

ICAM-1-suPAR-CD11b Axis Is a Novel Therapeutic Target for Metastatic Triple-Negative Breast Cancer
 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Human Blood Analyses
2.2. Animal Studies and TNBC Models
2.3. Cell Lines and Transfections
2.4. Mouse Neutrophil Isolation
2.5. In Vitro Tumor Cell and Neutrophil Binding Assay
2.6. Neutrophil Chemotaxis Assay
2.7. Extracellular H2O2 Production Measurement and Nicotinamide Adenine Dinucleotide Phosphate (NADPH) Oxidase Activity Assay
2.8. Generation of Conditional Medium and Cytokine Array
2.9. Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. Real-Time Quantitative PCR (qRT-PCR)
2.11. Western Blotting
2.12. Immunohistochemistry Staining
2.13. Bioinformatics Analysis
2.14. Statistical Analysis
3. Results
3.1. ICAM-1 and CD11b Mediate Tumor Cell and Neutrophil Binding
3.2. CD11b Deficiency Inhibits TNBC Metastasis
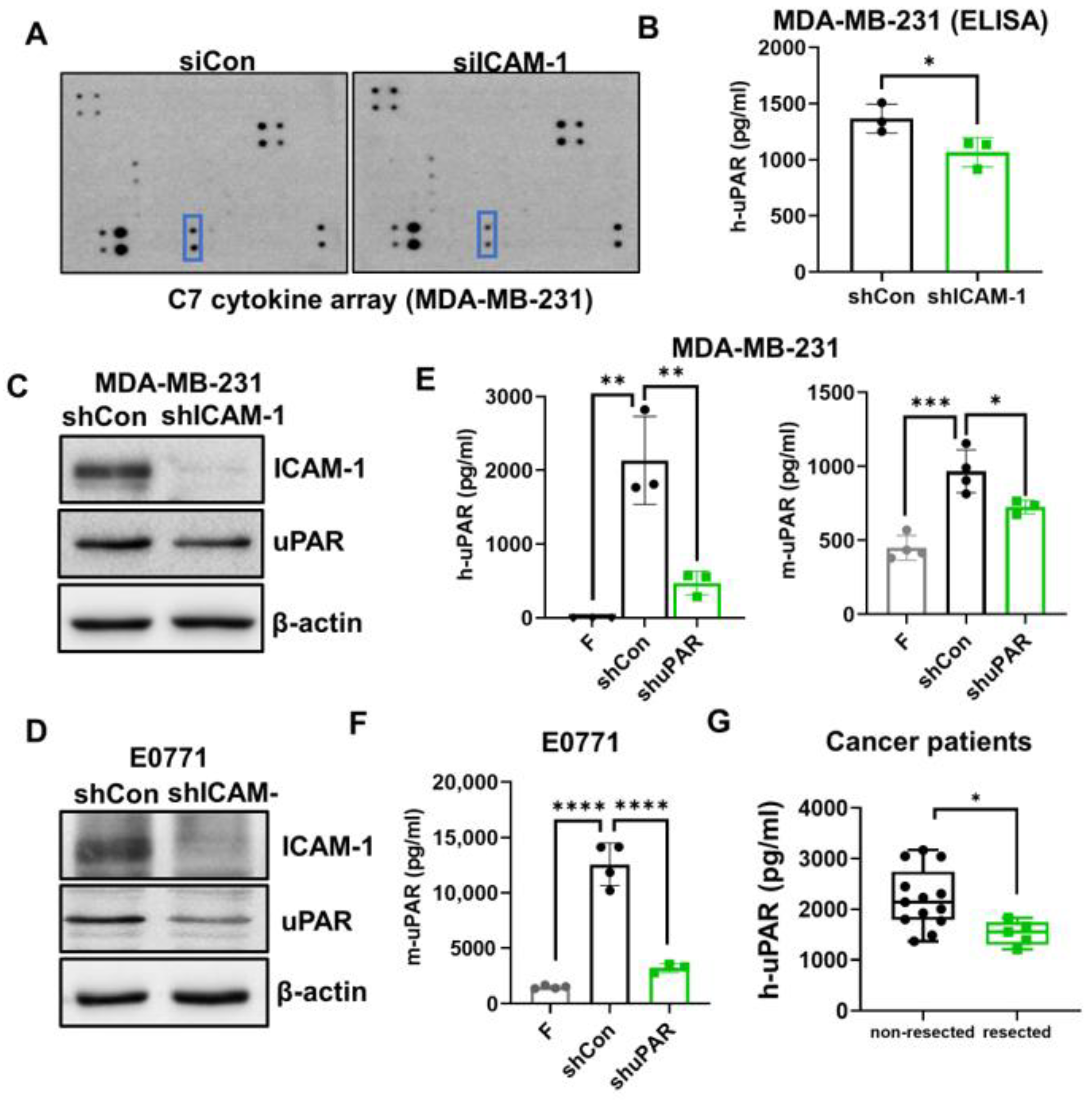
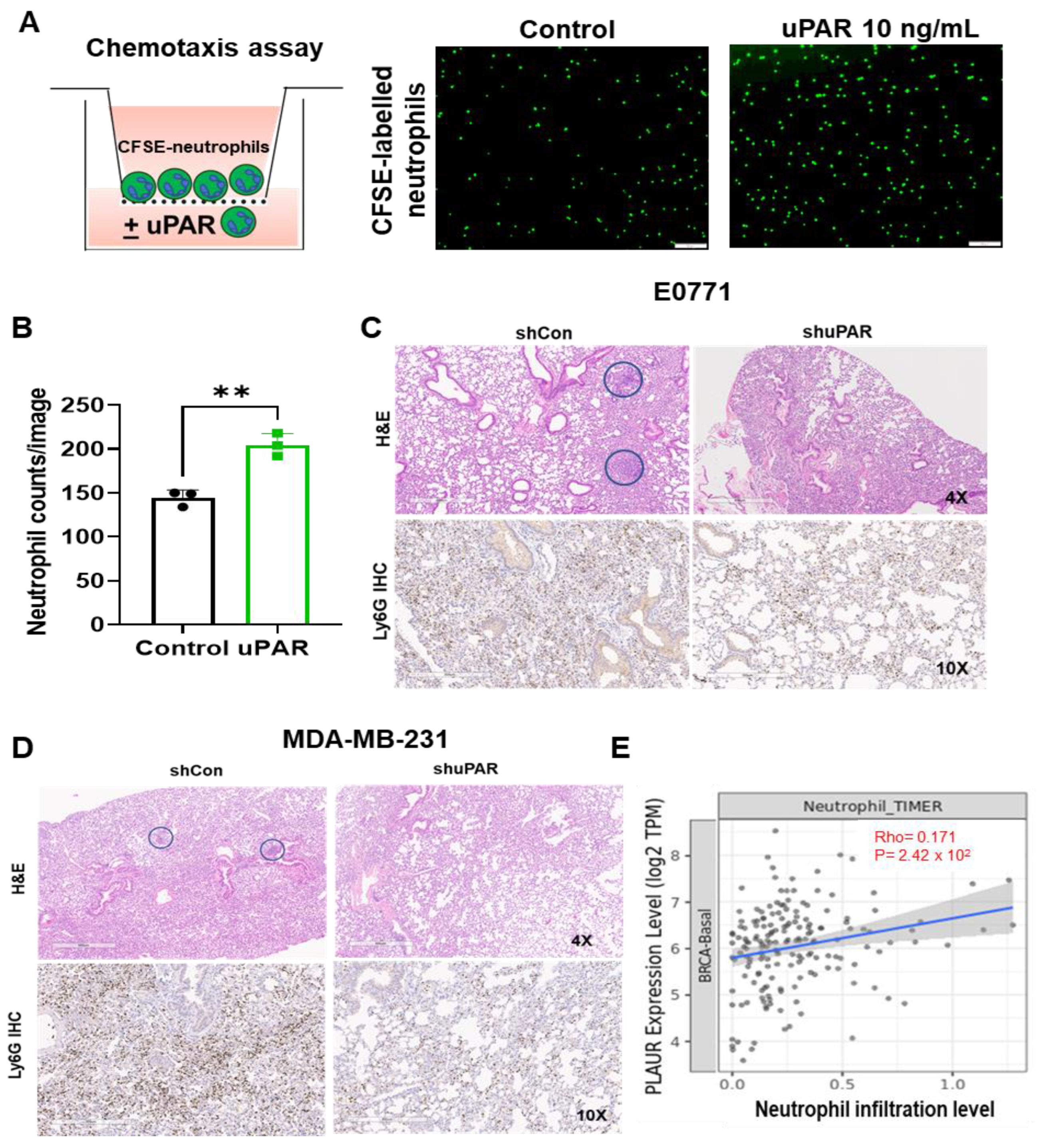
3.3. ICAM-1 Promotes TNBC Cells to Release suPAR, which Functions as a Chemoattractant for Neutrophils
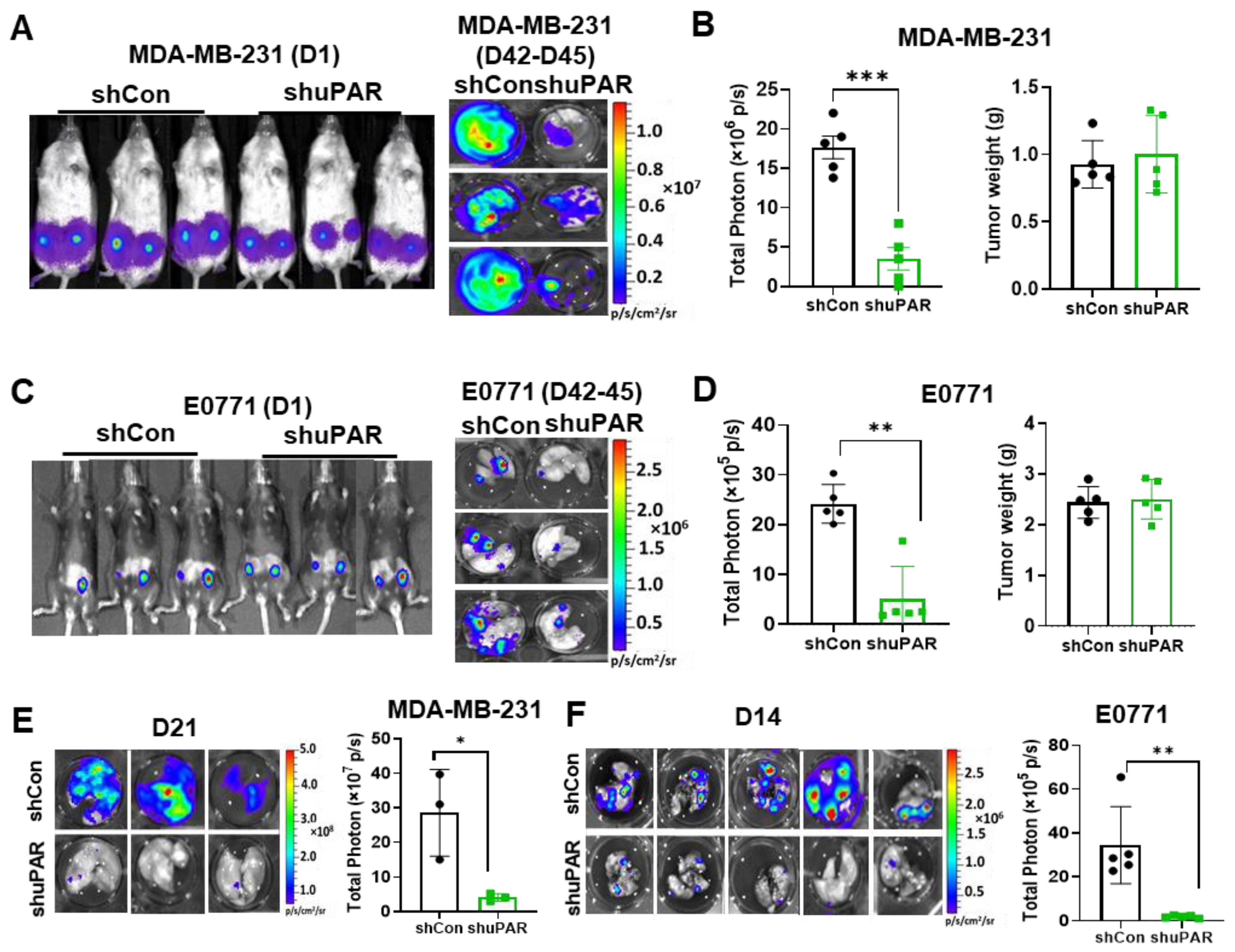
3.4. uPAR Knockdown Inhibits TNBC Metastasis
3.5. High uPAR Expression Correlates with Worse Breast Cancer Patient Outcome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Basal-Like Breast Cancer: A Critical Review. J. Clin. Oncol. 2008, 26, 2568–2581. [Google Scholar] [CrossRef] [PubMed]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Fadare, O.; Tavassoli, F.A. Clinical and pathologic aspects of basal-like breast cancers. Nat. Clin. Pract. Oncol. 2008, 5, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Kreike, B.; Van Kouwenhove, M.; Horlings, H.; Weigelt, B.; Peterse, H.; Bartelink, H.; Van De Vijver, M.J. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. 2007, 9, R65. [Google Scholar] [CrossRef] [PubMed]
- Korsching, E.; Jeffrey, S.S.; Meinerz, W.; Decker, T.; Boecker, W.; Buerger, H. Basal carcinoma of the breast revisited: An old entity with new interpretations. J. Clin. Pathol. 2008, 61, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef]
