
Extracellular Vesicles in Triple–Negative Breast Cancer: Immune Regulation, Biomarkers, and Immunotherapeutic Potential
 , , ,
, , ,  , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Extracellular Vesicles: More Than Cellular Dust
1.2. Microparticles
1.3. Exosomes
1.4. Autophagic EVs
1.5. Apoptotic Bodies (ApoBDs)
2. EVs and Diseases
3. Breast Cancer and Its Subtypes
3.1. Breast Cancer Subtypes
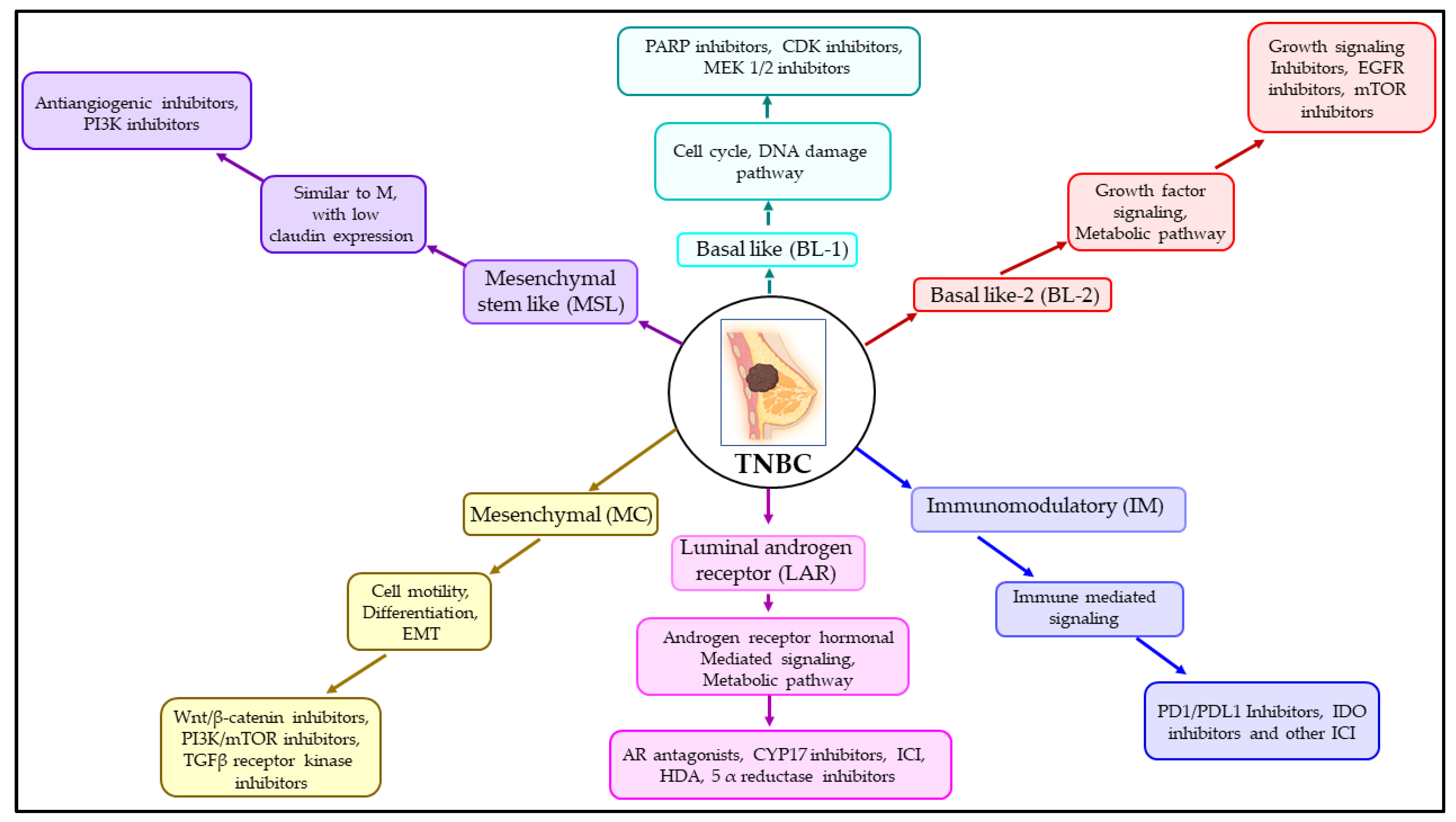
3.2. Subtypes of TNBC and Their Treatment Measures
4. The Role of EVs in the Progression of TNBC
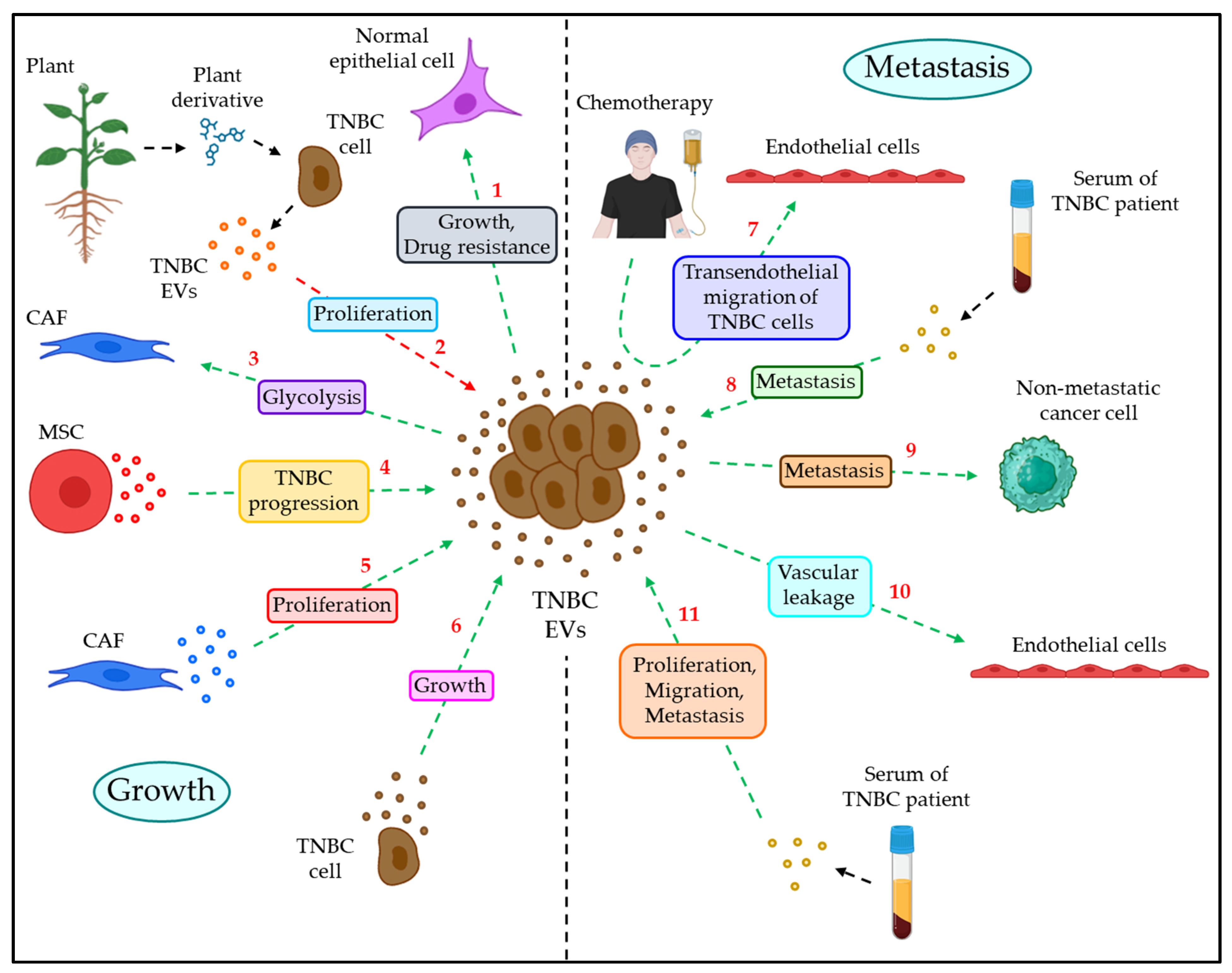
4.1. EVs in TNBC Growth
4.2. EVs in TNBC Metastasis
5. Blood Coagulation Is Associated with TNBC Progression
6. Role of EVs as a Biomarker for TNBC
6.1. EVs’ Proteins as a TNBC Biomarker
6.2. EVs’ miRNAs as a Biomarker for TNBC
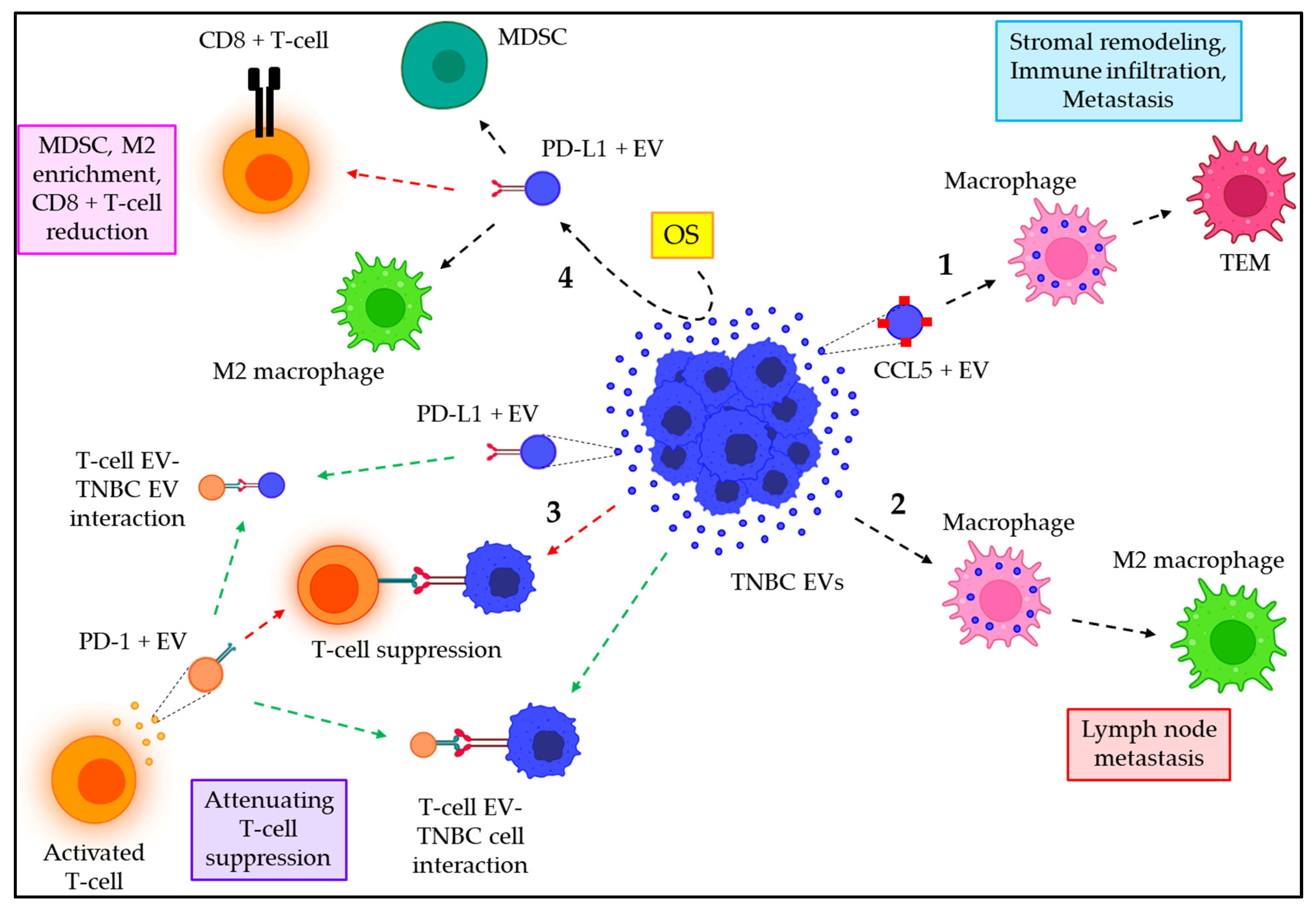
7. Emerging Role of EVs in TNBC Immune Regulation
8. Emerging Role of EVs in TNBC Immunotherapy
8.1. Immunotherapeutic Approaches against Human TNBC
8.2. EVs in TNBC Immunotherapy
9. EVs in Clinical Trials: The Translational Significance in Cancer
10. Conclusions, Future Direction and Challenges
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EV | extracellular vesicle |
| miR | microRNA |
| MP | microparticle |
| MV | microvesicle |
| PS | phosphatidylserine |
| HSP | heat–shock protein |
| MVB | multivesicular body |
| ESCRT | endosomal sorting complexes required for transport |
| TSG101 | tumor susceptibility gene 101 |
| Alix | ALG–2–interacting protein X |
| HSC70 | heat–shock cognate 70 kDa protein |
| LC3 | microtubule–associated proteins 1A/1B light chain 3B |
| CD | cluster of differentiation |
| GRP78 | glucose–regulated protein 78 |
| UTI | urinary tract infection |
| TF | tissue factor |
| ICAM–1 | intercellular adhesion molecule 1 |
| AKI | acute kidney injury |
| AQP1 | aquaporin 1 |
| HFF | human follicular fluid |
| PCOS | polycystic ovary syndrome |
| CSF | cerebrospinal fluid |
| PD | Parkinson’s disease |
| CM | congenital myopathies |
| NPC | nasopharyngeal carcinoma |
| TNBC | triple–negative breast cancer |
| PTEN | phosphatase and tensin homolog |
| STAT3 | signal transducer and activator of transcription 3 |
| M2 | type 2 macrophage |
| HCC | hepatocellular carcinoma |
| TXNIP | thioredoxin–interacting protein |
| IHC | immunohistochemistry |
| ER | estrogen receptor |
| PR | progesterone receptor |
| HER2 | human epidermal growth factor receptor–2 |
| EGFR | extracellular growth factor receptor |
| CK | cytokeratin |
| BRCA1 | breast cancer gene 1 |
| EpCAM | epithelial cellular adhesion molecule |
| EMT | epithelial to mesenchymal transition |
| BL | basal–like |
| IM | immunomodulatory |
| LAR | luminal androgen receptor |
| M | mesenchymal |
| MSL | mesenchymal stem like |
| NK | natural killer |
| BCR | B–cell receptor |
| PI3K | phosphoinositide 3–kinase |
| mTOR | mammalian target of rapamycin |
| Wnt | wingless/integrated |
| TGFβ | transforming growth factor β |
| MAPK | mitogen–activated protein kinase |
| HIF1A | hypoxia inducible factor 1 subunit α |
| DET | deoxyelephantopin |
| DETD–35 | DET derivative 35 |
| CAF | cancer–associated fibroblast |
| TME | tumor microenvironment |
| ITGB4 | integrin β4 |
| BNI3PL | bcl2 interacting protein 3 like |
| MSC | mesenchymal stem cell |
| HAND2–AS1 | heart and neural crest derivatives expressed 2 antisense RNA 1 |
| FOSL1 | Fos like 1 |
| SPANXB1 | sperm protein associated with the nucleus on the X chromosome (SPANX) family member B1 |
| SH3GL2 | SH3 domain containing GRB2 like 2 |
| DUSP14 | dual specificity phosphatase 14 |
| VE cadherin | vascular endothelial cadherin |
| PSMA1 | proteasome 20S subunit α1 |
| FVIIa | activated factor VII |
| PAR2 | protease–activated receptor 2 |
| MK2 | MAPK–activated protein kinase 2 |
| MEK | MAPK kinase |
| ERK | extracellular signal–regulated kinase |
| MLCK | myosin light chain kinase |
| MLC2 | myosin light chain 2 |
| Rab | Ras–associated binding |
| NF–ĸB | nuclear factor ĸB |
| E–cadherin | epithelial cadherin |
| N–cadherin | neuronal cadherin |
| AREG | amphiregulin |
| UCHL1 | ubiquitin C–terminal hydrolase 1 |
| SMAD2 | mothers against decapentaplegic homolog 2 |
| PTX | paclitaxel |
| P–gp | P–glycoprotein |
| Mef2c | myocyte–specific enhancer factor 2C |
| HOXD10 | homeobox D10 |
| KLF4 | Krüppel–like factor 4 |
| ZO–1 | zonula occludens 1 |
| CCL | chemokine (C–C motif) ligand |
| TEM | tumor EV–educated macrophage |
| IFNG | interferon γ |
| CXCL1 | CXC motif chemokine ligand 1 |
| OPN | osteopontin |
| CTLA–4 | cytotoxic T–lymphocyte–associated protein 4 |
| HGF | hepatocyte growth factor |
| LN | lymph node |
| PD–1 | programmed death 1 |
| PD–L1 | programmed death ligand 1 |
| OS | oscillatory strain |
| MDSC | myeloid–derived suppressor cell |
| CAR–T | chimeric antigen receptor T |
| FDA | Food and Drug Administration |
| TROP2 | tumor–associated calcium signal transducer 2 |
| TIL | tumor–infiltrating lymphocyte |
| MHC | major histocompatibility complex |
| FasL | Fas ligand |
| AS1411 | 26–mer G–rich DNA oligonucleotide |
| Ver–A | verrucarin A |
| NSCLC | non–small cell lung cancer |
| CRC | colorectal cancer |
| BALF | bronchoalveolar fluid |
| TP53 | tumor protein p53 |
| UBC | ubiquitin C |
| HRAS | Harvey Rat sarcoma virus |
| RPS27A | ribosomal protein S27A |
| GRB2 | growth factor receptor–bound protein 2 |
| UBA52 | ubiquitin A–52 residue ribosomal protein fusion product 1 |
| SRC | Src family kinase |
References
- Chargaff, E.; West, R. The biological significance of the thromboplastic protein of blood. J. Biol. Chem. 1946, 166, 189–197. [Google Scholar] [CrossRef]
- Wolf, P. The nature and significance of platelet products in human plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef] [PubMed]
- Harding, C.; Heuser, J.; Stahl, P. Endocytosis and intracellular processing of transferrin and colloidal gold-transferrin in rat reticulocytes: Demonstration of a pathway for receptor shedding. Eur. J. Cell Biol. 1984, 35, 256–263. [Google Scholar] [PubMed]
- Yanez-Mo, M.; Siljander, P.R.; Andreu, Z.; Zavec, A.B.; Borras, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef]
- Stranford, D.M.; Leonard, J.N. Delivery of Biomolecules via Extracellular Vesicles: A Budding Therapeutic Strategy. Adv. Genet. 2017, 98, 155–175. [Google Scholar] [PubMed]
- Couch, Y.; Buzas, E.I.; Di Vizio, D.; Gho, Y.S.; Harrison, P.; Hill, A.F.; Lotvall, J.; Raposo, G.; Stahl, P.D.; Thery, C.; et al. A brief history of nearly Everything—The rise and rise of extracellular vesicles. J. Extracell. Vesicles 2021, 10, e12144. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Keshava, S.; Ansari, S.A.; Kondreddy, V.; Esmon, C.T.; Griffin, J.H.; Pendurthi, U.R.; Rao, L.V.M. Factor VIIa induces extracellular vesicles from the endothelium: A potential mechanism for its hemostatic effect. Blood 2021, 137, 3428–3442. [Google Scholar] [CrossRef]
