
Pan-Cancer Analysis of Patient Tumor Single-Cell Transcriptomes Identifies Promising Selective and Safe Chimeric Antigen Receptor Targets in Head and Neck Cancer
 and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Trial Curation
2.2. Single-Cell Transcriptomics Data Collection
2.3. Single-Cell Data Analysis
2.4. Reference Atlas Analysis: Computing Safety Scores
2.5. DepMap Essentiality (Dependency) Scores
2.6. TCGA Primary Tumor vs. Matched Normal Analysis
3. Results
3.1. Overview of the Analysis
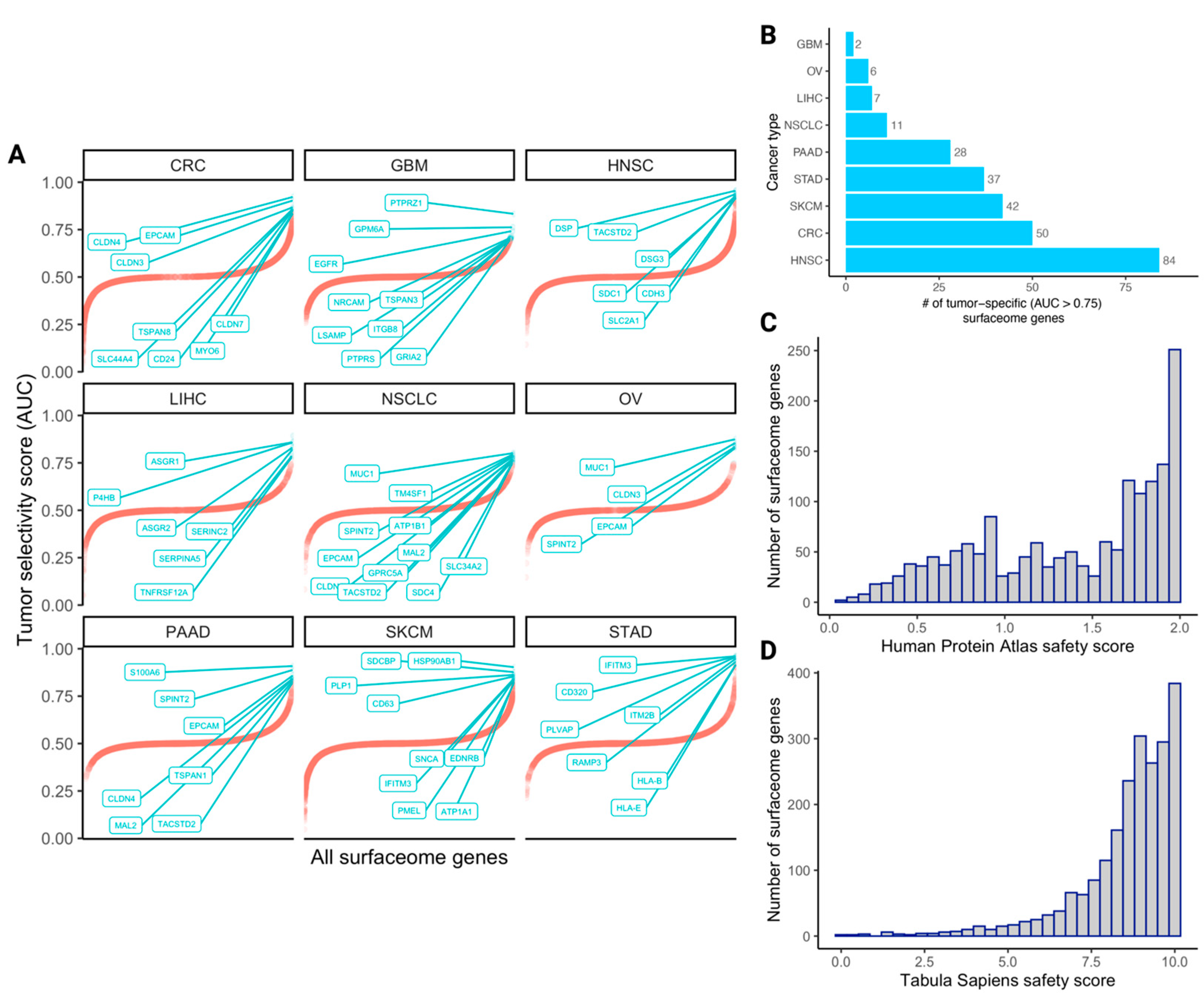
3.2. The Quantified Tumor Selectivity and Safety Landscape of the Human Surfaceome
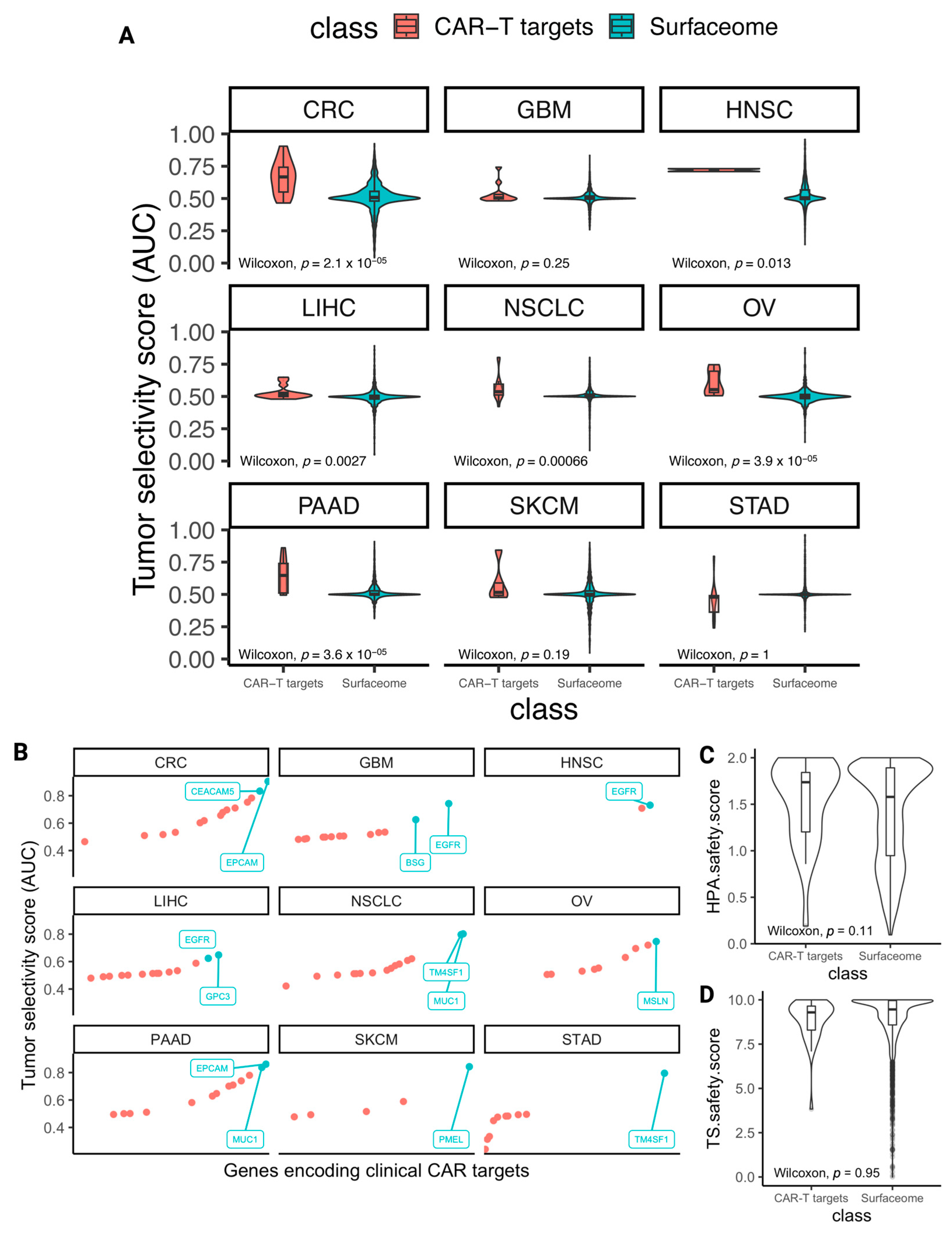
3.3. The Selectivity and Safety Landscape of Targets of Approved and Currently Studied CARs
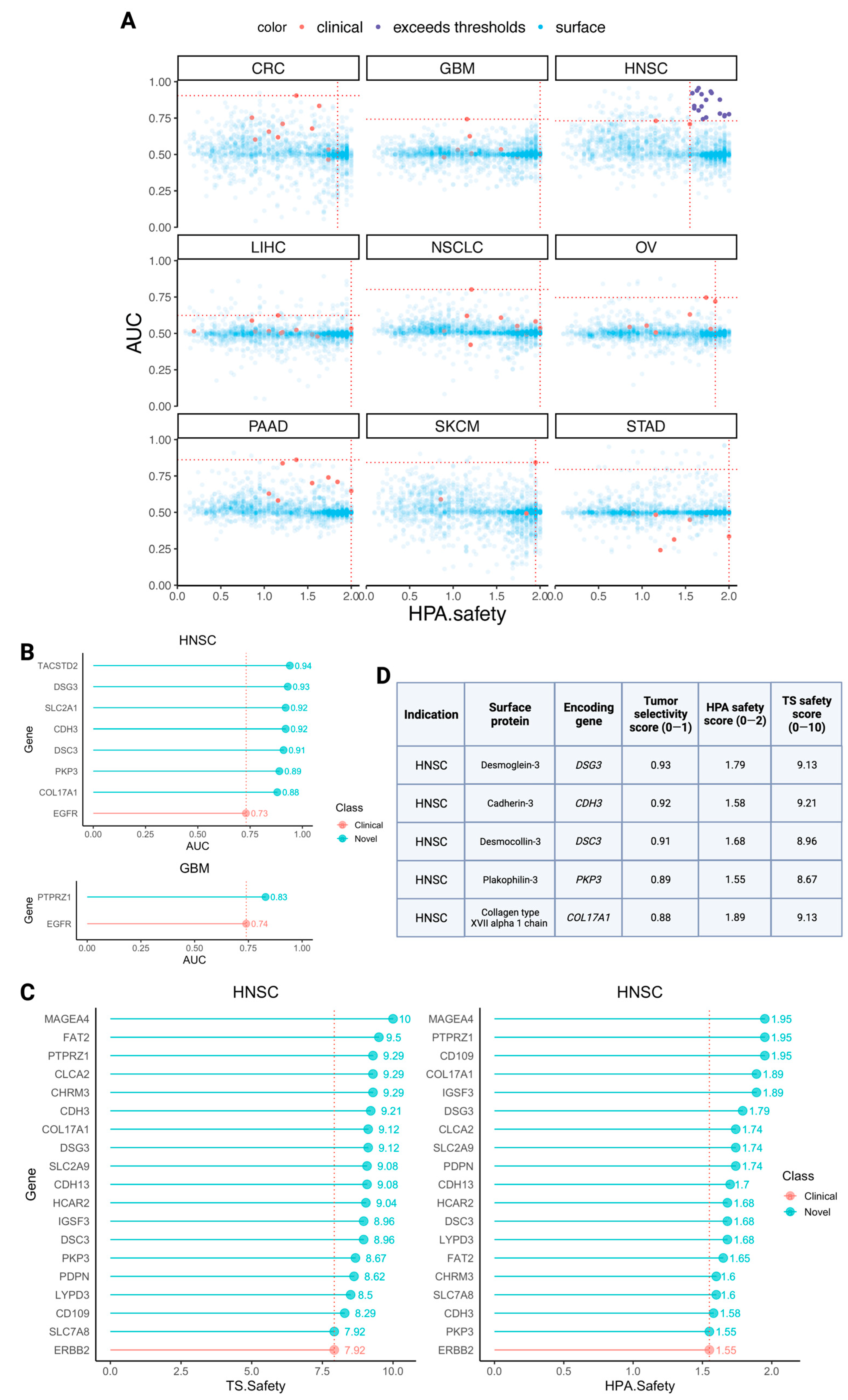
3.4. Identifying Novel CAR Targets with Higher Estimated Selectivity and Safety Scores
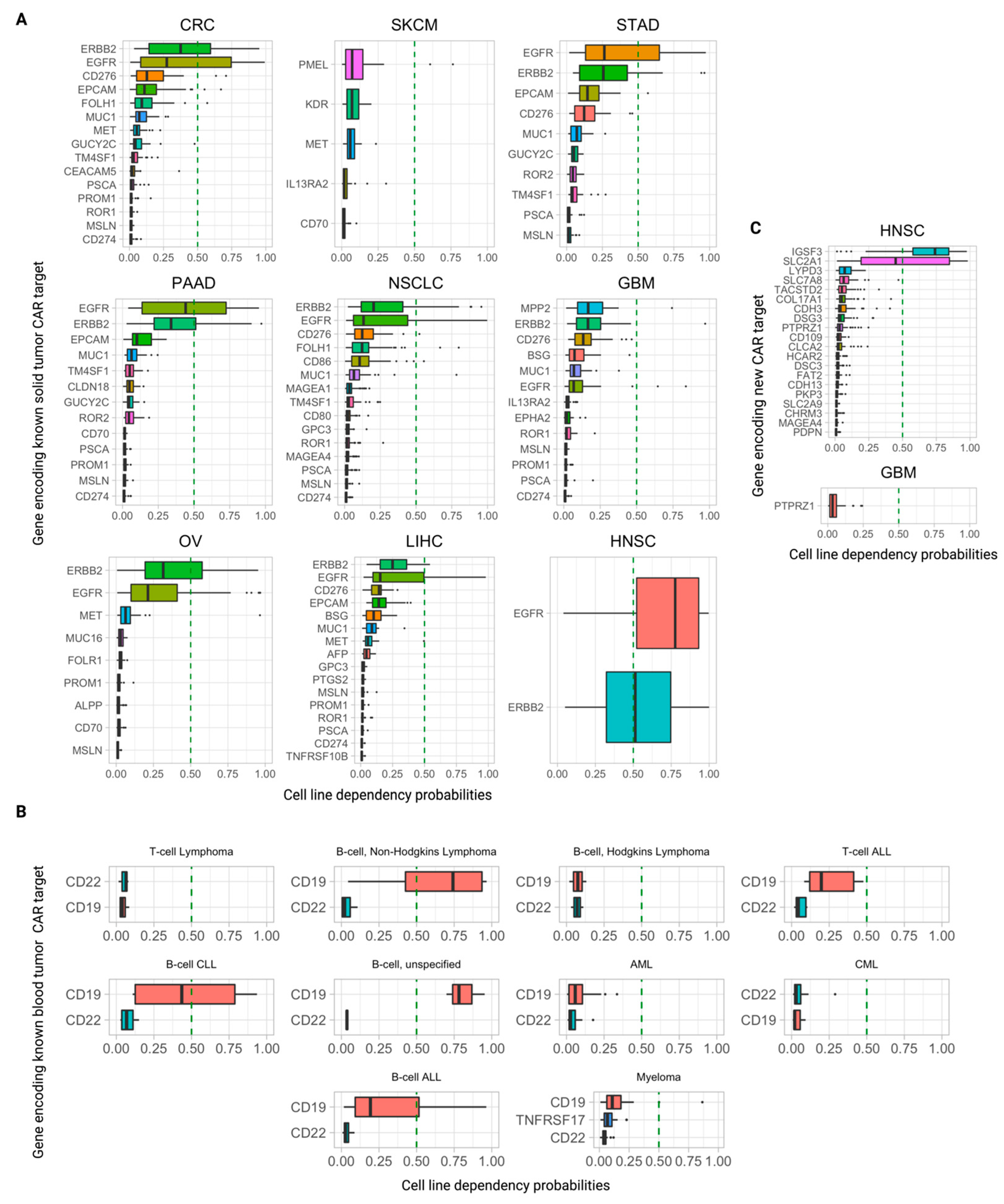
3.5. Further Ranking Top Predicted CAR Targets by Their Essentiality (Dependency) Scores in Tumor Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-long leukaemia remissions with persistence of CD4+ CAR T cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Stergiopoulos, S.; Getz, K.A.; Blazynski, C. Evaluating the completeness of ClinicalTrials.gov. Ther. Innov. Regul. Sci. 2019, 53, 307–317. [Google Scholar] [CrossRef]
- Milone, M.C.; Xu, J.; Chen, S.J.; Collins, M.A.; Zhou, J.; Powell, D.J.; Melenhorst, J.J. Engineering-enhanced CAR T cells for improved cancer therapy. Nat. Cancer 2021, 2, 780–793. [Google Scholar] [PubMed]
- Jing, Y.; Liu, Y.; Li, Q.; Ye, Y.; Diao, L.; Huang, Y.; Zhou, Y.; Green, M.R.; Mills, G.B.; Han, L. Expression of chimeric antigen receptor therapy targets detected by single-cell sequencing of normal cells may contribute to off-tumor toxicity. Cancer Cell 2021, 39, 1558–1559. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-cell analyses identify brain mural cells expressing CD19 as potential off-tumor targets for CAR-T immunotherapies. Cell 2020, 183, 126–142.e17. [Google Scholar]