- Kilgour, E.; Rothwell, D.G.; Brady, G.; Dive, C. Liquid Biopsy-Based Biomarkers of Treatment Response and Resistance. Cancer Cell 2020, 37, 485–495. [Google Scholar] [CrossRef]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Baeuerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef]
- Liu, X.; Taftaf, R.; Kawaguchi, M.; Chang, Y.-F.; Chen, W.; Entenberg, D.; Zhang, Y.; Gerratana, L.; Huang, S.; Patel, D.B.; et al. Homophilic CD44 Interactions Mediate Tumor Cell Aggregation and Polyclonal Metastasis in Patient-Derived Breast Cancer Models. Cancer Discov. 2019, 9, 96–113. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating Tumor Cell Clusters Are Oligoclonal Precursors of Breast Cancer Metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Dashzeveg, N.; Cao, Y.; Jia, Y.; Liu, X.; Shen, Y.; Liu, H. Extracellular Domains I and II of cell-surface glycoprotein CD44 mediate its trans-homophilic dimerization and tumor cluster aggregation. J. Biol. Chem. 2020, 295, 2640–2649. [Google Scholar] [CrossRef]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Luo, Q.; Wang, C.; Peng, B.; Pu, X.; Cai, L.; Liao, H.; Chen, K.; Zhang, C.; Cheng, Y.; Pan, M. Circulating Tumor-Cell-Associated White Blood Cell Clusters in Peripheral Blood Indicate Poor Prognosis in Patients with Hepatocellular Carcinoma. Front. Oncol. 2020, 10, 1758. [Google Scholar] [CrossRef]
- Iriondo, O.; Yu, M. Unexpected Friendship: Neutrophils Help Tumor Cells En Route to Metastasis. Dev. Cell 2019, 49, 308–310. [Google Scholar] [CrossRef]
- Smith, H.W.; Marshall, C.J. Regulation of cell signalling by uPAR. Nat. Rev. Mol. Cell Biol. 2010, 11, 23–36. [Google Scholar] [CrossRef]
- Noh, H.; Hong, S.; Huang, S. Role of Urokinase Receptor in Tumor Progression and Development. Theranostics 2013, 3, 487–495. [Google Scholar] [CrossRef]
- Ulisse, S.; Baldini, E.; Sorrenti, S.; D’Armiento, M. The Urokinase Plasminogen Activator System: A Target for Anti-Cancer Therapy. Curr. Cancer Drug Targets 2009, 9, 32–71. [Google Scholar] [CrossRef]
- Huber, M.C.; Mall, R.; Braselmann, H.; Feuchtinger, A.; Molatore, S.; Lindner, K.; Walch, A.; Gross, E.; Schmitt, M.; Falkenberg, N.; et al. uPAR enhances malignant potential of triple-negative breast cancer by directly interacting with uPA and IGF1R. BMC Cancer 2016, 16, 615. [Google Scholar] [CrossRef]