- Das, K.; Keshava, S.; Pendurthi, U.R.; Rao, L.V.M. Factor VIIa suppresses inflammation and barrier disruption through the release of EEVs and transfer of microRNA 10a. Blood 2022, 139, 118–133. [Google Scholar] [CrossRef]
- Das, K.; Pendurthi, U.R.; Manco-Johnson, M.; Martin, E.J.; Brophy, D.F.; Rao, L.V.M. Factor VIIa treatment increases circulating extracellular vesicles in hemophilia patients: Implications for the therapeutic hemostatic effect of FVIIa. J. Thromb. Haemost. 2022, 20, 1928–1933. [Google Scholar] [CrossRef]
- Das, K.; Rao, L.V.M. The Role of microRNAs in Inflammation. Int. J. Mol. Sci. 2022, 23, 15479. [Google Scholar] [CrossRef]
- Das, K.; Paul, S.; Singh, A.; Ghosh, A.; Roy, A.; Ansari, S.A.; Prasad, R.; Mukherjee, A.; Sen, P. Triple-negative breast cancer-derived microvesicles transfer microRNA221 to the recipient cells and thereby promote epithelial-to-mesenchymal transition. J. Biol. Chem. 2019, 294, 13681–13696. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.K.; Upadhya, R. Extracellular Vesicles in Health and Disease. Aging Dis. 2021, 12, 1358–1362. [Google Scholar] [CrossRef] [PubMed]
- Akbar, N.; Azzimato, V.; Choudhury, R.P.; Aouadi, M. Extracellular vesicles in metabolic disease. Diabetologia 2019, 62, 2179–2187. [Google Scholar] [CrossRef]
- Das, K.; Mukherjee, T.; Shankar, P. The Role of Extracellular Vesicles in the Pathogenesis of Hematological Malignancies: Interaction with Tumor Microenvironment; a Potential Biomarker and Targeted Therapy. Biomolecules 2023, 13, 897. [Google Scholar] [CrossRef] [PubMed]
- Buzas, E.I.; Gyorgy, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Paul, S.; Mukherjee, T.; Ghosh, A.; Sharma, A.; Shankar, P.; Gupta, S.; Keshava, S.; Parashar, D. Beyond Macromolecules: Extracellular Vesicles as Regulators of Inflammatory Diseases. Cells 2023, 12, 1963. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Prasad, R.; Roy, S.; Mukherjee, A.; Sen, P. The Protease Activated Receptor2 Promotes Rab5a Mediated Generation of Pro-metastatic Microvesicles. Sci. Rep. 2018, 8, 7357. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Prasad, R.; Singh, A.; Bhattacharya, A.; Roy, A.; Mallik, S.; Mukherjee, A.; Sen, P. Protease-activated receptor 2 promotes actomyosin dependent transforming microvesicles generation from human breast cancer. Mol. Carcinog. 2018, 57, 1707–1722. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef]
- Bonsergent, E.; Grisard, E.; Buchrieser, J.; Schwartz, O.; Thery, C.; Lavieu, G. Quantitative characterization of extracellular vesicle uptake and content delivery within mammalian cells. Nat. Commun. 2021, 12, 1864. [Google Scholar] [CrossRef]
- van Niel, G.; Carter, D.R.F.; Clayton, A.; Lambert, D.W.; Raposo, G.; Vader, P. Challenges and directions in studying cell-cell communication by extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2022, 23, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Bodega, G.; Alique, M.; Puebla, L.; Carracedo, J.; Ramirez, R.M. Microvesicles: ROS scavengers and ROS producers. J. Extracell. Vesicles 2019, 8, 1626654. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.P.; Mardini, O.; Ericsson, M.; Prabhakar, S.; Maguire, C.; Chen, J.W.; Tannous, B.A.; Breakefield, X.O. Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano 2014, 8, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Miller, V.M.; Li, Y.; Jayachandran, M. Microvesicles at the crossroads between infection and cardiovascular diseases. J. Cardiovasc. Pharmacol. 2012, 59, 124–132. [Google Scholar] [CrossRef] [PubMed]
- Escola, J.M.; Kleijmeer, M.J.; Stoorvogel, W.; Griffith, J.M.; Yoshie, O.; Geuze, H.J. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J. Biol. Chem. 1998, 273, 20121–20127. [Google Scholar] [CrossRef] [PubMed]
- Zoller, M. Tetraspanins: Push and pull in suppressing and promoting metastasis. Nat. Rev. Cancer 2009, 9, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Di Vizio, D.; Morello, M.; Dudley, A.C.; Schow, P.W.; Adam, R.M.; Morley, S.; Mulholland, D.; Rotinen, M.; Hager, M.H.; Insabato, L.; et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am. J. Pathol. 2012, 181, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Heijnen, H.F.; Schiel, A.E.; Fijnheer, R.; Geuze, H.J.; Sixma, J.J. Activated platelets release two types of membrane vesicles: Microvesicles by surface shedding and exosomes derived from exocytosis of multivesicular bodies and alpha-granules. Blood 1999, 94, 3791–3799. [Google Scholar] [CrossRef]
- Morello, M.; Minciacchi, V.R.; de Candia, P.; Yang, J.; Posadas, E.; Kim, H.; Griffiths, D.; Bhowmick, N.; Chung, L.W.; Gandellini, P.; et al. Large oncosomes mediate intercellular transfer of functional microRNA. Cell Cycle 2013, 12, 3526–3536. [Google Scholar] [CrossRef]
- Kowal, J.; Tkach, M.; Thery, C. Biogenesis and secretion of exosomes. Curr. Opin. Cell Biol. 2014, 29, 116–125. [Google Scholar] [CrossRef]
- Gupta, P.; Kadamberi, I.P.; Mittal, S.; Tsaih, S.W.; George, J.; Kumar, S.; Vijayan, D.K.; Geethadevi, A.; Parashar, D.; Topchyan, P.; et al. Tumor Derived Extracellular Vesicles Drive T Cell Exhaustion in Tumor Microenvironment through Sphingosine Mediated Signaling and Impacting Immunotherapy Outcomes in Ovarian Cancer. Adv. Sci. 2022, 9, e2104452. [Google Scholar] [CrossRef] [PubMed]
- Parashar, D.; Geethadevi, A.; McAllister, D.; Ebben, J.; Peterson, F.C.; Jensen, D.R.; Bishop, E.; Pradeep, S.; Volkman, B.F.; Dwinell, M.B.; et al. Targeted biologic inhibition of both tumor cell-intrinsic and intercellular CLPTM1L/CRR9-mediated chemotherapeutic drug resistance. NPJ Precis. Oncol. 2021, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Geminard, C.; De Gassart, A.; Blanc, L.; Vidal, M. Degradation of AP2 during reticulocyte maturation enhances binding of hsc70 and Alix to a common site on TFR for sorting into exosomes. Traffic 2004, 5, 181–193. [Google Scholar] [CrossRef] [PubMed]
- van Niel, G.; Porto-Carreiro, I.; Simoes, S.; Raposo, G. Exosomes: A common pathway for a specialized function. J. Biochem. 2006, 140, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Fader, C.M.; Colombo, M.I. Autophagy and multivesicular bodies: Two closely related partners. Cell Death Differ. 2009, 16, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Krylova, S.V.; Feng, D. The Machinery of Exosomes: Biogenesis, Release, and Uptake. Int. J. Mol. Sci. 2023, 24, 1337. [Google Scholar] [CrossRef]
- Brunel, A.; Bégaud, G.; Auger, C.; Durand, S.; Battu, S.; Bessette, B.; Verdier, M. Autophagy and Extracellular Vesicles, Connected to rabGTPase Family, Support Aggressiveness in Cancer Stem Cells. Cells 2021, 10, 1330. [Google Scholar] [CrossRef]