- Ahmadi, S.; Sukprasert, P.; Vegesna, R.; Sinha, S.; Schischlik, F.; Artzi, N.; Khuller, S.; Schäffer, A.A.; Ruppin, E. The landscape of receptor-mediated precision cancer combination therapy via a single-cell perspective. Nat. Commun. 2022, 13, 1613. [Google Scholar]
- Hu, Z.; Yuan, J.; Long, M.; Jiang, J.; Zhang, Y.; Zhang, T.; Xu, M.; Fan, Y.; Tanyi, J.L.; Montone, K.T.; et al. The Cancer Surfaceome Atlas integrates genomic, functional and drug response data to identify actionable targets. Nat. Cancer 2021, 2, 1406–1422. [Google Scholar]
- Lareau, C.A.; Parker, K.R.; Satpathy, A.T. Charting the tumor antigen maps drawn by single-cell genomics. Cancer Cell 2021, 39, 1553–1557. [Google Scholar] [CrossRef]
- Kwon, J.; Kang, J.; Jo, A.; Seo, K.; An, D.; Baykan, M.Y.; Lee, J.H.; Kim, N.; Eum, H.H.; Hwang, S.; et al. Single-cell mapping of combinatorial target antigens for CAR switches using logic gates. Nat. Biotechnol. 2023, in press. [Google Scholar] [CrossRef]
- MacKay, M.; Afshinnekoo, E.; Rub, J.; Hassan, C.; Khunte, M.; Baskaran, N.; Owens, B.; Liu, L.; Roboz, G.J.; Guzman, M.L.; et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat. Biotechnol. 2020, 38, 233–244. [Google Scholar]
- Sun, D.; Wang, J.; Han, Y.; Dong, X.; Ge, J.; Zheng, R.; Shi, X.; Wang, B.; Li, Z.; Ren, P.; et al. TISCH: A comprehensive web resource enabling interactive single-cell transcriptome visualization of tumor microenvironment. Nucleic Acids Res. 2021, 49, D1420–D1430. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- The Tabula Sapiens Consortium. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science 2022, 376, eabl4896. [Google Scholar]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef] [PubMed]
- DepMap 22Q2 Public. Figshare. 2022. Available online: https://figshare.com/articles/dataset/DepMap_22Q2_Public/19700056/2 (accessed on 27 March 2023).