- Asano, S.; Seishima, M.; Kitajima, Y. Phosphatidylinositol-specific-phospholipase c cleaves urokinase plasminogen activator receptor from the cell surface and leads to inhibition of pemphigus-igg-induced acantholysis in djm-1 cells, a squamous cell carcinoma line. Clin. Exp. Dermatol. 2001, 26, 289–295. [Google Scholar] [CrossRef]
- Stephens, R.W.; Nielsen, H.J.; Christensen, I.J.; Thorlacius-Ussing, O.; Sørensen, S.; Danø, K.; Brünner, N. Plasma urokinase receptor levels in patients with colorectal cancer: Relationship to prognosis. J. Natl. Cancer Inst. 1999, 91, 869–874. [Google Scholar] [CrossRef]
- Langkilde, A.; Hansen, T.W.; Ladelund, S.; Linneberg, A.; Andersen, O.; Haugaard, S.B.; Jeppesen, J.; Eugen-Olsen, J. Increased Plasma Soluble uPAR Level Is a Risk Marker of Respiratory Cancer in Initially Cancer-Free Individuals. Cancer Epidemiol. Biomark. Prev. 2011, 20, 609–618. [Google Scholar] [CrossRef]
- Shariat, S.F.; Roehrborn, C.G.; McConnell, J.D.; Park, S.; Alam, N.; Wheeler, T.M.; Slawin, K.M. Association of the Circulating Levels of the Urokinase System of Plasminogen Activation with the Presence of Prostate Cancer and Invasion, Progression, and Metastasis. J. Clin. Oncol. 2007, 25, 349–355. [Google Scholar] [CrossRef]
- Sier, C.; Stephens, R.W.; Bizik, J.; Mariani, A.; Bassan, M.; Pedersen, N.; Frigerio, L.; Ferrari, A.; Danø, K.; Brünner, N.; et al. The level of urokinase-type plasminogen activator receptor is increased in serum of ovarian cancer patients. Cancer Res. 1998, 58, 1843–1849. [Google Scholar]
- Riisbro, R.; Christensen, I.J.; Piironen, T.; Greenall, M.; Larsen, B.; Stephens, R.W.; Han, C.; Høyer-Hansen, G.; Smith, K.; Brünner, N.; et al. Prognostic significance of soluble urokinase plasminogen activator receptor in serum and cytosol of tumor tissue from patients with primary breast cancer. Clin. Cancer Res. 2002, 8, 1132–1141. [Google Scholar]
- Todd, R.F., 3rd. The continuing saga of complement receptor type 3 (CR3). J. Clin. Investig. 1996, 98, 1–2. [Google Scholar] [CrossRef]
- Solovjov, D.A.; Pluskota, E.; Plow, E.F. Distinct roles for the alpha and beta subunits in the functions of integrin alphambeta2. J. Biol. Chem. 2005, 280, 1336–1345. [Google Scholar] [CrossRef]