- Park, J.S.; Perl, A. Endosome Traffic Modulates Pro-Inflammatory Signal Transduction in CD4(+) T Cells-Implications for the Pathogenesis of Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2023, 24, 10749. [Google Scholar] [CrossRef] [PubMed]
- Arya, S.B.; Collie, S.P.; Parent, C.A. The ins-and-outs of exosome biogenesis, secretion, and internalization. Trends Cell Biol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Doyle, L.M.; Wang, M.Z. Overview of Extracellular Vesicles, Their Origin, Composition, Purpose, and Methods for Exosome Isolation and Analysis. Cells 2019, 8, 727. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.; Ughetto, S.; Mahjoum, S.; Nair, A.V.; Breakefield, X.O. Uptake, functionality, and re-release of extracellular vesicle-encapsulated cargo. Cell Rep. 2022, 39, 110651. [Google Scholar] [CrossRef] [PubMed]
- Montecalvo, A.; Larregina, A.T.; Shufesky, W.J.; Stolz, D.B.; Sullivan, M.L.; Karlsson, J.M.; Baty, C.J.; Gibson, G.A.; Erdos, G.; Wang, Z.; et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood 2012, 119, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef] [PubMed]
- Borges, F.T.; Reis, L.A.; Schor, N. Extracellular vesicles: Structure, function, and potential clinical uses in renal diseases. Braz. J. Med. Biol. Res. 2013, 46, 824–830. [Google Scholar] [CrossRef]
- Wickman, G.; Julian, L.; Olson, M.F. How apoptotic cells aid in the removal of their own cold dead bodies. Cell Death Differ. 2012, 19, 735–742. [Google Scholar] [CrossRef]
- Thery, C.; Boussac, M.; Veron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef]
- Escrevente, C.; Keller, S.; Altevogt, P.; Costa, J. Interaction and uptake of exosomes by ovarian cancer cells. BMC Cancer 2011, 11, 108. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Zhang, Q.; Franklin, J.L.; Coffey, R.J. Extracellular vesicles and nanoparticles: Emerging complexities. Trends Cell Biol. 2023, 33, 667–681. [Google Scholar] [CrossRef] [PubMed]
- Rother, N.; Yanginlar, C.; Pieterse, E.; Hilbrands, L.; van der Vlag, J. Microparticles in Autoimmunity: Cause or Consequence of Disease? Front. Immunol. 2022, 13, 822995. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Fenix, A.M.; Franklin, J.L.; Higginbotham, J.N.; Zhang, Q.; Zimmerman, L.J.; Liebler, D.C.; Ping, J.; Liu, Q.; Evans, R.; et al. Reassessment of Exosome Composition. Cell 2019, 177, 428–445.e18. [Google Scholar] [CrossRef]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, M.; Carmo, N.B.; Krumeich, S.; Fanget, I.; Raposo, G.; Savina, A.; Moita, C.F.; Schauer, K.; Hume, A.N.; Freitas, R.P.; et al. Rab27a and Rab27b control different steps of the exosome secretion pathway. Nat. Cell Biol. 2010, 12, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Dieudé, M.; Bell, C.; Turgeon, J.; Beillevaire, D.; Pomerleau, L.; Yang, B.; Hamelin, K.; Qi, S.; Pallet, N.; Béland, C.; et al. The 20S proteasome core, active within apoptotic exosome-like vesicles, induces autoantibody production and accelerates rejection. Sci. Transl. Med. 2015, 7, 318ra200. [Google Scholar] [CrossRef] [PubMed]
- Hristov, M.; Erl, W.; Linder, S.; Weber, P.C. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood 2004, 104, 2761–2766. [Google Scholar] [CrossRef]
- Woei, A.J.F.J.; van der Starre, W.E.; Tesselaar, M.E.; Garcia Rodriguez, P.; van Nieuwkoop, C.; Bertina, R.M.; van Dissel, J.T.; Osanto, S. Procoagulant tissue factor activity on microparticles is associated with disease severity and bacteremia in febrile urinary tract infections. Thromb. Res. 2014, 133, 799–803. [Google Scholar] [CrossRef]
- Tans, G.; Rosing, J.; Thomassen, M.C.; Heeb, M.J.; Zwaal, R.F.; Griffin, J.H. Comparison of anticoagulant and procoagulant activities of stimulated platelets and platelet-derived microparticles. Blood 1991, 77, 2641–2648. [Google Scholar] [CrossRef] [PubMed]
- Rautou, P.E.; Leroyer, A.S.; Ramkhelawon, B.; Devue, C.; Duflaut, D.; Vion, A.C.; Nalbone, G.; Castier, Y.; Leseche, G.; Lehoux, S.; et al. Microparticles from human atherosclerotic plaques promote endothelial ICAM-1-dependent monocyte adhesion and transendothelial migration. Circ. Res. 2011, 108, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhou, X.; Zhang, H.; Yao, Q.; Liu, Y.; Dong, Z. Extracellular vesicles in diagnosis and therapy of kidney diseases. Am. J. Physiol. Ren. Physiol. 2016, 311, F844–F851. [Google Scholar] [CrossRef] [PubMed]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thurmann, L.; Kurth, F.; Volker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.; Djaldetti, R.; Mollenhauer, B.; Offen, D. CSF-derived extracellular vesicles from patients with Parkinson’s disease induce symptoms and pathology. Brain 2023, 146, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.; Withrow, J.; Hunter, M.; Liu, Y.; Tang, Y.L.; Fulzele, S.; Hamrick, M.W. Emerging role of extracellular vesicles in musculoskeletal diseases. Mol. Asp. Med. 2018, 60, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Shin, T.S.; Kim, J.S.; Jee, Y.K.; Kim, Y.K. A new horizon of precision medicine: Combination of the microbiome and extracellular vesicles. Exp. Mol. Med. 2022, 54, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Liu, Y.; Wang, Z.; Wu, S. miR-144 delivered by nasopharyngeal carcinoma-derived EVs stimulates angiogenesis through the FBXW7/HIF-1alpha/VEGF-A axis. Mol. Ther. Nucleic Acids 2021, 24, 1000–1011. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Hung, J.Y.; Chang, W.A.; Jian, S.F.; Lin, Y.S.; Pan, Y.C.; Wu, C.Y.; Kuo, P.L. Hypoxic Lung-Cancer-Derived Extracellular Vesicle MicroRNA-103a Increases the Oncogenic Effects of Macrophages by Targeting PTEN. Mol. Ther. 2018, 26, 568–581. [Google Scholar] [CrossRef]
- Yang, Y.; Mao, F.; Guo, L.; Shi, J.; Wu, M.; Cheng, S.; Guo, W. Tumor cells derived-extracellular vesicles transfer miR-3129 to promote hepatocellular carcinoma metastasis by targeting TXNIP. Dig. Liver Dis. 2021, 53, 474–485. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, Z.; Gao, Q. Transfer of microRNA-25 by colorectal cancer cell-derived extracellular vesicles facilitates colorectal cancer development and metastasis. Mol. Ther. Nucleic Acids 2021, 23, 552–564. [Google Scholar] [CrossRef] [PubMed]
- Smolarz, B.; Nowak, A.Z.; Romanowicz, H. Breast Cancer-Epidemiology, Classification, Pathogenesis and Treatment (Review of Literature). Cancers 2022, 14, 2569. [Google Scholar] [CrossRef] [PubMed]
- Jagadish, N.; Gupta, N.; Agarwal, S.; Parashar, D.; Sharma, A.; Fatima, R.; Topno, A.P.; Kumar, V.; Suri, A. Sperm-associated antigen 9 (SPAG9) promotes the survival and tumor growth of triple-negative breast cancer cells. Tumour Biol. J. Int. Soc. Oncodevelopmental Biol. Med. 2016, 37, 13101–13110. [Google Scholar] [CrossRef] [PubMed]