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Tsujikawa, M.; Kurahashi, H.; Tanaka, T.; Nishida, K.; Shimomura, Y.; Tano, Y.; Nakamura, Y. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nat. Genet. 1999, 21, 420–423. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab Govitecan in metastatic triple-negative breast cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- Chen, Y.J.; Chang, J.T.; Lee, L.; Wang, H.M.; Liao, C.T.; Chiu, C.C.; Chen, P.-J.; Cheng, A.-J. DSG3 is overexpressed in head neck cancer and is a potential molecular target for inhibition of oncogenesis. Oncogene 2007, 26, 467–476. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, S.E.; Park, H.S.; Lee, S.H.; Lee, S.E.; Kim, S.C. A homozygous nonsense mutation in the DSG3 gene causes acantholytic blisters in the oral and laryngeal mucosa. J. Investig. Dermatol. 2019, 139, 1187–1190. [Google Scholar] [CrossRef]
- Amagai, M.; Klaus-Kovtun, V.; Stanley, J.R. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell 1991, 67, 869–877. [Google Scholar] [CrossRef]
- Dasgupta, S.; Tripathi, P.K.; Qin, H.; Bhattacharya-Chatterjee, M.; Valentino, J.; Chatterjee, S.K. Identification of molecular targets for immunotherapy of patients with head and neck squamous cell carcinoma. Oral Oncol. 2006, 42, 306–316. [Google Scholar] [CrossRef]
- Keck, M.K.; Zuo, Z.; Khattri, A.; Stricker, T.P.; Brown, C.D.; Imanguli, M.; Rieke, D.; Endhardt, K.; Fang, P.; Brägelmann, J.; et al. Integrative analysis of head and neck cancer identifies two biologically distinct HPV and three non-HPV Subtypes. Clin. Cancer Res. 2015, 21, 870–881. [Google Scholar] [CrossRef]
- Wu, T.; Xiao, Z.; Li, Y.; Jiao, Z.; Liang, X.; Zhang, Y.; Liu, H.; Yang, A. CDH3 is associated with poor prognosis by promoting the malignancy and chemoresistance in oral squamous cell carcinoma. Asian J. Surg. 2022, 45, 2651–2658. [Google Scholar] [CrossRef]
