
Anticarcinogenic Potency of EF24: An Overview of Its Pharmacokinetics, Efficacy, Mechanism of Action, and Nanoformulation for Drug Delivery
 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Structurally Related Mechanisms of Action
1.2. Lipophilic Properties
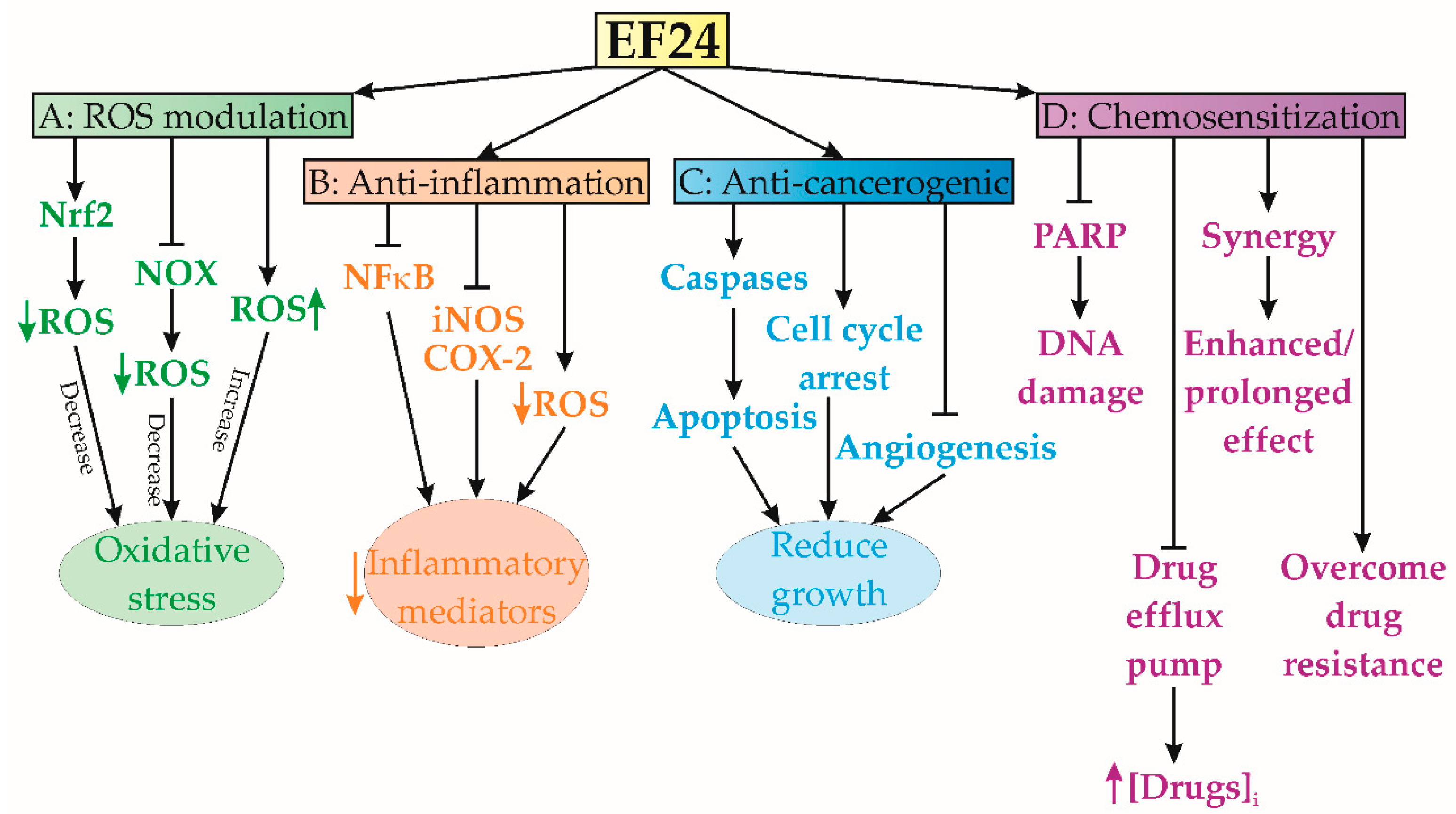
1.3. EF24-Mediated ROS Modulation
1.4. Anti-Inflammatory Effects
1.5. Anticarcinogenic Activity
1.6. Chemosensitization Characteristics
2. Pharmacokinetics of EF24
2.1. Absorption of EF24
2.2. Distribution of EF24
2.3. Metabolism of EF24
2.4. Elimination of EF24
2.5. Cytotoxicity of EF24
3. Antitumorigenic Effects and Mechanisms
3.1. Adrenocortical Carcinoma
3.2. Oral Squamous Cell Carcinoma
3.3. Nasopharyngeal Carcinoma
3.4. Breast Cancer
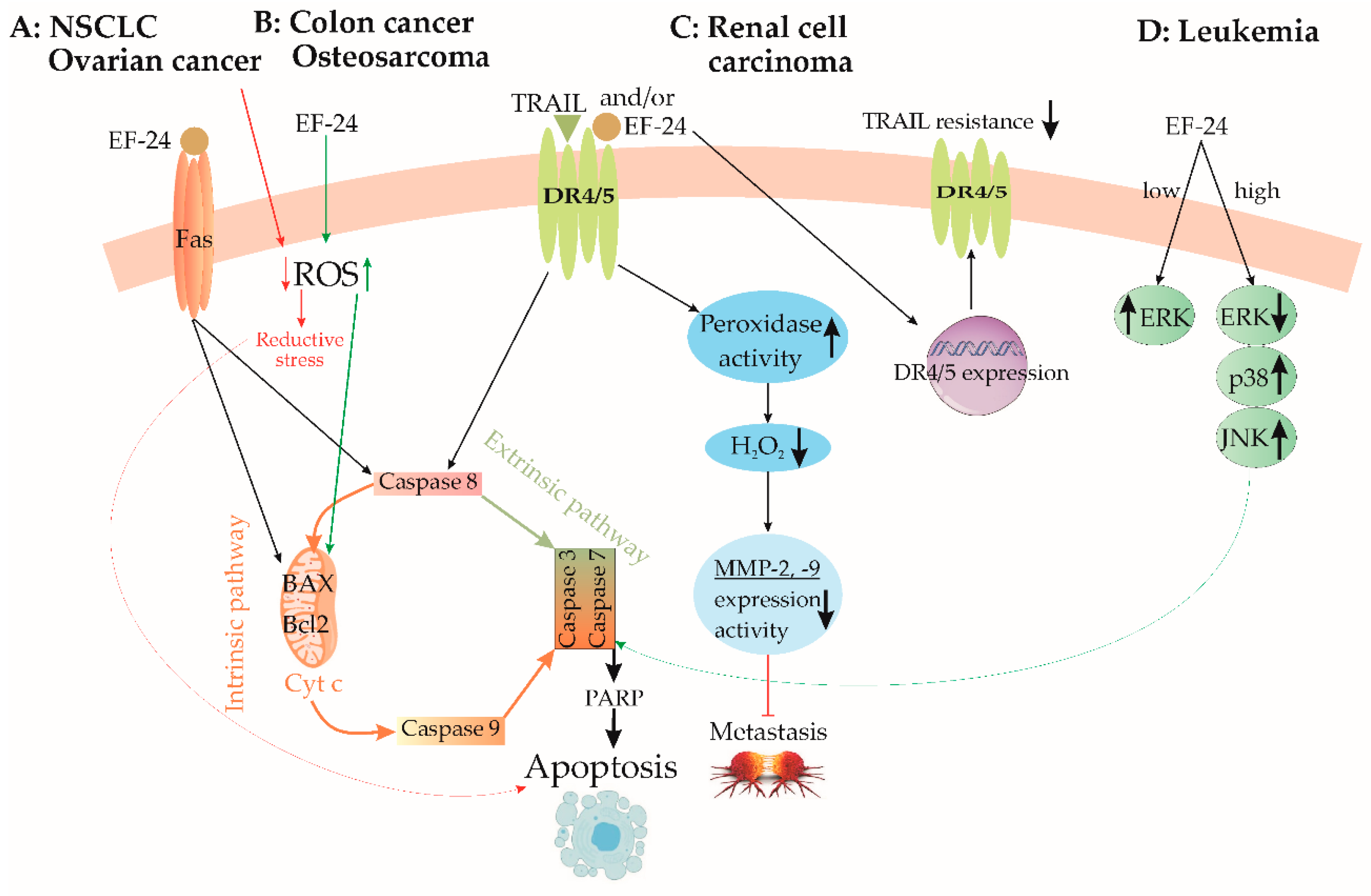
3.5. Lung Cancer
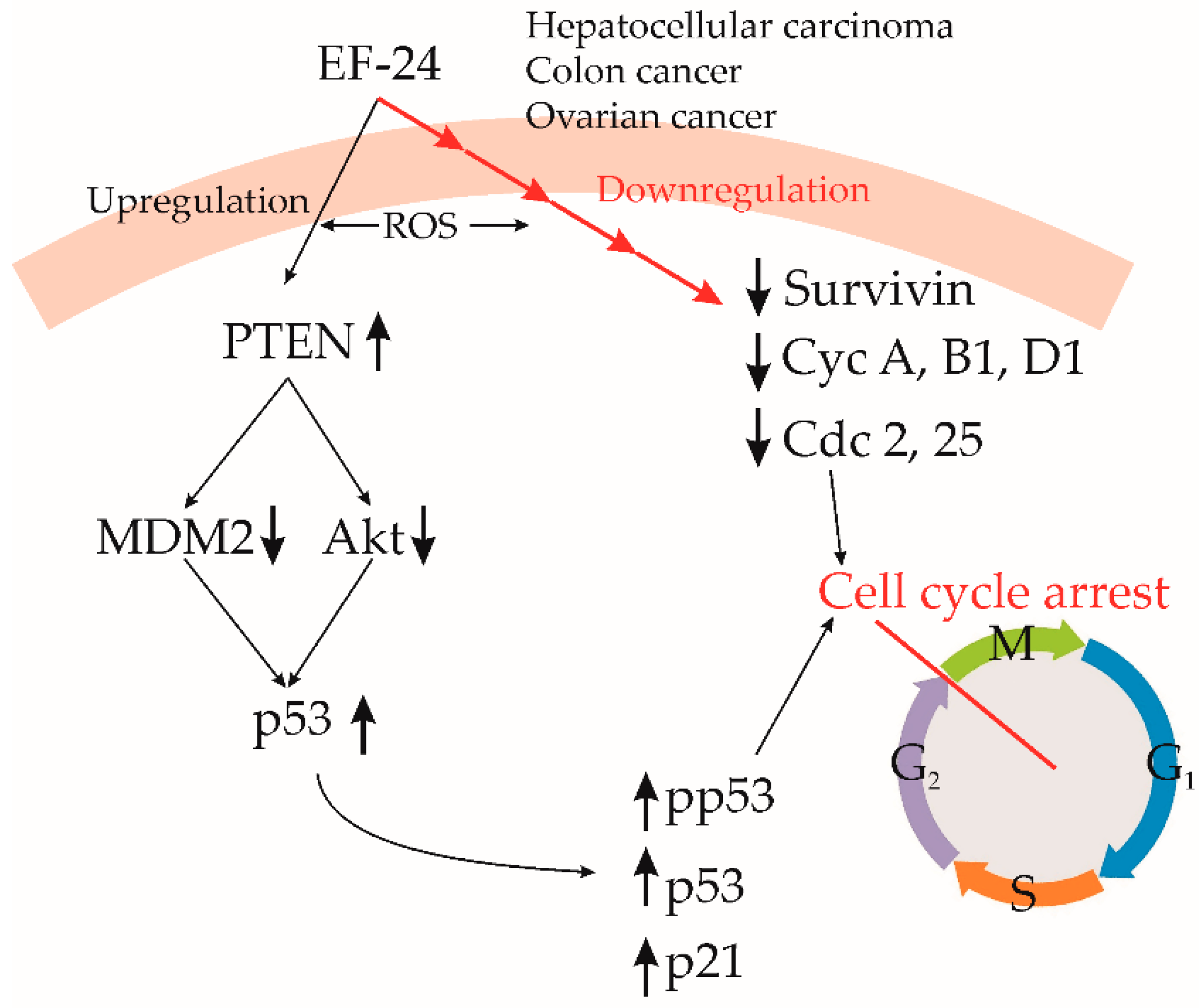
3.6. Hepatocellular Carcinoma
3.7. Gastric Cancer
3.8. Colon Cancer
3.9. Renal Cell Carcinoma
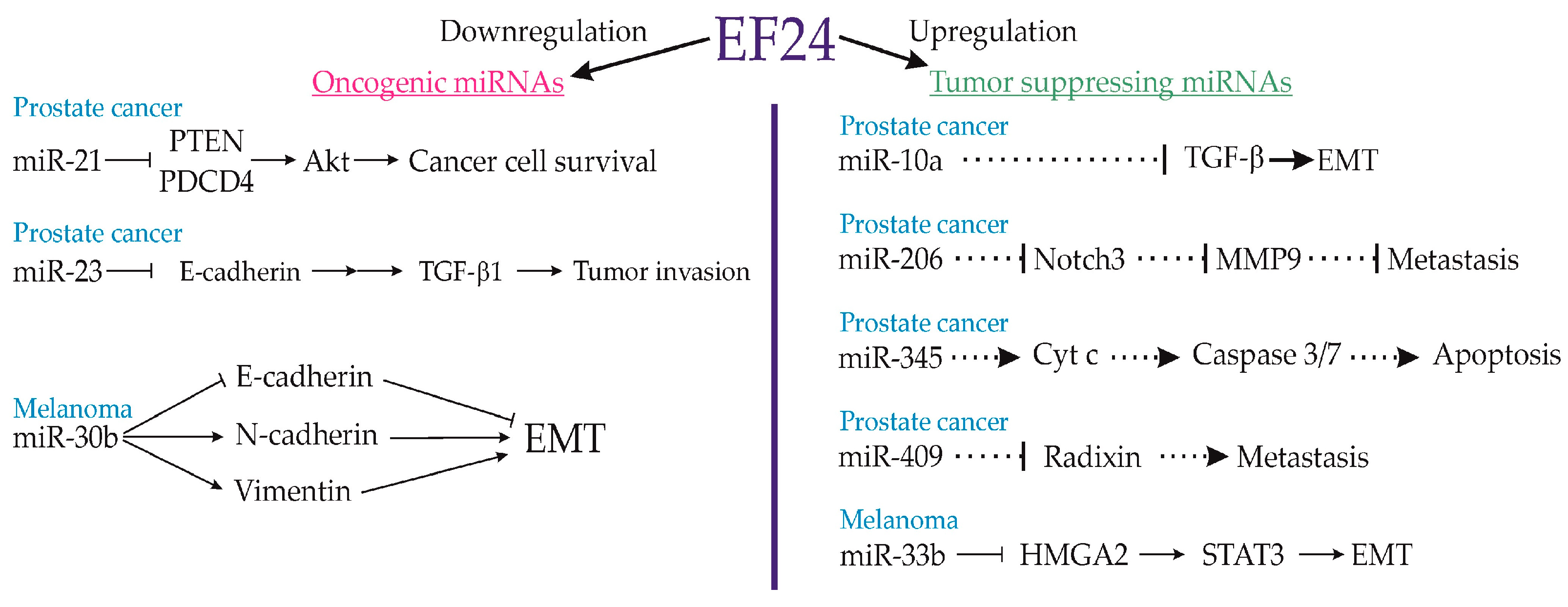
3.10. Prostate Cancer
3.11. Thyroid Carcinoma
3.12. Ovarian Cancer
3.13. Osteosarcoma
3.14. Neuroblastoma
3.15. Leukemia
3.16. Melanoma
3.17. Antiangiogenic Effects of EF24
4. EF24 Derivatives and Drug Delivery Systems
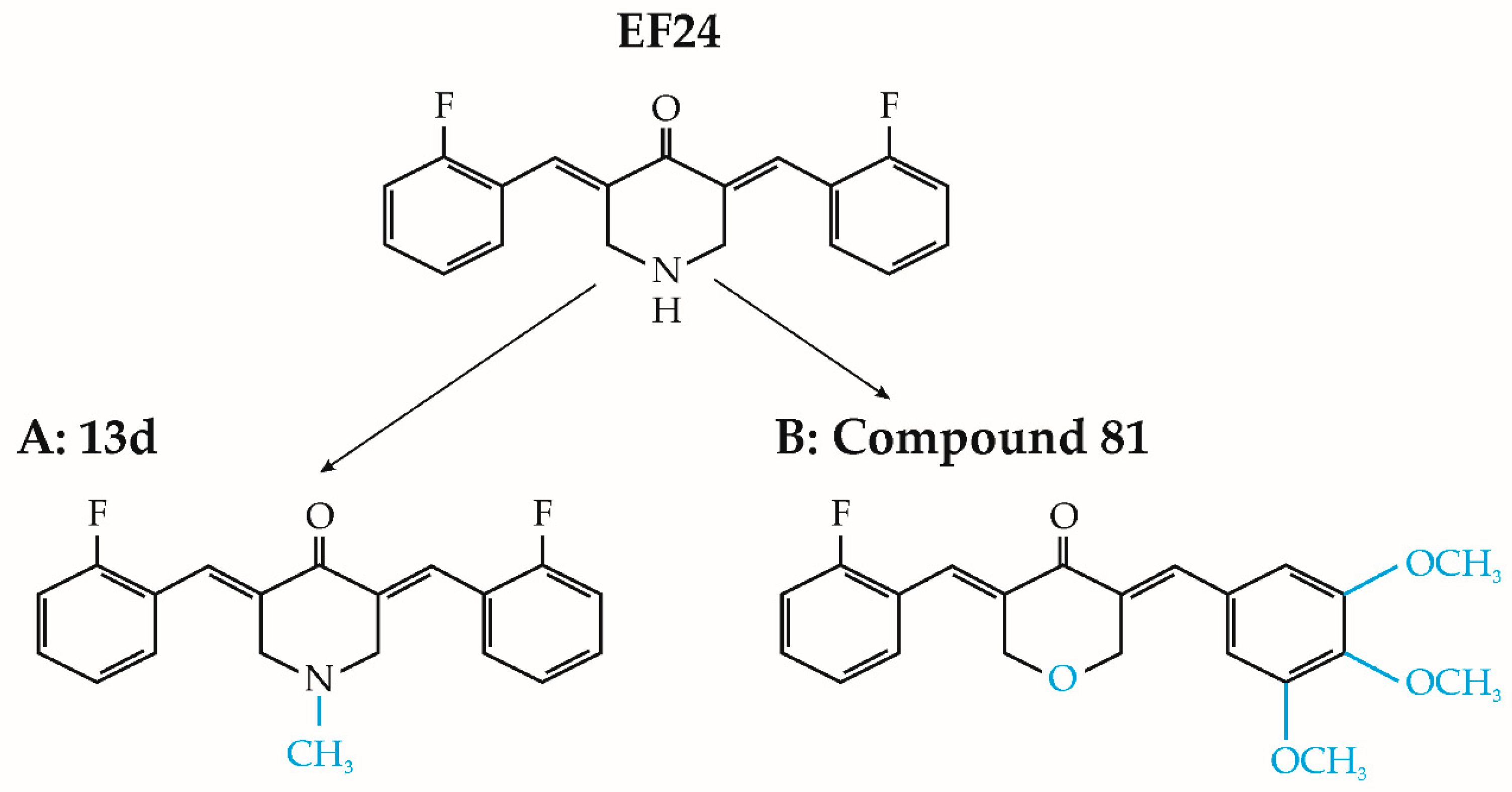
4.1. EF24 Derivatives
4.2. EF24 Drug Delivery Systems
5. Data and Notes from Preclinical Studies Pertaining to EF24
- EF24 has been designed to enhance its bioavailability, making it easier for the body to absorb and utilize.
- EF24 is designed to be more stable than CUR, ensuring a longer shelf life and improved effectiveness in various formulations.
- EF24 may exhibit a higher degree of specificity towards cancer cells, leading to reduced damage to healthy cells.
- EF24 has been reported to have superior anti-inflammatory effects in some studies.
- EF24 is known for its potent inhibition of the NF-κB signaling pathway, which is associated with inflammation, immunity, and cancer.
- EF24 may enhance the effectiveness of other cancer treatments when used in combination with other drugs.
- EF24 can be incorporated into drug delivery systems to further improve its delivery to specific tissues or cells.
- Researchers can modify the structure of EF24 to target specific conditions or diseases, making it a versatile compound for drug development.
- EF24 faces challenges due to its poor aqueous solubility and quick degradation in biological environments.
6. Future Directions
7. Conclusions
Limitations
Author Contributions
Funding
Conflicts of Interest
References
- He, Y.; Li, W.; Hu, G.; Sun, H.; Kong, Q. Bioactivities of Ef24, a Novel Curcumin Analog: A Review. Front. Oncol. 2018, 8, 614. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.K.; Cai, J.; Armstrong, J.; Herold, M.; Lu, Y.J.; Sun, A.; Snyder, J.P.; Liotta, D.C.; Jones, D.P.; Shoji, M. EF24, a Novel Synthetic Curcumin Analog, Induces Apoptosis in Cancer Cells via a Redox-Dependent Mechanism. Anti-Cancer Drugs 2005, 16, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Lu, Y.J.; Hu, H.; Shoji, M.; Liotta, D.C.; Snyder, J.P. Curcumin Analog Cytotoxicity against Breast Cancer Cells: Exploitation of a Redox-Dependent Mechanism. Bioorganic Med. Chem. Lett. 2009, 19, 6627–6631. [Google Scholar] [CrossRef]
- Hadzi-Petrushev, N.; Angelovski, M.; Rebok, K.; Mitrokhin, V.; Kamkin, A.; Mladenov, M. Antioxidant and Anti-inflammatory Effects of the Monocarbonyl Curcumin Analogs B2BRBC and C66 in Monocrotaline-induced Right Ventricular Hypertrophy. J. Biochem. Mol. Tox. 2019, 33, e22353. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, M.; Bogdanov, J.; Bogdanov, B.; Hadzi-Petrushev, N.; Kamkin, A.; Stojchevski, R.; Avtanski, D. Efficacy of Monocarbonyl Curcumin Analogs C66 in the reduction od diabetes associated cardiovascular and kidney complications. Mol. Med. 2022, 28, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Josifovska, S.; Panov, S.; Hadzi-Petrushev, N.; Mitrokhin, V.; Kamkin, A.; Stojchevski, R.; Avtanski, D.; Mladenov, M. Positive Tetrahydrocurcumin-Associated Brain-Related Metabolomic Implications. Molecules 2023, 28, 3734. [Google Scholar] [CrossRef]
- Zhang, L.; Li, C.; Wang, S.; Avtanski, D.; Hadzi-Petrushev, N.; Mitrokhin, V.; Mladenov, M.; Wang, F. Tetrahydrocurcumin-Related Vascular Protection: An Overview of the Findings from Animal Disease Models. Molecules 2022, 27, 5100. [Google Scholar] [CrossRef]