- Sinha, A.; Agarwal, S.; Parashar, D.; Verma, A.; Saini, S.; Jagadish, N.; Ansari, A.S.; Lohiya, N.K.; Suri, A. Down regulation of SPAG9 reduces growth and invasive potential of triple-negative breast cancer cells: Possible implications in targeted therapy. J. Exp. Clin. Cancer Res. CR 2013, 32, 69. [Google Scholar] [CrossRef] [PubMed]
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Vallejos, C.S.; Gomez, H.L.; Cruz, W.R.; Pinto, J.A.; Dyer, R.R.; Velarde, R.; Suazo, J.F.; Neciosup, S.P.; Leon, M.; de la Cruz, M.A.; et al. Breast cancer classification according to immunohistochemistry markers: Subtypes and association with clinicopathologic variables in a peruvian hospital database. Clin. Breast Cancer 2010, 10, 294–300. [Google Scholar] [CrossRef]
- Urruticoechea, A.; Smith, I.E.; Dowsett, M. Proliferation marker Ki-67 in early breast cancer. J. Clin. Oncol. 2005, 23, 7212–7220. [Google Scholar] [CrossRef]
- Abd El-Rehim, D.M.; Pinder, S.E.; Paish, C.E.; Bell, J.; Blamey, R.W.; Robertson, J.F.; Nicholson, R.I.; Ellis, I.O. Expression of luminal and basal cytokeratins in human breast carcinoma. J. Pathol. 2004, 203, 661–671. [Google Scholar] [CrossRef]
- Yersal, O.; Barutca, S. Biological subtypes of breast cancer: Prognostic and therapeutic implications. World J. Clin. Oncol. 2014, 5, 412–424. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Ellis, M.J.; Tao, Y.; Luo, J.; A’Hern, R.; Evans, D.B.; Bhatnagar, A.S.; Chaudri Ross, H.A.; von Kameke, A.; Miller, W.R.; Smith, I.; et al. Outcome prediction for estrogen receptor-positive breast cancer based on postneoadjuvant endocrine therapy tumor characteristics. J. Natl. Cancer Inst. 2008, 100, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef] [PubMed]
- von Minckwitz, G.; Procter, M.; de Azambuja, E.; Zardavas, D.; Benyunes, M.; Viale, G.; Suter, T.; Arahmani, A.; Rouchet, N.; Clark, E.; et al. Investigators, Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N. Engl. J. Med. 2017, 377, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, T.O.; Hsu, F.D.; Jensen, K.; Cheang, M.; Karaca, G.; Hu, Z.; Hernandez-Boussard, T.; Livasy, C.; Cowan, D.; Dressler, L.; et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin. Cancer Res. 2004, 10, 5367–5374. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.C.; Martyniak, A.; Kandel, M.J.; Stadler, Z.K.; Masciari, S.; Miron, A.; Richardson, A.L.; Schnitt, S.J.; Garber, J.E. Basal cytokeratin and epidermal growth factor receptor expression are not predictive of BRCA1 mutation status in women with triple-negative breast cancers. Am. J. Surg. Pathol. 2009, 33, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Kanapathy Pillai, S.K.; Tay, A.; Nair, S.; Leong, C.O. Triple-negative breast cancer is associated with EGFR, CK5/6 and c-KIT expression in Malaysian women. BMC Clin. Pathol. 2012, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Cheang, M.C.; Voduc, D.; Bajdik, C.; Leung, S.; McKinney, S.; Chia, S.K.; Perou, C.M.; Nielsen, T.O. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin. Cancer Res. 2008, 14, 1368–1376. [Google Scholar] [CrossRef]
- Rakha, E.A.; Elsheikh, S.E.; Aleskandarany, M.A.; Habashi, H.O.; Green, A.R.; Powe, D.G.; El-Sayed, M.E.; Benhasouna, A.; Brunet, J.S.; Akslen, L.A.; et al. Triple-negative breast cancer: Distinguishing between basal and nonbasal subtypes. Clin. Cancer Res. 2009, 15, 2302–2310. [Google Scholar] [CrossRef]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef]
- Nounou, M.I.; ElAmrawy, F.; Ahmed, N.; Abdelraouf, K.; Goda, S.; Syed-Sha-Qhattal, H. Breast Cancer: Conventional Diagnosis and Treatment Modalities and Recent Patents and Technologies. Breast Cancer 2015, 9 (Suppl. 2), 17–34. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Gilcrease, M.Z.; Krishnamurthy, S.; Lee, J.S.; Fridlyand, J.; Sahin, A.; Agarwal, R.; Joy, C.; et al. Characterization of a naturally occurring breast cancer subset enriched in epithelial-to-mesenchymal transition and stem cell characteristics. Cancer Res. 2009, 69, 4116–4124. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.H.; Herschkowitz, J.I.; Komurov, K.; Zhou, A.Y.; Gupta, S.; Yang, J.; Hartwell, K.; Onder, T.T.; Gupta, P.B.; Evans, K.W.; et al. Core epithelial-to-mesenchymal transition interactome gene-expression signature is associated with claudin-low and metaplastic breast cancer subtypes. Proc. Natl. Acad. Sci. USA 2010, 107, 15449–15454. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef]
- De Soto, J.A.; Wang, X.; Tominaga, Y.; Wang, R.H.; Cao, L.; Qiao, W.; Li, C.; Xu, X.; Skoumbourdis, A.P.; Prindiville, S.A.; et al. The inhibition and treatment of breast cancer with poly (ADP-ribose) polymerase (PARP-1) inhibitors. Int. J. Biol. Sci. 2006, 2, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.C.; Zhang, J.; Xu, B.H.; Cai, L.; Ragaz, J.; Wang, Z.H.; Wang, B.Y.; Teng, Y.E.; Tong, Z.S.; Pan, Y.Y.; et al. Cisplatin plus gemcitabine versus paclitaxel plus gemcitabine as first-line therapy for metastatic triple-negative breast cancer (CBCSG006): A randomised, open-label, multicentre, phase 3 trial. Lancet Oncol. 2015, 16, 436–446. [Google Scholar] [CrossRef] [PubMed]
- O’Shaughnessy, J.; Schwartzberg, L.; Danso, M.A.; Miller, K.D.; Rugo, H.S.; Neubauer, M.; Robert, N.; Hellerstedt, B.; Saleh, M.; Richards, P.; et al. Phase III study of iniparib plus gemcitabine and carboplatin versus gemcitabine and carboplatin in patients with metastatic triple-negative breast cancer. J. Clin. Oncol. 2014, 32, 3840–3847. [Google Scholar] [CrossRef]
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Mayer, E.L.; Esteva, F.J.; Ma, C.X.; Liu, M.C.; Storniolo, A.M.; Rimawi, M.F.; Forero-Torres, A.; et al. TBCRC 001: Randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 2615–2623. [Google Scholar] [CrossRef]
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef]
- Gucalp, A.; Traina, T.A. Triple-negative breast cancer: Role of the androgen receptor. Cancer J. 2010, 16, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A.; et al. Phase II trial of bicalutamide in patients with androgen receptor-positive, estrogen receptor-negative metastatic Breast Cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef]
- Herschkowitz, J.I.; Simin, K.; Weigman, V.J.; Mikaelian, I.; Usary, J.; Hu, Z.; Rasmussen, K.E.; Jones, L.P.; Assefnia, S.; Chandrasekharan, S.; et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007, 8, R76. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Prosperi, J.R.; Choudhury, N.; Olopade, O.I.; Goss, K.H. beta-Catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS ONE 2015, 10, e0117097. [Google Scholar]
- Basho, R.K.; Gilcrease, M.; Murthy, R.K.; Helgason, T.; Karp, D.D.; Meric-Bernstam, F.; Hess, K.R.; Herbrich, S.M.; Valero, V.; Albarracin, C.; et al. Targeting the PI3K/AKT/mTOR Pathway for the Treatment of Mesenchymal Triple-Negative Breast Cancer: Evidence From a Phase 1 Trial of mTOR Inhibition in Combination With Liposomal Doxorubicin and Bevacizumab. JAMA Oncol. 2017, 3, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sanchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-beta inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef]
