
Real-World Data on Chronic Myelomonocytic Leukemia: Clinical and Molecular Characteristics, Treatment, Emerging Drugs, and Patient Outcomes

 , , , , , , , , ,
, , , , , , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Cytogenetics
2.3. Gene Mutations
2.4. Treatment and Response
2.5. Statistical Analyses
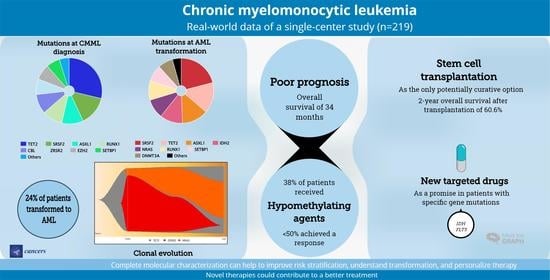
3. Results
3.1. Patient Characteristics
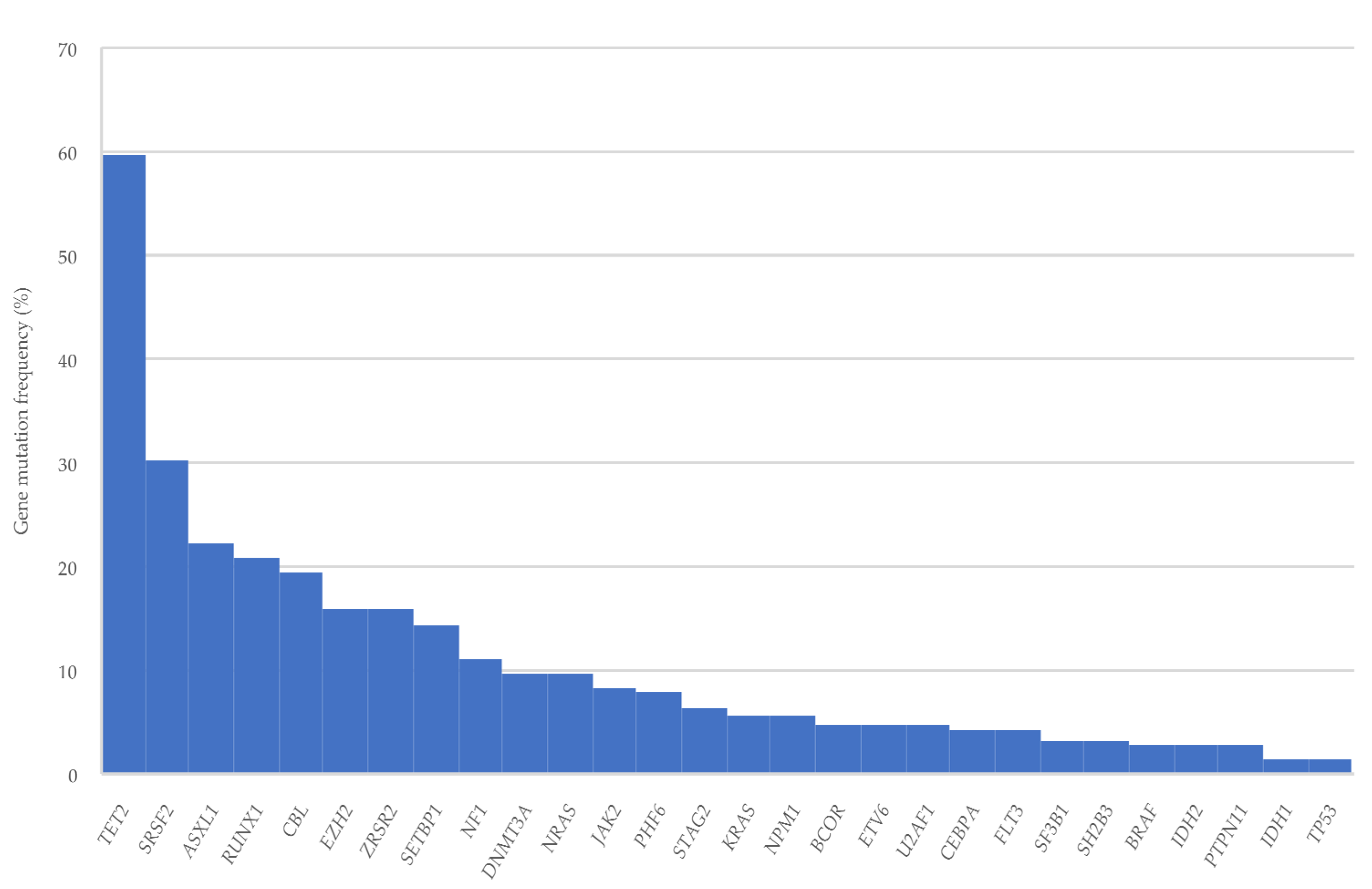
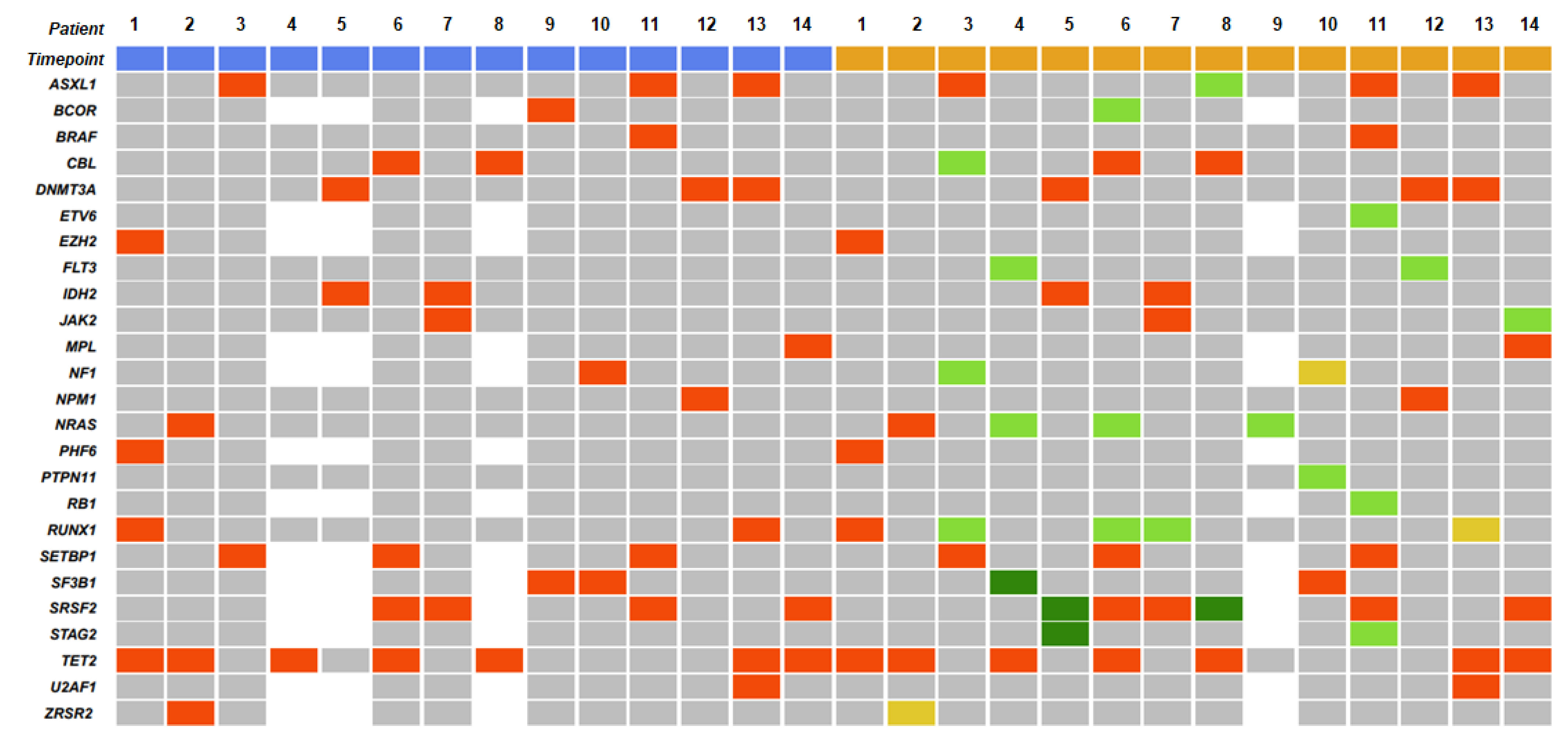
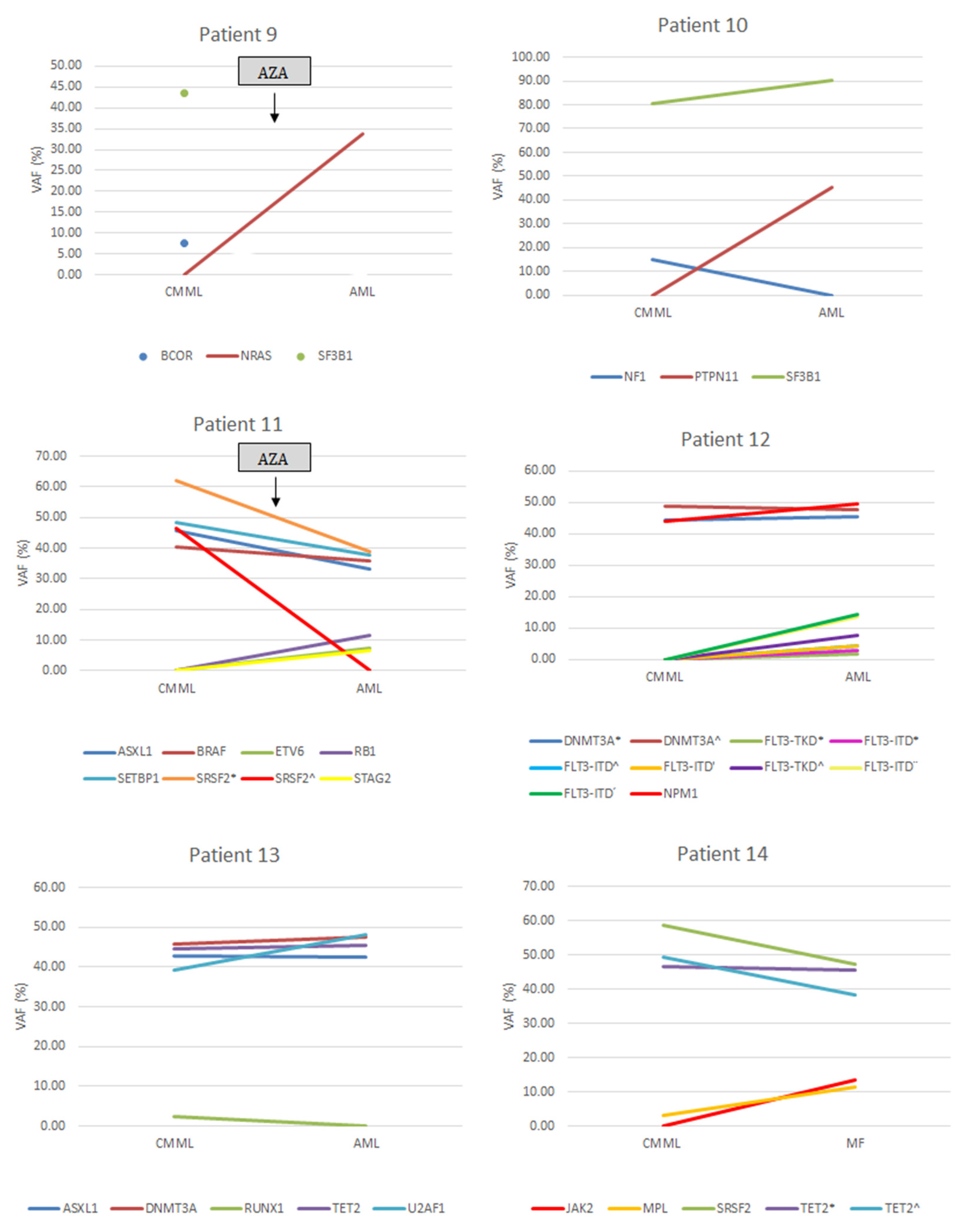
3.2. Gene Mutations
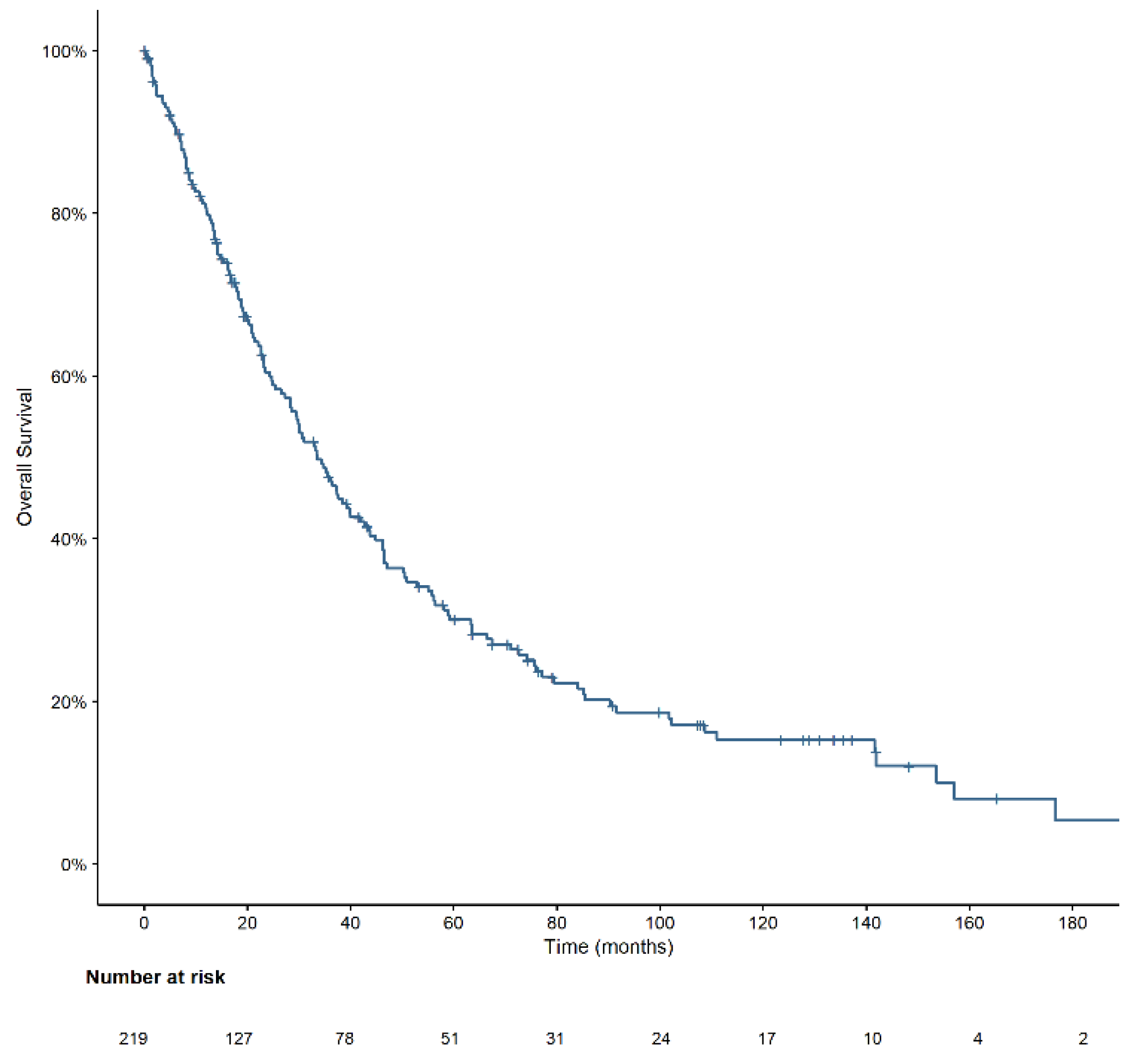
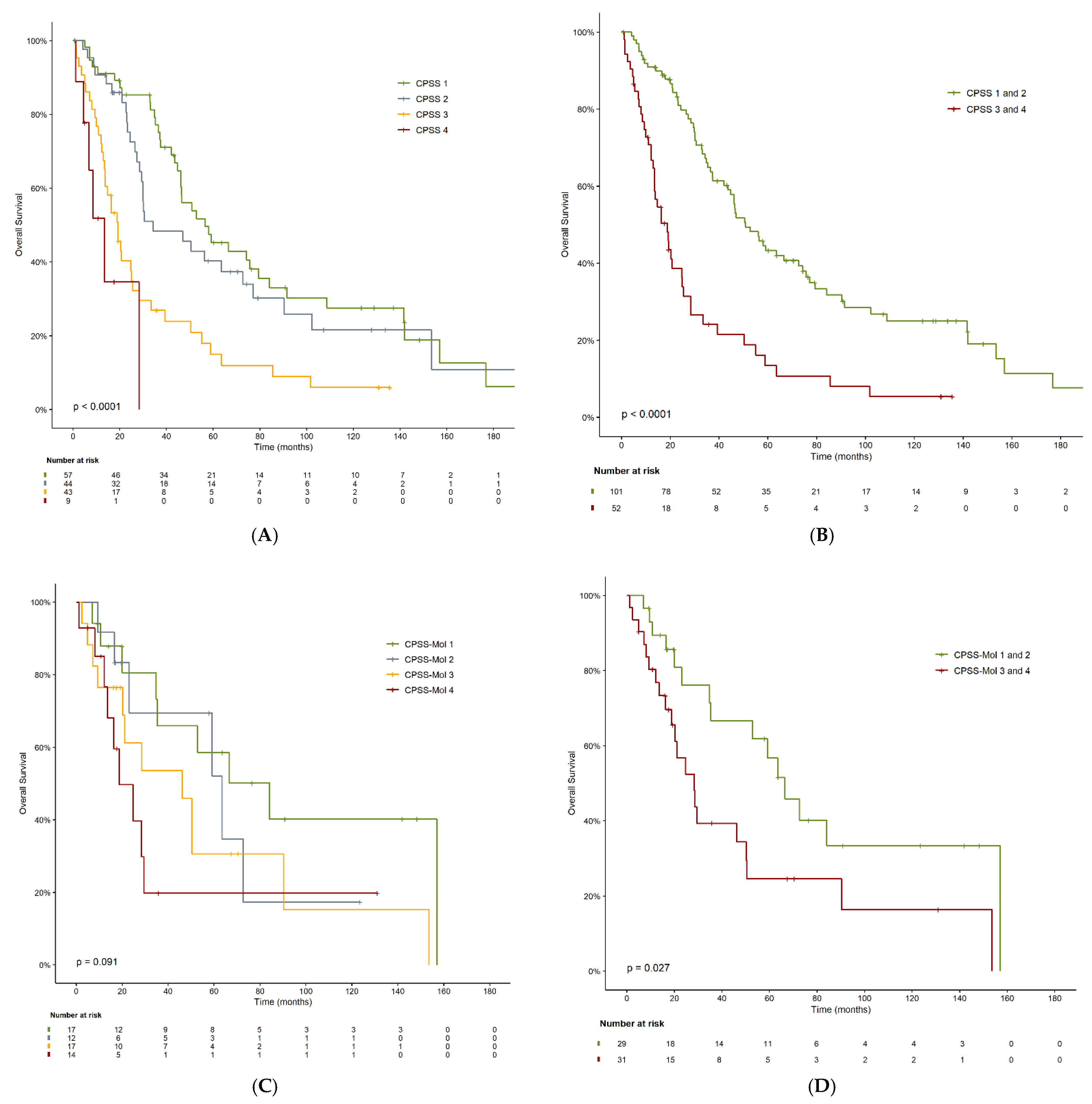
3.3. Overall Survival
3.4. Response to Treatment
3.5. AlloSCT
3.6. Targeted Therapies
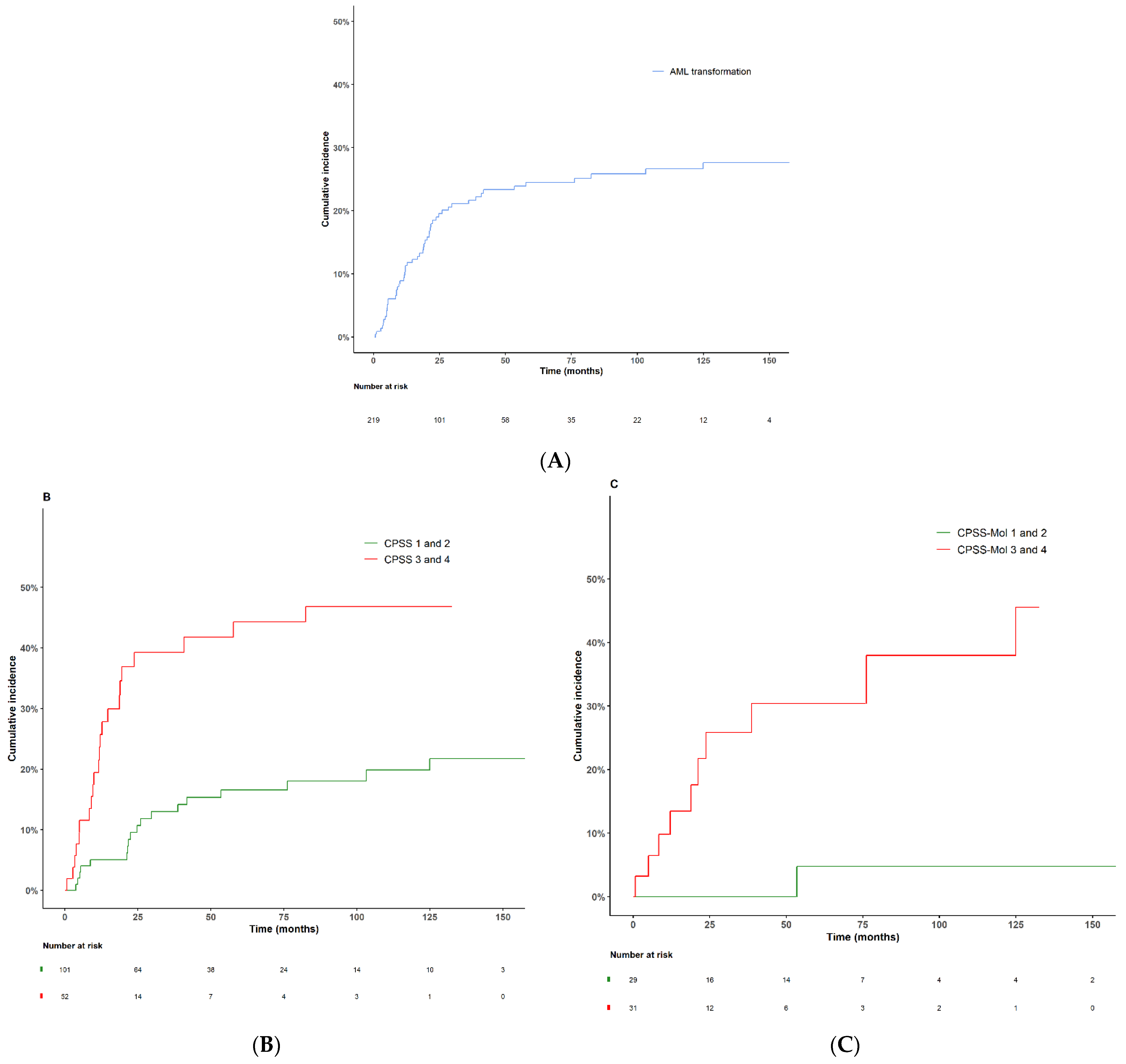
3.7. AML Transformation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Arber, D.A.; Hasserjian, R.P.; Le Beau, M.M. WHO Classification of Tumors of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017; Volume 2, pp. 82–86. [Google Scholar]
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposals for the classification of the myelodysplastic syndromes. Br. J. Haematol. 1982, 51, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Dinmohamed, A.G.; Brink, M.; Visser, O.; Sonneveld, P.; van de Loosdrecht, A.A.; Jongen-Lavrencic, M.; de Greef, G.E. Trends in Incidence, Primary Treatment and Survival of Chronic Myelomonocytic Leukemia: A Nationwide Population-Based Study Among 1359 Patients Diagnosed in the Netherlands from 1989 to 2012. Blood 2014, 124, 4645. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Tefferi, A. Chronic Myelomonocytic leukemia: 2020 update on diagnosis, risk stratification and management. Am. J. Hematol. 2020, 95, 97–115. [Google Scholar] [CrossRef]
- Zahid, M.F.; Barraco, D.; Lasho, T.L.; Finke, C.; Ketterling, R.P.; Gangat, N.; Hanson, C.A.; Tefferi, A.; Patnaik, M.M. Spectrum of autoimmune diseases and systemic inflammatory syndromes in patients with chronic myelomonocytic leukemia. Leuk. Lymphoma 2017, 58, 1488–1493. [Google Scholar] [CrossRef] [PubMed]
- Chan, O.; Renneville, A.; Padron, E. Chronic myelomonocytic leukemia diagnosis and management. Leukemia 2021, 35, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Tefferi, A. Cytogenetic and molecular abnormalities in chronic myelomonocytic leukemia. Blood Cancer J 2016, 6, e393. [Google Scholar] [CrossRef]
- Geissler, K.; Jager, E.; Barna, A.; Gurbisz, M.; Marschon, R.; Graf, T.; Graf, E.; Borjan, B.; Jilch, R.; Geissler, C.; et al. The Austrian biodatabase for chronic myelomonocytic leukemia (ABCMML): A representative and useful real-life data source for further biomedical research. Wien. Klin. Wochenschr. 2019, 131, 410–418. [Google Scholar] [CrossRef]
- Such, E.; Cervera, J.; Costa, D.; Sole, F.; Vallespi, T.; Luno, E.; Collado, R.; Calasanz, M.J.; Hernandez-Rivas, J.M.; Cigudosa, J.C.; et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica 2011, 96, 375–383. [Google Scholar] [CrossRef]
- Elena, C.; Galli, A.; Such, E.; Meggendorfer, M.; Germing, U.; Rizzo, E.; Cervera, J.; Molteni, E.; Fasan, A.; Schuler, E.; et al. Integrating clinical features and genetic lesions in the risk assessment of patients with chronic myelomonocytic leukemia. Blood 2016, 128, 1408–1417. [Google Scholar] [CrossRef]
