

KRAS Mutations Are Associated with Shortened Survival in Patients with Epithelioid Malignant Pleural Mesothelioma
 , and
, and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Tissue Samples
2.2. Genomic DNA Extraction
2.3. Next-Generation Sequencing and Variant Calling
2.4. Statistical Analysis
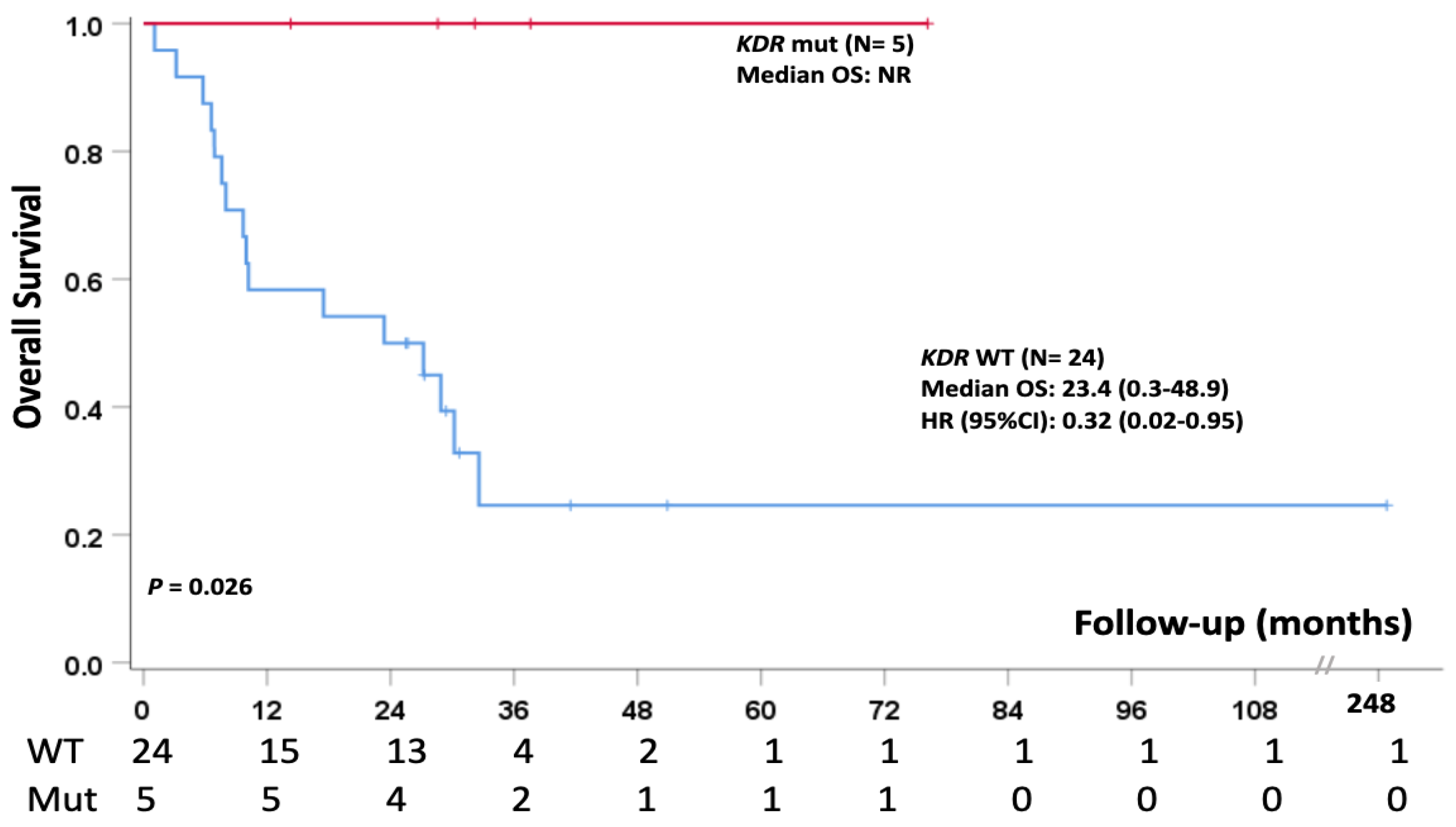
3. Results
3.1. Patients Characteristics and Clinical Outcomes
3.2. Somatic Mutations Landscape in MM
3.3. Identification of Novel EGFR Mutations
3.4. TP53 Mutations
3.5. KRAS Mutations
3.6. Single Nucleotide Polymorphism (SNP)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Travis, W.D.; Brambilla, E.; Nicholson, A.G.; Yatabe, Y.; Austin, J.H.M.; Beasley, M.B.; Chirieac, L.R.; Dacic, S.; Duhig, E.; Flieder, D.B.; et al. The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification. J. Thorac. Oncol. 2015, 10, 1243–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paajanen, J.; Laaksonen, S.; Ilonen, I.; Vehmas, T.; Mäyränpää, M.I.; Sutinen, E.; Kettunen, E.; Salo, J.A.; Räsänen, J.; Wolff, H.; et al. Clinical Features in Patients with Malignant Pleural Mesothelioma With 5-Year Survival and Evaluation of Original Diagnoses. Clin. Lung Cancer 2020, 21, e633–e639. [Google Scholar] [CrossRef] [PubMed]
- Furuya, S.; Chimed-Ochir, O.; Takahashi, K.; David, A.; Takala, J. Global Asbestos Disaster. Int. J. Environ. Res. Public Health 2018, 15, 1000. [Google Scholar] [CrossRef] [Green Version]
- International Agency for Research on Cancer. IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Man: Asbestos; International Agency for Research on Cancer: Lyon, France, 1977; Volume 14, pp. 1–106. [Google Scholar]
- Sauter, J.L.; Dacic, S.; Galateau-Salle, F.; Attanoos, R.L.; Butnor, K.J.; Churg, A.; Husain, A.N.; Kadota, K.; Khoor, A.; Nicholson, A.G.; et al. The 2021 WHO classification of tumors of the pleura: Advances since the 2015 classification. J. Thorac. Oncol. 2021, 17, 608–622. [Google Scholar] [CrossRef]
- van Meerbeeck, J.P.; Gaafar, R.; Manegold, C.; Van Klaveren, R.J.; Van Marck, E.A.; Vincent, M.; Legrand, C.; Bottomley, A.; Debruyne, C.; Giaccone, G. Randomized phase III study of cisplatin with or without raltitrexed in patients with malignant pleural mesothelioma: An intergroup study of the European Organisation for Research and Treatment of Cancer Lung Cancer Group and the National Cancer Institute of Canada. J. Clin. Oncol. 2005, 23, 6881–6889. [Google Scholar]
- Verma, V.; Ahern, C.A.; Berlind, C.G.; Lindsay, W.D.; Shabason, J.; Sharma, S.; Culligan, M.J.; Grover, S.; Friedberg, J.S.; Simone, C.B., 2nd. Survival by Histologic Subtype of Malignant Pleural Mesothelioma and the Impact of Surgical Resection on Overall Survival. Clin. Lung Cancer 2018, 19, e901–e912. [Google Scholar] [CrossRef] [PubMed]