- Yang, L.; Froio, R.M.; Sciuto, T.E.; Dvorak, A.M.; Alon, R.; Luscinskas, F.W. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-α-activated vascular endothelium under flow. Blood 2005, 106, 584–592. [Google Scholar] [CrossRef]
- Taftaf, R.; Liu, X.; Singh, S.; Jia, Y.; Dashzeveg, N.K.; Hoffmann, A.D.; El-Shennawy, L.; Ramos, E.K.; Adorno-Cruz, V.; Schuster, E.J.; et al. ICAM1 initiates CTC cluster formation and trans-endothelial migration in lung metastasis of breast cancer. Nat. Commun. 2021, 12, 4867. [Google Scholar] [CrossRef]
- Li, P.; Lu, M.; Shi, J.; Hua, L.; Gong, Z.; Li, Q.; Shultz, L.D.; Ren, G. Dual roles of neutrophils in metastatic colonization are governed by the host NK cell status. Nat. Commun. 2020, 11, 4387. [Google Scholar] [CrossRef]
- Liu, H.; Patel, M.R.; Prescher, J.A.; Patsialou, A.; Qian, D.; Lin, J.; Wen, S.; Chang, Y.-F.; Bachmann, M.H.; Shimono, Y.; et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl. Acad. Sci. USA 2010, 107, 18115–18120. [Google Scholar] [CrossRef]
- Li, T.; Fu, J.; Zeng, Z.; Cohen, D.; Li, J.; Chen, Q.; Li, B.; Liu, X.S. TIMER2.0 for analysis of tumor-infiltrating immune cells. Nucleic Acids Res. 2020, 48, W509–W514. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Mizuno, H.; Kitada, K.; Nakai, K.; Sarai, A. PrognoScan: A new database for meta-analysis of the prognostic value of genes. BMC Med. Genom. 2009, 2, 18. [Google Scholar] [CrossRef]
- Diamond, M.S.; Springer, T.A. A subpopulation of Mac-1 (CD11b/CD18) molecules mediates neutrophil adhesion to ICAM-1 and fibrinogen. J. Cell Biol. 1993, 120, 545–556. [Google Scholar] [CrossRef]
- Diamond, M.S.; Staunton, D.E.; Marlin, S.D.; Springer, T.A. Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell 1991, 65, 961–971. [Google Scholar] [CrossRef]
- Diamond, M.S.; Staunton, D.E.; De Fougerolles, A.R.; Stacker, S.; Garcia-Aguilar, J.; Hibbs, M.L.; Springer, T.A. ICAM-1 (CD54): A counter-receptor for Mac-1 (CD11b/CD18). J. Cell Biol. 1990, 111, 3129–3139. [Google Scholar] [CrossRef]
- Bui, T.M.; Wiesolek, H.L.; Sumagin, R. ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J. Leukoc. Biol. 2020, 108, 787–799. [Google Scholar] [CrossRef]