- Such, E.; Germing, U.; Malcovati, L.; Cervera, J.; Kuendgen, A.; Della Porta, M.G.; Nomdedeu, B.; Arenillas, L.; Luno, E.; Xicoy, B.; et al. Development and validation of a prognostic scoring system for patients with chronic myelomonocytic leukemia. Blood 2013, 121, 3005–3015. [Google Scholar] [CrossRef] [Green Version]
- Geissler, K.; Jager, E.; Barna, A.; Gurbisz, M.; Marschon, R.; Graf, T.; Nosslinger, T.; Pfeilstocker, M.; Machherndl-Spandl, S.; Stauder, R.; et al. Multistep pathogenesis of chronic myelomonocytic leukemia in patients. Eur. J. Haematol. 2022, 109, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, M.M.; Pierola, A.A.; Vallapureddy, R.; Yalniz, F.F.; Kadia, T.M.; Jabbour, E.J.; Lasho, T.; Hanson, C.A.; Ketterling, R.P.; Kantarjian, H.M.; et al. Blast phase chronic myelomonocytic leukemia: Mayo-MDACC collaborative study of 171 cases. Leukemia 2018, 32, 2512–2518. [Google Scholar] [CrossRef]
- Carr, R.M.; Vorobyev, D.; Lasho, T.; Marks, D.L.; Tolosa, E.J.; Vedder, A.; Almada, L.L.; Yurcheko, A.; Padioleau, I.; Alver, B.; et al. RAS mutations drive proliferative chronic myelomonocytic leukemia via a KMT2A-PLK1 axis. Nat. Commun. 2021, 12, 2901. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Zhou, F.; Wang, Z.; Hua, H.; Qin, W.; Jia, Z.; Cai, X.; Chen, M.; Liu, J.; Chao, H.; et al. Mutational landscape of chronic myelomonocytic leukemia and its potential clinical significance. Int. J. Hematol. 2022, 115, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.J.; Przychodzen, B.; Thota, S.; Radivoyevitch, T.; Visconte, V.; Kuzmanovic, T.; Clemente, M.; Hirsch, C.; Morawski, A.; Souaid, R.; et al. Genomic determinants of chronic myelomonocytic leukemia. Leukemia 2017, 31, 2815–2823. [Google Scholar] [CrossRef]
- Gagelmann, N.; Badbaran, A.; Beelen, D.W.; Salit, R.B.; Stolzel, F.; Rautenberg, C.; Becker, H.; Radujkovic, A.; Panagiota, V.; Bogdanov, R.; et al. A prognostic score including mutation profile and clinical features for patients with CMML undergoing stem cell transplantation. Blood Adv. 2021, 5, 1760–1769. [Google Scholar] [CrossRef]
- Pophali, P.; Matin, A.; Mangaonkar, A.A.; Carr, R.; Binder, M.; Al-Kali, A.; Begna, K.H.; Reichard, K.K.; Alkhateeb, H.; Shah, M.V.; et al. Prognostic impact and timing considerations for allogeneic hematopoietic stem cell transplantation in chronic myelomonocytic leukemia. Blood Cancer J. 2020, 10, 121. [Google Scholar] [CrossRef]
- Ocheni, S.; Kroger, N.; Zabelina, T.; Zander, A.R.; Bacher, U. Outcome of allo-SCT for chronic myelomonocytic leukemia. Bone Marrow Transplant. 2009, 43, 659–661. [Google Scholar] [CrossRef]
- Elliott, M.A.; Tefferi, A.; Hogan, W.J.; Letendre, L.; Gastineau, D.A.; Ansell, S.M.; Dispenzieri, A.; Gertz, M.A.; Hayman, S.R.; Inwards, D.J.; et al. Allogeneic stem cell transplantation and donor lymphocyte infusions for chronic myelomonocytic leukemia. Bone Marrow Transplant. 2006, 37, 1003–1008. [Google Scholar] [CrossRef]