- Sinn, K.; Mosleh, B.; Hoda, M.A. Malignant pleural mesothelioma: Recent developments. Curr. Opin. Oncol. 2021, 33, 80–86. [Google Scholar] [CrossRef]
- Janes, S.M.; Alrifai, D.; Fennell, D.A. Perspectives on the Treatment of Malignant Pleural Mesothelioma. N. Engl. J. Med. 2021, 385, 1207–1218. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): A multicentre, randomised, open-label, phase 3 trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Mielgo-Rubio, X.; Cardeña Gutiérrez, A.; Sotelo Peña, V.; Sánchez Becerra, M.V.; González López, A.M.; Rosero, A.; Trujillo-Reyes, J.C.; Couñago, F. Tsunami of immunotherapy reaches mesothelioma. World J. Clin. Oncol. 2022, 13, 267–275. [Google Scholar] [CrossRef]
- Fennell, D.A.; Dulloo, S.; Harber, J. Immunotherapy approaches for malignant pleural mesothelioma. Nat. Rev. Clin. Oncol. 2022, 19, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Tranchant, R.; Montagne, F.; Jaurand, M.C.; Jean, D. Molecular heterogeneity of malignant pleural mesotheliomas. Bull. Cancer 2018, 105, 35–45. [Google Scholar] [CrossRef]
- Panou, V.; Roe, O.D. Inherited Genetic Mutations and Polymorphisms in Malignant Mesothelioma: A Comprehensive Review. Int. J. Mol. Sci. 2020, 21, 4327. [Google Scholar] [CrossRef] [PubMed]
- Nymark, P.; Lindholm, P.M.; Korpela, M.V.; Lahti, L.; Ruosaari, S.; Kaski, S.; Hollmen, J.; Anttila, S.; Kinnula, V.L.; Knuutila, S. Gene expression profiles in asbestos-exposed epithelial and mesothelial lung cell lines. BMC Genom. 2007, 8, 62. [Google Scholar] [CrossRef] [Green Version]
- Wadowski, B.; De Rienzo, A.; Bueno, R. The Molecular Basis of Malignant Pleural Mesothelioma. Thorac. Surg. Clin. 2020, 30, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.C.; Kim, H.K.; Lee, S.; Mendez, P.; Kim, J.W.; Woodard, G.; Yoon, J.H.; Jen, K.Y.; Fang, L.T.; Jones, K.; et al. Whole exome and targeted deep sequencing identify genome-wide allelic loss and frequent SETDB1 mutations in malignant pleural mesotheliomas. Oncotarget 2016, 7, 8321–8331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, G.; Chmielecki, J.; Goparaju, C.; Heguy, A.; Dolgalev, I.; Carbone, M.; Seepo, S.; Meyerson, M.; Pass, H.I. Whole-exome sequencing reveals frequent genetic alterations in BAP1, NF2, CDKN2A, and CUL1 in malignant pleural mesothelioma. Cancer Res. 2015, 75, 264–269. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Sekido, Y. NF2/Merlin Inactivation and Potential Therapeutic Targets in Mesothelioma. Int. J. Mol. Sci. 2018, 19, 988. [Google Scholar] [CrossRef] [Green Version]
- Hmeljak, J.; Sanchez-Vega, F.; Hoadley, K.A.; Shih, J.; Stewart, C.; Heiman, D.; Tarpey, P.; Danilova, L.; Drill, E.; Gibb, E.A.; et al. Integrative Molecular Characterization of Malignant Pleural Mesothelioma. Cancer Discov. 2018, 8, 1548–1565. [Google Scholar] [CrossRef] [Green Version]