- Hadzi-Petrushev, N.; Bogdanov, J.; Krajoska, J.; Ilievska, J.; Bogdanova-Popov, B.; Gjorgievska, E.; Mitrokhin, V.; Sopi, R.; Gagov, H.; Kamkin, A.; et al. Comparative Study of the Antioxidant Properties of Monocarbonyl Curcumin Analogues C66 and B2BrBC in Isoproteranol Induced Cardiac Damage. Life Sci. 2018, 197, 10–18. [Google Scholar] [CrossRef]
- Tatsuzaki, J.; Bastow, K.F.; Nakagawa-Goto, K.; Nakamura, S.; Itokawa, H.; Lee, K.-H. Dehydrozingerone, Chalcone, and Isoeugenol Analogues as in Vitro Anticancer Agents. J. Nat. Prod. 2006, 69, 1445–1449. [Google Scholar] [CrossRef]
- Subramaniam, D.; May, R.; Sureban, S.M.; Lee, K.B.; George, R.; Kuppusamy, P.; Ramanujam, R.P.; Hideg, K.; Dieckgraefe, B.K.; Houchen, C.W.; et al. Diphenyl Difluoroketone: A Curcumin Derivative with Potent In Vivo Anticancer Activity. Cancer Res. 2008, 68, 1962–1969. [Google Scholar] [CrossRef]
- Abd El-Hack, M.E.; El-Saadony, M.T.; Swelum, A.A.; Arif, M.; Abo Ghanima, M.M.; Shukry, M.; Noreldin, A.; Taha, A.E.; El-Tarabily, K.A. Curcumin, the Active Substance of Turmeric: Its Effects on Health and Ways to Improve Its Bioavailability. J. Sci. Food Agric. 2021, 101, 5747–5762. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-Y.; Chen, C.-W.; Chen, L.-K.; Chou, H.-Y.; Chang, C.-L.; Juan, C.-C. Curcumin Attenuates Adipogenesis by Inducing Preadipocyte Apoptosis and Inhibiting Adipocyte Differentiation. Nutrients 2019, 11, 2307. [Google Scholar] [CrossRef] [PubMed]
- Reid, J.M.; Buhrow, S.A.; Gilbert, J.A.; Jia, L.; Shoji, M.; Snyder, J.P.; Ames, M.M. Mouse Pharmacokinetics and Metabolism of the Curcumin Analog, 4-Piperidinone,3,5-Bis[(2-Fluorophenyl)Methylene]-Acetate(3E,5E) (EF-24; NSC 716993). Cancer Chemother. Pharmacol. 2014, 73, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Bisht, S.; Schlesinger, M.; Rupp, A.; Schubert, R.; Nolting, J.; Wenzel, J.; Holdenrieder, S.; Brossart, P.; Bendas, G.; Feldmann, G. A Liposomal Formulation of the Synthetic Curcumin Analog EF24 (Lipo-EF24) Inhibits Pancreatic Cancer Progression: Towards Future Combination Therapies. J. Nanobiotechnol. 2016, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Shang, M.; Yuan, F.; Guo, W.; Wang, C. EF24 Exerts Cytotoxicity against NSCLC via Inducing ROS Accumulation. Cancer Cell Int. 2021, 21, 531. [Google Scholar] [CrossRef]
- Ibáñez Gaspar, V.; McMorrow, T. The Curcuminoid EF24 in Combination with TRAIL Reduces Human Renal Cancer Cell Migration by Decreasing MMP-2/MMP-9 Activity through a Reduction in H2O2. Int. J. Mol. Sci. 2023, 24, 1043. [Google Scholar] [CrossRef]
- Yadav, V.R.; Sahoo, K.; Roberts, P.R.; Awasthi, V. Pharmacologic Suppression of Inflammation by a Diphenyldifluoroketone, EF24, in a Rat Model of Fixed-Volume Hemorrhage Improves Survival. J. Pharmacol. Exp. Ther. 2013, 347, 346–356. [Google Scholar] [CrossRef]
- Yousefian, M.; Shakour, N.; Hosseinzadeh, H.; Hayes, A.W.; Hadizadeh, F.; Karimi, G. The Natural Phenolic Compounds as Modulators of NADPH Oxidases in Hypertension. Phytomedicine 2019, 55, 200–213. [Google Scholar] [CrossRef]
- Roy, D.; Kabiraj, P.; Pal, R. EF24 Prevents Rotenone-Induced Estrogenic Status Alteration in Breast Cancer: Effects of Rotenone and EF24 in Breast Cancer Model. Cell Biol. Int. 2014, 38, 511–519. [Google Scholar] [CrossRef]
- Tan, X.; Sidell, N.; Mancini, A.; Huang, R.-P.; Wang, S.; Horowitz, I.R.; Liotta, D.C.; Taylor, R.N.; Wieser, F. Multiple Anticancer Activities of EF24, a Novel Curcumin Analog, on Human Ovarian Carcinoma Cells. Reprod. Sci. 2010, 17, 931–940. [Google Scholar] [CrossRef]
- Bakalova, R.; Zhelev, Z.; Shibata, S.; Nikolova, B.; Aoki, I.; Higashi, T. Impressive Suppression of Colon Cancer Growth by Triple Combination SN38/EF24/Melatonin: “Oncogenic” Versus “Onco-Suppressive” Reactive Oxygen Species. Anticancer Res. 2017, 37, 5449–5458. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Xia, Y.; Chen, W.; Chen, X.; Ying, S.; Feng, Z.; Chen, T.; Ye, Q.; Wang, Z.; Qiu, C.; et al. EF24 Induces ROS-Mediated Apoptosis via Targeting Thioredoxin Reductase 1 in Gastric Cancer Cells. Oncotarget 2016, 7, 18050–18064. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zou, P.; Zhao, Z.; Chen, X.; Fan, X.; Vinothkumar, R.; Cui, R.; Wu, F.; Zhang, Q.; Liang, G.; et al. Synergistic Antitumor Activity of Rapamycin and EF24 via Increasing ROS for the Treatment of Gastric Cancer. Redox Biol. 2016, 10, 78–89. [Google Scholar] [CrossRef]
