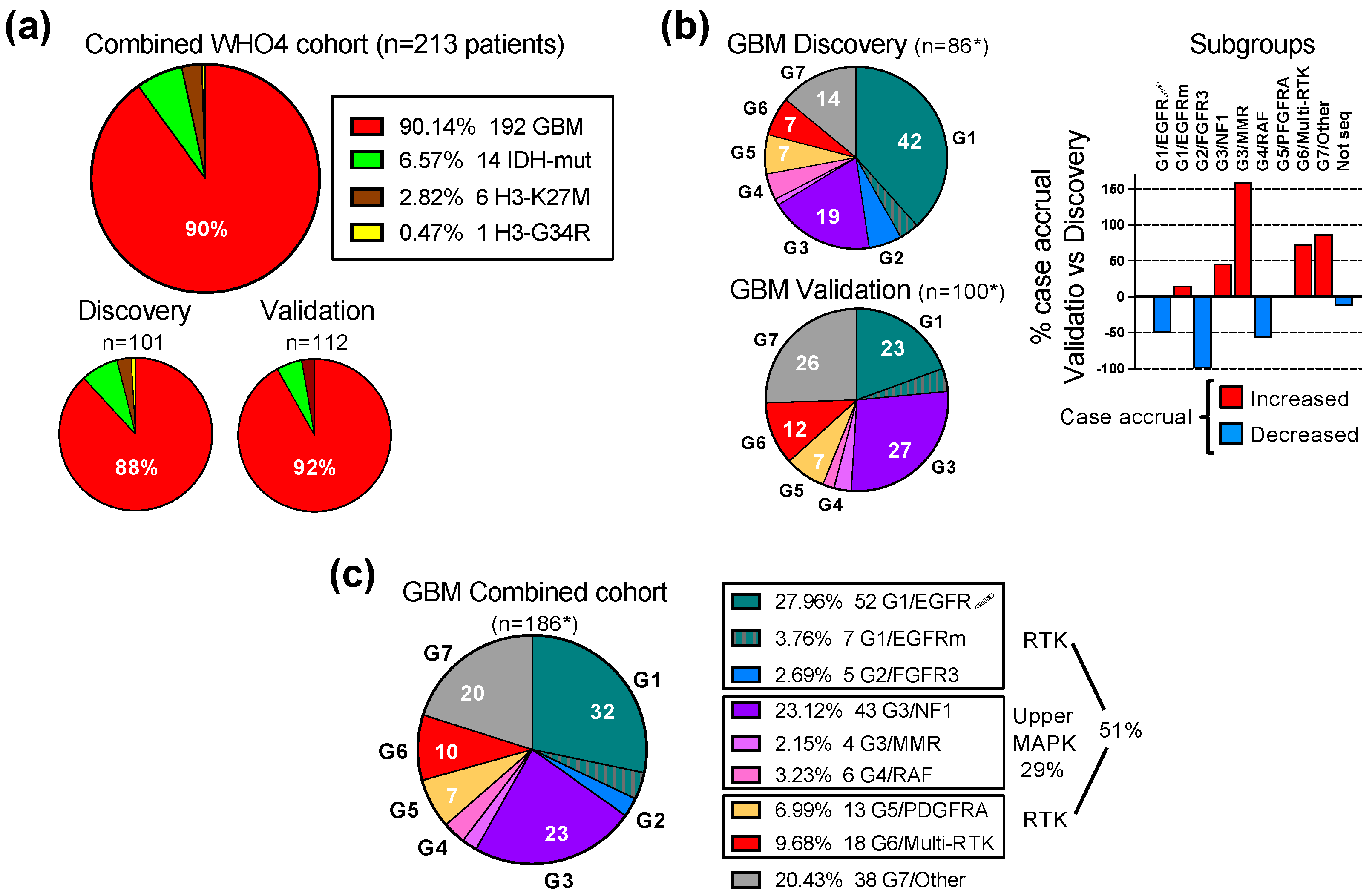
3.1. Validation of the G1-G7 Subgroup Classification of Glioblastoma
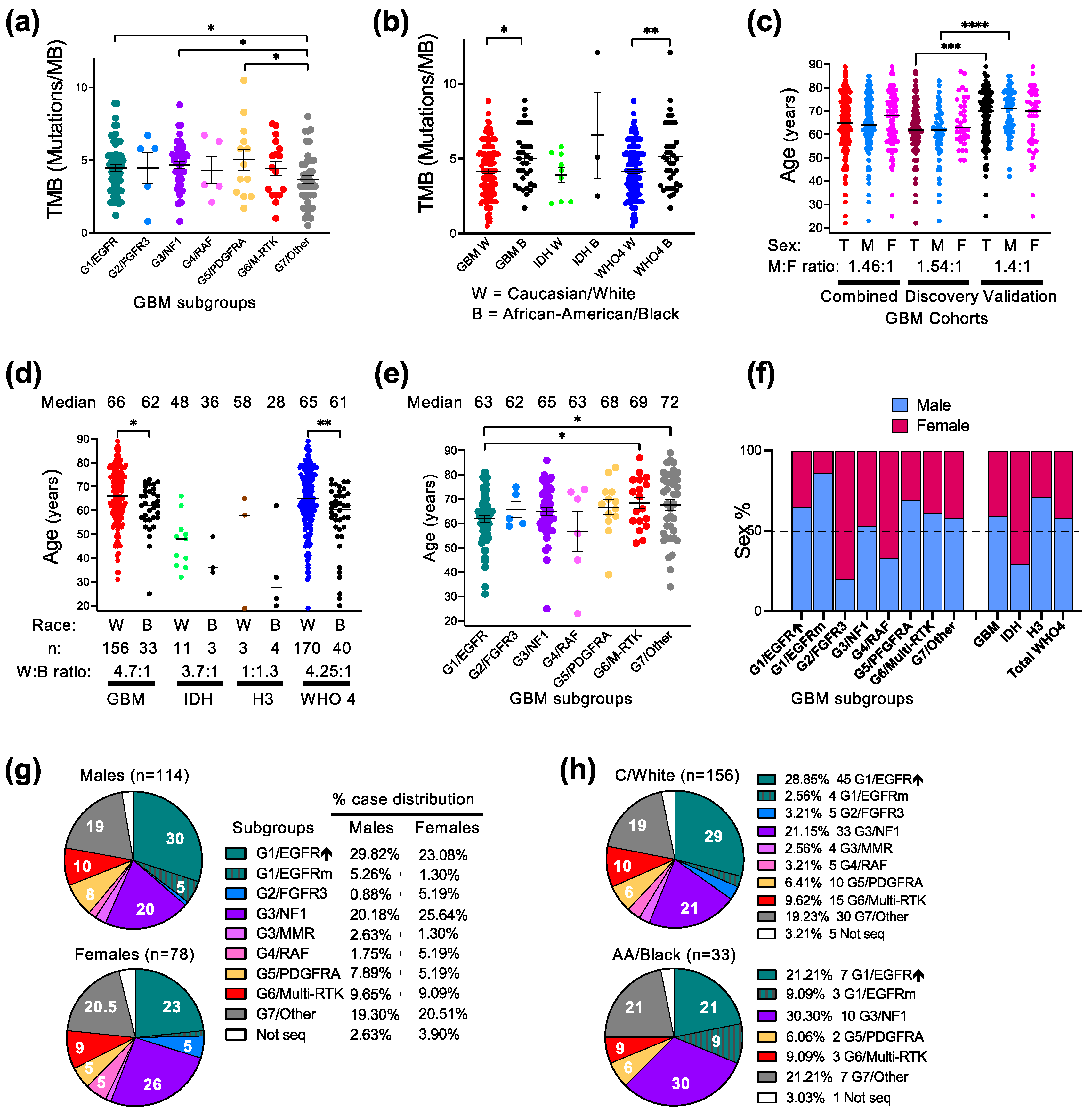
3.2. Molecular–Demographic Correlations in Glioblastoma
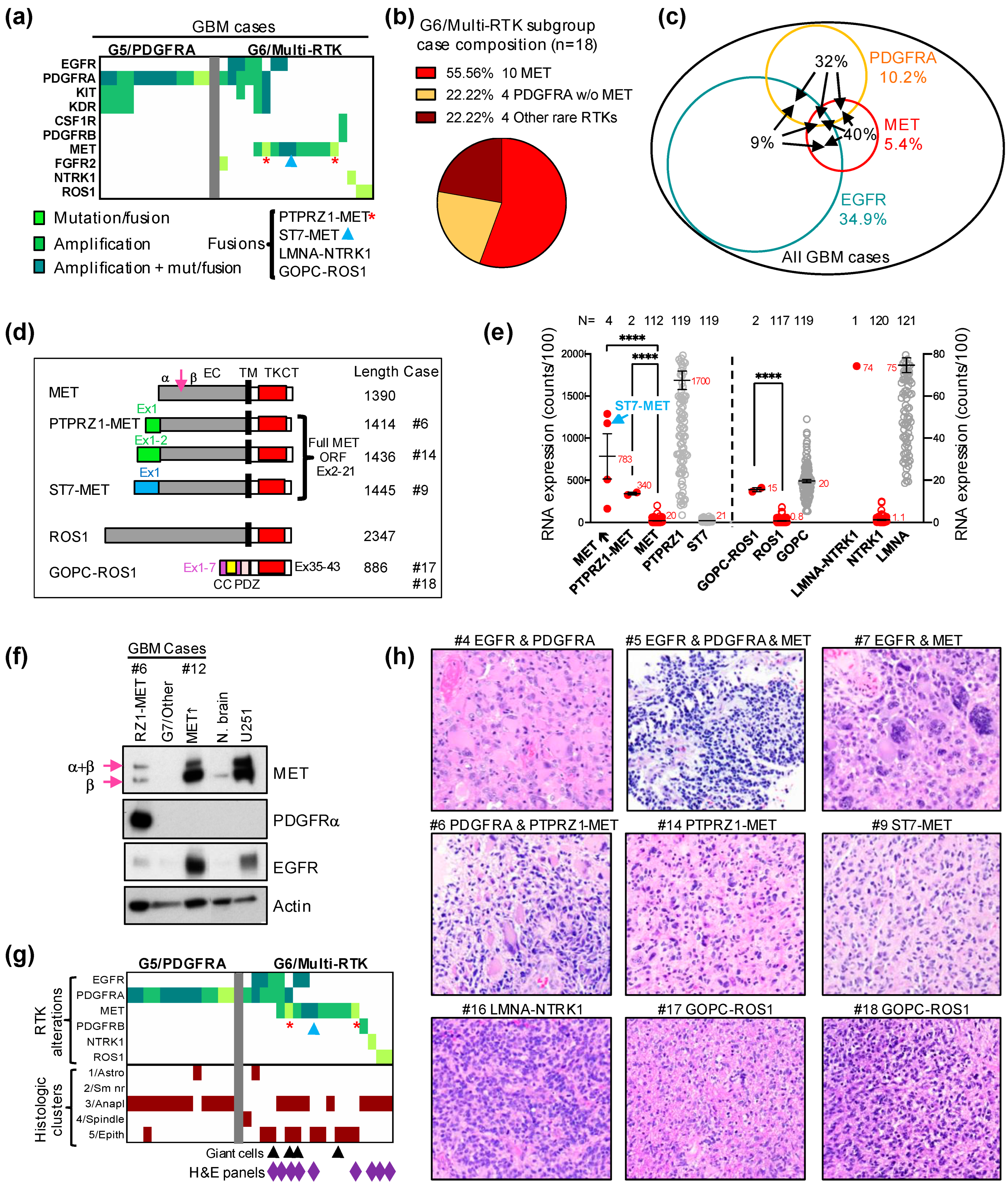
3.3. Characterization of the G6/Multi-RTK Subgroup of Glioblastoma
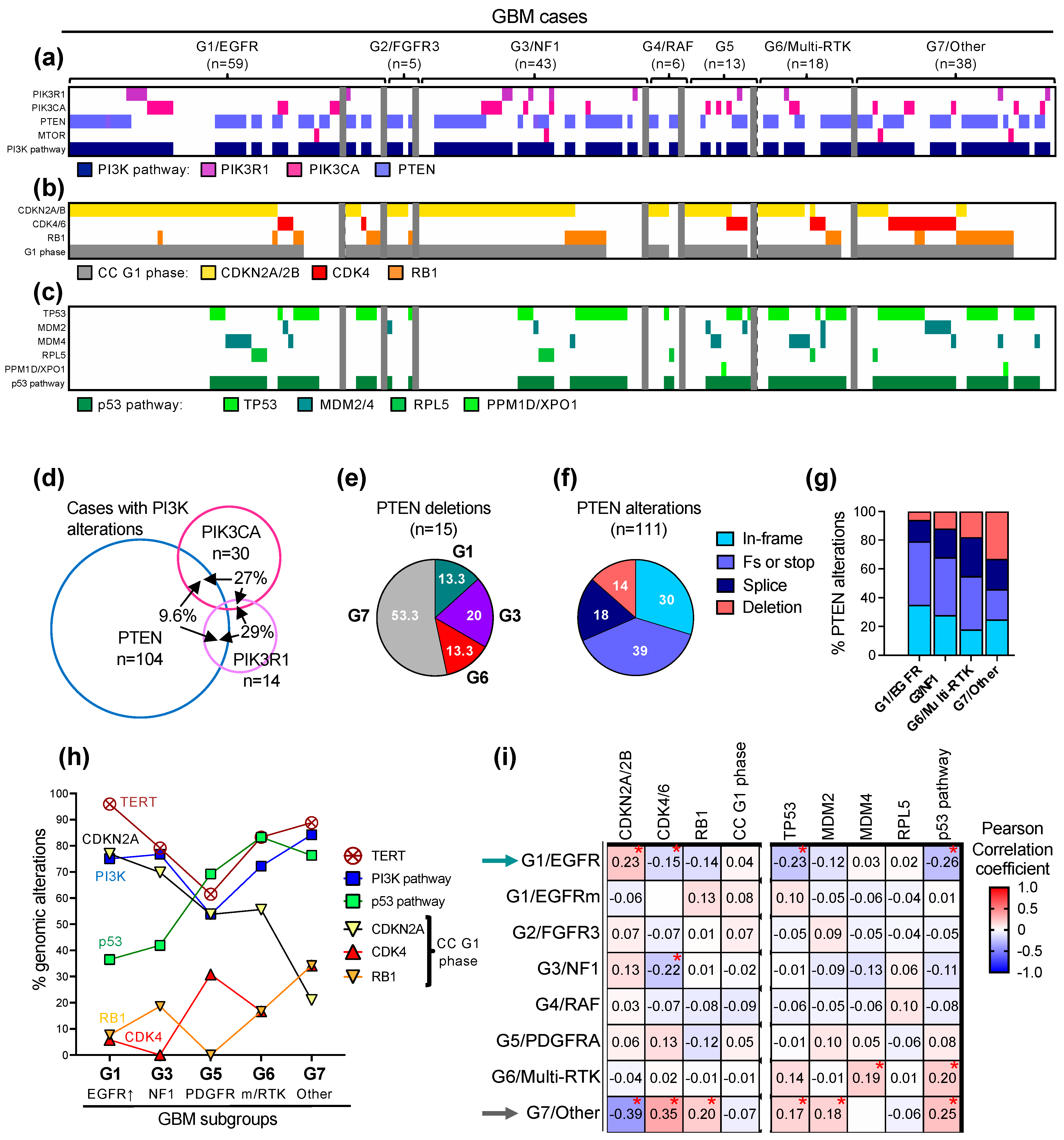
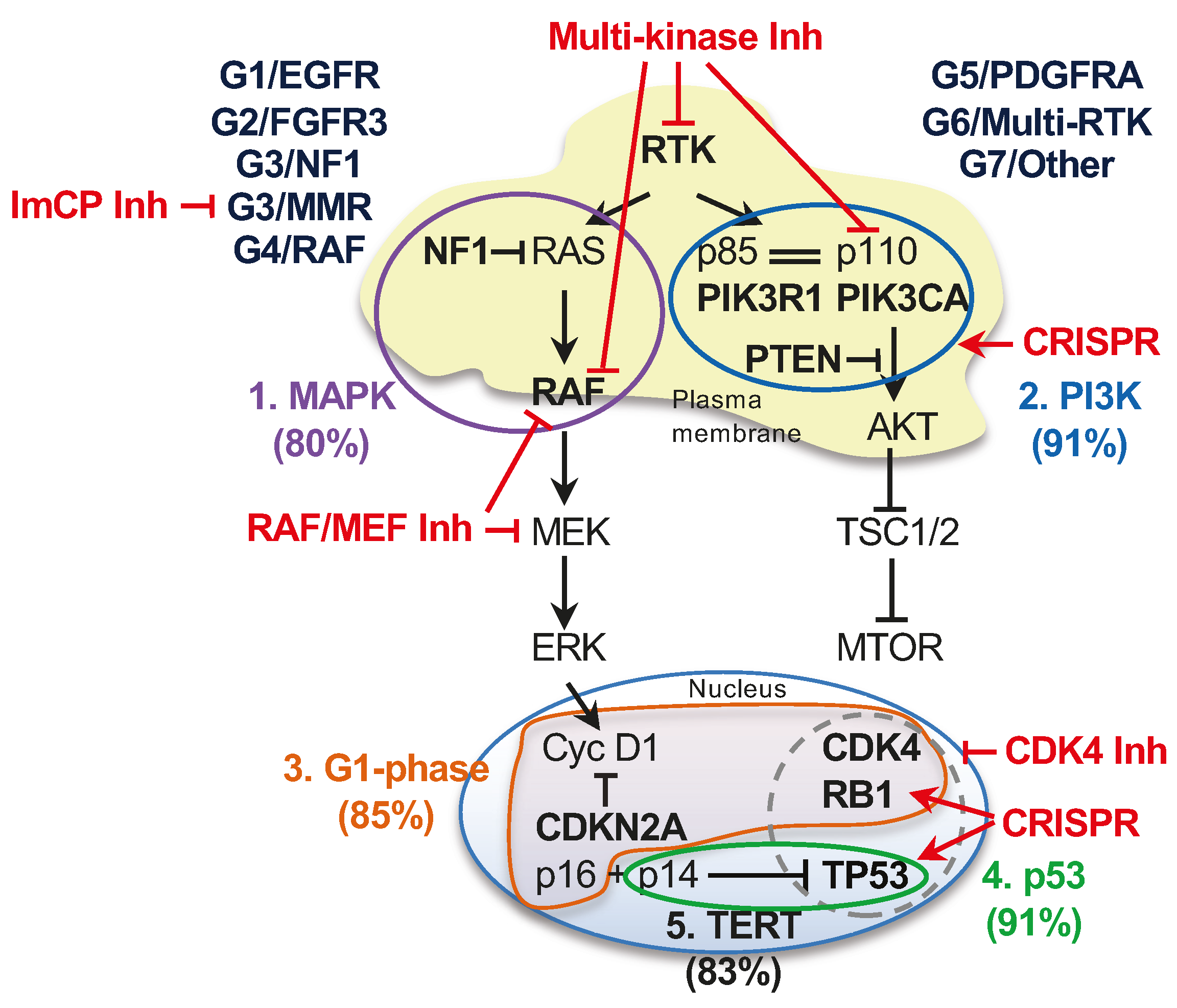
3.4. The Landscape of the Major Oncogenic Pathways in the G1-G7 Molecular Subgroups
3.5. Implementation into Practice of the G1-G7 Molecular Subgroup Classification of Glioblastoma
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Truitt, G.; Gittleman, H.; Brat, D.J.; Kruchko, C.; Wilson, R.; Barnholtz-Sloan, J.S. Relative survival after diagnosis with a primary brain or other central nervous system tumor in the National Program of Cancer Registries, 2004 to 2014. Neurooncol. Pract. 2020, 7, 306–312. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumors Editorial Board. Central Nervous System Tumors, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2021. [Google Scholar]
- Shirahata, M.; Ono, T.; Stichel, D.; Schrimpf, D.; Reuss, D.E.; Sahm, F.; Koelsche, C.; Wefers, A.; Reinhardt, A.; Huang, K.; et al. Novel, improved grading system(s) for IDH-mutant astrocytic gliomas. Acta Neuropathol. 2018, 136, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.M. Multi-Platform Classification of IDH-Wild-Type Glioblastoma Based on ERK/MAPK Pathway: Diagnostic, Prognostic and Therapeutic Implications. Cancers 2021, 13, 4532. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.M. PTEN Tumor Suppressor Network in PI3K-Akt Pathway Control. Genes Cancer 2010, 1, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.M.; Li, Y.; Islam, M.Z.; Notarianni, C.; Sun, H.; Olar, A.; Fuller, G.N. Mutations of the MAPK/TSC/mTOR pathway characterize periventricular glioblastoma with epithelioid SEGA-like morphology-morphological and therapeutic implications. Oncotarget 2019, 10, 4038–4052. [Google Scholar] [CrossRef]
