
Plasma-Based Measurements of Tumor Heterogeneity Correlate with Clinical Outcomes in Metastatic Colorectal Cancer

 , ,
, ,
Abstract
:Simple Summary
Abstract
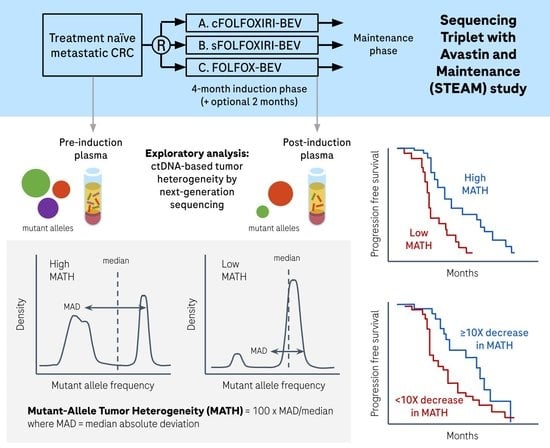
1. Introduction
2. Materials and Methods
2.1. Samples and Sequencing
2.2. Variant Calling
2.3. Calculation of MATH
2.4. Statistical Analysis
3. Results
3.1. Cohort Characteristics
3.2. MATH in Pre-Induction Plasma
3.3. MATH in Post-Induction Plasma
3.4. Change in MATH from Pre-Induction to Post-Induction
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A






References
- Blank, A.; Roberts, D.E.; Dawson, H.; Zlobec, I.; Lugli, A. Tumor Heterogeneity in Primary Colorectal Cancer and Corresponding Metastases. Does the Apple Fall Far from the Tree? Front. Med. 2018, 5, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinari, C.; Marisi, G.; Passardi, A.; Matteucci, L.; De Maio, G.; Ulivi, P. Heterogeneity in Colorectal Cancer: A Challenge for Personalized Medicine? Int. J. Mol. Sci. 2018, 19, 3733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [Green Version]
- Uchi, R.; Takahashi, Y.; Niida, A.; Shimamura, T.; Hirata, H.; Sugimachi, K.; Sawada, G.; Iwaya, T.; Kurashige, J.; Shinden, Y.; et al. Integrated Multiregional Analysis Proposing a New Model of Colorectal Cancer Evolution. PLoS Genet. 2016, 12, e1005778. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litchfield, K.; Stanislaw, S.; Spain, L.; Gallegos, L.L.; Rowan, A.; Schnidrig, D.; Rosenbaum, H.; Harle, A.; Au, L.; Hill, S.M.; et al. Representative Sequencing: Unbiased Sampling of Solid Tumor Tissue. Cell Rep. 2020, 31, 107550. [Google Scholar] [CrossRef]
- Strickler, J.H.; Loree, J.M.; Ahronian, L.G.; Parikh, A.R.; Niedzwiecki, D.; Pereira, A.A.L.; McKinney, M.; Michael Korn, W.; Atreya, C.E.; Banks, K.C.; et al. Genomic Landscape of Cell-Free DNA in Patients with Colorectal Cancer. Cancer Discov. 2018, 8, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Xu, Y.; Chang, L.; Gong, Y.; Li, L.; Mo, X.; Zhang, X.; Lin, G.; Zhou, J.; Liu, D.; et al. Genotyping of Circulating Tumor DNA Reveals the Clinically Actionable Mutation Landscape of Advanced Colorectal Cancer. Mol. Cancer Ther. 2019, 18, 1158–1167. [Google Scholar] [CrossRef] [Green Version]
- Parikh, A.R.; Leshchiner, I.; Elagina, L.; Goyal, L.; Levovitz, C.; Siravegna, G.; Livitz, D.; Rhrissorrakrai, K.; Martin, E.E.; Van Seventer, E.E.; et al. Liquid versus Tissue Biopsy for Detecting Acquired Resistance and Tumor Heterogeneity in Gastrointestinal Cancers. Nat. Med. 2019, 25, 1415–1421. [Google Scholar] [CrossRef]