- Kröger, N.; Zabelina, T.; Guardiola, P.; Runde, V.; Sierra, J.; Van Biezen, A.; Niederwieser, D.; Zander, A.R.; De Witte, T. Allogeneic stem cell transplantation of adult chronic myelomonocytic leukaemia. A report on behalf of the Chronic Leukaemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Br. J. Haematol. 2002, 118, 67–73. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Tefferi, A. Chronic myelomonocytic leukemia: 2022 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2022, 97, 352–372. [Google Scholar] [CrossRef] [PubMed]
- Solary, E.; Itzykson, R. How I treat chronic myelomonocytic leukemia. Blood 2017, 130, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Renneville, A.; Patnaik, M.M.; Chan, O.; Padron, E.; Solary, E. Increasing recognition and emerging therapies argue for dedicated clinical trials in chronic myelomonocytic leukemia. Leukemia 2021, 35, 2739–2751. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.N.; Litzow, M.R.; Gangat, N.; Al-Kali, A.; Foran, J.M.; Hogan, W.J.; Palmer, J.M.; Mangaonkar, A.A.; Tefferi, A.; Patnaik, M.M. Outcomes of venetoclax-based therapy in chronic phase and blast transformed chronic myelomonocytic leukemia. Am. J. Hematol. 2021, 96, E433–E436. [Google Scholar] [CrossRef]
- Montalban-Bravo, G.; Hammond, D.; DiNardo, C.D.; Konopleva, M.; Borthakur, G.; Short, N.J.; Ramos-Perez, J.; Guerra, V.; Kanagal-Shamanna, R.; Naqvi, K.; et al. Activity of venetoclax-based therapy in chronic myelomonocytic leukemia. Leukemia 2021, 35, 1494–1499. [Google Scholar] [CrossRef] [PubMed]
- Chifotides, H.T.; Masarova, L.; Alfayez, M.; Daver, N.; Alvarado, Y.; Jabbour, E.; Konopleva, M.; Kantarjian, H.M.; Patel, K.P.; DiNardo, C.D.; et al. Outcome of patients with IDH1/2-mutated post-myeloproliferative neoplasm AML in the era of IDH inhibitors. Blood Adv. 2020, 4, 5336–5342. [Google Scholar] [CrossRef] [PubMed]
- Ramos Perez, J.; Montalban-Bravo, G. Emerging drugs for the treatment of chronic myelomonocytic leukemia. Expert Opin. Emerg. Drugs 2020, 25, 515–529. [Google Scholar] [CrossRef]
- McGowan-Jordan, J.; Hastings, R.; Moore, S. Re: International System for Human Cytogenetic or Cytogenomic Nomenclature (ISCN): Some Thoughts, by T. Liehr. Cytogenet. Genome Res. 2021, 161, 225–226. [Google Scholar] [CrossRef]
- Gorello, P.; Cazzaniga, G.; Alberti, F.; Dell’Oro, M.G.; Gottardi, E.; Specchia, G.; Roti, G.; Rosati, R.; Martelli, M.F.; Diverio, D.; et al. Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (NPM1) gene mutations. Leukemia 2006, 20, 1103–1108. [Google Scholar] [CrossRef]
- Pratcorona, M.; Brunet, S.; Nomdedeu, J.; Ribera, J.M.; Tormo, M.; Duarte, R.; Escoda, L.; Guardia, R.; Queipo de Llano, M.P.; Salamero, O.; et al. Favorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: Relevance to post-remission therapy. Blood 2013, 121, 2734–2738. [Google Scholar] [CrossRef] [Green Version]
- Savona, M.R.; Malcovati, L.; Komrokji, R.; Tiu, R.V.; Mughal, T.I.; Orazi, A.; Kiladjian, J.J.; Padron, E.; Solary, E.; Tibes, R.; et al. An international consortium proposal of uniform response criteria for myelodysplastic/myeloproliferative neoplasms (MDS/MPN) in adults. Blood 2015, 125, 1857–1865. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L.; Della Porta, M.G.; Strupp, C.; Ambaglio, I.; Kuendgen, A.; Nachtkamp, K.; Travaglino, E.; Invernizzi, R.; Pascutto, C.; Lazzarino, M.; et al. Impact of the degree of anemia on the outcome of patients with myelodysplastic syndrome and its integration into the WHO classification-based Prognostic Scoring System (WPSS). Haematologica 2011, 96, 1433–1440. [Google Scholar] [CrossRef] [PubMed]