- Chen, X.; Dai, X.; Zou, P.; Chen, W.; Rajamanickam, V.; Feng, C.; Zhuge, W.; Qiu, C.; Ye, Q.; Zhang, X.; et al. Curcuminoid EF24 Enhances the Anti-tumour Activity of Akt Inhibitor MK-2206 through ROS-mediated Endoplasmic Reticulum Stress and Mitochondrial Dysfunction in Gastric Cancer. Br. J. Pharmacol. 2017, 174, 1131–1146. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Feng, C.; Vinothkumar, R.; Chen, W.; Dai, X.; Chen, X.; Ye, Q.; Qiu, C.; Zhou, H.; Wang, Y.; et al. Curcumin Analog EF24 Induces Apoptosis via ROS-Dependent Mitochondrial Dysfunction in Human Colorectal Cancer Cells. Cancer Chemother. Pharmacol. 2016, 78, 1151–1161. [Google Scholar] [CrossRef]
- Liu, H.; Liang, Y.; Wang, L.; Tian, L.; Song, R.; Han, T.; Pan, S.; Liu, L. In Vivo and In Vitro Suppression of Hepatocellular Carcinoma by EF24, a Curcumin Analog. PLoS ONE 2012, 7, e48075. [Google Scholar] [CrossRef]
- Yang, C.H.; Yue, J.; Sims, M.; Pfeffer, L.M. The Curcumin Analog EF24 Targets NF-κB and miRNA-21, and Has Potent Anticancer Activity In Vitro and In Vivo. PLoS ONE 2013, 8, e71130. [Google Scholar] [CrossRef]
- Vilekar, P.; Awasthi, S.; Natarajan, A.; Anant, S.; Awasthi, V. EF24 Suppresses Maturation and Inflammatory Response in Dendritic Cells. Int. Immunol. 2012, 24, 455–464. [Google Scholar] [CrossRef]
- Lin, H.; Chen, X.; Zhang, C.; Yang, T.; Deng, Z.; Song, Y.; Huang, L.; Li, F.; Li, Q.; Lin, S.; et al. EF24 Induces Ferroptosis in Osteosarcoma Cells through HMOX1. Biomed. Pharmacother. 2021, 136, 111202. [Google Scholar] [CrossRef]
- Selvendiran, K.; Tong, L.; Vishwanath, S.; Bratasz, A.; Trigg, N.J.; Kutala, V.K.; Hideg, K.; Kuppusamy, P. EF24 Induces G2/M Arrest and Apoptosis in Cisplatin-Resistant Human Ovarian Cancer Cells by Increasing PTEN Expression. J. Biol. Chem. 2007, 282, 28609–28618. [Google Scholar] [CrossRef]
- Yin, D.; Liang, Y.; Zheng, T.; Song, R.; Wang, J.; Sun, B.; Pan, S.; Qu, L.; Liu, J.; Jiang, H.; et al. EF24 Inhibits Tumor Growth and Metastasis via Suppressing NF-kappaB Dependent Pathways in Human Cholangiocarcinoma. Sci. Rep. 2016, 6, 32167. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Tu, C.; Ma, Y.; Ye, P.; Shao, X.; Yang, Z.; Fang, Y. Curcumin Analog EF24 Induces Apoptosis and Downregulates the Mitogen Activated Protein Kinase/Extracellular Signal-Regulated Signaling Pathway in Oral Squamous Cell Carcinoma. Mol. Med. Rep. 2017, 16, 4927–4933. [Google Scholar] [CrossRef] [PubMed]
- Bertazza, L.; Barollo, S.; Mari, M.E.; Faccio, I.; Zorzan, M.; Redaelli, M.; Rubin, B.; Armanini, D.; Mian, C.; Pezzani, R. Biological Effects of EF24, a Curcumin Derivative, Alone or Combined with Mitotane in Adrenocortical Tumor Cell Lines. Molecules 2019, 24, 2202. [Google Scholar] [CrossRef]
- Schmitt, F.; Gold, M.; Begemann, G.; Andronache, I.; Biersack, B.; Schobert, R. Fluoro and Pentafluorothio Analogs of the Antitumoral Curcuminoid EF24 with Superior Antiangiogenic and Vascular-Disruptive Effects. Bioorganic Med. Chem. 2017, 25, 4894–4903. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Sun, A.; Kisiel, W.; Lu, Y.J.; Shim, H.; McCarey, B.E.; Nichols, C.; Parker, E.T.; Pohl, J.; Mosley, C.A.; et al. Targeting Tissue Factor-Expressing Tumor Angiogenesis and Tumors with EF24 Conjugated to Factor VIIa. J. Drug Target. 2008, 16, 185–197. [Google Scholar] [CrossRef]
- Lee, C.; Ho, Y.; Lin, C.; Hsin, M.; Wang, P.; Tang, Y.; Yang, S.; Hsiao, Y. EF-24 Inhibits TPA-induced Cellular Migration and MMP-9 Expression through the P38 Signaling Pathway in Cervical Cancer Cells. Environ. Toxicol. 2023, 38, 451–459. [Google Scholar] [CrossRef]
- Su, S.C.; Hsin, C.H.; Lu, Y.T.; Chuang, C.Y.; Ho, Y.T.; Yeh, F.L.; Yang, S.F.; Lin, C.W. EF-24, a Curcumin Analog, Inhibits Cancer Cell Invasion in Human Nasopharyngeal Carcinoma through Transcriptional Suppression of Matrix Metalloproteinase-9 Gene Expression. Cancers 2023, 15, 1552. [Google Scholar] [CrossRef]
- Liang, Y.; Zheng, T.; Song, R.; Wang, J.; Yin, D.; Wang, L.; Liu, H.; Tian, L.; Fang, X.; Meng, X.; et al. Hypoxia-Mediated Sorafenib Resistance Can Be Overcome by EF24 through Von Hippel-Lindau Tumor Suppressor-Dependent HIF-1α Inhibition in Hepatocellular Carcinoma. Hepatology 2013, 57, 1847–1857. [Google Scholar] [CrossRef]