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L.; et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov. 2016, 6, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Dasari, A.; Morris, V.K.; Allegra, C.J.; Atreya, C.; Benson, A.B.; Boland, P.; Chung, K.; Copur, M.S.; Corcoran, R.B.; Deming, D.A.; et al. CtDNA Applications and Integration in Colorectal Cancer: An NCI Colon and Rectal–Anal Task Forces Whitepaper. Nat. Rev. Clin. Oncol. 2020, 17, 757–770. [Google Scholar] [CrossRef] [PubMed]
- Abécassis, J.; Hamy, A.S.; Laurent, C.; Sadacca, B.; Bonsang-Kitzis, H.; Reyal, F.; Vert, J.P. Assessing Reliability of Intra-Tumor Heterogeneity Estimates from Single Sample Whole Exome Sequencing Data. PLoS ONE 2019, 14, e0224143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, H.I.; Tan, B.R.; Reeves, J.A.; Xiong, H.; Somer, B.; Lenz, H.; Hochster, H.S.; Scappaticci, F.; Palma, J.F.; Price, R.; et al. Phase II Randomized Trial of Sequential or Concurrent FOLFOXIRI-Bevacizumab Versus FOLFOX-Bevacizumab for Metastatic Colorectal Cancer (STEAM). Oncologist 2019, 24, 921–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deveson, I.W.; Gong, B.; Lai, K.; LoCoco, J.S.; Richmond, T.A.; Schageman, J.; Zhang, Z.; Novoradovskaya, N.; Willey, J.C.; Jones, W.; et al. Evaluating the Analytical Validity of Circulating Tumor DNA Sequencing Assays for Precision Oncology. Nat. Biotechnol. 2021, 39, 1115–1128. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An Ultrasensitive Method for Quantitating Circulating Tumor DNA with Broad Patient Coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Newman, A.M.; Lovejoy, A.F.; Klass, D.M.; Kurtz, D.M.; Chabon, J.J.; Scherer, F.; Stehr, H.; Liu, C.L.; Bratman, S.V.; Say, C.; et al. Integrated Digital Error Suppression for Improved Detection of Circulating Tumor DNA. Nat. Biotechnol. 2016, 34, 547–555. [Google Scholar] [CrossRef]
- Yao, L.; Lal, P.; Fang, L.-T.; Lee, J.; Palma, J.; Rosenthal, A.; Hinzmann, B.; Lovejoy, A.; Lam, H.Y.K. Abstract 5293: A Method to Identify Somatic Mutations from Tumor Samples in the Absence of Matched Normal Tissue. Cancer Res. 2018, 78, 5293. [Google Scholar] [CrossRef]
- Mroz, E.A.; Rocco, J.W. MATH, a Novel Measure of Intratumor Genetic Heterogeneity, Is High in Poor-Outcome Classes of Head and Neck Squamous Cell Carcinoma. Oral Oncol. 2013, 49, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Mroz, E.A.; Tward, A.D.; Pickering, C.R.; Myers, J.N.; Ferris, R.L.; Rocco, J.W. High Intratumor Genetic Heterogeneity Is Related to Worse Outcome in Patients with Head and Neck Squamous Cell Carcinoma. Cancer 2013, 119, 3034–3042. [Google Scholar] [CrossRef]
- McDonald, K.A.; Kawaguchi, T.; Qi, Q.; Peng, X.; Asaoka, M.; Young, J.; Opyrchal, M.; Yan, L.; Patnaik, S.; Otsuji, E.; et al. Tumor Heterogeneity Correlates with Less Immune Response and Worse Survival in Breast Cancer Patients. Ann. Surg. Oncol. 2019, 26, 2191–2199. [Google Scholar] [CrossRef]