- Iacono, M.L.; Monica, V.; Righi, L.; Grosso, F.; Libener, R.; Vatrano, S.; Bironzo, P.; Novello, S.; Musmeci, L.; Volante, M.; et al. Targeted next-generation sequencing of cancer genes in advanced stage malignant pleural mesothelioma: A retrospective study. J. Thorac. Oncol. 2015, 10, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Harrison, P.T.; Vyse, S.; Huang, P.H. Rare epidermal growth factor receptor (EGFR) mutations in non small cell lung cancer. Semin. Cancer Biol. 2020, 61, 167–179. [Google Scholar] [CrossRef]
- Barta, J.A.; Pauley, K.; Kossenkov, A.V.; McMahon, S.B. The lung-enriched p53 mutants V157F and R158L/P regulate a gain of function transcriptome in lung cancer. Carcinogenesis 2020, 41, 67–77. [Google Scholar] [CrossRef]
- de Assis, L.V.; Isoldi, M.C. The function, mechanisms, and role of the genes PTEN and TP53 and the effects of asbestos in the development of malignant mesothelioma: A review focused on the genes’ molecular mechanisms. Tumour. Biol. 2014, 35, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Sementino, E.; Menges, C.W.; Kadariya, Y.; Peri, S.; Xu, J.; Liu, Z.; Wilkes, R.G.; Cai, K.Q.; Rauscher, F.J., 3rd; Klein-Szanto, A.J.; et al. Inactivation of Tp53 and Pten drives rapid development of pleural and peritoneal malignant mesotheliomas. J. Cell. Physiol. 2018, 233, 8952–8961. [Google Scholar] [CrossRef] [PubMed]
- Tian, K.; Bakker, E.; Hussain, M.; Guazzelli, A.; Alhebshi, H.; Meysami, P.; Demonacos, C.; Schwartz, J.M.; Mutti, L.; Krstic-Demonacos, M. p53 modeling as a route to mesothelioma patients stratification and novel therapeutic identification. J. Transl. Med. 2018, 16, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hylebos, M.; Van Camp, G.; van Meerbeeck, J.P.; Op de Beeck, K. The Genetic Landscape of Malignant Pleural Mesothelioma: Results from Massively Parallel Sequencing. J. Thorac. Oncol. 2016, 11, 1615–1626. [Google Scholar] [CrossRef] [Green Version]
- Forde, P.M.; Anagnostou, V.; Sun, Z.; Dahlberg, S.E.; Kindler, H.L.; Niknafs, N.; Purcell, T.; Santana-Davila, R.; Dudek, A.Z.; Borghaei, H.; et al. Durvalumab with platinum-pemetrexed for unresectable pleural mesothelioma: Survival, genomic and immunologic analyses from the phase 2 PrE0505 trial. Nat. Med. 2021, 27, 1910–1920. [Google Scholar] [CrossRef] [PubMed]
- Hiltbrunner, S.; Fleischmann, Z.; Sokol, E.S.; Zoche, M.; Felley-Bosco, E.; Curioni-Fontecedro, A. Genomic landscape of pleural and peritoneal mesothelioma tumours. Br. J. Cancer. 2022, 127, 1997–2005. [Google Scholar] [CrossRef]
- Pagano, M.; Ceresoli, L.; Zucali, P.; Pasello, G.; Garassino, M.; Grosso, F.; Tiseo, M.; Parra, H.S.; Zanelli, F.; Cappuzzo, F.; et al. Mutational Profile of Malignant Pleural Mesothelioma (MPM) in the Phase II RAMES Study. Cancers 2020, 12, 2948. [Google Scholar] [CrossRef]