- Georgescu, M.M.; Islam, M.Z.; Li, Y.; Traylor, J.; Nanda, A. Novel targetable FGFR2 and FGFR3 alterations in glioblastoma associate with aggressive phenotype and distinct gene expression programs. Acta Neuropathol. Commun. 2021, 9, 69. [Google Scholar] [CrossRef]
- Georgescu, M.M.; Whipple, S.G.; Notarianni, C.M. Novel neoplasms associated with syndromic pediatric medulloblastoma: Integrated pathway delineation for personalized therapy. Cell Commun. Signal 2022, 20, 123. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.M.; Islam, M.Z.; Li, Y.; Circu, M.L.; Traylor, J.; Notarianni, C.M.; Kline, C.N.; Burns, D.K. Global activation of oncogenic pathways underlies therapy resistance in diffuse midline glioma. Acta Neuropathol. Commun. 2020, 8, 111. [Google Scholar] [CrossRef]
- Molina, J.R.; Hayashi, Y.; Stephens, C.; Georgescu, M.M. Invasive glioblastoma cells acquire stemness and increased Akt activation. Neoplasia 2010, 12, 453–463. [Google Scholar] [CrossRef]
- Radu, A.; Neubauer, V.; Akagi, T.; Hanafusa, H.; Georgescu, M.M. PTEN induces cell cycle arrest by decreasing the level and nuclear localization of cyclin D1. Mol. Cell Biol. 2003, 23, 6139–6149. [Google Scholar] [CrossRef]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Bielle, F.; Di Stefano, A.L.; Meyronet, D.; Picca, A.; Villa, C.; Bernier, M.; Schmitt, Y.; Giry, M.; Rousseau, A.; Figarella-Branger, D.; et al. Diffuse gliomas with FGFR3-TACC3 fusion have characteristic histopathological and molecular features. Brain Pathol. 