- Mao, H. Clinical Relevance of Mutant-Allele Tumor Heterogeneity and Lung Adenocarcinoma. Ann. Transl. Med. 2019, 7, 432. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, X.; Ye, H.; Tian, Y.; Ji, Z. Lower Mutant-Allele Tumor Heterogeneity Is a Biomarker in FGFR3-Mutant Bladder Cancer for Better Prognosis. World J. Surg. Oncol. 2020, 18, 310. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhao, G.; Wang, J.; Wang, L.; Jin, G.; Lin, R.; Yang, Y. Integration of TMB and Tumor Heterogeneity Identify an Immunologic Subtype of Melanoma with Favorable Survival. J. Clin. Oncol. 2020, 38, e15183. [Google Scholar] [CrossRef]
- Hou, Y.; Li, T.; Gan, W.; Lv, S.; Zeng, Z.; Yan, Z.; Wang, W.; Yang, M. Prognostic Significance of Mutant-Allele Tumor Heterogeneity in Uterine Corpus Endometrial Carcinoma. Ann. Transl. Med. 2020, 8, 339. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.G.; Oh, B.Y.; Hong, H.K.; Al-Khalidi, H.; Al-Alem, F.; Lee, H.O.; Bae, J.S.; Kim, J.; Cha, H.U.; Alotaibi, M.; et al. Tumor Heterogeneity Predicts Metastatic Potential in Colorectal Cancer. Clin. Cancer Res. 2017, 23, 7209–7216. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yan, S.; Liu, X.; Gan, L.; Wu, Z.; Gong, Y.; Huang, M.; Zhang, X.; Guo, W. Gender-Related Prognostic Value and Genomic Pattern of Intra-Tumor Heterogeneity in Colorectal Cancer. Carcinogenesis 2017, 38, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Rajput, A.; Bocklage, T.; Greenbaum, A.; Lee, J.-H.; Ness, S.A. Mutant-Allele Tumor Heterogeneity Scores Correlate With Risk of Metastases in Colon Cancer. Clin. Colorectal Cancer 2017, 16, e165–e170. [Google Scholar] [CrossRef] [Green Version]
- Greenbaum, A.; Martin, D.R.; Bocklage, T.; Lee, J.H.; Ness, S.A.; Rajput, A. Tumor Heterogeneity as a Predictor of Response to Neoadjuvant Chemotherapy in Locally Advanced Rectal Cancer. Clin. Colorectal Cancer 2019, 18, 102–109. [Google Scholar] [CrossRef] [Green Version]
- Perdigones, N.; Murtaza, M. Capturing Tumor Heterogeneity and Clonal Evolution in Solid Cancers Using Circulating Tumor DNA Analysis. Pharmacol. Ther. 2017, 174, 22–26. [Google Scholar] [CrossRef]
- Miura, S.; Vu, T.; Deng, J.; Buturla, T.; Oladeinde, O.; Choi, J.; Kumar, S. Power and Pitfalls of Computational Methods for Inferring Clone Phylogenies and Mutation Orders from Bulk Sequencing Data. Sci. Rep. 2020, 10, 3498. [Google Scholar] [CrossRef] [Green Version]
- Moser, T.; Waldispuehl-Geigl, J.; Belic, J.; Weber, S.; Zhou, Q.; Hasenleithner, S.O.; Graf, R.; Terzic, J.A.; Posch, F.; Sill, H.; et al. On-Treatment Measurements of Circulating Tumor DNA during FOLFOX Therapy in Patients with Colorectal Cancer. NPJ Precis. Oncol. 