- Sprecher, E.; Bergman, R.; Richard, G.; Lurie, R.; Shalev, S.; Petronius, D.; Shalata, A.; Anbinder, Y.; Leibu, R.; Perlman, I.; et al. Hypotrichosis with juvenile macular dystrophy is caused by a mutation in CDH3, encoding P-cadherin. Nat. Genet. 2001, 29, 134–136. [Google Scholar] [CrossRef]
- Kjaer, K.W.; Hansen, L.; Schwabe, G.C.; Marques-de-Faria, A.P.; Eiberg, H.; Mundlos, S.; Tommerup, N.; Rosenberg, T. Distinct CDH3 mutations cause ectodermal dysplasia, ectrodactyly, macular dystrophy (EEM syndrome). J. Med. Genet. 2005, 42, 292–298. [Google Scholar] [CrossRef]
- Wang, L.; Liu, T.; Wang, Y.; Cao, L.; Nishioka, M.; Aguirre, R.L.; Ishikawa, A.; Geng, L.; Okada, N. Altered expression of desmocollin 3, desmoglein 3, and beta-catenin in oral squamous cell carcinoma: Correlation with lymph node metastasis and cell proliferation. Virchows Arch. 2007, 451, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Onoufriadis, A.; Ahmed, N.; Bessar, H.; Guy, A.; Liu, L.; Marantzidis, A.; Kesidou, E.; Papanikolaou, M.; Simpson, M.A.; Mellerio, J.E.; et al. Homozygous nonsense mutation in DSC3 resulting in skin fragility and hypotrichosis. J. Investig. Dermatol. 2020, 140, 1285–1288. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.Q.; Zou, H.Y.; Xie, J.J.; Fang, W.K. Paradoxical roles of desmosomal components in head and neck cancer. Biomolecules 2021, 11, 914. [Google Scholar] [CrossRef] [PubMed]
- Jones, V.A.; Patel, P.M.; Gibson, F.T.; Cordova, A.; Amber, K.T. The role of collagen XVII in cancer: Squamous cell carcinoma and beyond. Front. Oncol. 2020, 10, 352. [Google Scholar] [CrossRef] [PubMed]
- Gatalica, B.; Pulkkinen, L.; Li, K.; Kuokkanen, K.; Ryynänen, M.; McGrath, J.A.; Uitto, J. Cloning of the human type XVII collagen gene (COL17A1), and detection of novel mutations in generalized atrophic benign epidermolysis bullosa. Am. J. Hum. Genet. 1997, 60, 352–365. [Google Scholar] [PubMed]