- Liang, Y.; Yin, D.; Hou, L.; Zheng, T.; Wang, J.; Meng, X.; Lu, Z.; Song, X.; Pan, S.; Jiang, H.; et al. Diphenyl Difluoroketone: A Potent Chemotherapy Candidate for Human Hepatocellular Carcinoma. PLoS ONE 2011, 6, e23908. [Google Scholar] [CrossRef]
- Lagisetty, P.; Subramaniam, D.; Sahoo, K.; Anant, S.; Awasthi, V. Anticancer Activity of an Imageable Curcuminoid 1-[2-Aminoethyl-(6-Hydrazinopyridine-3-Carbamidyl)-3,5-Bis-(2-Fluorobenzylidene)-4-Piperidone (EFAH): Imageable Curcuminoid EFAH. Chem. Biol. Drug Des. 2012, 79, 194–201. [Google Scholar] [CrossRef]
- Lagisetty, P.; Powell, D.R.; Awasthi, V. Synthesis and Structural Determination of 3,5-Bis(2-Fluorobenzylidene)-4-Piperidone Analogs of Curcumin. J. Mol. Struct. 2009, 936, 23–28. [Google Scholar] [CrossRef]
- Robinson, T.P.; Hubbard, R.B.; Ehlers, T.J.; Arbiser, J.L.; Goldsmith, D.J.; Bowen, J.P. Synthesis and Biological Evaluation of Aromatic Enones Related to Curcumin. Bioorganic Med. Chem. 2005, 13, 4007–4013. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Shoji, M.; Lu, Y.J.; Liotta, D.C.; Snyder, J.P. Synthesis of EF24−Tripeptide Chloromethyl Ketone: A Novel Curcumin-Related Anticancer Drug Delivery System. J. Med. Chem. 2006, 49, 3153–3158. [Google Scholar] [CrossRef]
- Mdzinarishvili, A.; Houson, H.; Hedrick, A.; Awasthi, V. Evaluation of Anti-Inflammatory Diphenyldihaloketone EF24 in Transient Ischemic Stroke Model. Brain Inj. 2022, 36, 279–286. [Google Scholar] [CrossRef]
- Monroe, J.D.; Hodzic, D.; Millay, M.H.; Patty, B.G.; Smith, M.E. Anti-Cancer and Ototoxicity Characteristics of the Curcuminoids, CLEFMA and EF24, in Combination with Cisplatin. Molecules 2019, 24, 3889. [Google Scholar] [CrossRef] [PubMed]
- Skoupa, N.; Dolezel, P.; Ruzickova, E.; Mlejnek, P. Apoptosis Induced by the Curcumin Analogue EF-24 Is Neither Mediated by Oxidative Stress-Related Mechanisms nor Affected by Expression of Main Drug Transporters ABCB1 and ABCG2 in Human Leukemia Cells. Int. J. Mol. Sci. 2017, 18, 2289. [Google Scholar] [CrossRef]
- Mosley, C.A.; Liotta, D.C.; Snyder, J.P. Highly active anticancer curcumin analogs. Adv. Exp. Med. Biol. 2007, 595, 77–103. [Google Scholar] [CrossRef]
- Adams, B.K.; Ferstl, E.M.; Davis, M.C.; Herold, M.; Kurtkaya, S.; Camalier, R.F.; Hollingshead, M.G.; Kaur, G.; Sausville, E.A.; Rickles, F.R.; et al. Synthesis and Biological Evaluation of Novel Curcumin Analogs as Anti-Cancer and Anti-Angiogenesis Agents. Bioorganic Med. Chem. 2004, 12, 3871–3883. [Google Scholar] [CrossRef]
- Rubin, B.; Regazzo, D.; Redaelli, M.; Mucignat, C.; Citton, M.; Iacobone, M.; Scaroni, C.; Betterle, C.; Mantero, F.; Fassina, A.; et al. Investigation of N-Cadherin/β-Catenin Expression in Adrenocortical Tumors. Tumor Biol. 2016, 37, 13545–13555. [Google Scholar] [CrossRef]
- Thomas, S.L.; Zhao, J.; Li, Z.; Lou, B.; Du, Y.; Purcell, J.; Snyder, J.P.; Khuri, F.R.; Liotta, D.; Fu, H. Activation of the P38 Pathway by a Novel Monoketone Curcumin Analog, EF24, Suggests a Potential Combination Strategy. Biochem. Pharmacol. 2010, 80, 1309–1316. [Google Scholar] [CrossRef]
- Lu, Z.; Xu, S. ERK1/2 MAP Kinases in Cell Survival and Apoptosis. IUBMB Life Int. Union Biochem. Mol. Biol. Life 2006, 58, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Ali, K. Oral Cancer—The Fight Must Go on against All Odd. Evid. Based Dent. 2022, 23, 4–5. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK Signal Pathways in the Regulation of Cell Proliferation in Mammalian Cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Mukherjee, D.; Krishnan, A. Therapeutic Potential of Curcumin and Its Nanoformulations for Treating Oral Cancer. World J. Methodol. 2023, 13, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Chen, J.; Shi, Y.; Fang, X.; Tang, Z. MAPK Signaling Pathway in Oral Squamous Cell Carcinoma: Biological Function and Targeted Therapy. Cancers 2022, 14, 4625. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Chen, H.; Ling, M.; Zhang, S.; Ma, F.; Zhang, H.; Lv, X. The Curcumin Analog EF24 Inhibits Proliferation and Invasion of Triple-Negative Breast Cancer Cells by Targeting the Long Noncoding RNA HCG11/Sp1 Axis. Mol. Cell. Biol. 2022, 42, e00163-21. [Google Scholar] [CrossRef]