- Schemper, M.; Smith, T. A note on quantifying follow-up in studies of failure time. Control Clin. Trials 1996, 17, 343–346. [Google Scholar] [CrossRef]
- Fine, J.; Gray, R. A Proportional Hazards Model for the Subdistribution of a Competing Risk. J. Am. Stat. Assoc. 1999, 94, 496–509. [Google Scholar] [CrossRef]
- Iacobelli, S.; Committee, E.S. Suggestions on the use of statistical methodologies in studies of the European Group for Blood and Marrow Transplantation. Bone Marrow Transplant. 2013, 48 (Suppl. S1), S1–S37. [Google Scholar] [CrossRef]
- Machherndl-Spandl, S.; Jager, E.; Barna, A.; Gurbisz, M.; Marschon, R.; Graf, T.; Graf, E.; Geissler, C.; Hoermann, G.; Nosslinger, T.; et al. Impact of age on the cumulative risk of transformation in patients with chronic myelomonocytic leukaemia. Eur. J. Haematol. 2021, 107, 265–274. [Google Scholar] [CrossRef]
- Pleyer, L.; Leisch, M.; Kourakli, A.; Padron, E.; Maciejewski, J.P.; Xicoy Cirici, B.; Kaivers, J.; Ungerstedt, J.; Heibl, S.; Patiou, P.; et al. Outcomes of patients with chronic myelomonocytic leukaemia treated with non-curative therapies: A retrospective cohort study. Lancet Haematol. 2021, 8, e135–e148. [Google Scholar] [CrossRef]
- Palomo, L.; Meggendorfer, M.; Hutter, S.; Twardziok, S.; Ademà, V.; Fuhrmann, I.; Fuster-Tormo, F.; Xicoy, B.; Zamora, L.; Acha, P.; et al. Molecular landscape and clonal architecture of adult myelodysplastic/myeloproliferative neoplasms. Blood 2020, 136, 1851–1862. [Google Scholar] [CrossRef]
- Calvo, X.; Nomdedeu, M.; Santacruz, R.; Martinez, N.; Costa, D.; Pereira, A.; Estrada, N.; Xicoy, B.; Esteve, J.; Nomdedeu, B. Comparison of three prognostic scoring systems in a series of 146 cases of chronic myelomonocytic leukemia (CMML): MD Anderson prognostic score (MDAPS), CMML-specific prognostic scoring system (CPSS) and Mayo prognostic model. A detailed review of prognostic factors in CMML. Leuk. Res. 2015, 39, 1146–1153. [Google Scholar] [CrossRef]
- Xicoy, B.; Triguero, A.; Such, E.; García, O.; Jiménez, M.-J.; Arnán, M.; Bernal, T.; Diaz-Beya, M.; Valcárcel, D.; Pedro, C.; et al. The division of chronic myelomonocytic leukemia (CMML)-1 into CMML-0 and CMML-1 according to 2016 World Health Organization (WHO) classification has no impact in outcome in a large series of patients from the Spanish group of MDS. Leuk. Res. 2018, 70, 34–36. [Google Scholar] [CrossRef]
- Itzykson, R.; Kosmider, O.; Renneville, A.; Gelsi-Boyer, V.; Meggendorfer, M.; Morabito, M.; Berthon, C.; Ades, L.; Fenaux, P.; Beyne-Rauzy, O.; et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J. Clin. Oncol. 2013, 31, 2428–2436. [Google Scholar] [CrossRef]
- Itzykson, R.; Duchmann, M.; Lucas, N.; Solary, E. CMML: Clinical and molecular aspects. Int. J. Hematol. 2017, 105, 711–719. [Google Scholar] [CrossRef]