- Jonsson, F.; Byström, B.; Davidson, A.E.; Backman, L.J.; Kellgren, T.G.; Tuft, S.J.; Koskela, T.; Rydén, P.; Sandgren, O.; Danielson, P.; et al. Mutations in collagen, type XVII, Alpha 1 (COL17A1) cause epithelial recurrent erosion dystrophy (ERED). Hum Mutat. 2015, 36, 463–473. [Google Scholar] [CrossRef]
- Cuffel, C.; Rivals, J.P.; Zaugg, Y.; Salvi, S.; Seelentag, W.; Speiser, D.E.; Liénard, D.; Monnier, P.; Romero, P.; Bron, L.; et al. Pattern and clinical significance of cancer-testis gene expression in head and neck squamous cell carcinoma. Int. J. Cancer 2011, 128, 2625–2634. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, M.; Kageyama, S.; Miyahara, Y.; Ishikawa, T.; Ueda, S.; Soga, N.; Naota, H.; Mukai, K.; Harada, N.; Ikeda, H.; et al. MAGE-A4, NY-ESO-1 and SAGE mRNA expression rates and co-expression relationships in solid tumours. BMC Cancer 2020, 20, 606. [Google Scholar] [CrossRef]
- Melo, D.H.; Mamede, R.C.M.; Neder, L.; Saggioro, F.P.; Figueiredo, D.L.A.; Araújo da Silva, W., Jr.; Jungbluth, A.A.; Zago, M.A. Expression of mage-A4 and mage-C1 tumor-associated antigen in benign and malignant thyroid diseases. Head Neck 2011, 33, 1426–1432. [Google Scholar] [CrossRef]
- Montoro JR de, M.C.; Mamede, R.C.M.; Neder Serafini, L.; Saggioro, F.P.; Figueiredo, D.L.A.; da Silva, W.A., Jr.; Jungbluth, A.A.; Spagnoli, G.C.; Zago, M.A. Expression of cancer-testis antigens MAGE-A4 and MAGE-C1 in oral squamous cell carcinoma. Head Neck 2012, 34, 1123–1128. [Google Scholar] [CrossRef]
- Prasad, M.L.; Jungbluth, A.A.; Patel, S.G.; Iversen, K.; Hoshaw-Woodard, S.; Busam, K.J. Expression and significance of cancer testis antigens in primary mucosal melanoma of the head and neck. Head Neck 2004, 26, 1053–1057. [Google Scholar] [CrossRef]
- Williams, J.Z.; Allen, G.M.; Shah, D.; Sterin, I.S.; Kim, K.H.; Garcia, V.P.; Shavey, G.E.; Yu, W.; Puig-Saus, C.; Tsoi, J.; et al. Precise T cell recognition programs designed by transcriptionally linking multiple receptors. Science 2020, 370, 1099–1104. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tissue Group | Tissue Types |