- Ozawa, P.M.M.; Alkhilaiwi, F.; Cavalli, I.J.; Malheiros, D.; de Souza Fonseca Ribeiro, E.M.; Cavalli, L.R. Extracellular vesicles from triple-negative breast cancer cells promote proliferation and drug resistance in non-tumorigenic breast cells. Breast Cancer Res. Treat. 2018, 172, 713–723. [Google Scholar] [CrossRef]
- Shiau, J.Y.; Chang, Y.Q.; Nakagawa-Goto, K.; Lee, K.H.; Shyur, L.F. Phytoagent Deoxyelephantopin and Its Derivative Inhibit Triple Negative Breast Cancer Cell Activity through ROS-Mediated Exosomal Activity and Protein Functions. Front. Pharmacol. 2017, 8, 398. [Google Scholar] [CrossRef]
- Sung, J.S.; Kang, C.W.; Kang, S.; Jang, Y.; Chae, Y.C.; Kim, B.G.; Cho, N.H. ITGB4-mediated metabolic reprogramming of cancer-associated fibroblasts. Oncogene 2020, 39, 664–676. [Google Scholar] [CrossRef]
- Xing, L.; Tang, X.; Wu, K.; Huang, X.; Yi, Y.; Huan, J. LncRNA HAND2-AS1 suppressed the growth of triple negative breast cancer via reducing secretion of MSCs derived exosomal miR-106a-5p. Aging 2020, 13, 424–436. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, B.G.; Jang, Y.; Kang, S.; Lee, J.H.; Cho, N.H. The stromal loss of miR-4516 promotes the FOSL1-dependent proliferation and malignancy of triple negative breast cancer. Cancer Lett. 2020, 469, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Wang, X.; Li, C.; Zhang, H.; Liu, Y.; Han, D.; Li, Y.; Li, Z.; Luo, D.; Zhang, N.; et al. CircHIF1A regulated by FUS accelerates triple-negative breast cancer progression by modulating NFIB expression and translocation. Oncogene 2021, 40, 2756–2771. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yang, Z.; Zhou, P.; Yu, M.; Li, B.; Liu, Y.; Jin, J.; Liu, W.; Jing, H.; Du, J.; et al. Phosphatidylserine-exposing tumor-derived microparticles exacerbate coagulation and cancer cell transendothelial migration in triple-negative breast cancer. Theranostics 2021, 11, 6445–6460. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, X.; Yang, S.; Pi, H.; Li, Z.; Yao, P.; Zhang, Q.; Wang, Q.; Shen, P.; Li, X.; et al. Proteomic Landscape of Exosomes Reveals the Functional Contributions of CD151 in Triple-Negative Breast Cancer. Mol. Cell Proteom. 2021, 20, 100121. [Google Scholar] [CrossRef] [PubMed]
- Kannan, A.; Philley, J.V.; Hertweck, K.L.; Ndetan, H.; Singh, K.P.; Sivakumar, S.; Wells, R.B.; Vadlamudi, R.K.; Dasgupta, S. Cancer Testis Antigen Promotes Triple Negative Breast Cancer Metastasis and is Traceable in the Circulating Extracellular Vesicles. Sci. Rep. 2019, 9, 11632. [Google Scholar] [CrossRef] [PubMed]
- Kia, V.; Paryan, M.; Mortazavi, Y.; Biglari, A.; Mohammadi-Yeganeh, S. Evaluation of exosomal miR-9 and miR-155 targeting PTEN and DUSP14 in highly metastatic breast cancer and their effect on low metastatic cells. J. Cell. Biochem. 2019, 120, 5666–5676. [Google Scholar] [CrossRef] [PubMed]
- Di Modica, M.; Regondi, V.; Sandri, M.; Iorio, M.V.; Zanetti, A.; Tagliabue, E.; Casalini, P.; Triulzi, T. Breast cancer-secreted miR-939 downregulates VE-cadherin and destroys the barrier function of endothelial monolayers. Cancer Lett. 2017, 384, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Wang, D.D.; Zhong, S.L.; Chen, W.Q.; Wang, F.L.; Zhang, J.; Xu, W.X.; Xu, D.; Zhang, Q.; Li, J.; et al. Tumor-derived exosomal circPSMA1 facilitates the tumorigenesis, metastasis, and migration in triple-negative breast cancer (TNBC) through miR-637/Akt1/beta-catenin (cyclin D1) axis. Cell Death Dis. 2021, 12, 420. [Google Scholar] [CrossRef]
- Abdol Razak, N.B.; Jones, G.; Bhandari, M.; Berndt, M.C.; Metharom, P. Cancer-Associated Thrombosis: An Overview of Mechanisms, Risk Factors, and Treatment. Cancers 2018, 10, 380. [Google Scholar] [CrossRef]
- Das, K.; Prasad, R.; Ansari, S.A.; Roy, A.; Mukherjee, A.; Sen, P. Matrix metalloproteinase-2: A key regulator in coagulation proteases mediated human breast cancer progression through autocrine signaling. Biomed. Pharmacother. 2018, 105, 395–406. [Google Scholar] [CrossRef]
- Higginbotham, J.N.; Demory Beckler, M.; Gephart, J.D.; Franklin, J.L.; Bogatcheva, G.; Kremers, G.J.; Piston, D.W.; Ayers, G.D.; McConnell, R.E.; Tyska, M.J.; et al. Amphiregulin exosomes increase cancer cell invasion. Curr. Biol. 2011, 21, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gonzalez-Prieto, R.; Zhang, M.; Geurink, P.P.; Kooij, R.; Iyengar, P.V.; van Dinther, M.; Bos, E.; Zhang, X.; Le Devedec, S.E.; et al. Deubiquitinase Activity Profiling Identifies UCHL1 as a Candidate Oncoprotein That Promotes TGFbeta-Induced Breast Cancer Metastasis. Clin. Cancer Res. 2020, 26, 1460–1473. [Google Scholar] [CrossRef] [PubMed]
- Kreger, B.T.; Johansen, E.R.; Cerione, R.A.; Antonyak, M.A. The Enrichment of Survivin in Exosomes from Breast Cancer Cells Treated with Paclitaxel Promotes Cell Survival and Chemoresistance. Cancers 2016, 8, 111. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhang, Y.; Ye, M.; Wu, J.; Ma, L.; Chen, H. Cisplatin-resistant MDA-MB-231 Cell-derived Exosomes Increase the Resistance of Recipient Cells in an Exosomal miR-423-5p-dependent Manner. Curr. Drug Metab. 2019, 20, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Chen, J.; Su, F.; Yu, B.; Su, F.; Lin, L.; Liu, Y.; Huang, J.D.; Song, E. Microvesicles secreted by macrophages shuttle invasion-potentiating microRNAs into breast cancer cells. Mol. Cancer 2011, 10, 117. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Pochampally, R.; Watabe, K.; Lu, Z.; Mo, Y.Y. Exosome-mediated transfer of miR-10b promotes cell invasion in breast cancer. Mol. Cancer 2014, 13, 256. [Google Scholar] [CrossRef]
- Zhou, W.; Fong, M.Y.; Min, Y.; Somlo, G.; Liu, L.; Palomares, M.R.; Yu, Y.; Chow, A.; O’Connor, S.T.; Chin, A.R.; et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell 2014, 25, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Parashar, D.; Geethadevi, A.; Aure, M.R.; Mishra, J.; George, J.; Chen, C.; Mishra, M.K.; Tahiri, A.; Zhao, W.; Nair, B.; et al. miRNA551b-3p Activates an Oncostatin Signaling Module for the Progression of Triple-Negative Breast Cancer. Cell Rep. 2019, 29, 4389–4406.e10. [Google Scholar] [CrossRef]
- Rabe, D.C.; Walker, N.D.; Rustandy, F.D.; Wallace, J.; Lee, J.; Stott, S.L.; Rosner, M.R. Tumor Extracellular Vesicles Regulate Macrophage-Driven Metastasis through CCL5. Cancers 2021, 13, 3459. [Google Scholar] [CrossRef]
- Piao, Y.J.; Kim, H.S.; Hwang, E.H.; Woo, J.; Zhang, M.; Moon, W.K. Breast cancer cell-derived exosomes and macrophage polarization are associated with lymph node metastasis. Oncotarget 2018, 9, 7398–7410. [Google Scholar] [CrossRef]