- Mason, C.C.; Khorashad, J.S.; Tantravahi, S.K.; Kelley, T.W.; Zabriskie, M.S.; Yan, D.; Pomicter, A.D.; Reynolds, K.R.; Eiring, A.M.; Kronenberg, Z.; et al. Age-related mutations and chronic myelomonocytic leukemia. Leukemia 2016, 30, 906–913. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Itzykson, R.; Lasho, T.L.; Kosmider, O.; Finke, C.M.; Hanson, C.A.; Knudson, R.A.; Ketterling, R.P.; Tefferi, A.; Solary, E. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: A two-center study of 466 patients. Leukemia 2014, 28, 2206–2212. [Google Scholar] [CrossRef]
- Patnaik, M.M.; Vallapureddy, R.; Lasho, T.L.; Hoversten, K.P.; Finke, C.M.; Ketterling, R.; Hanson, C.; Gangat, N.; Tefferi, A. EZH2 mutations in chronic myelomonocytic leukemia cluster with ASXL1 mutations and their co-occurrence is prognostically detrimental. Blood Cancer J. 2018, 8, 12. [Google Scholar] [CrossRef]
- Jian, J.; Qiao, Y.; Li, Y.; Guo, Y.; Ma, H.; Liu, B. Mutations in chronic myelomonocytic leukemia and their prognostic relevance. Clin. Transl. Oncol. 2021, 23, 1731–1742. [Google Scholar] [CrossRef]
- Coltro, G.; Mangaonkar, A.A.; Lasho, T.L.; Finke, C.M.; Pophali, P.; Carr, R.; Gangat, N.; Binder, M.; Pardanani, A.; Fernandez-Zapico, M.; et al. Clinical, molecular, and prognostic correlates of number, type, and functional localization of TET2 mutations in chronic myelomonocytic leukemia (CMML)-a study of 1084 patients. Leukemia 2020, 34, 1407–1421. [Google Scholar] [CrossRef]
- Heibl, S.; Gisslinger, B.; Jager, E.; Barna, A.; Gurbisz, M.; Stegemann, M.; Bettelheim, P.; Machherndl-Spandl, S.; Pfeilstocker, M.; Nosslinger, T.; et al. Clinical, Hematologic, Biologic and Molecular Characteristics of Patients with Myeloproliferative Neoplasms and a Chronic Myelomonocytic Leukemia-Like Phenotype. Cancers 2020, 12, 1891. [Google Scholar] [CrossRef]
- Belotserkovskaya, E.; Demidov, O. Mouse Models of CMML. Int. J. Mol. Sci. 2021, 22, 11510. [Google Scholar] [CrossRef]
- Montalban-Bravo, G.; Kanagal-Shamanna, R.; Sasaki, K.; Patel, K.; Ganan-Gomez, I.; Jabbour, E.; Kadia, T.; Ravandi, F.; DiNardo, C.; Borthakur, G.; et al. NPM1 mutations define a specific subgroup of MDS and MDS/MPN patients with favorable outcomes with intensive chemotherapy. Blood Adv. 2019, 3, 922–933. [Google Scholar] [CrossRef] [Green Version]
- Itzykson, R.; Santini, V.; Chaffaut, C.; Lionel, A.; Thepot, S.; Giagounidis, A.; Morabito, M.; Droin, N.; Luebbert, M.; Sapena, R.; et al. Decitabine Versus Hydroxyurea for Advanced Proliferative CMML: Results of the Emsco Randomized Phase 3 Dacota Trial. Blood 2020, 136, 53–54. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Jiang, L.; Yang, W.; Luo, Y.; Mei, C.; Zhou, X.; Xu, G.; Xu, W.; Ye, L.; Ren, Y.; et al. Real-world data of chronic myelomonocytic leukemia: A chinese single-center retrospective study. Cancer Med. 2021, 10, 1715–1725. [Google Scholar] [CrossRef] [PubMed]
- Wedge, E.; Hansen, J.W.; Dybedal, I.; Creignou, M.; Ejerblad, E.; Lorenz, F.; Werlenius, O.; Ungerstedt, J.; Holm, M.S.; Nilsson, L.; et al. Allogeneic Hematopoietic Stem Cell Transplantation for Chronic Myelomonocytic Leukemia: Clinical and Molecular Genetic Prognostic Factors in a Nordic Population. Transplant. Cell Ther. 2021, 27, 991.e1. [Google Scholar] [CrossRef]