|---|---|
| Adipose and soft | adipose tissue and soft tissue |
| Endocrine | adrenal gland, parathyroid gland, thymus, and thyroid gland |
| Nervous system | caudate, cerebellum, cerebral cortex, choroid plexus, hippocampus, hypothalamus, pituitary gland, substantia nigra, and dorsal raphe |
| Circulatory | heart muscle |
| Immune tissue | appendix, lymph node, spleen, and tonsil |
| Immune cells | bone marrow |
| Female tissue | breast, cervix, endometrium, fallopian tube, lactating breast, ovary, placenta, and vagina |
| Lungs | bronchus, lung, and nasopharynx |
| Colon | colon |
| Gastrointestinal | duodenum, oral mucosa, and rectum |
| Male tissue | epididymis, prostate, seminal vesicle, and testis |
| Esophagus | esophagus |
| Eye | eye, retina |
| Gallbladder | gallbladder |
| Skin | hair, skin, and sole of foot |
| Urinary | Kidney and urinary bladder |
| Liver | liver |
| Pancreas | pancreas |
| Salivary gland | salivary gland |
| Muscle | skeletal muscle and smooth muscle |
| Small intestine | small intestine |
| Stomach | stomach |
| Cancer Type | Genes Encoding Clinical CAR Targets |
|---|---|
| CRC | MUC1, CD274, PROM1, ROR1, PSCA, MET, EPCAM, EGFR, FOLH1, ERBB2, MSLN, CD276, TM4SF1, CEACAM5, and GUCY2C |
| GBM | MUC1, CD274, PROM1, ROR1, MPP2, EPHA2, PSCA, IL13RA2, EGFR, ERBB2, MSLN, CD276, and BSG |
| LIHC | MUC1, CD274, PROM1, ROR1, AFP, TNFRSF10B, PTGS2, PSCA, MET, EPCAM, EGFR, GPC3, ERBB2, MSLN, CD276, and BSG |
| NSCLC | MUC1, CD274, ROR1, CD80, CD86, MAGEA1, MAGEA4, PSCA, EGFR, FOLH1, GPC3, ERBB2, MSLN, CD276, and TM4SF1 |
| PAAD | MUC1, CD274, PROM1, CD70, ROR2, CLDN18, PSCA, EPCAM, EGFR, ERBB2, MSLN, TM4SF1, and GUCY2C |
| STAD | MUC2, ROR2, PSCA, EPCAM, EGFR, ERBB2, MSLN, CD276, TM4SF1, and GUCY2C |
| OV | PROM1, CD70, FOLR1, MUC16, MET, EGFR, ERBB2, MSLN, and ALPP |