- Qiu, Y.; Yang, Y.; Yang, R.; Liu, C.; Hsu, J.M.; Jiang, Z.; Sun, L.; Wei, Y.; Li, C.W.; Yu, D.; et al. Activated T cell-derived exosomal PD-1 attenuates PD-L1-induced immune dysfunction in triple-negative breast cancer. Oncogene 2021, 40, 4992–5001. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Goliwas, K.F.; Severino, P.E.; Hough, K.P.; Van Vessem, D.; Wang, H.; Tousif, S.; Koomullil, R.P.; Frost, A.R.; Ponnazhagan, S.; et al. Mechanical strain induces phenotypic changes in breast cancer cells and promotes immunosuppression in the tumor microenvironment. Lab. Invest. 2020, 100, 1503–1516. [Google Scholar] [CrossRef]
- Naran, K.; Nundalall, T.; Chetty, S.; Barth, S. Principles of Immunotherapy: Implications for Treatment Strategies in Cancer and Infectious Diseases. Front. Microbiol. 2018, 9, 3158. [Google Scholar] [CrossRef] [PubMed]
- Koury, J.; Lucero, M.; Cato, C.; Chang, L.; Geiger, J.; Henry, D.; Hernandez, J.; Hung, F.; Kaur, P.; Teskey, G.; et al. Immunotherapies: Exploiting the Immune System for Cancer Treatment. J. Immunol. Res. 2018, 2018, 9585614. [Google Scholar] [CrossRef] [PubMed]
- Nasiri, F.; Kazemi, M.; Mirarefin, S.M.J.; Mahboubi Kancha, M.; Ahmadi Najafabadi, M.; Salem, F.; Dashti Shokoohi, S.; Evazi Bakhshi, S.; Safarzadeh Kozani, P.; Safarzadeh Kozani, P. CAR-T cell therapy in triple-negative breast cancer: Hunting the invisible devil. Front. Immunol. 2022, 13, 1018786. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Cruz, C.; Eder, J.P.; Braiteh, F.; Chung, C.; Tolaney, S.M.; Kuter, I.; Nanda, R.; Cassier, P.A.; Delord, J.P.; et al. Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer: A Phase 1 Study. JAMA Oncol. 2019, 5, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.; Gligorov, J.; Andre, F.; Cameron, D.; Schneeweiss, A.; Barrios, C.; Xu, B.; Wardley, A.; Kaen, D.; Andrade, L.; et al. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann. Oncol. 2021, 32, 994–1004. [Google Scholar] [CrossRef]
- Weiss, J.; Glode, A.; Messersmith, W.A.; Diamond, J. Sacituzumab govitecan: Breakthrough targeted therapy for triple-negative breast cancer. Expert. Rev. Anticancer. Ther. 2019, 19, 673–679. [Google Scholar] [CrossRef]
- Bardia, A.; Mayer, I.A.; Diamond, J.R.; Moroose, R.L.; Isakoff, S.J.; Starodub, A.N.; Shah, N.C.; O’Shaughnessy, J.; Kalinsky, K.; Guarino, M.; et al. Efficacy and Safety of Anti-Trop-2 Antibody Drug Conjugate Sacituzumab Govitecan (IMMU-132) in Heavily Pretreated Patients With Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2017, 35, 2141–2148. [Google Scholar] [CrossRef]
- Lyons, T.G. Targeted Therapies for Triple-Negative Breast Cancer. Curr. Treat. Options Oncol. 2019, 20, 82. [Google Scholar] [CrossRef]
- Rosenberg, J.; Huang, J. CD8(+) T Cells and NK Cells: Parallel and Complementary Soldiers of Immunotherapy. Curr. Opin. Chem. Eng. 2018, 19, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed]
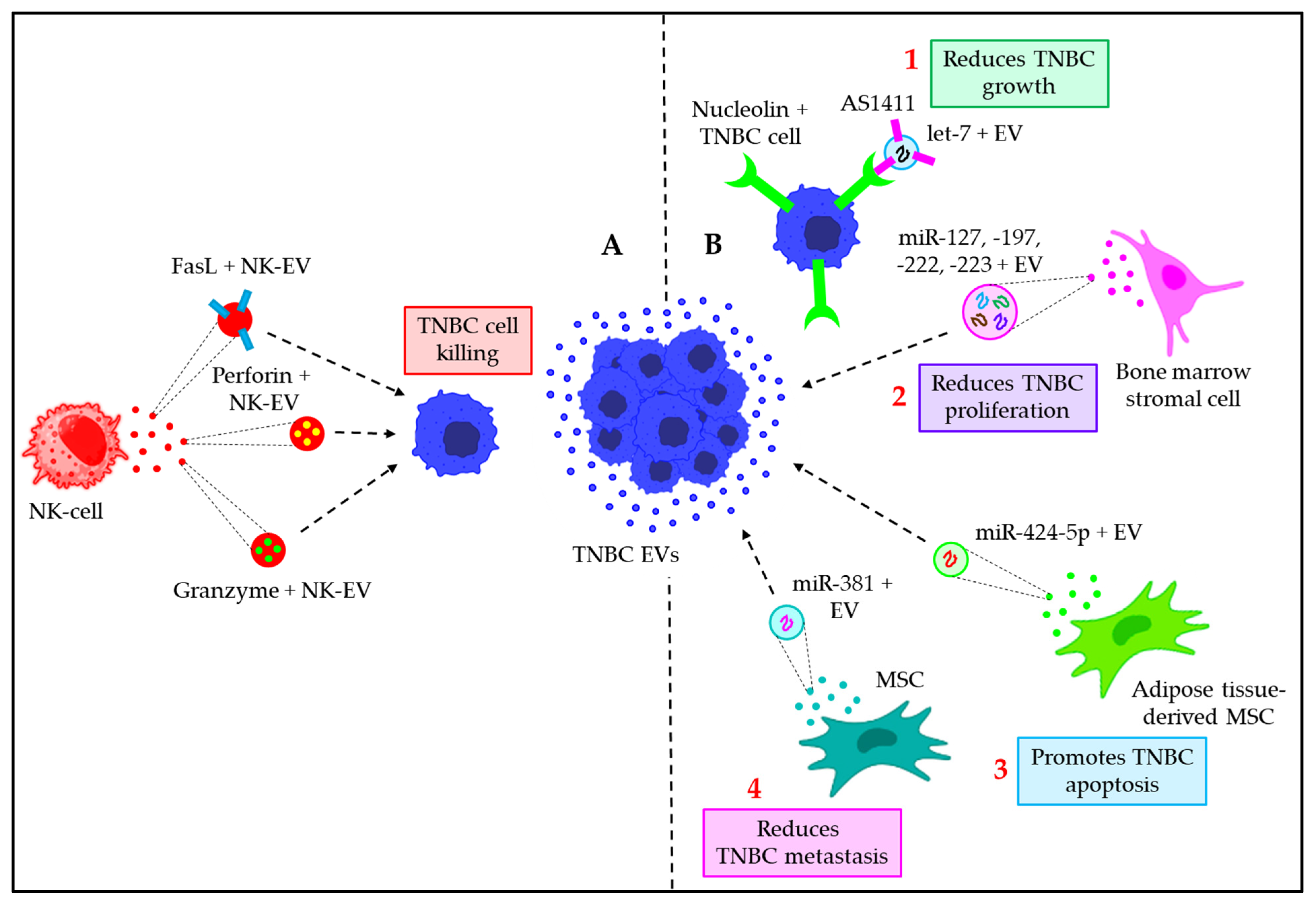
- Batista, I.A.; Quintas, S.T.; Melo, S.A. The Interplay of Exosomes and NK Cells in Cancer Biology. Cancers 2021, 13, 473. [Google Scholar] [CrossRef] [PubMed]
- Lugini, L.; Cecchetti, S.; Huber, V.; Luciani, F.; Macchia, G.; Spadaro, F.; Paris, L.; Abalsamo, L.; Colone, M.; Molinari, A.; et al. Immune surveillance properties of human NK cell-derived exosomes. J. Immunol. 2012, 189, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Kalimuthu, S.; Gangadaran, P.; Oh, J.M.; Lee, H.W.; Baek, S.H.; Jeong, S.Y.; Lee, S.W.; Lee, J.; Ahn, B.C. Exosomes Derived From Natural Killer Cells Exert Therapeutic Effect in Melanoma. Theranostics 2017, 7, 2732–2745. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Li, J.; Li, L.; Sun, J.; Fabbri, M.; Wayne, A.S.; Seeger, R.C.; Jong, A.Y. Extracellular vesicles derived from natural killer cells use multiple cytotoxic proteins and killing mechanisms to target cancer cells. J. Extracell. Vesicles 2019, 8, 1588538. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, X.; Tian, B.; Liu, J.; Yang, L.; Zeng, L.; Chen, T.; Hong, A.; Wang, X. Nucleolin-targeted Extracellular Vesicles as a Versatile Platform for Biologics Delivery to Breast Cancer. Theranostics 2017, 7, 1360–1372. [Google Scholar] [CrossRef]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef]
- Zhou, Y.; Yamamoto, Y.; Takeshita, F.; Yamamoto, T.; Xiao, Z.; Ochiya, T. Delivery of miR-424-5p via Extracellular Vesicles Promotes the Apoptosis of MDA-MB-231 TNBC Cells in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 844. [Google Scholar] [CrossRef]
- Shojaei, S.; Hashemi, S.M.; Ghanbarian, H.; Sharifi, K.; Salehi, M.; Mohammadi-Yeganeh, S. Delivery of miR-381-3p Mimic by Mesenchymal Stem Cell-Derived Exosomes Inhibits Triple Negative Breast Cancer Aggressiveness; an In Vitro Study. Stem Cell Rev. Rep. 2021, 17, 1027–1038. [Google Scholar] [CrossRef]
- Hadla, M.; Palazzolo, S.; Corona, G.; Caligiuri, I.; Canzonieri, V.; Toffoli, G.; Rizzolio, F. Exosomes increase the therapeutic index of doxorubicin in breast and ovarian cancer mouse models. Nanomedicine 2016, 11, 2431–2441. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, L.; Zheng, M.; Zhu, C.; Wang, G.; Xia, Y.; Blumenthal, E.J.; Mao, W.; Wan, Y. Engineered extracellular vesicles for concurrent Anti-PDL1 immunotherapy and chemotherapy. Bioact. Mater. 2022, 9, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Si, Y.; Chen, K.; Ngo, H.G.; Guan, J.S.; Totoro, A.; Zhou, Z.; Kim, S.; Kim, T.; Zhou, L.; Liu, X. Targeted EV to Deliver Chemotherapy to Treat Triple-Negative Breast Cancers. Pharmaceutics 2022, 14, 146. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, A.; Tutrone, R.; Alter, J.; Berg, E.; Fischer, C.; Kumar, S.; Torkler, P.; Tadigotla, V.; Donovan, M.; Sant, G.; et al. Pre-diagnosis urine exosomal RNA (ExoDx EPI score) is associated with post-prostatectomy pathology outcome. World J. Urol. 2022, 40, 983–989. [Google Scholar] [CrossRef]
- Panigrahi, A.R.; Srinivas, L.; Panda, J. Exosomes: Insights and therapeutic applications in cancer. Transl. Oncol. 2022, 21, 101439. [Google Scholar] [CrossRef]
- Buscail, E.; Alix-Panabières, C.; Quincy, P.; Cauvin, T.; Chauvet, A.; Degrandi, O.; Caumont, C.; Verdon, S.; Lamrissi, I.; Moranvillier, I.; et al. High Clinical Value of Liquid Biopsy to Detect Circulating Tumor Cells and Tumor Exosomes in Pancreatic Ductal Adenocarcinoma Patients Eligible for Up-Front Surgery. Cancers 2019, 11, 1656. [Google Scholar] [CrossRef]
- Aheget, H.; Mazini, L.; Martin, F.; Belqat, B.; Marchal, J.A.; Benabdellah, K. Exosomes: Their Role in Pathogenesis, Diagnosis and Treatment of Diseases. Cancers 2020, 13, 84. [Google Scholar] [CrossRef]
- Janockova, J.; Slovinska, L.; Harvanova, D.; Spakova, T.; Rosocha, J. New therapeutic approaches of mesenchymal stem cells-derived exosomes. J. Biomed. Sci. 2021, 28, 39. [Google Scholar] [CrossRef]