- Zhang, J.; Qiao, X.; Shi, H.; Han, X.; Liu, W.; Tian, X.; Zeng, X. Circulating tumor-associated neutrophils (cTAN) contribute to circulating tumor cell survival by suppressing peripheral leukocyte activation. Tumor Biol. 2016, 37, 5397–5404. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, A.; Brooks, M.W.; Houshyar, S.; Reinhardt, F.; Ardolino, M.; Fessler, E.; Chen, M.B.; Krall, J.A.; Decock, J.; Zervantonakis, I.K.; et al. Neutrophils Suppress Intraluminal NK Cell–Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discov. 2016, 6, 630–649. [Google Scholar] [CrossRef]
- Hanna, N.; Fidler, I.J. Role of Natural Killer Cells in the Destruction of Circulating Tumor Emboli. J. Natl. Cancer Inst. 1980, 65, 801–809. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.C.; Xu, Z.; Kim, I.S.; Pingel, B.; Aguirre, S.; Kodali, S.; Liu, J.; Zhang, W.; Muscarella, A.M.; Hein, S.M.; et al. Resistance to natural killer cell immunosurveillance confers a selective advantage to polyclonal metastasis. Nat. Cancer 2020, 1, 709–722. [Google Scholar] [CrossRef] [PubMed]
- Husemann, J.; Obstfeld, A.; Febbraio, M.; Kodama, T.; Silverstein, S.C. Cd11b/cd18 mediates production of reactive oxygen species by mouse and human macrophages adherent to matrixes containing oxidized ldl. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1301–1305. [Google Scholar] [CrossRef]
- Kerkhoff, C.; Nacken, W.; Benedyk, M.; Dagher, M.C.; Sopalla, C.; Doussiere, J. The arachidonic acid-binding protein s100a8/a9 promotes nadph oxidase activation by interaction with p67phox and rac-2. FASEB J. 2005, 19, 467–469. [Google Scholar] [CrossRef]
- Bouzidi, F.; Doussiere, J. Binding of arachidonic acid to myeloid-related proteins (s100a8/a9) enhances phagocytic nadph oxidase activation. Biochem. Biophys. Res. Commun. 2004, 325, 1060–1065. [Google Scholar] [CrossRef]
- Petri, B.; Sanz, M.J. Neutrophil chemotaxis. Cell Tissue Res. 2018, 371, 425–436. [Google Scholar] [CrossRef]
- Metzemaekers, M.; Gouwy, M.; Proost, P. Neutrophil chemoattractant receptors in health and disease: Double-edged swords. Cell. Mol. Immunol. 2020, 17, 433–450. [Google Scholar] [CrossRef]