- Liu, H.D.; Ahn, K.W.; Hu, Z.H.; Hamadani, M.; Nishihori, T.; Wirk, B.; Beitinjaneh, A.; Rizzieri, D.; Grunwald, M.R.; Sabloff, M.; et al. Allogeneic Hematopoietic Cell Transplantation for Adult Chronic Myelomonocytic Leukemia. Biol. Blood Marrow Transplant. 2017, 23, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Phase I First-in-Human Dose Escalation Study of the oral SF3B1 modulator H3B-8800 in myeloid neoplasms. Leukemia 2021, 35, 3542–3550. [Google Scholar] [CrossRef]
- Rioux, N.; Smith, S.; Colombo, F.; Kim, A.; Lai, W.G.; Nix, D.; Siu, Y.A.; Schindler, J.; Smith, P.G. Metabolic disposition of H3B-8800, an orally available small-molecule splicing modulator, in rats, monkeys, and humans. Xenobiotica 2020, 50, 1101–1114. [Google Scholar] [CrossRef]
- Steensma, D.P.; Wermke, M.; Klimek, V.M.; Greenberg, P.L.; Font, P.; Komrokji, R.S.; Yang, J.; Brunner, A.M.; Carraway, H.E.; Ades, L.; et al. Results of a Clinical Trial of H3B-8800, a Splicing Modulator, in Patients with Myelodysplastic Syndromes (MDS), Acute Myeloid Leukemia (AML) or Chronic Myelomonocytic Leukemia (CMML). Blood 2019, 134, 673. [Google Scholar] [CrossRef]
- Seiler, M.; Yoshimi, A.; Darman, R.; Chan, B.; Keaney, G.; Thomas, M.; Agrawal, A.A.; Caleb, B.; Csibi, A.; Sean, E.; et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018, 24, 497–504. [Google Scholar] [CrossRef]
- Kloos, A.; Mintzas, K.; Winckler, L.; Gabdoulline, R.; Alwie, Y.; Jyotsana, N.; Kattre, N.; Schottmann, R.; Scherr, M.; Gupta, C.; et al. Effective drug treatment identified by in vivo screening in a transplantable patient-derived xenograft model of chronic myelomonocytic leukemia. Leukemia 2020, 34, 2951–2963. [Google Scholar] [CrossRef]
- Desikan, S.P.; Ravandi, F.; Pemmaraju, N.; Konopleva, M.; Loghavi, S.; Borthakur, G.; Jabbour, E.J.; Daver, N.; Jain, N.; Chien, K.S.; et al. A Phase II Study of Azacitidine, Venetoclax and Trametinib in Relapsed/Refractory AML Harboring a Ras Pathway-Activating Mutation. Blood 2021, 138, 4436. [Google Scholar] [CrossRef]
- Borthakur, G.; Popplewell, L.; Boyiadzis, M.; Foran, J.; Platzbecker, U.; Vey, N.; Walter, R.B.; Olin, R.; Raza, A.; Giagounidis, A.; et al. Activity of the oral mitogen-activated protein kinase kinase inhibitor trametinib in RAS-mutant relapsed or refractory myeloid malignancies. Cancer 2016, 122, 1871–1879. [Google Scholar] [CrossRef]
- Mill, C.P.; Fiskus, W.; DiNardo, C.D.; Birdwell, C.; Davis, J.A.; Kadia, T.M.; Takahashi, K.; Short, N.; Daver, N.; Ohanian, M.; et al. Effective therapy for AML with RUNX1 mutation by cotreatment with inhibitors of protein translation and BCL2. Blood 2022, 139, 907–921. [Google Scholar] [CrossRef]
- Liapis, K.; Kotsianidis, I. Approaching First-Line Treatment in Patients With Advanced CMML: Hypomethylating Agents or Cytotoxic Treatment? Front. Oncol. 2021, 11, 801524. [Google Scholar] [CrossRef]
- Geissler, K. Molecular Pathogenesis of Chronic Myelomonocytic Leukemia and Potential Molecular Targets for Treatment Approaches. Front. Oncol. 2021, 11, 751668. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics at Diagnosis | Patients (n = 219) |
|---|---|
| Age, years—median (range) | 74.1 (28–99) |
| Men—n (%) | 144 (65.8) |
| WBC count—median (range) | 9.47 (3–119) × 109/L |
| Neutrophil count—median (range) | 4.7 (0.49–57.12) × 109/L |