2018, 28, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Little, S.E.; Popov, S.; Jury, A.; Bax, D.A.; Doey, L.; Al-Sarraj, S.; Jurgensmeier, J.M.; Jones, C. Receptor tyrosine kinase genes amplified in glioblastoma exhibit a mutual exclusivity in variable proportions reflective of individual tumor heterogeneity. Cancer Res. 2012, 72, 1614–1620. [Google Scholar] [CrossRef] [PubMed]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef] [PubMed]
- Szerlip, N.J.; Pedraza, A.; Chakravarty, D.; Azim, M.; McGuire, J.; Fang, Y.; Ozawa, T.; Holland, E.C.; Huse, J.T.; Jhanwar, S.; et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proc. Natl. Acad. Sci. USA 2012, 109, 3041–3046. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.S.; Chen, H.M.; Yang, M.Y.; Zhang, C.B.; Yu, K.; Ye, W.L.; Hu, B.Q.; Yan, W.; Zhang, W.; Akers, J.; et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Genome Res. 2014, 24, 1765–1773. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Yoda, S.; Lennerz, J.K.; Langenbucher, A.; Lin, J.J.; Rooney, M.M.; Prutisto-Chang, K.; Oh, A.; Adams, N.A.; Yeap, B.Y.; et al. MET Alterations Are a Recurring and Actionable Resistance Mechanism in ALK-Positive Lung Cancer. Clin. Cancer Res. 2020, 26, 2535–2545. [Google Scholar] [CrossRef]
- Charest, A.; Lane, K.; McMahon, K.; Park, J.; Preisinger, E.; Conroy, H.; Housman, D. Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21). Genes Chromosomes Cancer 2003, 37, 58–71. [Google Scholar] [CrossRef]
- Torre, M.; Vasudevaraja, V.; Serrano, J.; DeLorenzo, M.; Malinowski, S.; Blandin, A.F.; Pages, M.; Ligon, A.H.; Dong, F.; Meredith, D.M.; et al. Molecular and clinicopathologic features of gliomas harboring NTRK fusions. Acta Neuropathol. Commun. 2020, 8, 107. [Google Scholar] [CrossRef]
- Charest, A.; Kheifets, V.; Park, J.; Lane, K.; McMahon, K.; Nutt, C.L.; Housman, D. Oncogenic targeting of an activated tyrosine kinase to the Golgi apparatus in a glioblastoma. Proc. Natl. Acad. Sci. USA 2003, 100, 916–921. [Google Scholar] [CrossRef]