| SKCM | CD70, PMEL, KDR, MET, and IL13RA2 |
| HNSC | EGFR and ERBB2 |
| Cancer Type | Gene with Highest Tumor Selectivity Score (0–1) | Gene with Highest HPA Safety Score (0–2) | Gene with Highest TS Safety Score (0–10) |
|---|---|---|---|
| STAD | TM4SF1 (0.80) | PSCA (2.0) | GUCY2C (9.83) |
| HNSC | EGFR (0.73) | ERBB2 (1.55) | ERBB2 (7.92) |
| OV | MSLN (0.75) | MUC16 (1.84) | ALPP (10.0) |
| SKCM | PMEL (0.84) | PMEL (1.95) | PMEL (9.79) |
| CRC | EPCAM (0.90) | PSCA (2.0) | GUCY2C (9.83) |
| GBM | EGFR (0.74) | PSCA (2.0) | IL13RA2 (9.79) |
| PAAD | EPCAM (0.86) | PSCA (2.0) | CLDN18 (9.83) |
| LIHC | GPC3 (0.65) | AFP (2.0) | AFP (9.79) |
| NSCLC | MUC1 (0.80) | PSCA (2.0) | MAGEA1 (10.0) |
| Row | Cancer Type | Surface Protein | Encoding Gene | Protein Family/Group(s) |
|---|---|---|---|---|
| 1 | HNSC | Tumor-associated calcium signal transducer 2; Trop-2; epithelial glycoprotein-1 antigen | TACSTD2 | Epithelial cell adhesion molecule family; transmembrane glycoprotein |
| 2 | HNSC | Desmoglein-3 | DSG3 | Desmoglein family;cadherin superfamily;transmembrane glycoprotein |
| 3 | HNSC | Cadherin-3 | CDH3 | Classical cadherin;cadherin superfamily |
| 4 | HNSC | Glucose transporter 1; solute carrier family 2 | SLC2A1 | Solute carrier 2A family |
| 5 | HNSC | Desmocollin 3 | DSC3 | Desmosomal cadherin; cadherin superfamily |
| 6 | HNSC | Plakophilin 3 | PKP3 | Plakophilin family;arm-repeat (armadillo) family; anddesmosomal protein |
| 7 | HNSC | Collagen type XVII alpha 1 chain | COL17A1 | Collagen family; hemidesmosomal protein |
| 8 | HNSC, GBM | Protein Tyrosine Phosphatase Receptor Type Z1 | PTPRZ1 | Receptor-type tyrosine-protein phosphatase family |
| 9 | HNSC | Melanoma-associated antigen 4 | MAGEA4 | MAGE family of tumor-associated antigens |
| 10 | HNSC | FAT Atypical Cadherin 2 | FAT2 | Protocadherin;FAT family |
| 11 | HNSC | Chloride Channel Accessory 2 | CLCA2 | Calcium-activated chloride channel (CLCA) family |