- Zhang, P.; Bai, H.; Liu, G.; Wang, H.; Chen, F.; Zhang, B.; Zeng, P.; Wu, C.; Peng, C.; Huang, C.; et al. MicroRNA-33b, Upregulated by EF24, a Curcumin Analog, Suppresses the Epithelial-to-Mesenchymal Transition (EMT) and Migratory Potential of Melanoma Cells by Targeting HMGA2. Toxicol. Lett. 2015, 234, 151–161. [Google Scholar] [CrossRef]
- Lan, Y.; Liu, B.; Guo, H. The Role of M6A Modification in the Regulation of Tumor-Related lncRNAs. Mol. Ther.-Nucleic Acids 2021, 24, 768–779. [Google Scholar] [CrossRef]
- Huang, W.; Chen, T.-Q.; Fang, K.; Zeng, Z.-C.; Ye, H.; Chen, Y.-Q. N6-Methyladenosine Methyltransferases: Functions, Regulation, and Clinical Potential. J. Hematol. Oncol. 2021, 14, 117. [Google Scholar] [CrossRef]
- Vizcaíno, C.; Mansilla, S.; Portugal, J. Sp1 Transcription Factor: A Long-Standing Target in Cancer Chemotherapy. Pharmacol. Ther. 2015, 152, 111–124. [Google Scholar] [CrossRef]
- Luo, C.-W.; Hou, M.-F.; Huang, C.-W.; Wu, C.-C.; Ou-Yang, F.; Li, Q.-L.; Wu, C.-C.; Pan, M.-R. The CDK6-c-Jun-Sp1-MMP-2 Axis as a Biomarker and Therapeutic Target for Triple-Negative Breast Cancer. Am. J. Cancer Res. 2020, 10, 4325–4341. [Google Scholar] [PubMed]
- Hull, R.; Mbita, Z.; Dlamini, Z. Long Non-Coding RNAs (LncRNAs), Viral Oncogenomics, and Aberrant Splicing Events: Therapeutics Implications. Am. J. Cancer Res. 2021, 11, 866–883. [Google Scholar] [PubMed]
- Zhang, T.; Beeharry, M.K.; Wang, Z.; Zhu, Z.; Li, J.; Li, C. YY1-Modulated Long Non-Coding RNA SNHG12 Promotes Gastric Cancer Metastasis by Activating the miR-218-5p/YWHAZ Axis. Int. J. Biol. Sci. 2021, 17, 1629–1643. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.H.; Raulston, J.E.; Wyrick, P.B. Protein Disulfide Isomerase, a Component of the Estrogen Receptor Complex, Is Associated with Chlamydia Trachomatis Serovar E Attached to Human Endometrial Epithelial Cells. Infect. Immun. 2002, 70, 3413–3418. [Google Scholar] [CrossRef] [PubMed]
- Onen, H.; Yilmaz, A.; Alp, E.; Celik, A.; Demiroz, S.; Konac, E.; Kurul, I.; Menevse, E. EF24 and RAD001 Potentiates the Anticancer Effect of Platinum-Based Agents in Human Malignant Pleural Mesothelioma (MSTO-211H) Cells and Protects Nonmalignant Mesothelial (MET-5A) Cells. Hum. Exp. Toxicol. 2015, 34, 117–126. [Google Scholar] [CrossRef]
- Kasinski, A.L.; Du, Y.; Thomas, S.L.; Zhao, J.; Sun, S.-Y.; Khuri, F.R.; Wang, C.-Y.; Shoji, M.; Sun, A.; Snyder, J.P.; et al. Inhibition of IκB Kinase-Nuclear Factor-κB Signaling Pathway by 3,5-Bis(2-Flurobenzylidene)Piperidin-4-One (EF24), a Novel Monoketone Analog of Curcumin. Mol. Pharmacol. 2008, 74, 654–661. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, Q.; Zheng, Z.; Xie, J.; Lin, X.; Jiang, C.; Xu, H.; Wu, X.; Wu, J.; Zhang, H. Design and Optimize N-substituted EF 24 as Effective and Low Toxicity NF-κB Inhibitor for Lung Cancer Therapy via Apoptosis-to-pyroptosis Switch. Chem. Biol. Drug Des. 2019, 94, 1368–1377. [Google Scholar] [CrossRef]
- Wu, J.; Wu, S.; Shi, L.; Zhang, S.; Ren, J.; Yao, S.; Yun, D.; Huang, L.; Wang, J.; Li, W.; et al. Design, Synthesis, and Evaluation of Asymmetric EF24 Analogues as Potential Anti-Cancer Agents for Lung Cancer. Eur. J. Med. Chem. 2017, 125, 1321–1331. [Google Scholar] [CrossRef]
- Hou, Z.; Liu, J.; Jin, Z.; Qiu, G.; Xie, Q.; Mi, S.; Huang, J. Use of Chemotherapy to Treat Hepatocellular Carcinoma. BST 2022, 16, 31–45. [Google Scholar] [CrossRef]
- Agarwal, M.L.; Agarwal, A.; Taylor, W.R.; Stark, G.R. P53 Controls Both the G2/M and the G1 Cell Cycle Checkpoints and Mediates Reversible Growth Arrest in Human Fibroblasts. Proc. Natl. Acad. Sci. USA 1995, 92, 8493–8497. [Google Scholar] [CrossRef]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The Release of Cytochrome c from Mitochondria: A Primary Site for Bcl-2 Regulation of Apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Rennefahrt, U.; Janakiraman, M.; Öllinger, R.; Troppmair, J. Stress Kinase Signal. Cancer: Fact or Fiction? Cancer Lett. 2005, 217, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Tin, L.; Zhang, Y.; Wu, Y.; Jin, Y.; Jin, X.; Zhang, F.; Li, X. EF24 Suppresses Invasion and Migration of Hepatocellular Carcinoma Cells In Vitro via Inhibiting the Phosphorylation of Src. BioMed Res. Int. 2016, 2016, 8569684. [Google Scholar] [CrossRef] [PubMed]