- Campbell, R.B.; Liu, F.; Ross, A.H. Allosteric activation of PTEN phosphatase by phosphatidylinositol 4,5-bisphosphate. J. Biol. Chem. 2003, 278, 33617–33620. [Google Scholar] [CrossRef] [PubMed]
- Georgescu, M.M.; Kirsch, K.H.; Akagi, T.; Shishido, T.; Hanafusa, H. The tumor-suppressor activity of PTEN is regulated by its carboxyl-terminal region. Proc. Natl. Acad. Sci. USA 1999, 96, 10182–10187. [Google Scholar] [CrossRef]
- Georgescu, M.M.; Kirsch, K.H.; Kaloudis, P.; Yang, H.; Pavletich, N.P.; Hanafusa, H. Stabilization and productive positioning roles of the C2 domain of PTEN tumor suppressor. Cancer Res. 2000, 60, 7033–7038. [Google Scholar] [PubMed]
- Iijima, M.; Devreotes, P. Tumor suppressor PTEN mediates sensing of chemoattractant gradients. Cell 2002, 109, 599–610. [Google Scholar] [CrossRef]
- Higa, N.; Akahane, T.; Yokoyama, S.; Yonezawa, H.; Uchida, H.; Takajo, T.; Kirishima, M.; Hamada, T.; Matsuo, K.; Fujio, S.; et al. A tailored next-generation sequencing panel identified distinct subtypes of wildtype IDH and TERT promoter glioblastomas. Cancer Sci. 2020, 111, 3902–3911. [Google Scholar] [CrossRef]
- Dobbelstein, M.; Levine, A.J. Mdm2: Open questions. Cancer Sci. 2020, 111, 2203–2211. [Google Scholar] [CrossRef]
- Bailey, M.L.; O’Neil, N.J.; van Pel, D.M.; Solomon, D.A.; Waldman, T.; Hieter, P. Glioblastoma cells containing mutations in the cohesin component STAG2 are sensitive to PARP inhibition. Mol. Cancer Ther. 2014, 13, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Cruz Da Silva, E.; Mercier, M.C.; Etienne-Selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef]