2020, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Samuel, S.; Gaur, P.; Lu, J.; Dallas, N.A.; Xia, L.; Bose, D.; Ramachandran, V.; Ellis, L.M. Chronic Exposure of Colorectal Cancer Cells to Bevacizumab Promotes Compensatory Pathways That Mediate Tumour Cell Migration. Br. J. Cancer 2011, 104, 1270–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Perakis, S.O.; Ulz, P.; Mohan, S.; Riedl, J.M.; Talakic, E.; Lax, S.; Tötsch, M.; Hoefler, G.; Bauernhofer, T.; et al. Cell-Free DNA Analysis Reveals POLR1D-Mediated Resistance to Bevacizumab in Colorectal Cancer. Genome Med. 2020, 12, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Kraak, L.; Goel, G.; Ramanan, K.; Kaltenmeier, C.; Zhang, L.; Normolle, D.P.; Freeman, G.J.; Tang, D.; Nason, K.S.; Davison, J.M.; et al. 5-Fluorouracil Upregulates Cell Surface B7-H1 (PD-L1) Expression in Gastrointestinal Cancers. J. Immunother. Cancer 2016, 4, 65. [Google Scholar] [CrossRef] [Green Version]
- Derakhshani, A.; Hashemzadeh, S.; Asadzadeh, Z.; Shadbad, M.A.; Rasibonab, F.; Safarpour, H.; Jafarlou, V.; Solimando, A.G.; Racanelli, V.; Singh, P.K.; et al. Cytotoxic T-Lymphocyte Antigen-4 in Colorectal Cancer: Another Therapeutic Side of Capecitabine. Cancers 2021, 13, 2414. [Google Scholar] [CrossRef]
- Lu, C.; Bera, K.; Wang, X.; Prasanna, P.; Xu, J.; Janowczyk, A.; Beig, N.; Yang, M.; Fu, P.; Lewis, J.; et al. A Prognostic Model for Overall Survival of Patients with Early-Stage Non-Small Cell Lung Cancer: A Multicentre, Retrospective Study. Lancet Digit. Health 2020, 2, e594–e606. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | All Patients (n = 280) | Pre-Induction BEP (n = 146) | Remaining ITT Population (n = 134) | p |
|---|---|---|---|---|
| Age, years (median, range) | 57.5 (23–75) | 57.0 (23–74) | 58.0 (25–75) | 0.7534 |
| Sex, n (%) | 0.9091 | |||
| Female | 118 (42.1) | 62 (42.5) | 56 (41.8) | |
| Male | 162 (57.9) | 84 (57.5) | 78 (58.2) | |
| ECOG performance status, n (%) | 0.2274 | |||
| 0 | 165 (58.9) | 91 (62.3) | 74 (55.2) | |
| 1 | 115 (41.1) | 55 (37.7) | 60 (44.8) | |
| Cancer type at initial diagnosis, n (%) | 0.9004 | |||
| Colon cancer | 208 (74.3) | 108 (74.0) | 100 (74.6) | |
| Rectal cancer | 72 (25.7) | 38 (26.0) | 34 (25.4) | |
| Prior cancer surgery, n (%) | 164 (58.6) | 81 (55.5) | 83 (61.9) | 0.2729 |
| Extent of metastatic disease, n (%) | 0.8501 | |||
| Liver-limited Disease | 83 (29.6) | 44 (30.1) | 39 (29.1) | |
| Non-Liver-limited Disease | 197 (70.4) | 102 (69.9) | 95 (70.9) | |
| Location of Primary Tumor, n (%) | 0.6969 | |||
| Left | 158 (56.4) | 84 (57.5) | 74 (55.2) | |
| Right | 122 (43.6) | 62 (42.5) | 60 (44.8) | |
| Treatment Arm, n (%) | 0.8125 | |||
| FOLFOXIRI/bevacizumab | 93 (33.2) | 50 (34.2) | 43 (32.1) | |
| Sequential FOLFOXIRI/bevacizumab | 92 (32.9) | 49 (33.6) | 43 (32.1) | |
| FOLFOX/bevacizumab | 95 (33.9) | 47 (32.2) | 48 (35.8) | |
| Liver resection rate in 1 L, n (%) | 18 (6.4) | 13 (8.9) | 5 (3.7) | 0.0779 |
| Characteristic | All Patients (n = 280) | Post-Induction BEP (n = 89) | Remaining ITT Population (n = 191) | p |