- Suraneni, P.; Rubinstein, B.; Unruh, J.R.; Durnin, M.; Hanein, D.; Li, R. The Arp2/3 Complex Is Required for Lamellipodia Extension and Directional Fibroblast Cell Migration. J. Cell Biol. 2012, 197, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.L.; Zhong, D.; Zhou, W.; Malik, S.; Liotta, D.; Snyder, J.P.; Hamel, E.; Giannakakou, P. EF24, a Novel Curcumin Analog, Disrupts the Microtubule Cytoskeleton and Inhibits HIF-1. Cell Cycle 2008, 7, 2409–2417. [Google Scholar] [CrossRef] [PubMed]
- Frame, M.C. Src in Cancer: Deregulation and Consequences for Cell Behaviour. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2002, 1602, 114–130. [Google Scholar] [CrossRef]
- Charpentier, M.S.; Whipple, R.A.; Vitolo, M.I.; Boggs, A.E.; Slovic, J.; Thompson, K.N.; Bhandary, L.; Martin, S.S. Curcumin Targets Breast Cancer Stem–like Cells with Microtentacles That Persist in Mammospheres and Promote Reattachment. Cancer Res. 2014, 74, 1250–1260. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef]
- Zinszner, H.; Kuroda, M.; Wang, X.; Batchvarova, N.; Lightfoot, R.T.; Remotti, H.; Stevens, J.L.; Ron, D. CHOP Is Implicated in Programmed Cell Death in Response to Impaired Function of the Endoplasmic Reticulum. Genes Dev. 1998, 12, 982–995. [Google Scholar] [CrossRef]
- Ramanathan, R.K.; McDonough, S.L.; Kennecke, H.F.; Iqbal, S.; Baranda, J.C.; Seery, T.E.; Lim, H.J.; Hezel, A.F.; Vaccaro, G.M.; Blanke, C.D. Phase 2 Study of MK-2206, an Allosteric Inhibitor of AKT, as Second-Line Therapy for Advanced Gastric and Gastroesophageal Junction Cancer: A SWOG Cooperative Group Trial (S1005): Phase 2 Study of MK-2206. Cancer 2015, 121, 2193–2197. [Google Scholar] [CrossRef]
- Zou, P.; Chen, M.; Ji, J.; Chen, W.; Chen, X.; Ying, S.; Zhang, J.; Zhang, Z.; Liu, Z.; Yang, S.; et al. Auranofin Induces Apoptosis by ROS-Mediated ER Stress and Mitochondrial Dysfunction and Displayed Synergistic Lethality with Piperlongumine in Gastric Cancer. Oncotarget 2015, 6, 36505–36521. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Zhang, J.; Xia, Y.; Kanchana, K.; Guo, G.; Chen, W.; Huang, Y.; Wang, Z.; Yang, S.; Liang, G. ROS Generation Mediates the Anti-Cancer Effects of WZ35 via Activating JNK and ER Stress Apoptotic Pathways in Gastric Cancer. Oncotarget 2015, 6, 5860–5876. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.E.; Harris, G.T. Renal Cell Carcinoma: Diagnosis and Management. Am. Fam. Physician. 2019, 99, 179–184. [Google Scholar] [PubMed]
- Chow, W.-H.; Dong, L.M.; Devesa, S.S. Epidemiology and Risk Factors for Kidney Cancer. Nat. Rev. Urol. 2010, 7, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Lenis, A.T.; Donin, N.M.; Johnson, D.C.; Faiena, I.; Salmasi, A.; Drakaki, A.; Belldegrun, A.; Pantuck, A.; Chamie, K. Adjuvant Therapy for High Risk Localized Kidney Cancer: Emerging Evidence and Future Clinical Trials. J. Urol. 2018, 199, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Daniels, R.A.; Turley, H.; Kimberley, F.C.; Liu, X.S.; Mongkolsapaya, J.; Ch’En, P.; Xu, X.N.; Jin, B.; Pezzella, F.; Screaton, G.R. Expression of TRAIL and TRAIL Receptors in Normal and Malignant Tissues. Cell Res. 2005, 15, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Almasan, A.; Ashkenazi, A. Apo2L/TRAIL: Apoptosis Signaling, Biology, and Potential for Cancer Therapy. Cytokine Growth Factor Rev. 2003, 14, 337–348. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, X.; Xu, T.; Kong, Q.; Zhang, Y.; Shen, Y.; Wei, Y.; Wang, G.; Chang, K.-J. Overcoming Resistance to TRAIL-Induced Apoptosis in Solid Tumor Cells by Simultaneously Targeting Death Receptors, c-FLIP and IAPs. Int. J. Oncol. 2016, 49, 153–163. [Google Scholar] [CrossRef]
- Ibáñez Gaspar, V.; McCaul, J.; Cassidy, H.; Slattery, C.; McMorrow, T. Effects of Curcumin Analogues DMC and EF24 in Combination with the Cytokine TRAIL against Kidney Cancer. Molecules 2021, 26, 6302. [Google Scholar] [CrossRef]
- Waas, E.T.; Wobbes, T.; Lomme, R.M.L.M.; DeGroot, J.; Ruers, T.; Hendriks, T. Matrix Metalloproteinase 2 and 9 Activity in Patients with Colorectal Cancer Liver Metastasis. Br. J. Surg. 2003, 90, 1556–1564. [Google Scholar] [CrossRef]
- Tochigi, M.; Inoue, T.; Suzuki-Karasaki, M.; Ochiai, T.; Ra, C.; Suzuki-Karasaki, Y. Hydrogen Peroxide Induces Cell Death in Human TRAIL-Resistant Melanoma through Intracellular Superoxide Generation. Int. J. Oncol. 2013, 42, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Bertazza, L.; Sensi, F.; Cavedon, E.; Watutantrige-Fernando, S.; Censi, S.; Manso, J.; Vianello, F.; Casal Ide, E.; Iacobone, M.; Pezzani, R.; et al. EF24 (a Curcumin Analog) and ZSTK474 Emphasize the Effect of Cabozantinib in Medullary Thyroid Cancer. Endocrinology 2018, 159, 2348–2360. [Google Scholar] [CrossRef] [PubMed]