- Capucetti, A.; Albano, F.; Bonecchi, R. Multiple Roles for Chemokines in Neutrophil Biology. Front. Immunol. 2020, 11, 1259. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Wculek, S.K.; Malanchi, I. Neutrophils support lung colonization of metastasis-initiating breast cancer cells. Nature 2015, 528, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.C.; Khan, S.Q.; Kaneda, M.M.; Pathria, P.; Shepard, R.; Louis, T.L.; Anand, S.; Woo, G.; Leem, C.; Faridi, M.H.; et al. Integrin CD11b activation drives anti-tumor innate immunity. Nat. Commun. 2018, 9, 5379. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.; Leitch, A.E.; Duffin, R.; Haslett, C.; Rossi, A.G. Neutrophil Apoptosis: Relevance to the Innate Immune Response and Inflammatory Disease. J. Innate Immun. 2010, 2, 216–227. [Google Scholar] [CrossRef]
- Lahoz-Beneytez, J.; Elemans, M.; Zhang, Y.; Ahmed, R.; Salam, A.; Block, M.; Niederalt, C.; Asquith, B.; Macallan, D. Human neutrophil kinetics: Modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood 2016, 127, 3431–3438. [Google Scholar] [CrossRef]
- Pfirschke, C.; Engblom, C.; Gungabeesoon, J.; Lin, Y.; Rickelt, S.; Zilionis, R.; Messemaker, M.; Siwicki, M.; Gerhard, G.M.; Kohl, A.; et al. Tumor-promoting ly-6g(+) siglecf(high) cells are mature and long-lived neutrophils. Cell Rep. 2020, 32, 108164. [Google Scholar] [CrossRef]
- Sody, S.; Uddin, M.; Grüneboom, A.; Görgens, A.; Giebel, B.; Gunzer, M.; Brandau, S. Distinct Spatio-Temporal Dynamics of Tumor-Associated Neutrophils in Small Tumor Lesions. Front. Immunol. 2019, 10, 1419. [Google Scholar] [CrossRef]
- Behrendt, N.; Ronne, E.; Dano, K. Domain interplay in the urokinase receptor. Requirement for the third domain in high affinity ligand binding and demonstration of ligand contact sites in distinct receptor domains. J. Biol. Chem. 1996, 271, 22885–22894. [Google Scholar]
- Wilhelm, O.G.; Wilhelm, S.; Escott, G.M.; Lutz, V.; Magdolen, V.; Schmitt, M.; Rifkin, D.B.; Wilson, E.L.; Graeff, H.; Brunner, G. Cellular glycosylphosphatidylinositol-specific phospholipase d regulates urokinase receptor shedding and cell surface expression. J. Cell. Physiol. 1999, 180, 225–235. [Google Scholar] [CrossRef]