|---|---|---|---|---|
| Age, years (median, range) | 57.5 (23–75) | 58.0 (23–74) | 57.0 (25–75) | 0.5441 |
| Sex, n (%) | 0.2414 | |||
| Female | 118 (42.1) | 33 (37.1) | 85 (44.5) | |
| Male | 162 (57.9) | 56 (62.9) | 106 (55.5) | |
| ECOG performance status, n (%) | 0.0257 | |||
| 0 | 165 (58.9) | 61 (68.5) | 104 (54.5) | |
| 1 | 115 (41.1) | 28 (31.5) | 87 (45.5) | |
| Cancer type at initial diagnosis, n (%) | 0.2539 | |||
| Colon cancer | 208 (74.3) | 70 (78.7) | 138 (72.3) | |
| Rectal cancer | 72 (25.7) | 19 (21.3) | 53 (27.7) | |
| Prior cancer surgery, n (%) | 164 (58.6) | 48 (53.9) | 116 (60.7) | 0.2821 |
| Extent of metastatic disease, n (%) | 0.6493 | |||
| Liver-limited Disease | 83 (29.6) | 28 (31.5) | 55 (28.8) | |
| Non-Liver-limited Disease | 197 (70.4) | 61 (68.5) | 136 (71.2) | |
| Location of Primary Tumor, n (%) | 0.4720 | |||
| Left | 158 (56.4) | 53 (59.6) | 105 (55.0) | |
| Right | 122 (43.6) | 36 (40.4) | 86 (45.0) | |
| Treatment Arm, n (%) | 0.9754 | |||
| FOLFOXIRI/bevacizumab | 93 (33.2) | 29 (32.6) | 64 (33.5) | |
| Sequential FOLFOXIRI/bevacizumab | 92 (32.9) | 29 (32.6) | 63 (33.0) | |
| FOLFOX/bevacizumab | 95 (33.9) | 31 (34.8) | 64 (33.5) | |
| Liver resection rate in 1 L, n (%) | 18 (6.4) | 8 (9.0) | 10 (5.2) | 0.2331 |
| Variable | Unadjusted for Clinical Factors | Adjusted for Clinical Factors | ||
|---|---|---|---|---|
| HR (95% CI) | p | HR (95% CI) | p | |
| MATH (unit of 5) | 1.03 (1.01, 1.05) | 0.0128 | 1.02 (1.00, 1.04) | 0.1341 |
| MATH (unit of 5), | ||||
| adjusted for mean AF | 1.02 (0.99,1.04) | 0.1329 | 1.01 (0.98, 1.03) | 0.6691 |
| adjusted for mean MMPM | 1.02 (1.00, 1.05) | 0.0297 | 1.02 (0.99, 1.04) | 0.0785 |
| adjusted for number of somatic mutations | 1.02 (1.00, 1.04) | 0.0474 | 1.02 (0.99, 1.04) | 0.1742 |
| Model with MATH | ||||
|---|---|---|---|---|
| Unadjusted | Adjusted for Clinical Factors | |||
| MATH Variable | HR (95% CI) | p | HR (95% CI) | p |
| unit of 5 | 1.06 (1.03, 1.09) | <0.0001 | 1.05 (1.02,1.08) | 0.0008 |
| High vs. Low | 3.23 (1.85, 5.63) | <0.0001 | 3.34 (1.90, 5.86) | <0.0001 |
| Low vs. Undefined | 1.50 (0.79, 2.85) | 0.0001 | 1.24 (0.65, 2.40) | 0.0001 |
| High vs. Undefined | 4.14 (2.06, 8.35) | 3.83 (1.89, 7.76) | ||
| Model with MATH and Mean AF | ||||
| Adjusted for Mean AF | Adjusted for Mean AF and Clinical Factors | |||
| MATH Variable | HR (95% CI) | p | HR (95% CI) | p |
| unit of 5 | 1.06 (1.03, 1.09) | < 0.0001 | 1.05 (1.02, 1.09) | 0.0009 |
| High vs. Low | 3.51 (1.93, 6.39) | < 0.0001 | 3.69 (2.02, 6.73) | <0.0001 |
| Low vs. Undefined | 1.46 (0.76, 2.78) | 0.0002 | 1.18 (0.61, 2.29) | 0.0001 |
| High vs. Undefined | 4.38 (2.12, 9.08) | 4.09 (1.96, 8.50) | ||
| Model with MATH and Mean MMPM | ||||