- McKiernan, J.; Donovan, M.J.; Margolis, E.; Partin, A.; Carter, B.; Brown, G.; Torkler, P.; Noerholm, M.; Skog, J.; Shore, N.; et al. A Prospective Adaptive Utility Trial to Validate Performance of a Novel Urine Exosome Gene Expression Assay to Predict High-grade Prostate Cancer in Patients with Prostate-specific Antigen 2-10ng/ml at Initial Biopsy. Eur. Urol. 2018, 74, 731–738. [Google Scholar] [CrossRef]
- Zhao, Z.; Fan, J.; Hsu, Y.S.; Lyon, C.J.; Ning, B.; Hu, T.Y. Extracellular vesicles as cancer liquid biopsies: From discovery, validation, to clinical application. Lab. A Chip 2019, 19, 1114–1140. [Google Scholar] [CrossRef]
- Chanteloup, G.; Cordonnier, M.; Isambert, N.; Bertaut, A.; Marcion, G.; Garrido, C.; Gobbo, J. Membrane-bound exosomal HSP70 as a biomarker for detection and monitoring of malignant solid tumours: A pilot study. Pilot. Feasibility Stud. 2020, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Casella, G.; Zhang, Y.; Rostami, A.; Li, X. Potential roles of extracellular vesicles in the pathophysiology, diagnosis, and treatment of autoimmune diseases. Int. J. Biol. Sci. 2020, 16, 620–632. [Google Scholar] [CrossRef]
- Kumar, P.; Kumawat, R.K.; Uttam, V.; Behera, A.; Rani, M.; Singh, N.; Barwal, T.S.; Sharma, U.; Jain, A. The imminent role of microRNAs in salivary adenoid cystic carcinoma. Transl. Oncol. 2023, 27, 101573. [Google Scholar] [CrossRef] [PubMed]
- Snyder, M.; Iraola-Guzmán, S.; Saus, E.; Gabaldón, T. Discovery and Validation of Clinically Relevant Long Non-Coding RNAs in Colorectal Cancer. Cancers 2022, 14, 3866. [Google Scholar] [CrossRef] [PubMed]
- Mo, Z.; Cheong, J.Y.A.; Xiang, L.; Le, M.T.N.; Grimson, A.; Zhang, D.X. Extracellular vesicle-associated organotropic metastasis. Cell Prolif. 2021, 54, e12948. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.; Amreddy, N.; Pareek, V.; Chinnappan, M.; Ahmed, R.; Mehta, M.; Razaq, M.; Munshi, A.; Ramesh, R. Progress in extracellular vesicle biology and their application in cancer medicine. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2020, 12, e1621. [Google Scholar] [CrossRef] [PubMed]
- Du, R.; Wang, C.; Zhu, L.; Yang, Y. Extracellular Vesicles as Delivery Vehicles for Therapeutic Nucleic Acids in Cancer Gene Therapy: Progress and Challenges. Pharmaceutics 2022, 14, 2236. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of EVs | Marker/s | Size (Diameter) | Biogenetic Mechanism | Reference |
|---|---|---|---|---|
| Microparticles (MPs) | Tetraspanins, CD41, CD63 | 100 nm–1 µm | Evagination of the plasma membrane followed by pinching to release the MPs outside the cells. | [53,54] |
| Exosomes | Alix, TSG101, HSP90β, HSC70, CD63, Syntenin 1 | 30–150 nm | Conventional mechanism of exosome biogenesis includes the invagination of the early endosomal membrane to form exosomes, which fuse with the plasma membrane to release the exosomes from the cells. | [55,56,57] |
| Apoptotic bodies | HSP60, GRP78, Annexin V (phosphatidylserine) | 50 nm–5 µm | Cellular contraction is induced during apoptosis, which produces a hydrodynamic force that triggers the separation of the plasma membrane from the cytoskeleton, leading to the release of apoptotic bodies from the cells. | [55,58,59] |
| Subtype | Immunohistochemistry Profile | Other Features | References |
|---|---|---|---|
| Luminal A | ER+, PR+, HER2− | Ki–67− | [76] |
| Luminal B | ER+, PR+, HER2+ | Ki–67+ | [76] |
| HER2 high | ER−, PR−, HER2+ | Ki–67+ | [81] |
| Normal–like | ER+, PR+, HER2− | Ki–67− | [81] |
| Basal–like (TNBC) | ER−, PR−, HER2−, basal markers | Ki–67+, (EGFR, CK5/6) | [76] |
| Claudin low (TNBC) | ER−, PR−, HER2− | Low Ki–67, E–cadherin, Claudin 3, 4, 7 | [93] |
| TNBC Property | Mechanism of Action | References |
|---|---|---|
| Growth | HCC1806–EVs promote the growth and drug resistance to MCF10A cells via the involvement of PI3K/AKT, MAPK, and HIF1A pathways. | [107] |
| DET– and DETD–35–treated MDA–MB–231–derived EVs inhibit the proliferation of MDA–MB–231 cells by downregulating cell adhesion, migration, and angiogenesis. | [108] | |
| ITGB4 + EVs from MDA–MB–231 cells trigger the glycolytic pathway of CAFs via BNI3PL–dependent mitophagy and lactate production, which, in turn, promote tumor growth. | [109] | |
| MSC–EVs transfer miR–106a–5p to the TNBC tumor and induce tumor progression via downregulating HAND2–AS1. | [110] | |
| CAF–EVs show a relatively lower expression of miR–4516, which promotes the proliferation of TNBC cells by upregulating the miR–4516 target, namely, FOSL1 expression. | [111] | |
| CircHIF1A + EVs from human TNBC promote tumor growth via the upregulation of the PI3K/AKT pathway while downregulating p21. | [112] | |
| Metastasis | In response to chemotherapy, TNBC cells release a significant number of PS + EVs, which induce endothelial barrier permeability, hence helping with the transendothelial migration of cancer cells, contributing to TNBC metastasis. | [113] |
| Serum EVs of TNBC patients are enriched with CD151, which triggers the migration and invasion of TNBC cells. | [114] | |
| Circulating EVs of TNBC patients are enriched with SPANXB1, which downregulates SH3GL2 expression in TNBC cells, and upregulates Rac1, FAK, and α–actinin expression, leading to TNBC metastasis. | [115] | |
| MDA–MB–231–EVs are abundant with miR–9 and miR–155, which target PTEN and DUSP14 in MCF–7, thereby inducing the metastatic potential of recipient MCF–7 cells. | [116] | |
| TNBC–EVs are enriched with miR–393, which targets VE–cadherin in endothelial cells, leading to enhanced vascular leakage, and thereby facilitating transendothelial migration of tumor cells. | [117] | |
| Serum EVs of TNBC patients are enriched with circPSMA1, which absorbs miR–637 to release the inhibitory function on AKT1 and leads to the downstream activation of β–catenin and cyclin D1 to trigger the proliferation, migration, and metastasis of TNBC cells. | [118] |
| EVs’ Cargo Type | Name of the Cargo | Origin | Function | References |
|---|---|---|---|---|
| Protein | EGFR ligands | MDA–MB–231 | EVs’ EGFR ligands, such as AREG, promote the invasiveness of recipient breast cancer cells. | [121] |
| UCHL1 | TNBC cells, TNBC plasma | EV–carried UCHL1 protects the TGFβ type I receptor and SMAD2 from ubiquitination, stimulating the migration and extravasation of the breast cancer cells. | [122] | |
| Survivin | MDA–MB–231 | PTX–treated TNBC–derived EVs are enriched with Survivin, which promotes the growth and survivability of PTX–exposed fibroblasts and other breast cancer cells. | [123] | |
| CD151 | TNBC serum | EVs from TNBC serum are enriched with CD151, which promotes the migration and invasion of TNBC cells. | [114] | |
| miRNA | miR–423–5p | MDA–MB–231 | EVs from cisplatin–resistant TNBC cells are positive for miR–423–5p, which induces P–gp expression, migration, invasion, and anti–apoptosis in other breast cancer cells. | [124] |