- Montuori, N.; Visconte, V.; Rossi, G.; Ragno, P. Soluble and cleaved forms of the urokinase-receptor: Degradation products or active molecules? Thromb. Haemost. 2005, 93, 192–198. [Google Scholar] [CrossRef]
- Kiss, Z. Regulation of phospholipase D by protein kinase C. Chem. Phys. Lipids 1996, 80, 81–102. [Google Scholar] [CrossRef] [PubMed]
- Conricode, K.; Brewer, K.; Exton, J. Activation of phospholipase D by protein kinase C. Evidence for a phosphorylation-independent mechanism. J. Biol. Chem. 1992, 267, 7199–7202. [Google Scholar] [CrossRef] [PubMed]
- Abdala-Valencia, H.; Berdnikovs, S.; Cook-Mills, J.M. Vitamin e isoforms differentially regulate intercellular adhesion molecule-1 activation of pkcalpha in human microvascular endothelial cells. PLoS ONE 2012, 7, e41054. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, L.J.H.; Petersen, J.E.V.; Eugen-Olsen, J. Soluble Urokinase Plasminogen Activator Receptor (suPAR) as a Biomarker of Systemic Chronic Inflammation. Front. Immunol. 2021, 12, 780641. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.-A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wang, L.; Zhang, J.; Zhou, H.; Yang, Z.; Wang, Y.; Cai, W.; Wen, F.; Jiang, X.; Zhang, T.; et al. Therapeutic strategies targeting uPAR potentiate anti–PD-1 efficacy in diffuse-type gastric cancer. Sci. Adv. 2022, 8, eabn3774. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, D.; Hemati, H.; Park, Y.; Taftaf, R.; Zhang, Y.; Liu, J.; Cristofanilli, M.; Liu, X. ICAM-1-suPAR-CD11b Axis Is a Novel Therapeutic Target for Metastatic Triple-Negative Breast Cancer. Cancers 2023, 15, 2734. https://doi.org/10.3390/cancers15102734
Li D, Hemati H, Park Y, Taftaf R, Zhang Y, Liu J, Cristofanilli M, Liu X. ICAM-1-suPAR-CD11b Axis Is a Novel Therapeutic Target for Metastatic Triple-Negative Breast Cancer. Cancers. 2023; 15(10):2734. https://doi.org/10.3390/cancers15102734
Chicago/Turabian StyleLi, Dong, Hami Hemati, Younhee Park, Rokana Taftaf, Youbin Zhang, Jinpeng Liu, Massimo Cristofanilli, and Xia Liu. 2023. "ICAM-1-suPAR-CD11b Axis Is a Novel Therapeutic Target for Metastatic Triple-Negative Breast Cancer" Cancers 15, no. 10: 2734. https://doi.org/10.3390/cancers15102734