| Adjusted for Mean MMPM | Adjusted for Mean MMPM and Clinical Factors | |||
| MATH Variable | HR (95% CI) | p | HR (95% CI) | p |
| unit of 5 | 1.06 (1.03, 1.10) | <0.0001 | 1.06 (1.02, 1.09) | 0.0005 |
| High vs. Low | 3.54 (1.93, 6.47) | <0.0001 | 4.25 (2.27, 7.94) | <0.0001 |
| Low vs. Undefined | 1.49 (0.78, 2.83) | 0.0001 | 1.19 (0.62, 2.30) | <0.0001 |
| High vs. Undefined | 4.50 (2.15, 9.40) | 4.72 (2.23, 9.98) | ||
| Model with MATH and Number of Somatic Mutations | ||||
| Adjusted for Number of Somatic Mutations | Adjusted for Number of Somatic Mutations and Clinical Factors | |||
| MATH Variable | HR (95% CI) | p | HR (95% CI) | p |
| unit of 5 | 1.05 (1.02, 1.08) | 0.0023 | 1.04 (1.01, 1.08) | 0.0110 |
| High vs. Low | 2.78 (1.48, 5.23) | 0.0016 | 3.21 (1.66, 6.20) | 0.0005 |
| Low vs. Undefined | 1.39 (0.71, 2.74) | 0.0037 | 1.24 (0.63, 2.42) | 0.0018 |
| High vs. Undefined | 3.58 (1.58, 8.10) | 3.76 (1.65, 8.57) | ||
| Assessment | Low MATH (n = 66) | High MATH (n = 23) |
|---|---|---|
| ORR (CR or PR) | ||
| n (%) | 58 (87.9) | 13 (56.5) |
| p-value | 0.0026 | - |
| Odds Ratio, unadjusted | ||
| Low vs. High (95% CI) | 5.58 (1.84, 16.88) | - |
| p-value | 0.0023 | - |
| Odds Ratio, adjusted | ||
| Low vs. High (95% CI) | 5.94 (1.87, 18.90) | - |
| p-value | 0.0026 | - |
| Model | MATH (<10 Fold vs. >/= 10 Fold Drop) | |
|---|---|---|
| HR (95% CI) | p | |
| Unadjusted | 2.18 (1.21, 3.91) | 0.0093 |
| Adjusted | 1.65 (0.90, 3.05) | 0.1064 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yaung, S.J.; Ju, C.; Gattam, S.; Nicholas, A.; Sommer, N.; Bendell, J.C.; Hurwitz, H.I.; Lee, J.J.; Casey, F.; Price, R.; et al. Plasma-Based Measurements of Tumor Heterogeneity Correlate with Clinical Outcomes in Metastatic Colorectal Cancer. Cancers 2022, 14, 2240. https://doi.org/10.3390/cancers14092240
Yaung SJ, Ju C, Gattam S, Nicholas A, Sommer N, Bendell JC, Hurwitz HI, Lee JJ, Casey F, Price R, et al. Plasma-Based Measurements of Tumor Heterogeneity Correlate with Clinical Outcomes in Metastatic Colorectal Cancer. Cancers. 2022; 14(9):2240. https://doi.org/10.3390/cancers14092240
Chicago/Turabian StyleYaung, Stephanie J., Christine Ju, Sandeep Gattam, Alan Nicholas, Nicolas Sommer, Johanna C. Bendell, Herbert I. Hurwitz, John J. Lee, Fergal Casey, Richard Price, and et al. 2022. "Plasma-Based Measurements of Tumor Heterogeneity Correlate with Clinical Outcomes in Metastatic Colorectal Cancer" Cancers 14, no. 9: 2240. https://doi.org/10.3390/cancers14092240
APA StyleYaung, S. J., Ju, C., Gattam, S., Nicholas, A., Sommer, N., Bendell, J. C., Hurwitz, H. I., Lee, J. J., Casey, F., Price, R., & Palma, J. F. (2022). Plasma-Based Measurements of Tumor Heterogeneity Correlate with Clinical Outcomes in Metastatic Colorectal Cancer. Cancers, 14(9), 2240. https://doi.org/10.3390/cancers14092240