- Chia, P.L.; Scott, A.M.; John, T. Epidermal growth factor receptor (EGFR)-targeted therapies in mesothelioma. Expert Opin. Drug Deliv. 2019, 16, 441–451. [Google Scholar] [CrossRef]
- Agama, N.; Yasuda, Y.; Ozasa, H. Malignant Pleural Mesothelioma Harboring Both G719C and S768I Mutations of EGFR Successfully Treated with Afatinib. J. Thorac. Oncol. 2017, 12, e141–e143. [Google Scholar]
- Tallet, A.; Nault, J.-C.; Renier, A.; Hysi, I.; Galateau-Sallé, F.; Cazes, A.; Copin, M.-C.; Hofman, P.; Andujar, P.; Le Pimpec-Barthes, F.; et al. Overexpression and promoter mutation of the TERT gene in malignant pleural mesothelioma. Oncogene 2014, 33, 3748–3752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cakiroglu, E.; Senturk, S. Genomics and Functional Genomics of Malignant Pleural Mesothelioma. Int. J. Mol. Sci. 2020, 21, 6342. [Google Scholar] [CrossRef] [PubMed]
- Na, B.; Yu, X.; Withers, T.; Gilleran, J.; Yao, M.; Foo, T.K.; Chen, C.; Moore, D.; Lin, Y.; Kimball, S.D.; et al. Therapeutic targeting of BRCA1 and TP53 mutant breast cancer through mutant p53 reactivation. NPJ. Breast Cancer 2019, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Destro, A.; Ceresoli, G.L.; Falleni, M.; Zucali, P.A.; Morenghi, E.; Bianchi, P.; Pellegrini, C.; Cordani, N.; Vaira, V.; Alloisio, M.; et al. EGFR overexpression in malignant pleural mesothelioma. An immunohistochemical and molecular study with clinico-pathological correlations. Lung Cancer 2006, 51, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Mezzapelle, R.; Miglio, U.; Rena, O.; Paganotti, A.; Allegrini, S.; Antona, J.; Molinari, F.; Frattini, M.; Monga, G.; Alabiso, O.; et al. Mutation analysis of the EGFR gene and downstream signalling pathway in histologic samples of malignant pleural mesothelioma. Br. J. Cancer 2013, 108, 1743–1749. [Google Scholar] [CrossRef] [Green Version]
- Govindan, R.; Kratzke, R.A.; Herndon, J.E., 2nd; Niehans, G.A.; Vollmer, R.; Watson, D.; Green, M.R.; Kindler, H.L.; Cancer and Leukemia Group B. Gefitinib in patients with malignant mesothelioma: A phase II study by the Cancer and Leukemia Group B. Clin. Cancer Res. 2005, 11, 2300–2304. [Google Scholar] [CrossRef] [Green Version]
- Schildgen, V.; Pabst, O.; Tillmann, R.L.; Lusebrink, J.; Schildgen, O.; Ludwig, C.; Brockmann, M.; Stoelben, E. Low frequency of EGFR mutations in pleural mesothelioma patients, Cologne, Germany. Appl. Immunohistochem. Mol. Morphol. 2015, 23, 118–125. [Google Scholar] [CrossRef]