- Umehara, T.; Arita, H.; Yoshioka, E.; Shofuda, T.; Kanematsu, D.; Kinoshita, M.; Kodama, Y.; Mano, M.; Kagawa, N.; Fujimoto, Y.; et al. Distribution differences in prognostic copy number alteration profiles in IDH-wild-type glioblastoma cause survival discrepancies across cohorts. Acta Neuropathol. Commun. 2019, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus Ipilimumab in Lung Cancer with a High Tumor Mutational Burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef] [PubMed]
- Ah-Pine, F.; Casas, D.; Menei, P.; Boisselier, B.; Garcion, E.; Rousseau, A. RNA-sequencing of IDH-wild-type glioblastoma with chromothripsis identifies novel gene fusions with potential oncogenic properties. Transl. Oncol. 2021, 14, 100884. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Guo, D. MET in glioma: Signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 2019, 38, 270. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, M.; Rabushko, E.; Rozenberg, J.M.; Mohammad, T.; Seryakov, A.; Sekacheva, M.; Buzdin, A. Clinically relevant fusion oncogenes: Detection and practical implications. Ther. Adv. Med. Oncol. 2022, 14, 17588359221144108. [Google Scholar] [CrossRef] [PubMed]
- Dunn, D.B. Larotrectinib and Entrectinib: TRK Inhibitors for the Treatment of Pediatric and Adult Patients With NTRK Gene Fusion. J. Adv. Pract. Oncol. 2020, 11, 418–423. [Google Scholar] [PubMed]
- Akhavan, D.; Pourzia, A.L.; Nourian, A.A.; Williams, K.J.; Nathanson, D.; Babic, I.; Villa, G.R.; Tanaka, K.; Nael, A.; Yang, H.; et al. De-repression of PDGFRbeta transcription promotes acquired resistance to EGFR tyrosine kinase inhibitors in glioblastoma patients. Cancer Discov. 2013, 3, 534–547. [Google Scholar] [CrossRef]
- Kleczko, E.K.; Heasley, L.E. Mechanisms of rapid cancer cell reprogramming initiated by targeted receptor tyrosine kinase inhibitors and inherent therapeutic vulnerabilities. Mol. Cancer 2018, 17, 60. [Google Scholar] [CrossRef]