- Arora, T.; Mullangi, S.; Lekkala, M.R. Ovarian Cancer. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Guo, B.-Q.; Lu, W.-Q. The Prognostic Significance of High/Positive Expression of Tissue VEGF in Ovarian Cancer. Oncotarget 2018, 9, 30552–30560. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, B.A.; Markel, J.E.; Kleinerman, E.S. Osteosarcoma Overview. Rheumatol. Ther. 2017, 4, 25–43. [Google Scholar] [CrossRef]
- Martin, J.W.; Squire, J.A.; Zielenska, M. The Genetics of Osteosarcoma. Sarcoma 2012, 2012, 627254. [Google Scholar] [CrossRef]
- Prater, S.; McKeon, B. Osteosarcoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Jin, R.; Chen, Q.; Yao, S.; Bai, E.; Fu, W.; Wang, L.; Wang, J.; Du, X.; Wei, T.; Xu, H.; et al. Synthesis and Anti-Tumor Activity of EF24 Analogues as IKKβ Inhibitors. Eur. J. Med. Chem. 2018, 144, 218–228. [Google Scholar] [CrossRef]
- Yang, S.-J.; Lee, S.A.; Park, M.-G.; Kim, J.-S.; Yu, S.-K.; Kim, C.S.; Kim, J.-S.; Kim, S.-G.; Oh, J.-S.; Kim, H.-J.; et al. Induction of Apoptosis by Diphenyldifluoroketone in Osteogenic Sarcoma Cells Is Associated with Activation of Caspases. Oncol. Rep. 2014, 31, 2286–2292. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Speleman, F.; Park, J.R.; Henderson, T.O. Neuroblastoma: A Tough Nut to Crack. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, e548-557. [Google Scholar] [CrossRef]
- Aravindan, S.; Natarajan, M.; Awasthi, V.; Herman, T.S.; Aravindan, N. Novel Synthetic Monoketone Transmute Radiation-Triggered NFκB-Dependent TNFα Cross-Signaling Feedback Maintained NFκB and Favors Neuroblastoma Regression. PLoS ONE 2013, 8, e72464. [Google Scholar] [CrossRef]
- Castleberry, R.P.; Kun, L.E.; Shuster, J.J.; Altshuler, G.; Smith, I.E.; Nitschke, R.; Wharam, M.; McWilliams, N.; Joshi, V.; Hayes, F.A. Radiotherapy Improves the Outlook for Patients Older than 1 Year with Pediatric Oncology Group Stage C Neuroblastoma. J. Clin. Oncol. 1991, 9, 789–795. [Google Scholar] [CrossRef]
- Aravindan, N.; Madhusoodhanan, R.; Natarajan, M.; Herman, T.S. Alteration of Apoptotic Signaling Molecules as a Function of Time after Radiation in Human Neuroblastoma Cells. Mol. Cell Biochem. 2008, 310, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Aravindan, N.; Madhusoodhanan, R.; Ahmad, S.; Johnson, D.; Herman, T.S. Curcumin Inhibits NFκB Mediated Radioprotection and Modulate Apoptosis Related Genes in Human Neuroblastoma Cells. Cancer Biol. Ther. 2008, 7, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Madhusoodhanan, R.; Natarajan, M.; Veeraraghavan, J.; Herman, T.S.; Jamgade, A.; Singh, N.; Aravindan, N. NFκB Signaling Related Molecular Alterations in Human Neuroblastoma Cells after Fractionated Irradiation. JRR 2009, 50, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Aravindan, N.; Veeraraghavan, J.; Madhusoodhanan, R.; Herman, T.S.; Natarajan, M. Curcumin Regulates Low-Linear Energy Transfer γ-Radiation-Induced NFκB-Dependent Telomerase Activity in Human Neuroblastoma Cells. Int. J. Radiat. Oncol. *Biol. *Phys. 2011, 79, 1206–1215. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B. Nuclear Factor-κB. Cancer Cell 2004, 6, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Molecular Basis of the Keap1–Nrf2 System. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef]
- Hsiao, P.-C.; Chang, J.-H.; Lee, W.-J.; Ku, C.-C.; Tsai, M.-Y.; Yang, S.-F.; Chien, M.-H. The Curcumin Analogue, EF-24, Triggers P38 MAPK-Mediated Apoptotic Cell Death via Inducing PP2A-Modulated ERK Deactivation in Human Acute Myeloid Leukemia Cells. Cancers 2020, 12, 2163. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK Pathway in Cell Growth, Malignant Transformation and Drug Resistance. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2007, 1773, 1263–1284. [Google Scholar] [CrossRef]
- Lunghi, P.; Tabilio, A.; Dall’Aglio, P.P.; Ridolo, E.; Carlo-Stella, C.; Pelicci, P.G.; Bonati, A. Downmodulation of ERK Activity Inhibits the Proliferation and Induces the Apoptosis of Primary Acute Myelogenous Leukemia Blasts. Leukemia 2003, 17, 1783–1793. [Google Scholar] [CrossRef]