| 12 | HNSC | Cholinergic Receptor Muscarinic 3; M3 Muscarinic receptor | CHRM3 | Muscarinic acetylcholine receptors family |
| 13 | HNSC | Solute carrier family 2 member 9 | SLC2A9 | Solute carrier 2A family |
| 14 | HNSC | Cadherin-13 | CDH13 | Non-classical cadherin; cadherin superfamily |
| 15 | HNSC | Hydroxycarboxylic Acid Receptor 2 | HCAR2 | Hydroxycarboxylic acid receptor family;G-protein coupled receptor 1 family |
| 16 | HNSC | Immunoglobulin Superfamily Member 3 | IGSF3 | Immunoglobulin superfamily |
| 17 | HNSC | Podoplanin | PDPN | Podoplanin family; transmembrane receptor glycoprotein |
| 18 | HNSC | LY6/PLAUR Domain Containing 3 | LYPD3 | LY6/PLAUR domain-containing family;GPI-anchored metastasis-associated protein |
| 19 | HNSC | Cluster of differentiation 109 | CD109 | Alpha-2-macroglobulin family |
| 20 | HNSC | Solute carrier family 7 member 8 | SLC7A8 | Solute carrier 7A family |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madan, S.; Sinha, S.; Chang, T.; Gutkind, J.S.; Cohen, E.E.W.; Schäffer, A.A.; Ruppin, E. Pan-Cancer Analysis of Patient Tumor Single-Cell Transcriptomes Identifies Promising Selective and Safe Chimeric Antigen Receptor Targets in Head and Neck Cancer. Cancers 2023, 15, 4885. https://doi.org/10.3390/cancers15194885
Madan S, Sinha S, Chang T, Gutkind JS, Cohen EEW, Schäffer AA, Ruppin E. Pan-Cancer Analysis of Patient Tumor Single-Cell Transcriptomes Identifies Promising Selective and Safe Chimeric Antigen Receptor Targets in Head and Neck Cancer. Cancers. 2023; 15(19):4885. https://doi.org/10.3390/cancers15194885
Chicago/Turabian StyleMadan, Sanna, Sanju Sinha, Tiangen Chang, J. Silvio Gutkind, Ezra E. W. Cohen, Alejandro A. Schäffer, and Eytan Ruppin. 2023. "Pan-Cancer Analysis of Patient Tumor Single-Cell Transcriptomes Identifies Promising Selective and Safe Chimeric Antigen Receptor Targets in Head and Neck Cancer" Cancers 15, no. 19: 4885. https://doi.org/10.3390/cancers15194885