- Ware, K.E.; Marshall, M.E.; Heasley, L.R.; Marek, L.; Hinz, T.K.; Hercule, P.; Helfrich, B.A.; Doebele, R.C.; Heasley, L.E. Rapidly acquired resistance to EGFR tyrosine kinase inhibitors in NSCLC cell lines through de-repression of FGFR2 and FGFR3 expression. PLoS ONE 2010, 5, e14117. [Google Scholar] [CrossRef]
- Strumberg, D.; Schultheis, B. Regorafenib for cancer. Expert. Opin. Investig. Drugs 2012, 21, 879–889. [Google Scholar] [CrossRef]
- Lombardi, G.; De Salvo, G.L.; Brandes, A.A.; Eoli, M.; Ruda, R.; Faedi, M.; Lolli, I.; Pace, A.; Daniele, B.; Pasqualetti, F.; et al. Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): A multicentre, open-label, randomised, controlled, phase 2 trial. Lancet Oncol. 2019, 20, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell 2007, 12, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Lan, K.H.; Zhou, X.; Tan, M.; Esteva, F.J.; Sahin, A.A.; Klos, K.S.; Li, P.; Monia, B.P.; Nguyen, N.T.; et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell 2004, 6, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Sayed, S.; Sidorova, O.A.; Hennig, A.; Augsburg, M.; Cortes Vesga, C.P.; Abohawya, M.; Schmitt, L.T.; Surun, D.; Stange, D.E.; Mircetic, J.; et al. Efficient Correction of Oncogenic KRAS and TP53 Mutations through CRISPR Base Editing. Cancer Res. 2022, 82, 3002–3015. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, Y.; Mendonca, C.A.; Yukselen, O.; Muneeruddin, K.; Ren, L.; Liang, J.; Zhou, C.; Xie, J.; Li, J.; et al. AAV-delivered suppressor tRNA overcomes a nonsense mutation in mice. Nature 2022, 604, 343–348. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GBM Cohorts | Age 1 | Age M 1 | Age F 1 | Age W 1,2 | Age B 1,2 | M:F Ratio | W:B Ratio 2 | W M:F Ratio | B M:F Ratio |