- Shukuya, T.; Serizawa, M.; Watanabe, M.; Akamatsu, H.; Abe, M.; Imai, H.; Tokito, T.; Ono, A.; Taira, T.; Kenmotsu, H.; et al. Identification of actionable mutations in malignant pleural mesothelioma. Lung Cancer 2014, 86, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, Z.; Wang, J.; Zhang, M.; Li, Z.; Wang, S.; Dong, B.; Zhang, C.; Gao, J.; Shen, L. Mouse avatar models of esophageal squamous cell carcinoma proved the potential for EGFR-TKI afatinib and uncovered Src family kinases involved in acquired resistance. J. Hematol. Oncol. 2018, 11, 109. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Kim, D.; Hong, Y.S.; Kim, K.P.; Yoon, Y.K.; Lee, D.H.; Kim, S.W.; Chun, S.M.; Jang, S.J.; Kim, T.W. Mutational Profiling of Malignant Mesothelioma Revealed Potential Therapeutic Targets in EGFR and NRAS. Transl. Oncol. 2018, 11, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Trassl, L.; Stathopoulos, G.T. KRAS Pathway Alterations in Malignant Pleural Mesothelioma: An Underestimated Player. Cancers 2022, 14, 4303. [Google Scholar] [CrossRef]
- Marazioti, A.; Krontira, A.C.; Behrend, S.J.; Giotopoulou, G.A.; Ntaliarda, G.; Blanquart, C.; Bayram, H.; Iliopoulou, M.; Vreka, M.; Trassl, L.; et al. KRAS signaling in malignant pleural mesothelioma. EMBO Mol. Med. 2022, 14, e13631. [Google Scholar] [CrossRef]
- Duzkopru, Y.; Oruc, Z.; Kaplan, M.A.; Ulku, R.; Tanrikulu, C.; Esmer, D.; Birak, A.; Kucukoner, M.; Urakci, Z.; Isikdogan, A. The importance of serum and pleural fluid level of vascular endothelial growth factor (VEGF) and VEGF fluid/serum ratio in the differential diagnosis of malignant mesothelioma-related pleural effusion. Contemp. Oncol. 2017, 21, 213–217. [Google Scholar]
- Hirayama, N.; Tabata, C.; Tabata, R.; Maeda, R.; Yasumitsu, A.; Yamada, S.; Kuribayashi, K.; Fukuoka, K.; Nakano, T. Pleural effusion VEGF levels as a prognostic factor of malignant pleural mesothelioma. Respir. Med. 2011, 105, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Catalano, A.; Lazzarini, R.; Di Nuzzo, S.; Orciari, S.; Procopio, A. The plexin-A1 receptor activates vascular endothelial growth factor-receptor 2 and nuclear factor-kappaB to mediate survival and anchorage-independent growth of malignant mesothelioma cells. Cancer Res. 2009, 69, 1485–1493. [Google Scholar] [CrossRef] [Green Version]
- Geng, N.; Su, J.; Liu, Z.; Ding, C.; Xie, S.; Hu, W. The Influence of KDR Genetic Variation on the Efficacy and Safety of Patients with Advanced NSCLC Receiving First-Line Bevacizumab Plus Chemotherapy Regimen. Technol Cancer Res. Treat 2021, 20, 15330338211019433. [Google Scholar] [CrossRef]
- Bai, M.; Li, Z.G.; Ba, Y. Influence of KDR Genetic Variation on the Efficacy and Safety of Patients with Chemotherapy Refractory Metastatic CRC Who Received Apatinib Treatment. Int. J. Gen. Med. 2021, 14, 1041–1055. [Google Scholar] [CrossRef]
- Zheng, Y.B.; Huang, J.W.; Zhan, M.X.; Zhao, W.; Liu, B.; He, X.; Li, Y.; Hu, B.S.; Lu, L.G. Genetic variants in the KDR gene is associated with the prognosis of transarterial chemoembolization treated hepatocellular carcinoma. Tumour. Biol. 2014, 35, 11473–11481. [Google Scholar] [CrossRef] [PubMed]