| miR–223 | MDA–MB–231 | TNBC–associated macrophages release miR–223–containing EVs, which promote the invasion of breast cancer. | [125] | |
| miR–10b | TNBC cells | miR–10b + TNBC–EVs target HOXD10 and KLF4 in non–malignant cells to promote cell invasion. | [126] | |
| miR–105 | MDA–MB–231 | miR–105 + EVs target ZO–1 in endothelial cells, leading to increased vascular permeability and facilitating the metastasis of TNBC. | [127] |
| Donor Cells | Recipient Cells | EVs’ Cargo | Function | References |
|---|---|---|---|---|
| TNBC | Macrophages | CCL5 | TNBC–EVs transport CCL5 to macrophages leading to the development of TEMs, which induce stromal remodeling and immune infiltration to facilitate TNBC metastasis. | [129] |
| TNBC | Macrophages | TNBC–EVs trigger macrophage polarization into M2 phenotypes, leading to LN metastasis. | [130] | |
| Activated T–cells | TNBC cells or TNBC–EVs | PD–1 | Activated T–cell–derived EVs release PD–1 + EVs, which bind to PD–L1 + TNBC cells or EVs, thereby preventing T–cell/TNBC cell interaction and attenuating immune suppression of T–cells by TNBC cells. | [131] |
| TNBC | MDSCs, M2– macrophages, CD8 + T–cells | PD–L1 | OS stimulates the release of PD–L1 + EVs from TNBC cells, which enriches MDSCs and M2 macrophages in the TME and reduces CD8 + T–cells. | [132] |
| Trial Name | NCT Number | Characteristics | Cancer Types | EVs’ Source | Status | Outcome Measures | Refs. |
|---|---|---|---|---|---|---|---|
| Clinical Validation of an Urinary Exosome Gene Signature in Men Presenting for Suspicion of Prostate Cancer | NCT02702856 | Duration: May 2014–June 2015 Population: 2000 men Age: >50 | Prostate cancer | Urine | Completed | The ExoDx Prostate IntelliScore (EPI) EPI score was associated with low–risk pathology post–RP, with potential implications on informing AS decisions. | [154,155] |
| Diagnostic Accuracy of Circulating Tumor Cells (CTCs) And Onco–exosome Quantification in the Diagnosis of Pancreatic Cancer—PANC–CTC (PANC–CTC) | NCT03032913 | Duration: February 2017–November 2017 Population: 20 with PDAC and 20 controls Age: 18 years and older | Pancreatic ductal adenocarcinoma | Circulation | Completed | [156] | |
| microRNAs Role in Pre–eclampsia Diagnosis | NCT03562715 | Duration: November 2016–December 2017 Population: 100 patients and 100 controls Females Age: 23 years to 35 years | Preeclampsia | Circulation | Completed | Liquid biopsy combining several biomarkers could provide a rapid, reliable, noninvasive decision–making tool in early, potentially curable pancreatic cancer. | [157,158] |
| Clinical Evaluation of the “ExoDx Prostate IntelliScore” (EPI) | NCT03031418 | Duration: September 2016–September 2018 Population: 532 Patients Age: above 50 years of age Males | Prostate cancer | Urine | Completed | The expression of miRNAs 136, 494, and 495 in exosomes of peripheral blood and UCMSCs conditioned media. | [159] |
| Olmutinib Trial in T790M(+)NSCLC Patients Detected by Liquid Biopsy Using BALF Extracellular Vesicular DNA | NCT03228277 | Duration: July 2017–July 2019 Population: 25 Males or females, aged at least 19 years. | NSCLC | BALF | Completed | ExoDx Prostate (IntelliScore) test can predict ≥ GG2 PCa at initial biopsy and defer unnecessary biopsies better than existing risk calculators and standard clinical data. | [160] |
| Pilot Study with the Aim to Quantify a Stress Protein in the Blood and in the Urine for the Monitoring and Early Diagnosis of Malignant Solid Tumors (EXODIAG) | NCT02662621 | Duration: December 2015–April 2019 Population: 71 patients Age: 18 years and older | Cancer | Circulation | Completed | Assess the anti–tumor efficacy via objective response rate (ORR), disease control rate (DCR), and progression–free survival (PFS). | [161] |
| Pimo Study: Extracellular Vesicle–based Liquid Biopsy to Detect cancer Hypoxia in Tumors | NCT03262311 | Duration: November 2017–September 2019 Population: 21 Age: ≥18 years | Hypoxia–induced cancer | Blood | Completed | HSP70 exosomes could be a powerful tool to diagnose cancer and guide. | [162] |
| The Sensitivity and Specificity of Using Salivary miRNAs in Detection of Malignant Transformation of Oral Lesions | NCT04913545 | Duration: January 2020–August 2020 Population: 18 Age: 35 years to 70 years | Oral premalignant lesions | Saliva | Completed | Clinicians in therapeutic decision–making, improving patient care. | [163] |
| Clinical Evaluation of ExoDx Prostate (IntelliScore) in Men Presenting for Initial Prostate Biopsy | NCT04720599 | Duration: June 2020–June 2021 Population: 120 males Age: 50 years and older | Urologic cancer | Urine | Completed | Not available. | |
| Serum Exosomal Long Noncoding RNAs as Potential Biomarkers for Lung Cancer Diagnosis | NCT03830619 | Duration: Jan 2017–July 2021 Population: 1000 Age: 18 years to 75 years Location: Hubei, China | Lung cancer | Serum | Completed | Measuring the sensitivity and specificity of using the salivary miRNAs (412,512) to detect the malignant transformation in potentially malignant lesions. | [164] |
| Identification of New Diagnostic Protein Markers for Colorectal Cancer (EXOSCOL01) | NCT03895216 | Duration: December 2018–December 2021 Population: 34 Age: 18 years and older | Bone metastasis | Plasma | Completed | EPI–CE provides information beyond standard clinical parameters and provides a better risk assessment prior to MRI of patients suspected of prostate cancer than the commonly used multiparametric risk calculators. | [165] |
| Exosomes Implication in PD1–PD–L1 Activation in OSAS (ExoSAS) | NCT03811600 | Duration: March 2019–October 2020 Population: 90 Age: 18 years and older | Cancer, obstructive sleep apnea | Plasma | Completed | No study results are posted on ClinicalTrials.gov for this study. | [166] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, K.; Paul, S.; Ghosh, A.; Gupta, S.; Mukherjee, T.; Shankar, P.; Sharma, A.; Keshava, S.; Chauhan, S.C.; Kashyap, V.K.; et al. Extracellular Vesicles in Triple–Negative Breast Cancer: Immune Regulation, Biomarkers, and Immunotherapeutic Potential. Cancers 2023, 15, 4879. https://doi.org/10.3390/cancers15194879
Das K, Paul S, Ghosh A, Gupta S, Mukherjee T, Shankar P, Sharma A, Keshava S, Chauhan SC, Kashyap VK, et al. Extracellular Vesicles in Triple–Negative Breast Cancer: Immune Regulation, Biomarkers, and Immunotherapeutic Potential. Cancers. 2023; 15(19):4879. https://doi.org/10.3390/cancers15194879
Chicago/Turabian StyleDas, Kaushik, Subhojit Paul, Arnab Ghosh, Saurabh Gupta, Tanmoy Mukherjee, Prem Shankar, Anshul Sharma, Shiva Keshava, Subhash C. Chauhan, Vivek Kumar Kashyap, and et al. 2023. "Extracellular Vesicles in Triple–Negative Breast Cancer: Immune Regulation, Biomarkers, and Immunotherapeutic Potential" Cancers 15, no. 19: 4879. https://doi.org/10.3390/cancers15194879