|---|---|---|---|---|---|---|---|---|---|
| Combined n = 192 | 65 | 64.5 | 68 | 66 | 62 | 1.46:1 | 4.7:1 | 1.4:1 | 1.75:1 |
| Discovery n = 89 | 62 | 62 | 63 | 62 | 63 | 1.5:1 | 4.9:1 | 1.4:1 | 4:1 |
| Validation n = 103 | 70 | 70.5 | 70 | 72 | 61 | 1.4:1 | 4.6:1 | 1.5:1 | 1:1 |
| GBM Subgroups | Cases | Age 2 | M:F Ratio | W:B 3 Ratio | W M:F Ratio | B M:F Ratio | TMB 4 |
|---|---|---|---|---|---|---|---|
| G1/EGFR | 59 | 63 (50) | 2.1:1 | 4.9:1 | 1.7:1 | 9:1 | 4.5 ± 0.2 |
| G2/FGFR3 | 5 | 62 (16) | 1:4 | W only | 1:4 | NA | 4.5 ± 1.1 |
| G3/NF1 | 43 | 65 (61) | 1.1:1 | 3.3:1 | 1.1:1 | 1.5:1 | 4.7 ± 0.2 |
| G3/MMR | 4 | 71 (17) | 3:1 | W only | 3:1 | NA | 23 ± 6.4 |
| G4/RAF | 6 | 63 (51) | 1:2 | W only | 1:2 | NA | 4.3 ± 0.9 |
| G5/PDGFRA | 13 | 68 (44) | 2.2:1 | 5:1 | 4:1 | 1:1 | 5.0 ± 0.7 |
| G6/Multi-RTK | 18 | 69 (35) | 1.6:1 | 5:1 | 1.5:1 | 2:1 | 4.4 ± 0.5 |
| G7/Other | 38 | 72 (55) | 1.4:1 | 4.3:1 | 1.5:1 | 0.75:1 | 3.7 ± 0.3 |
| GBM total sequenced 1 | 186 | 65 (66) | 1.5:1 | 4.7:1 | 1.4:1 | 1.9:1 | 4.4 ± 0.1 |
| Gene | GBM n = 186 | G1/EGFR ↑ n = 52 | G1/EGFRm n = 7 | G2/FGFR3 n = 5 | G3/NF1 n = 43 | G4/RAF n = 6 | G5/PDGFRA n = 13 | G6/Multi-RTK n = 18 | G7/Other n = 38 |
|---|---|---|---|---|---|---|---|---|---|
| TERT | 83.1 | 95.9 | 85.7 | 80.0 | 79.1 | 83.3 | 61.5 | 83.3 | 86.8 |
| PTEN | 55.9 | 51.9 | 57.1 | 80.0 | 55.8 | 66.7 | 30.8 | 61.1 | 60.5 |
| PIK3CA | 16.1 | 17.3 | 0 | 0 | 16.3 | 0 | 23.1 | 11.1 | 15.8 |
| PIK3R1 | 7.5 | 7.7 | 14.3 | 0 | 11.6 | 0 | 7.7 | 5.6 | 5.3 |
| PI3K/mTOR 2 | 75.3 | 75.0 | 57.1 | 80.0 | 76.7 | 66.7 | 53.8 | 72.2 | 84.2 |
| CDKN2A↓ | 57.8 | 76.9 * | 42.9 | 80.0 | 69.8 | 80 | 61.5 | 55.6 | 21.0 * |
| CDK4↑ 3 | 13.0 | 5.8 * | 14.3 | 0 | 0 * | 0 | 30.8 | 16.7 | 34.2 * |
| RB1 | 17.8 | 7.7 | 42.9 | 20.0 | 18.6 | 0 | 0 | 16.7 | 34.2 * |