- Tinhofer, I.; Stenzinger, A.; Eder, T.; Konschak, R.; Niehr, F.; Endris, V.; Distel, L.; Hautmann, M.G.; Mandic, R.; Stromberger, C.; et al. Targeted next-generation sequencing identifies molecular subgroups in squamous cell carcinoma of the head and neck with distinct outcome after concurrent chemoradiation. Ann. Oncol. 2016, 27, 2262–2268. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Jacobson, B.A.; De, A.; Frizelle, S.P.; Janne, P.; Thumma, S.C.; Whitson, B.A.; Farassati, F.; Kratzke, R.A. Ras pathway activation in malignant mesothelioma. J. Thorac. Oncol. 2007, 2, 789–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Patients. ID | Sex | Age | Alive at the Time of the Study | OS (Month) | Type of Specimen | Pathological Stage (pT) | Grading (Sec. WHO, 2021) |
|---|---|---|---|---|---|---|---|
| 1 | Male | 73 | No | 10 | Pleurectomy | pT1 | 2 |
| 2 | Male | 81 | No | 28.9 | Biopsy | pT1 | 1 |
| 3 | Male | 67 | No | 7.6 | Biopsy | pT1 | 1 |
| 4 | Female | 72 | Yes | 76.2 | Pleurectomy | pT1 | 1 |
| 5 | Male | 68 | No | 5.8 | Pleurectomy | pT1 | 2 |
| 6 | Male | 64 | Yes | 50.9 | Biopsy | pT1 | 2 |
| 7 | Male | 73 | No | 27.2 | Biopsy | pT1 | 1 |
| 8 | Male | 72 | No | 23.4 | Biopsy | pT1 | 2 |
| 9 | Male | 63 | No | 37.6 | Biopsy | pT1 | 1 |
| 10 | Male | 70 | Yes | 41.5 | Pleurectomy | pT1 | 1 |
| 11 | Female | 73 | No | 9.7 | Biopsy | pT1 | 3 |
| 12 | Male | 69 | No | 17.5 | Biopsy | pT1 | 1 |
| 13 | Male | 70 | No | 32.2 | Biopsy | pT1 | 1 |
| 14 | Male | 63 | No | 30.7 | Pleurectomy | pT1 | 1 |
| 15 | Female | 65 | Yes | 29.4 | Biopsy | pT1 | 2 |
| 16 | Male | 76 | No | 28.6 | Biopsy | pT1 | 1 |
| 17 | Male | 67 | No | 27.3 | Biopsy | pT1 | 2 |
| 18 | Male | 74 | Yes | 248.8 | Biopsy | pT1 | 1 |
| 19 | Male | 70 | No | 25.7 | Biopsy | pT1 | 1 |
| 20 | Male | 70 | No | 25.5 | Biopsy | pT1 | 1 |
| 21 | Male | 83 | No | 14.3 | Biopsy | pT1 | 2 |
| 22 | Male | 82 | Yes | 6.9 | Biopsy | pT1 | 3 |
| 23 | Male | 84 | No | 32.6 | Biopsy | pT1 | 1 |
| 24 | Male | 71 | No | 6.6 | Biopsy | pT1 | 1 |
| 25 | Male | 69 | Yes | 8 | Pleurectomy | pT1 | 2 |
| 26 | Female | 71 | Yes | 30.2 | Biopsy | pT1 | 1 |
| 27 | Male | 67 | No | 1.1 | Biopsy | pT1 | 3 |
| 28 | Male | 69 | No | 3.2 | Biopsy | pT1 | 1 |
| 29 | Male | 80 | No | 10.2 | Biopsy | pT1 | 2 |
| Gene | Exon | cHGVS | pHGVS | COSMIC_ID | VAF | Functions |
|---|---|---|---|---|---|---|
| KRAS | 2 | c.38G>C | p.G13A | COSM533 | 33.5 | missense |
| 2 | c.50G>A | p.S17N | COSM51382 | 6.36 | missense | |
| 3 | c.169G>A | p.D57N | COSM166779 | 4.6 | missense | |
| 3 | c.143G>A | p.G48E | 4.2 | missense | ||
| 3 | c.142G>A | p.G48R | 3.1 | missense | ||
| EGFR | 19 | c.2320G>A | p.V774M | COSM13006 | 13.5 | missense |