- Chudnovsky, Y.; Khavari, P.A.; Adams, A.E. Melanoma Genetics and the Development of Rational Therapeutics. J. Clin. Invest. 2005, 115, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Siwak, D.R.; Shishodia, S.; Aggarwal, B.B.; Kurzrock, R. Curcumin-Induced Antiproliferative and Proapoptotic Effects in Melanoma Cells Are Associated with Suppression of IκB Kinase and Nuclear Factor κB Activity and Are Independent of the B-Raf/Mitogen-Activated/Extracellular Signal-Regulated Protein Kinase Pathway and the Akt Pathway. Cancer 2005, 104, 879–890. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Song, E.; Hu, D.-N.; Chen, M.; Xue, C.; Rosen, R.; McCormick, S.A. Curcumin Induces Cell Death in Human Uveal Melanoma Cells through Mitochondrial Pathway. Curr. Eye Res. 2010, 35, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Bush, J.A.; Cheung, K.-J.J.; Li, G. Curcumin Induces Apoptosis in Human Melanoma Cells through a Fas Receptor/Caspase-8 Pathway Independent of P53. Exp. Cell Res. 2001, 271, 305–314. [Google Scholar] [CrossRef]
- Fusco, A.; Fedele, M. Roles of HMGA Proteins in Cancer. Nat. Rev. Cancer 2007, 7, 899–910. [Google Scholar] [CrossRef]
- Guo, L.; Chen, C.; Shi, M.; Wang, F.; Chen, X.; Diao, D.; Hu, M.; Yu, M.; Qian, L.; Guo, N. Stat3-Coordinated Lin-28–Let-7–HMGA2 and miR-200–ZEB1 Circuits Initiate and Maintain Oncostatin M-Driven Epithelial–Mesenchymal Transition. Oncogene 2013, 32, 5272–5282. [Google Scholar] [CrossRef]
- Folkman, J. Tumor Angiogenesis. In Advances in Cancer Research; Elsevier: Amsterdam, The Netherlands, 1985; Volume 43, pp. 175–203. ISBN 9780120066438. [Google Scholar]
- Pilat, M.J.; McCormick, J.; LoRusso, P.M. Vascular Targeting Agents. Curr. Oncol. Rep. 2004, 6, 103–110. [Google Scholar] [CrossRef]
- Contrino, J.; Hair, G.; Kreutzer, D.L.; Rickles, F.R. In Situ Detection of Tissue Factor in Vascular Endothelial Cells: Correlation with the Malignant Phenotype of Human Breast Disease. Nat. Med. 1996, 2, 209–215. [Google Scholar] [CrossRef]
- Shen, Y.; Sheng, R.; Guo, R. Application of Zebrafish as a Model for Anti-Cancer Activity Evaluation and Toxicity Testing of Natural Products. Pharmaceuticals 2023, 16, 827. [Google Scholar] [CrossRef]
- Xie, X.; Tu, J.; You, H.; Hu, B. Design, Synthesis, and Biological Evaluation of Novel EF24 and EF31 Analogs as Potential IκB Kinase β Inhibitors for the Treatment of Pancreatic Cancer. Drug Des. Devel. Ther. 2017, 11, 1439–1451. [Google Scholar] [CrossRef]
- Chen, L.; Li, Q.; Weng, B.; Wang, J.; Zhou, Y.; Cheng, D.; Sirirak, T.; Qiu, P.; Wu, J. Design, Synthesis, Anti-Lung Cancer Activity, and Chemosensitization of Tumor-Selective MCACs Based on ROS-Mediated JNK Pathway Activation and NF-κB Pathway Inhibition. Eur. J. Med. Chem. 2018, 151, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Callander, N.S.; Varki, N.; Vijaya Rao, L.M. Immunohistochemical Identification of Tissue Factor in Solid Tumors. Cancer 1992, 70, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Agashe, H.; Lagisetty, P.; Sahoo, K.; Bourne, D.; Grady, B.; Awasthi, V. Liposome-Encapsulated EF24-HPβCD Inclusion Complex: A Preformulation Study and Biodistribution in a Rat Model. J. Nanopart. Res. 2011, 13, 2609–2623. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sazdova, I.; Keremidarska-Markova, M.; Dimitrova, D.; Mitrokhin, V.; Kamkin, A.; Hadzi-Petrushev, N.; Bogdanov, J.; Schubert, R.; Gagov, H.; Avtanski, D.; et al. Anticarcinogenic Potency of EF24: An Overview of Its Pharmacokinetics, Efficacy, Mechanism of Action, and Nanoformulation for Drug Delivery. Cancers 2023, 15, 5478. https://doi.org/10.3390/cancers15225478
Sazdova I, Keremidarska-Markova M, Dimitrova D, Mitrokhin V, Kamkin A, Hadzi-Petrushev N, Bogdanov J, Schubert R, Gagov H, Avtanski D, et al. Anticarcinogenic Potency of EF24: An Overview of Its Pharmacokinetics, Efficacy, Mechanism of Action, and Nanoformulation for Drug Delivery. Cancers. 2023; 15(22):5478. https://doi.org/10.3390/cancers15225478
Chicago/Turabian StyleSazdova, Iliyana, Milena Keremidarska-Markova, Daniela Dimitrova, Vadim Mitrokhin, Andre Kamkin, Nikola Hadzi-Petrushev, Jane Bogdanov, Rudolf Schubert, Hristo Gagov, Dimiter Avtanski, and et al. 2023. "Anticarcinogenic Potency of EF24: An Overview of Its Pharmacokinetics, Efficacy, Mechanism of Action, and Nanoformulation for Drug Delivery" Cancers 15, no. 22: 5478. https://doi.org/10.3390/cancers15225478