| Monocyte count—median (range) | 1.91 (1–33.72) × 109/L |
| % monocytes—median (range) | 20 (10–60) |
| Platelets—median (range) | 126 (7–1037) × 109/L |
| Hemoglobin—median (range) | 11.6 (4.9–16.1) g/dl |
| Blast percentage 1—median (range) | 3% (0–19) |
| Transfusion dependence—n (%) | 53 (24.3) |
| Presentation—therapy related n (%) | 14 (7.3) |
| Cytogenetic risk—n (%) | n = 156 |
| low | 110 (70.6) |
| intermediate | 23 (14.7) |
| high | 23 (14.7) |
| Cytogenetic profiling—n (%) | n = 156 |
| normal | 106 (67.9) |
| abnormal | 50 (32.1) |
| FAB classification—n (%) | n = 218 |
| myelodysplastic | 148 (67.9) |
| myeloproliferative | 70 (32.1) |
| WHO classification—n (%) | n = 213 |
| CMML-0 | 151 (70.9) |
| CMML-1 | 35 (16.4) |
| CMML-2 | 27 (12.7) |
| CPSS classification—n (%) | n = 153 |
| low | 57 (37.5) |
| intermediate-1 | 44 (28.8) |
| intermediate-2 | 43 (28.1) |
| high | 9 (5.9) |
| CPSS-Mol classification—n (%) | n = 60 |
| low | 17 (28.3) |
| intermediate-1 | 12 (20) |
| intermediate-2 | 17 (28.3) |
| high | 14 (23.3) |
| Patients receiving disease-modifying treatment—n (%) 2 | n = 50 |
| Only HMA | 34 (68) |
| Only chemotherapy | 13 (26) |
| Both HMA and chemotherapy | 3 (6) |
| Patients receiving alloSCT—n (%) | 22 (10) |
| Patients progressing to AML—n (%) | 53 (24.2) |
| Gene Mutations | At CMML Diagnosis | At AML Transformation |
|---|---|---|
| Patients (n = 72) n (n/N, %) 1 | Patients (n = 23) n (n/N, %) 1 | |
| Number of mutated genes per patient | ||
| 0 | 3 (3/72, 4.2) | 1 (1/23, 4.3) |
| 1 | 13 (13/72, 18.1) | 1 (1/23, 4.3) |
| 2 | 18 (18/72, 25) | 3 (3/23, 13) |
| 3 | 14 (14/72, 19.4) | 3 (3/23, 13) |
| ≥4 | 24 (24/72, 33.3) | 15 (15/23, 65.1) |
| Most frequently mutated genes | ||
| TET2 | 43 (43/72, 59.7) | 8 (8/23, 34.8) |
| SRSF2 | 19 (19/63, 30.2) | 10 (10/21, 47.6) |
| ASXL1 | 16 (16/72, 22.2) | 7 (7/23, 30.4) |
| RUNX1 | 15 (15/72, 20.8) | 5 (5/23, 21.7) |
| CBL | 14 (14/72, 19.4) | 3 (3/23, 13) |
| ZRSR2 | 10 (10/63, 15.9) | 2 (2/21, 9.5) |
| EZH2 | 10 (10/63, 15.9) | 1 (1/21, 4.8) |
| SETBP1 | 9 (9/63, 14.3) | 4 (4/21, 19) |
| IDH2 | 2 (2/72, 2.8) | 6 (6/23, 26.1) |
| NRAS | 7 (7/72, 9.7) | 5 (5/23, 21.7) |
| DNMT3A | 7 (7/72, 9.7) | 4 (4/23, 17.4) |
| U2AF1 | 3 (3/63, 4.8) | 3 (3/21, 14.3) |
| SF3B1 | 2 (2/63, 3.2) | 3 (3/21, 14.3) |
| JAK2 | 6 (6/72, 8.3) | 3 (3/23, 13) |
| FLT3 | 3 (3/72, 4.2) | 3 (3/23, 13) |
| PHF6 | 5 (5/63, 7.9) | 2 (2/21, 9.5) |
| KRAS | 4 (4/72, 5.6) | 2 (2/23, 8.7) |
| Genes in signaling pathways | ||
| NRAS | 7 (7/72, 9.7) | 5 (5/23, 21.7) |
| KRAS | 4 (4/72, 5.6) | 2 (2/23, 8.7) |
| FLT3 | 3 (3/72, 4.2) | 3 (3/23, 13) |
| CSF3R | 0 (0/63, 0) | 1 (1/21, 4.8) |
| JAK2 | 6 (6/72, 8.3) | 3 (3/23, 13) |
| CBL | 14 (14/72, 19.4) | 3 (3/23, 13) |
| PTPN11 | 2 (2/71, 2.8) | 1 (1/23, 4.3) |
| Epigenetic regulators | ||
| TET2 | 43 (43/72, 59.7) | 8 (8/23, 34.8) |
| IDH2 | 2 (2/72, 2.8) | 6 (6/23, 26.1) |
| DNMT3A | 7 (7/72, 9.7) | 4 (4/23, 17.4) |
| ASXL1 | 16 (16/72, 22.2) | 7 (7/23, 30.4) |
| EZH2 | 10 (10/63, 15.9) | 1 (1/21, 4.8) |
| IDH1 | 1 (1/72, 1.4) | 1 (1/23, 4.3) |
| Transcription factors | ||
| RUNX1 | 15 (15/72, 20.8) | 5 (5/23, 21.7) |
| SETBP1 | 9 (9/63, 14.3) | 4 (4/21, 19) |
| CEBPA | 3 (3/72, 4.2) | 0 (0/23, 0) |