| G1 phase 4 | 84.9 | 86.5 | 100 | 100 | 83.7 | 80.0 | 92.3 | 88.9 | 78.9 |
| TP53 | 34.4 | 17.3 * | 57.1 | 20.0 | 30.2 | 16.7 | 30.8 | 50.0 | 50.0 * |
| MDM2↑ | 5.9 | 1.9 | 0 | 20.0 | 2.3 | 0 | 15.4 | 5.6 | 13.2 * |
| MDM4↑ | 8.6 | 11.5 | 0 | 0 | 2.3 | 0 | 15.4 | 27.8 * | 7.9 |
| RPL5 | 5.4 | 5.8 | 0 | 0 | 7.0 | 16.7 | 0 | 5.6 | 2.6 |
| TP53 path 5 | 54.8 | 36.5 * | 57.1 | 40.0 | 41.9 | 33.3 | 69.2 | 83.3 * | 76.3 * |
| DDR path 6 | 28.0 | 26.9 | 14.3 | 60 | 27.9 | 16.7 | 46.1 | 22.2 | 18.4 |
| SWI/SNF 7 | 10.2 | 11.5 | 0 | 0 | 16.3 | 16.7 | 7.7 | 11.1 | 0 |
| Other ChRm 8 | 23.1 | 21.1 | 0 | 80 | 20.9 | 0 | 23.1 | 27.8 | 21.0 |
| STAG2 | 9.1 | 13.5 | 28.6 | 20 | 9.3 | 16.7 | 7.7 | 5.6 | 0 |
| Survival 9 | 9 | 11.3 | 13 | 20 | 10 | 5.3 | 10 | 9 | 5.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Georgescu, M.-M. Translation into Clinical Practice of the G1-G7 Molecular Subgroup Classification of Glioblastoma: Comprehensive Demographic and Molecular Pathway Profiling. Cancers 2024, 16, 361. https://doi.org/10.3390/cancers16020361
Georgescu M-M. Translation into Clinical Practice of the G1-G7 Molecular Subgroup Classification of Glioblastoma: Comprehensive Demographic and Molecular Pathway Profiling. Cancers. 2024; 16(2):361. https://doi.org/10.3390/cancers16020361
Chicago/Turabian StyleGeorgescu, Maria-Magdalena. 2024. "Translation into Clinical Practice of the G1-G7 Molecular Subgroup Classification of Glioblastoma: Comprehensive Demographic and Molecular Pathway Profiling" Cancers 16, no. 2: 361. https://doi.org/10.3390/cancers16020361
APA StyleGeorgescu, M.-M. (2024). Translation into Clinical Practice of the G1-G7 Molecular Subgroup Classification of Glioblastoma: Comprehensive Demographic and Molecular Pathway Profiling. Cancers, 16(2), 361. https://doi.org/10.3390/cancers16020361