| 18 | c.2155G>A | p.G719S | COSM6252 | 4.4 | missense | |
| 18 | c.2105C>A | p.A702D | COSM3266631 | 5.5 | missense | |
| 19 | c.2252C>T | p.T751I | COSM13185 | 11.8 | missense | |
| 21 | c.2596G>A | p.E866K | COSM13198 | 9.5 | missense | |
| 19 | c.2255C>G | p.S752C | 7.7 | missense | ||
| 19 | c.2255C>A | p.S752Y | COSM13186 | 5.1 | missense | |
| 18 | c.2080C>T | p.P694T | COSM13179 | 5.6 | missense | |
| TP53 | 5 | c.524G>A | p.R175H | COSM10648 | 32.6 | missense |
| 5 | c.454C>T | p.P152S | COSM43582 | 9.6 | missense | |
| 6 | c.637C>T | p.R213* | COSM99618 | 5.7 | nonsense | |
| 6 | c.574C>T | p.Q192* | COSM10733 | 5.5 | nonsense | |
| 6 | c.644G>A | p.S215N | COSM744739 | 4.3 | missense | |
| 6 | c.672 + 1G>A | p.? | COSM6906 | 4.7 | stop-loss | |
| MET | 14 | c.3268G>A | p.G1090S | 5.2 | missense | |
| 3 | c.1113C>G | p.N371K | 40.2 | missense | ||
| CTNNB1 | 4 | c.100G>C | p.G34R | COSM5685 | 4.9 | missense |
| FGFR3 | 10 | c.1138G>C | p.G380R | COSM9276948 | 5.8 | missense |
| 15 | c.1921G>A | p.D641N | COSM6953958 | 6.2 | missense | |
| BRAF | 11 | c.1390G>A | p.G464R | COSM1111 | 8.5 | missense |
| STK11 | 4 | c.182G>A | p.G61D | COSM6191357 | 3.8 | missense |
| TSC2 | 23 | c.2714G>A | p.R905Q | COSM4736821 | 7.9 | missense |
| FLT3 | 20 | c.2464G>T | p.G822W | COSM3885171 | 5.1 | missense |
| AKT1 | 1 | c.152C>T | p.P51L | COSM4468165 | 13.2 | missense |
| HNF1A | 3 | c.599G>A | p.R200Q | COSM3457098 | 14.5 | missense |
| 3 | c.607C>T | p.R203C | COSM24915 | 5.9 | missense | |
| PTEN | 6 | c.733C>T | p.Q245* | COSM5159 | 6 | nonsense |
| RB1 | 20 | c.2048T>C | p.L683P | 4.7 | missense | |
| CDH1 | 7 | c.1081G>A | p.A361T | 5.2 | missense | |
| MAP2K1 | 7 | c.709G>T | p.G237W | 8.17 | missense | |
| KIT | 17 | c.2473G>A | p.V825I | COSM19110 | 12.5 | missense |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vannucchi, M.; Pennati, V.; Mencaroni, C.; Defraia, C.; Bardhi, L.; Castiglione, F.; Bellan, C.; Comin, C.E. KRAS Mutations Are Associated with Shortened Survival in Patients with Epithelioid Malignant Pleural Mesothelioma. Cancers 2023, 15, 2072. https://doi.org/10.3390/cancers15072072
Vannucchi M, Pennati V, Mencaroni C, Defraia C, Bardhi L, Castiglione F, Bellan C, Comin CE. KRAS Mutations Are Associated with Shortened Survival in Patients with Epithelioid Malignant Pleural Mesothelioma. Cancers. 2023; 15(7):2072. https://doi.org/10.3390/cancers15072072
Chicago/Turabian StyleVannucchi, Margherita, Veronica Pennati, Clelia Mencaroni, Chiara Defraia, Ledi Bardhi, Francesca Castiglione, Cristiana Bellan, and Camilla Eva Comin. 2023. "KRAS Mutations Are Associated with Shortened Survival in Patients with Epithelioid Malignant Pleural Mesothelioma" Cancers 15, no. 7: 2072. https://doi.org/10.3390/cancers15072072