| Spliceosome complex | ||
| SRSF2 | 19 (19/63, 30.2) | 10 (10/21, 47.6) |
| SF3B1 | 2 (2/63, 3.2) | 3 (3/21, 14.3) |
| ZRSR2 | 10 (10/63, 15.9) | 2 (2/21, 9.5) |
| U2AF1 | 3 (3/63, 4.8) | 3 (3/21, 14.3) |
| DNA damage response genes | ||
| TP53 | 1 (1/72, 1.4) | 1 (1/23, 4.3) |
| PHF6 | 5 (5/63, 7.9) | 2 (2/21, 9.5) |
| Others | ||
| NPM1 | 4 (4/72, 5.6) | 2 (2/23, 8.7) |
| Factor | No. Patients | OS (Months) | 95% CI | p |
|---|---|---|---|---|
| Age | 0.002 | |||
| ≥70 years | 142 | 28.9 | 21.0–37.3 | |
| <70 years | 77 | 51.0 | 37.9–64.1 | |
| Cytogenetic risk | 0.001 | |||
| Low | 110 | 51.0 | 40.7–61.4 | |
| Intermediate | 22 | 30.4 | 23.6–37.2 | |
| High | 24 | 19.4 | 9.1–29.8 | |
| FAB | 0.04 | |||
| MDS | 148 | 40.3 | 32.3–49.3 | |
| MPN | 70 | 21.3 | 14.7–27.8 | |
| WHO | 0.227 | |||
| CMML-0 | 151 | 37.7 | 29.4–45.9 | |
| CMML-1 | 35 | 25.0 | 19.1–30.9 | |
| CMML-2 | 27 | 18.3 | 11.3–25.4 | |
| CPSS | 0.01 | |||
| Low | 57 | 57.2 | 35.4–78.8 | |
| Intermediate-1 | 44 | 34.8 | 10.3–59.3 | |
| Intermediate-2 | 43 | 19.4 | 13.8–25.2 | |
| High | 9 | 13.7 | 5.6–21.8 | |
| Dichotomized CPSS | 0.01 | |||
| Low | 101 | 51.6 | 39.9–63.2 | |
| High | 52 | 19.1 | 13.3–24.9 | |
| CPSS-Mol | 0.09 | |||
| Low | 17 | 85.1 | 37.6–132.7 | |
| Intermediate-1 | 12 | 64.3 | 20.3–108.3 | |
| Intermediate-2 | 17 | 46.8 | 12.8–80.8 | |
| High | 14 | 19.1 | 6.7–31.5 | |
| Dichotomized CPSS-Mol | 0.03 | |||
| Low | 29 | 67.3 | 49.6–85.1 | |
| High | 31 | 28.7 | 17.2–40.0 | |
| No. gene mutations | 0.006 | |||
| <2 mutations | 16 | 85.2 | 51.3–119.0 | |
| ≥2 mutations | 56 | 25.0 | 17.6–32.5 | |
| RUNX1 mutation | 0.02 | |||
| Detected | 15 | 16.6 | 10.0–23.0 | |
| Not detected | 57 | 51.1 | 21.5–80.8 | |
| Mutations in transcription factors | 0.001 | |||
| ≥1 mutation detected | 25 | 21.3 | 12.0–30.4 | |
| No mutation detected | 41 | 64.3 | 31.8–96.9 | |
| ASXL1/EZH2 co-mutation | 0.001 | |||
| Detected | 3 | 2.5 | 0.3–4.7 | |
| Not detected | 68 | 35.2 | 10.3–60.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castaño-Díez, S.; López-Guerra, M.; Bosch-Castañeda, C.; Bataller, A.; Charry, P.; Esteban, D.; Guijarro, F.; Jiménez-Vicente, C.; Castillo-Girón, C.; Cortes, A.; et al. Real-World Data on Chronic Myelomonocytic Leukemia: Clinical and Molecular Characteristics, Treatment, Emerging Drugs, and Patient Outcomes. Cancers 2022, 14, 4107. https://doi.org/10.3390/cancers14174107
Castaño-Díez S, López-Guerra M, Bosch-Castañeda C, Bataller A, Charry P, Esteban D, Guijarro F, Jiménez-Vicente C, Castillo-Girón C, Cortes A, et al. Real-World Data on Chronic Myelomonocytic Leukemia: Clinical and Molecular Characteristics, Treatment, Emerging Drugs, and Patient Outcomes. Cancers. 2022; 14(17):4107. https://doi.org/10.3390/cancers14174107
Chicago/Turabian StyleCastaño-Díez, Sandra, Mónica López-Guerra, Cristina Bosch-Castañeda, Alex Bataller, Paola Charry, Daniel Esteban, Francesca Guijarro, Carlos Jiménez-Vicente, Carlos Castillo-Girón, Albert Cortes, and et al. 2022. "Real-World Data on Chronic Myelomonocytic Leukemia: Clinical and Molecular Characteristics, Treatment, Emerging Drugs, and Patient Outcomes" Cancers 14, no. 17: 4107. https://doi.org/10.3390/cancers14174107